Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1873-1882. doi:10.7150/ijbs.27746 This issue Cite
Research Paper
SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis
Jianle Wang1,2, Ke Wang1,2, Chongan Huang1,2, Dongdong Lin1,3, Yifei Zhou1,2,5, Yaosen Wu1,2,5, Naifeng Tian1,2,5, Pei Fan1, Xiangxiang Pan1,2, Daoliang Xu1, Jianing Hu5, Ying Zhou5, Xiangyang Wang1,2,5 ![]() , Xiaolei Zhang1,2,4,5
, Xiaolei Zhang1,2,4,5 ![]()
1. Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, Zhejiang, China
2. Zhejiang Provincial Key Laboratory of Orthopaedics, Wenzhou 325027, Zhejiang, China
3. Department of Neurosurgery Surgery, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, Zhejiang, China
4. Chinese Orthopaedic Regenerative Medicine Society
5. The Second School of Medicine, Wenzhou Medical University, Wenzhou Medical University, Wenzhou, China
Received 2018-6-8; Accepted 2018-8-30; Published 2018-10-20
Abstract

Mitochondrial dysfunction is an important contributor to the development of osteoarthritis (OA). Sirtuin 3 (SIRT3) regulates diverse mitochondrial proteins to maintain mitochondrial homeostasis, and dihydromyricetin (DHM) is reported as a potential SIRT3 activator. This study aims to explore the relevance of SIRT3 and OA, as well as the therapeutic effects of DHM on mitochondrial homeostasis in TNF-α-treated chondrocytes. The relationship between SIRT3 and OA was confirmed by detecting SIRT3 level in vitro and in vivo. Mitochondrial dysfunction was evaluated in chondrocytes with or without SIRT3 knockdown. Furthermore, the effects of DHM on mitochondrial homeostasis were performed in TNF-α-treated rat chondrocytes in vitro. In this study, our results showed that the SIRT3 level was decreased in mouse OA cartilage, corresponding to the reduced SIRT3 level in TNF-α-treated chondrocytes in vitro. SIRT3 knockdown induced mitochondrial dysfunction in chondrocytes. Moreover, our study demonstrated that DHM might activate SIRT3 to protect rat chondrocytes from TNF-α-induced degeneration and protective effects of DHM on mitochondrial homeostasis in chondrocytes attributed to enhanced SIRT3. Collectively, SIRT3 deficiency is implicated in OA development and DHM exerts anti-degeneration effect by maintaining mitochondrial homeostasis via a SIRT3-dependent manner in chondrocytes.
Keywords: Dihydromyricetin, osteoarthritis, mitochondria, SIRT3
Introduction
As one chronic and progressive debilitating disease, osteoarthritis (OA) leads to pain and disability in a huge proportion of the human population1. Tumor necrosis factor-α (TNF-α) is a well-recognized inflammatory cytokine, which increases significantly in synovial fluid with the progression of OA2. It is reported that TNF-α exacerbates the chondrocytes degeneration by disturbing the balance between anabolism and catabolism of extracellular matrix (ECM)3, 4. Also, TNF-α induces mitochondrial dysfunction by triggering more intracellular oxygen species (ROS) and more mitochondrial fragments5, 6.
Mitochondria, modulating various cellular activities such as ATP production and oxidative phosphorylation, will be the major source of excessive reactive oxygen species (ROS) once damaged7. Overproduction of ROS is able to impair the balance between anabolism and catabolism in chondrocytes8. Chondrocytes in OA show impaired mitochondrial membrane potential (ΔΨm) and reduced activity of complex I, II, and III compared to normal chondrocytes, indicating mitochondrial dysfunction is involved in degeneration of chondrocytes9. Therefore, it is necessary to enrich therapeutic strategy targeting maintaining mitochondrial homeostasis against OA.
Sirtuin 3 (SIRT3), localized in mitochondria, serves as a deacetylase of mitochondrial proteins. SIRT3 is considered to promote mitochondrial dynamics, anti-oxidation, and mitophagy to maintain mitochondrial dysfunction induced by H2O210, and also deacetylates manganese superoxide dismutase (MnSOD) enhancing it enzymatic activity in senescent cartilage, indicating that SIRT3 might play an important role in mitochondrial homeostasis in chondrocytes against oxidative stress11. However, there is still no study regarding SIRT3 as the therapeutic target against OA.
Till now flavonoids such as resveratrol extracted from traditional herbs has drawn increasing attention in academic community against degenerative diseases. With similar chemical structures as resveratrol, dihydromyricetin (DHM), also called ampelopsin, is reported to have properties of anti-inflammation, anti-oxidation and anti-tumor12. In addition, DHM has been confirmed to exert neuro-protective effects against oxidative stress via up-regulating SIRT3 level under hypobaric hypoxia, suggesting that DHM is a reliable SIRT3 agonist13.
In this study, we discovered the SIRT3 level decreases significantly in knee cartilage from OA mouse model compared with the sham group, and identified SIRT3 as a novel protective factor against OA by maintaining mitochondrial homeostasis. Moreover, we explored the effects and mechanism of DHM on TNF-α-induced matrix metabolism disorder and mitochondrial dysfunction in chondrocytes.
Materials and Methods
Ethics statement
All of animal surgical interventions, treatments and postoperative care procedures were performed in strict accordance with the Animal Care and Use Committee of Wenzhou Medical University (wydw2014-0129).
Reagents
Dihydromyricetin (purity >98%) and type II collagenase were purchased from Sigma-Aldrich (St Louis, MO, USA). TNF-α was purchased from Peprotech (Texas, USA). Compound C was acquired from Selleckchem (Huston, USA). The primary antibody against BRD4, ADAMTS-5, p53, DRP1, MFN2, NIX, BNIP3, LC3, β-actin and Alexa Fluor 488 or 594 labeled Goat Anti-Rabbit or Anti-Mouse IgG (H+L) second antibody were purchased from Abcam (Cambridge, UK). Primary antibodies of type II collagen, aggrecan, MMP-13, TOM70 were from Proteintech (Rosemont, USA). SIRT3 antibody was from Cell Signaling (Danvers, MA, USA). were from Jackson ImmunoResearch (West Grove, PA, USA).
Animal model
Fifty 10-week-old C57BL/6 mice (25 male and 25 female) were purchased from Animal Center of Chinese Academy of Sciences (Shanghai, China) and were divided randomly into the sham group and OA group. Osteoarthritis mouse model was established by surgical destabilization of the medial meniscus as previously described14. Arthrotomy without the transaction of medial collateral ligament was performed in the left knee joint of mice and the joint was used as sham group.
Histopathologic analysis
The mice were sacrificed by intraperitoneal injection of 10% chloral hydrate and the knee joint were harvested 2 months after surgery. The specimens were fixed in 4% (v/v) paraformaldehyde for 36 h and decalcified in 10% (v/v) ETDA for 1.5 month. After dehydrated, tissues were embedded in paraffin and cut into 5-μm sagittal sections. Slides of each joint were stained with safranin O-fast green (S-O). Mice in each group were evaluated by Osteoarthritis Research Society International (OARSI) scoring system for medial femoral condyle and medial tibial plateau as described previously15.
Human articular cartilage collection
Control human articular cartilages with no significant clinical and imaging features of OA were obtained from femoral condyles and tibial plateaus from 3 patients who suffered amputation, which were defined as relative normal. OA human articular cartilages were obtained from 5 patients undergoing total knee arthroplasty. The articular cartilages were collected for further experiments.
Primary chondrocytes culture
40 Sprague Dawley rats (20 male and 20 female, 4 weeks old) were euthanized with an overdose of sodium pentobarbital. Then, the knee cartilages of rats and human were collected and incubated with 2 mg/ml (0.1%) collagenase Ⅱ for 4 h at 37°C. When confluent, cells were harvested by using 0.25% Trypsin-EDTA (Gibco, Invitrogen) and replanted into 6 cm culture plates at the appropriate density. No significant changes exist in morphology of cells between passage 0 to 3. Cells were cultured in the incubator maintained at 5% CO2 and 37°C. The culture medium was changed every other day. Chondrocytes were used from passage 1 to 3.
Lentivirus and siRNA transfection
The scramble and SIRT3 lentiviral particals were chemically synthesized by GeneChem (Shanghai, China). Cells were transfected with lentivirus at multiplicity of infection (MOI) of 100, when reaching 30-50% confluence. After 12 h of transfection, culture medium was changed with DMEM/F12 culture medium with 10% FBS. All of cells were passaged for further experiments after three days. In addition, rat chondrocytes were transfected with PGC-1α siRNA and control siRNA (Ribobio, Guangzhou, China) according to the manufacturer's protocol. Western blot analysis was used to detect levels of SIRT3 and PGC-1α.
Cell viability assay
Cell viability assay was performed using cell counting kit-8 (CCK-8; Dojindo, Japan) following the manufacturer's protocol. Briefly, second passage chondrocytes were planted into the 96 well-plate (8000 cells/ well) and incubated in DMEM/F12 with 10% FBS at 37°C for 12 h. Then, chondrocytes were treated with different concentration of DHM as described above. Following treatment, chondrocytes were washed with 100µl phosphate-buffered saline (PBS), and subsequently 100µl of DMEM containing 10 µl of CCK-8 solution was added into each well, and the plate was incubated for an additional 1.5 h. The absorbance of samples was measured at 450 nm using a microplate reader (Thermo Scientific, USA).
Western blot assay
Rat chondrocytes were lysed in ice-cold RIPA with 1 mM PMSF (Phenylmethanesulfonyl fluoride, Beyotime). Protein concentrations of samples were measured by the BCA protein assay kit (Beyotime). Proteins of chondrocytes was separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were transfferred to polyvinylidene difluoride membrane (Millipore, USA) followed by blocking with 5% nonfat milk. After that, the bands were probed with primary antibodies specific to SIRT3 (1:1000, Cell Signaling Technology), MMP-3 (1:1000, Cell Signaling Technology), MMP-13 (1:1000, Cell Signaling Technology), ADAMTS-5 (1:1000, Cell Signaling Technology), aggrecan (1:1000, Proteintech), collagen II (1:1000, Proteintech), p-AMPK (1:500, Sigma), AMPK (1:1000, Sigma), PGC-1α (1:1000, Abcam), MFN2 (1:1000, Abcam), BNIP3 (1:1000, Abcam), NIX (1:1000, Santa cruz), LC3 (1:1000, Cell Signaling Technology), DRP1 (1:1000, Abcam) , FIS1 (1:1000, Genetex) and β-actin (1:1000, Abcam) overnight at 4°C, before incubated with respective secondary antibodies. Last, the intensity of these bands was quantified using Image Lab 3.0 software (Bio-Rad).
Immunofluorescence
Samples were blocked by 10% goat serum for 1 h at 37°C and probed using primary antibodies mouse anti-SIRT3 (1:200, Santa Cruz Biotechnology), p53 (1:1000, Abcam), Tom70 (1:100, Proteintech), rabbit anti-LC3 (1:100, Cell Signaling Technology) and NIX (Santa Cruz Biotechnology, 1:50) at 4°C overnight. Then slides were incubated with luorescein isothiocyanate- or tetramethyl rhodamine isothiocyanate-conjugated second antibodies for 1 h and labeled with 40, 6-diamidino-2-phenylindole (DAPI, Beyotime) for 5 mins. At last slides were observed in a fluorescence microscope (Olympus Inc., Tokyo, Japan) and a confocal fluorescence microscope (Nikon, Japan). In addition, fluorescence intensity was measured using Image J software 2.1 (Bethesda, MD, USA).
Transmission electron microscopy
After fixed in 2.5% glutaraldehyde overnight, cells were fixed in 2% osmium tetroxide for 1 h and stained with 2% uranyl acetate for 1 h. After dehydrated in an ascending series of acetone, samples were embedded into araldite and cut into semi-thin sections. Then semi-thin sections were stained with toluidine blue to locate cells and observed with a transmission electron microscope (Hitachi, Tokyo, Japan) and a Nikon ECLIPSE Ti microscope (Nikon, Tokyo, Japan).
Mitochondrial membrane potential assay
The mitochondrial transmembrane potential (MMP) was detected by using a green-fluorescent dye, MitoTracker Red CMXRos (Molecular ProbesTM, Thermo Fisher Scientific Inc.), which stains mitochondria in live cells and accumulates in a MMP-dependent manner at the concentration of 50 nM for 45 min at 37°C. After that, nuclei were stained with Hoechst 33342 dye for 15min at 37°C. Last, samples were observed in a fluorescence microscope (Olympus Inc., Tokyo, Japan) and fluorescence intensity was quantified using Image J software 2.1 (Bethesda, MD, USA).
Analysis of enzymatic activity of MnSOD
MnSOD activity was measured with Cu/ MnSOD and MnSOD assay kit (KeyGen, Nanjing, China) according to the manufacture's instruction. After collected, cells were lysed. Both Cu/ZnSOD and exteracellular SOD were inhibited by 3 mM potassium cyanide. Following samples and SOD standard were prepared, 20µl of diluted xanthine oxidase was added into 96-well plate. The plate was placed on a shaker for 30 min at room temperature. The OD values were acquired by using a spectrophotometer (Thermo Scientific, USA) at 450 nm. MnSOD activity of samples was calculated by using equation (LR, LR = Ablank1- Ablank2- Asample)/ (Ablank1- Ablank2) × 100%). One unit is stipulated as the content of enzyme needed to display 50% dismutation of oxygen free radical. Therefore, MnSOD activity (U) sample = LRsample /(1-LRsample) units.
ATP assay
Cell ATP levels of different groups were assessed by using the ATP-GloTM Bioluminometric cell viability assay (Biotium, CA, USA) following the manufacturer's protocol. Data were acquired from multiple replicate wells of each group.
MitoSOX assay
The content of intracellular superoxide anion was detected by using a red-fluorescent dye, MitoSOX (Life Technologies), which stains superoxide anion in live cells and accumulates as superoxide anion-dependent manner, at the concentration of 5 μM for 45 min at 37°C. Then, nuclei were stained with Hoechst 33324 dye for 10 min at 37°C. Last, samples were observed in a fluorescence microscope (Olympus Inc., Tokyo, Japan) and fluorescence intensity was quantified by using Image J software 2.1 (Bethesda, MD, USA).
Statistical analysis
The results were expressed as mean ± S.D. Raw statistical analyses were processed by using Graphpad Prism 6 (USA). Nonparametric data were analyzed by Mann Whitney U test. Data were analyzed by one-way analysis of variance (ANOVA) followed by the Tukey's test for comparison between different groups.
Results
SIRT3 level decreases in pathology of OA in vivo and in vitro
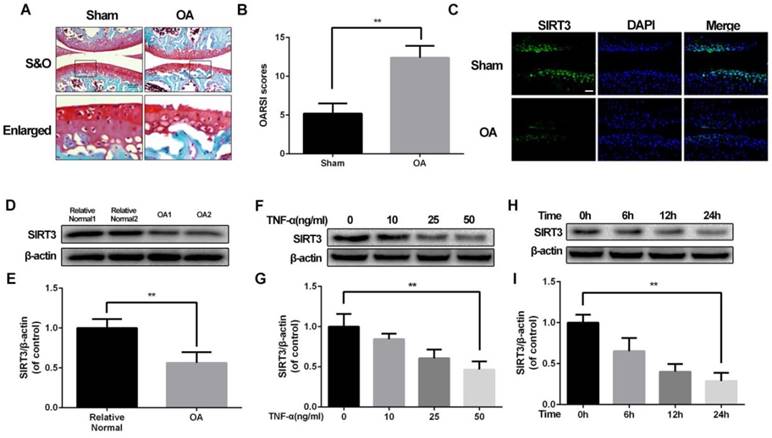
Our previous study discovered mitochondrial dysfunction is involved in chondrocytes degeneration16, implying that SIRT3 could participate in the development of OA. To confirm the level of SIRT3 in pathology of OA, we successfully establish destabilized medial meniscus mouse OA model (Figure 1A and 1B). Then we detected the level of SIRT3 in vivo and in vitro. As shown in figure 1C, the level of SIRT3 in the cartilage of OA group is significantly decreased, compared with the sham group. To further confirm our assumption, we detected SIRT3 level in cultured chondrocytes from human articular cartilage. Chondrocytes from relative normal groups displayed higher SIRT3 level than SIRT3 level in chondrocytes from OA groups (Figure 1D and 1E). Similarly, rat chondrocytes treated with increasing concentrations of TNF-α displayed the decreasing level of SIRT3 (Figure 1F and 1G), in accordance with chondrocytes treated with TNF-α increasing disposal time (Figure 1H and 1I). These data suggest that the level of SIRT3 decreases with degeneration of cartilage in vivo and in vitro.
SIRT3 decreases in mouse OA cartilage and TNF-α-treated rat chondrocytes (A, B) Double meniscus destabilization resulted in articular cartilage necrosis and thining. Scale bar 50 μM. (C) Immunofluorescence staining of SIRT3 in rat knee articular cartilage from sham group and OA group. (D, E) SIRT3 levels of chondrocytes from human cartilage. (F, G) The level of SIRT3 in chondrocytes treated with different concentrations of TNF-α for 24 h. (H, I) The level of SIRT3 in chondrocytes treated with TNF-α (50 ng/ml) for increasing time (0, 6 h, 12 h and 24 h). All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
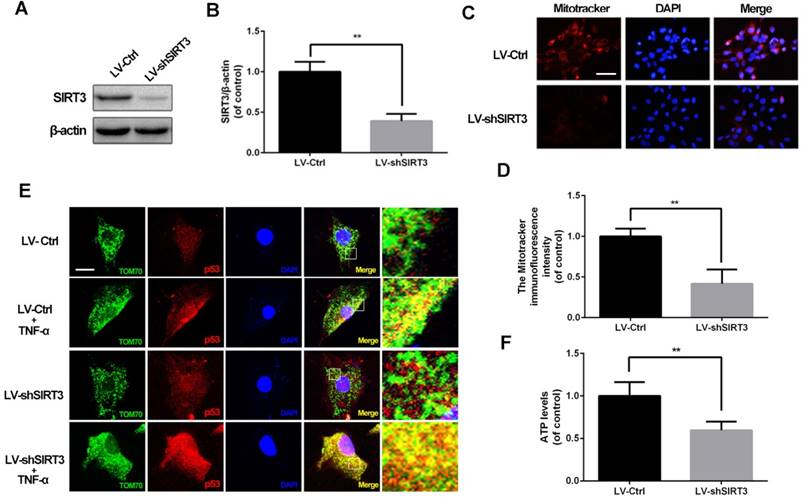
SIRT3 knockdown induces mitochondrial dysfunction in chondrocytes
Based on the phenomenon that the SIRT3 level decreases in chondrocytes treated with TNF-α, we evaluated SIRT3 deficiency on mitochondrial dysfunction in chondrocytes. As shown in figure 2A and 2B, primary rat chondrocytes transfected SIRT3 knockdown lentivirus (LV-shSIRT3) displayed lower level of SIRT3 compared with the scramble lentivirus group (LV-Ctrl). Mitochondrial membrane potential is a indicator for mitochondrial homeostasis. Mitotracker Red assay showed that SIRT3 knockdown induced lower fluorescence intensity in chondrocytes, indicating SIRT3 knockdown reduced the mitochondrial membrane potential in chondrocytes (Figure 2C and 2D). p53 mitochondrial translocation is used for detecting mitochondrial dysfunction, because SIRT3 reduction negatively regulates mitochondrial permeability transition pore17. As shown in figure 2E, SIRT3 knockdown enhanced the co-localization of tom70, one mitochondrial marker, and p53, in TNF-α-treated chondrocytes, indicating that SIRT3 played a critical role in maintaining mitochondrial membrane permeability. In addition, the SIRT3 knockdown decreased the ATP level in chondrocytes, indicating that loss of SIRT3 impaired energy production in chondrocytes (Figure 2F). Together, these results demonstrated that SIRT3 is a positive regulator in maintaining mitochondrial homeostasis.
SIRT3 knockdown impairs mitochondrial dysfunction induced by TNF-α in rat chondrocytes. (A, B) Rats chondrocytes was transfected with scramble lentivirus and SIRT3 knockdown lentivirus. Representative Western blots of and quantitative data for SIRT3 level in each group. (C, D) The effect of SIRT3 knockdown on the mitochondrial membrane potential in chondrocytes. (E) SIRT3 knockdown promoted the translocation of p53 to mitochondria in TNF-α-treated chondrocytes. (F) The effect of SIRT3 knockdown on ATP production in chondrocytes. All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
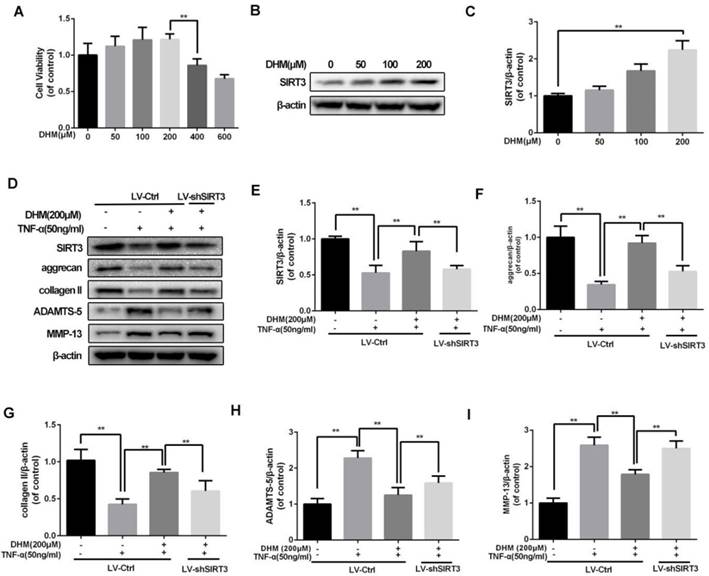
DHM protects chondrocytes from disorder of ECM synthesis induced by TNF-α
To confirm the safe concentration of DHM on rat chondrocytes, the results of CCK-8 showed that DHM was not cytotoxic to chondrocytes at concentration of 200µM after treatment of 24 h (Figure 3A). Chondrocytes treated with the increasing concentration of DHM displayed increasing SIRT3 level, suggesting that DHM was able to enhance SIRT3 in chondrocytes (Figure 3B and 3C). Then we detected the degeneration-related proteins of ECM in DHM-treated chondrocytes. As shown in figure 3D-3I, we found that TNF-α decreased levels of anabolic proteins (aggrecan and collagen II) and upregulated the catabolic markers (MMP-13 and ADAMTS-5), which were reversed partly with administration of DHM. Moreover, the protective effects of DHM against ECM imbalance were abolished by SIRT3 knockdown. These results suggest that DHM is able to improve chondrocytes degeneration via activating SIRT3.
DHM maintains the balance between the anabolism and catabolism of ECM in TNF-α-treated chondrocytes. (A) The effect of DHM on viability of rat chondrocytes. (B, C) DHM promotes the level of SIRT3 in chondrocytes. (D-I) Representative western blots of and quantitative data for SIRT3, aggrecan, collagen II, ADAMTS-5 and MMP-13 levels in each group. All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
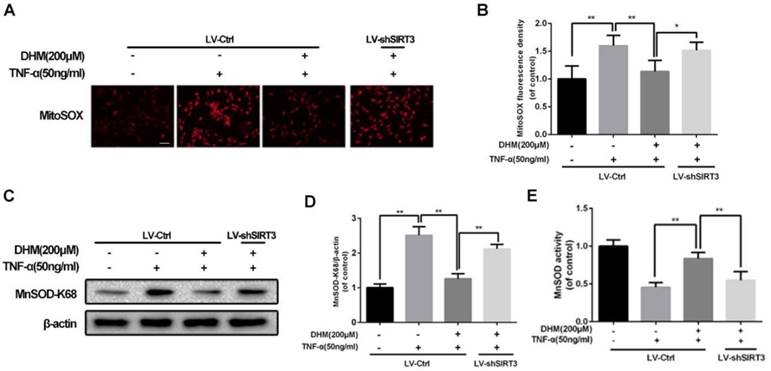
DHM improves the capacity of mitochondrial anti-oxidation via SIRT3 activation
For the capacity of up-regulation of SIRT3, DHM might be able to enhance the capacity of mitochondrial anti-oxidation in chondrocytes. To detect whether ROS can be scavenged by DHM, the intracellular ROS-dependent MitoTracker was used to assess the degree of oxidative stress. The results of MitoTracker assay showed that the red fluorescence was increased in chondrocytes treated with TNF-α and was negatively regulated by DHM, while SIRT3 knockdown abolished the protective effect of DHM on ROS (Figure 4A and 4B). MnSOD, as a major superoxide-scavenger in mitochondria, is inclined to be acetylated at K68 under stress18. The results of western blot assay displayed that the level of acetyl-k68 MnSOD was increased in chondrocytes treated with TNF-α, which was blocked partially by DHM via SIRT3 activation (Figure 4C and 4D). Also, with administration of DHM, the enzymatic anti-oxidation of MnSOD was enhanced, which was reversed partly by SIRT3 knockdown (Figure 4E). Therefore our results suggest that DHM exerts anti-oxidation property by promoting SIRT3-mediated deacetylation of MnSOD at K68.
DHM promotes the capacity of anti-oxidation via activating SIRT3 in rat chondrocytes. (A, B) Representative micrographs of MitoSOX assay showed the effects of DHM on the ROS level in chondrocytes against TNF-α. Scale bar 100 μM.(C, D) Representative western blots and results of densitometry analysis of acetyl-k68 MnSOD in rat chondrocytes treated as above. (E) DHM promoted the activity of MnSOD via enhancing SIRT3 in chondrocytes. All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
DHM alleviates the imbalance of mitochondrial dynamics induced by TNF-α as a SIRT3-dependent manner
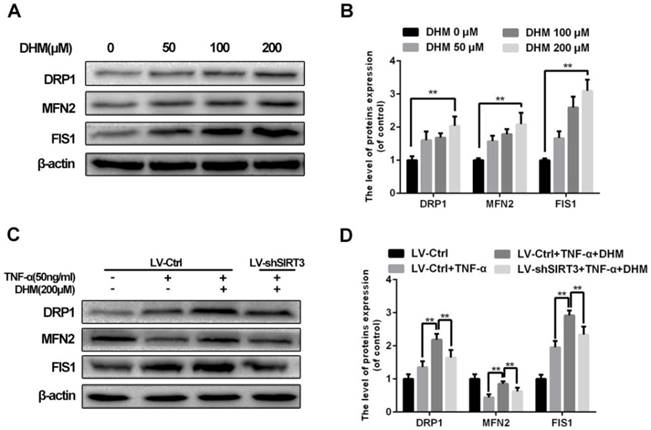
To abandon or rescue defective mitochondria under stress, it is essential to maintain the imbalance in mitochondrial fission and fusion in time. Enhanced SIRT3 increased the content of mitochondrial fission related protein, dynamin-related protein 1 (DRP1) and mitochondrial fission 1 protein (FIS1), as well as mitochondrial fusion related protein mitofusin 2 (MFN2)10. As a SIRT3 agonist, DHM promoted the levels of mitochondrial fission and fusion related proteins as a dose-dependent manner, indicating that mitochondrial dynamics became more active with treatment of DHM (Figure 5A and 5B). Moreover we found that DHM could compromised the reduction of MFN2 level in chondrocytes treated with TNF-α via promoting SIRT3. Therefore our results indicate that DHM can attenuate the mitochondrial dynamic imbalance under TNF-α-induced inflammatory microenvironment via enhancing SIRT3.
DHM alleviates the mitochondrial dynamic imbalance through enhancing SIRT3. (A, B) The effects of DHM on mitochondrial dynamics markers in chondrocytes. Representative western blots of and quantitative data for DRP1, MFN2 and FIS1 levels of chondrocytes in each group. (C, D) Activation of SIRT3 by DHM promoted mitochondrial fission and fusion against TNF-α. All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
SIRT3 knockdown inhibits mitophagy induced by DHM in rat chondrocytes
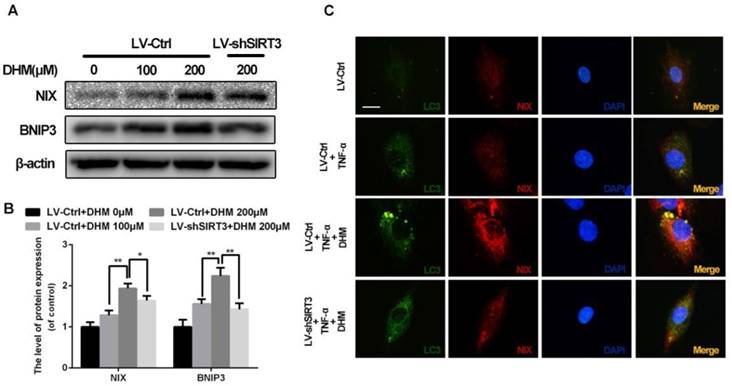
To maintain the mitochondrial homeostasis, damaged mitochondria could be detected and eliminated by mitophagy, the selected autophagy for mitochondria19. As shown in Figure 6A and 6B, the mitophagic receptors on outer mitochondrial membrane, NIX and BNIP3, were up-regulated by treatment of DHM, while SIRT3 knockdown reduced levels of NIX and BNIP3 relative to control. To determine whether impaired mitochondria were phagocytized by autophagic vacuoles, we performed the double-staining immunofluorescence for LC3 and NIX (Figure 6C). The results of immunofluorescence showed numerous and large double positive puncta were observed in the cells with administration of TNF-α, but the pro-mitophagy effect of DHM was partly abolished by SIRT3 knockdown. These data indicate that DHM is able to promote mitophagy as a SIRT3-dependent manner.
DHM promotes mitophagy via up-regulating SIRT3 in rat chondrocytes treated with TNF-α. (A, B) The effects of SIRT3 on levels of NIX and BNIP3. Representative western blots of and quantitative data for NIX and BNIP3 levels in each group. (C) DHM induced more co-localization of LC3 and NIX via activating SIRT3 in chondrocytes. Immunofluorescent signals were labeled by LC3 (green) and NIX (red). All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
AMPK-PGC-1α-SIRT3 signaling pathway is involved in activation of SIRT3 in rat chondrocytes treated with DHM
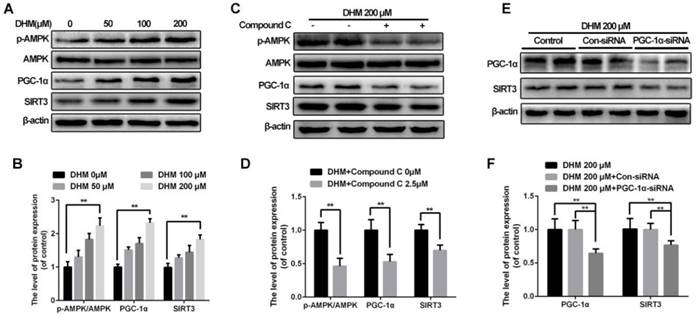
To reveal how SIRT3 is activated in chondrocytes treated with DHM, we explored the upstream pathway related to SIRT3 regulation. Our results showed that DHM could promote the ratio of p-AMPK and AMPK, and levels of PGC-1α and SIRT3 in a dose-dependent manner in chondrocytes (Figure 7A and 7B). Chondrocytes were administrated with compound C to inhibit AMPK before they receiving DHM. Compared with the control group, the compound C group showed the decreased the ratio of p-AMPK to AMPK as well as the band intensity of PGC-1α and SIRT3 (Figure 7C and 7D). Then we silenced PGC-1α to determine whether it could regulate SIRT3 in chondrocytes. We found that the level of SIRT3 was reduced in PGC-1α-siRNA group compared with the Con-siRNA group (Figure 7E and 7F). These data suggest that SIRT3 is up-regulated by DHM via activating AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes.
SIRT3 is regulated through AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes treated with DHM. (A, B) DHM up-regulated the ratio of p-AMPK to AMPK as well as levels of PGC-1α and SIRT3 as a dose-dependent manner. Representative western blots of and quantitative data for p-AMPK/AMPK, PGC-1α and SIRT3 levels in chondrocytes from each group. (C, D) Effects of compound C on levels of AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes treated with DHM. (E, F) Effects of PGC-1α on SIRT3 in chondrocytes treated with DHM. All experiments were performed as mean ± SD of 5 times in duplicates. *P < 0.05, **P < 0.01.
Discussion
SIRT3, thought to be a “master” regulator of mitochondria, maintains mitochondrial homeostasis against stress via deacetylating different substrates20, 21. Growing evidences support that mitochondrial dysfunction are implicated with the pathogenesis of OA16, but it remains elusive whether destabilized medial meniscus OA is relevant to SIRT3. The present study demonstrates the level of SIRT3 decreases significantly in degenerative knee articular cartilage and TNF-α treated chondrocytes. To identify the effects of SIRT3 deficiency in chondrocytes, we transfected rat chondrocytes with lentivirus to knockdown SIRT3. We found that SIRT3 deficiency was able to induce lower mitochondrial membrane potential, impaired mitochondrial membrane permeability and lower level of intracellular ATP, which are considered as the characters of mitochondrial dysfunction.
Conversely, SIRT3 activation protects cells from mitochondrial dysfunction by modulating mitochondrial dynamics and mitophagy10, indicating that SIRT3 might be a potential therapeutic target against chondrocytes degeneration and OA. DHM has been reported to activate SIRT3 in various tissues, including liver22, brain13 and muscle23. We found that DHM not only promoted the level of SIRT3, but also compromised the degeneration phenotypes such as MMP-13 and ADAMTS-5 and improved ECM anabolic markers such as aggrecan and collagen II via enhancing SIRT3 in TNF-α-treated rat chondrocytes.
On the basis of SIRT3-mediated protective effects of DHM against degeneration in chondrocytes, it is highly possible that DHM could protect chondrocytes from degeneration via attenuating mitochondrial dysfunction. The result of MitoSOX, one specific ROS-dependent probe, indicated that DHM scavenged TNF-α-induced high ROS level via activating SIRT3 in chondrocytes. Acetylation of Lys68 (K68) in MnSOD represents impaired enzymatic activity24, 25. In this study we found that DHM reduced the degree of oxidative stress, compromised the acetylation of MnSOD at K68 and promoted MnSOD enzymatic activity through enhancing SIRT3 in chondrocytes. Therefore DHM is able to improve the capacity of anti-oxidation via SIRT3 activation in chondrocytes.
It is well recognized that mitochondria, highly motile and plastic organelles, continually undergo activities of fusion and fission which alter the morphology of mitochondria26. Mitochondrial fusion and fission are essential for maintenance of mitochondrial homeostasis, which coordinates the diverse intracellular physiological activities against apoptosis induced by stress27, 28. Our finding suggested that TNF-α promoted levels of two fission markers (DRP1 and FIS1) of mitochondria, and reduced the level of a fusion-related marker (MFN2), indicating that mitochondria were inclined to separate damaged mitochondria but deficient in fusion to neutralize impaired mitochondrial membrane potential under TNF-α-induced stress. Also, besides promoting mitochondrial fission, DHM was able to enhance the capacity of mitochondrial fusion via enhancing SIRT3 in chondrocytes.
Damaged mitochondria would like to be the main source of ROS, once cannot be removed in time29. Mitophagy, capable of eliminating impaired mitochondria segregated by mitochondrial fission, plays a crucial role in maintaining mitochondrial homoeostasis. SIRT3 positively regulates BNIP3 and NIX, two mitophagic receptors, to promote mitophagy in human umbilical vein endothelial cells10. In our study, DHM was capable of up-regulating the levels mitophagic markers in chondrocytes. In addition, DHM promoted the interaction between mitochondria and autophagosomes in TNF-α-treated chondrocytes, whereas SIRT3 knockdown compressed mitophagy. Together, we think that DHM can promotes mitophagy via activating SIRT3.
Adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK) senses metabolic stress and harmonizes different physiological activities to support energy production30. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is able to regulate mitochondrial biogenesis and respiratory function31, 32. Recent studies found that AMPK-PGC-1α-SIRT3 is a classical signaling pathway to activate SIRT3 32, 33, 34. One previous study found the positive correlation between PGC-1α and SIRT3 activity in both muscle cells and hepatocytes, suggesting the PGC-1α might be the endogenous regulator of SIRT335, but it is still required to confirm whether SIRT3 could be up-regulated via AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes. Our findings indicate that DHM is able to promote SIRT3 via AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes.
In summary, SIRT3 level decreases in the pathology of OA mouse models compared with sham group. SIRT3 deficiency leads to mitochondrial dysfunction in chondrocytes. DHM is able to reverse chondrocytes degeneration via maintaining the SIRT3-mediated mitochondrial homeostasis. In addition, DHM can regulate SIRT3 via AMPK-PGC-1α-SIRT3 signaling pathway in chondrocytes. Our findings contribute to understand the mechanism of OA and enrich the therapeutic strategy against OA.
Acknowledgements
This study is supported by Zhejiang Provincial Natural Science Foundation of China (LY17H060010, LY15H060008, LY18H060012), Zhejiang Public Service Technology Research Program / Social Development (LGF18H060008), National Natural Science Foundation of China (81501907), Major Scientific and Technological Project of Medical and Health in Zhejiang Province (WKJ-ZJ-1527), Zhejiang Provincial Project for Medical and Health Science and Technology (2017KY463), and Zhejiang Undergraduate Talent Project (2018R413074).
Competing Interests
The authors have declared that no competing interest exists.
References
1. He BH, Christin M, Mouchbahani-Constance S, Davidova A, Sharif-Naeini R. Mechanosensitive ion channels in articular nociceptors drive mechanical allodynia in osteoarthritis. Osteoarthritis and cartilage. 2017
2. Larsson S, Englund M, Struglics A, Lohmander LS. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthritis & Cartilage. 2015;23(11):1906
3. Song W, Zhang Y, Wang J, Ma T, Hao L, Wang K. Antagonism of cysteinyl leukotriene receptor 1 (cysLTR1) by montelukast suppresses cell senescence of chondrocytes. Cytokine. 2018;103:83-89
4. Qu R, Chen X, Wang W, Qiu C, Ban M, Guo L. et al. Ghrelin protects against osteoarthritis through interplay with Akt and NF-kappaB signaling pathways. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2017 fj201700265R
5. Kim WS, Lee KS, Kim JH, Kim CK, Lee G, Choe J. et al. The caspase-8/Bid/cytochrome c axis links signals from death receptors to mitochondrial reactive oxygen species production. Free radical biology & medicine. 2017
6. Shen YL, Shi YZ, Chen GG, Wang LL, Zheng MZ, Jin HF. et al. TNF-alpha induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int J Mol Med. 2018;41(4):2317-2327
7. Hosseinzadeh A, Kamrava SK, Joghataei MT, Darabi R, Shakeri-Zadeh A, Shahriari M. et al. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. Journal of pineal research. 2016;61(4):411-425
8. Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S, Haqqi TM. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radical Biology & Medicine. 2017;106:288
9. Maneiro E, Martin MA, de Andres MC, Lopez-Armada MJ, Fernandez-Sueiro JL, del Hoyo P. et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis and rheumatism. 2003;48(3):700-708
10. Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radical Biology & Medicine. 2013;63(10):222
11. Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR. et al. Aging Promotes Sirtuin 3-Dependent Cartilage Superoxide Dismutase 2 Acetylation and Osteoarthritis. Arthritis & Rheumatology. 2016;68(8):1887-1898
12. Zhou Y, Liang X, Chang H, Shu F, Wu Y, Zhang T. et al. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Science. 2015;105(10):1279-1287
13. Liu P, Zou D, Chen K, Zhou Q, Gao Y, Huang Y. et al. Dihydromyricetin Improves Hypobaric Hypoxia-Induced Memory Impairment via Modulation of SIRT3 Signaling. Molecular Neurobiology. 2015;53(10):1-13
14. Chang TR, Tseng FJ, Chou YC, Chang HL, Wang TH, Chia WT. THE Surgical Destabilization of the Medial Meniscus (DMM) Model of Osteoarthritis in the BALA/C Mouse: MMP-9 as a Prognostic Maker: A Pilot Study. Journal of Orthopaedic Surgery Taiwan. 2009;26(1):7-12
15. Glasson S, Chambers M, Van Den Berg W, Little C. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil. 2010;18(Suppl 3):S17-23
16. Tang Q, Zheng G, Feng Z, Chen Y, Lou Y, Wang C. et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. 2017; 8(10): e3081.
17. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A. et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY). 2010;2(12):914-923
18. Lu J, Cheng K, Zhang B, Xu H, Cao Y, Guo F. et al. Novel mechanisms for superoxide-scavenging activity of human manganese superoxide dismutase determined by the K68 key acetylation site. Free radical biology & medicine. 2015;85:114-126
19. Kim HS, Patel K, Muldoonjacobs K, Bisht KS, Aykinburns N, Pennington JD. et al. SIRT3 is a Mitochondrial Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism During Stress. Cancer Cell. 2010;17(1):41
20. Dimozi A, Mavrogonatou E, Sklirou A, Kletsas D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. European cells & materials. 2015;30:89-102 discussion 103
21. Zhang M, Tang J, Li Y, Xie Y, Shan H, Chen M. et al. Curcumin attenuates skeletal muscle mitochondrial impairment in COPD rats: PGC-1alpha/SIRT3 pathway involved. Chem Biol Interact. 2017;277:168-175
22. Zeng X, Yang J, Hu O, Huang J, Ran L, Chen M. et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxidants & redox signaling. 2018
23. Shi L, Zhang T, Liang X, Hu Q, Huang J, Zhou Y. et al. Dihydromyricetin improves skeletal muscle insulin resistance by inducing autophagy via the AMPK signaling pathway. Molecular and cellular endocrinology. 2015;409:92-102
24. Qiu X, Brown K, Hirschey M, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12(6):662-667
25. Lu J, Zhang H, Chen X, Zou Y, Li J, Wang L. et al. A small molecule activator of SIRT3 promotes deacetylation and activation of manganese superoxide dismutase. Free Radic Biol Med. 2017;112:287-297
26. Seo AY, Joseph AM, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. Journal of Cell Science. 2010;123(Pt 15):2533
27. Zhang T, Ikejima T, Li L, Wu R, Yuan X, Zhao J. et al. Impairment of Mitochondrial Biogenesis and Dynamics Involved in Isoniazid-Induced Apoptosis of HepG2 Cells Was Alleviated by p38 MAPK Pathway. Front Pharmacol. 2017;8:753
28. Liu N, Jiang Z, Liu Y, Nie Y, Chen J, Ouyang B. et al. Human trypsin inhibitor reduces the apoptosis of lipopolysaccharide-induced human kidney-2 cells by promoting mitochondrial fusion. Molecular medicine reports. 2017;16(3):2899-2906
29. Dagda RK, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. Journal of Biological Chemistry. 2009;284(20):13843-13855
30. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metabolism. 2010;12(6):662
31. Zhang L, Zhou Y, Wu W, Hou L, Chen H, Zuo B. et al. Skeletal Muscle-Specific Overexpression of PGC-1α Induces Fiber-Type Conversion through Enhanced Mitochondrial Respiration and Fatty Acid Oxidation in Mice and Pigs. Int J Biol Sci. 2017;13(9):1152-1162
32. Ma CH, Chiua YC, Wu CH, Jou IM, Tu YK, Hung CH. et al. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: A possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018;15:504-512
33. Liu S, Wang Y, Zhang Y, He X, Ma T, Song W. et al. ZL006 protects spinal cord neurons against ischemia-induced oxidative stress through AMPK-PGC-1α-Sirt3 pathway. Neurochem Int. 2017;108:230-237
34. Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z. et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1α-SIRT3 signaling. Sci Rep. 2017;7:41337
35. Park S, Ahmad F, Philp A, Baar K, Williams T, Luo H. et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148(3):421-433
Author contact
![]() Corresponding authors: Xiaolei Zhang, Department of Orthopaedics, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou 325027, Zhejiang, China. E-mail: zhangxiaoleiedu.cn, phone: 0577-88002760 or Xiangyang Wang, Department of Orthopaedics, The Second Affiliated Hospital of Wenzhou Medical University, 109 Xueyuan Xi Road, Wenzhou 325027, Zhejiang, China. E-mail: xiangyangwangedu.cn, phone: 0577-88002815
Corresponding authors: Xiaolei Zhang, Department of Orthopaedics, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou 325027, Zhejiang, China. E-mail: zhangxiaoleiedu.cn, phone: 0577-88002760 or Xiangyang Wang, Department of Orthopaedics, The Second Affiliated Hospital of Wenzhou Medical University, 109 Xueyuan Xi Road, Wenzhou 325027, Zhejiang, China. E-mail: xiangyangwangedu.cn, phone: 0577-88002815