Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(14):1974-1984. doi:10.7150/ijbs.28050 This issue Cite
Research Paper
Nitric oxide prevents H2O2-induced apoptosis in SK-N-MC human neuroblastoma cells
Yeong-Min Yoo, Eui-Man Jung, Changhwan Ahn, Eui-Bae Jeung ![]()
Laboratory of Veterinary Biochemistry and Molecular Biology, College of Veterinary Medicine, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea
Received 2018-6-22; Accepted 2018-9-4; Published 2018-11-2
Abstract

Nitric oxide (NO) is a cellular signaling molecule in many physiological and pathological processes including neuroprotector. Here we examined the antiapoptotic effect of NO in SK-N-MC cells. H2O2 treatment (10-200 μM) induced cell death in a dose-dependent manner and pretreatment of cells with 100 μM S-nitroso-N-acetylpenicillamine (SNAP), an NO donor, attenuated the occurrence of H2O2-induced cell death. DAPI staining showed H2O2-induced nuclear fragmentation and NO treatment suppressed it. NO inhibited the proteolytic activation of caspase-3 and mitochondrial cytochrome c release. Treatment of soluble guanylyl cyclase inhibitor ODQ decreased the protective effect of SNAP on H2O2-treated cells and increased caspase 3-like enzyme activity and activation, cytochrome c release, PARP cleavage, and DNA fragmentation, indicating that cGMP is a key mediator in NO-mediated antiapoptosis. The cGMP analog 8-Br-cGMP blocked H2O2-induced apoptotic cell death; reduction of caspase-3 enzyme, cytochrome c release, and caspase-8 and -9. These preventive effects of SNAP and 8-Br-cGMP were suppressed by PKG inhibitor KT5823. Levels of PKGI, PKGII, and p-VASP proteins were increased by SNAP and 8-Br-cGMP and suppressed by KT5823 treatment. These results indicate that PKG is a downstream signal mediator in the suppression of apoptosis by NO and cGMP. Akt activation was inhibited the PI3K inhibitors LY294002 and Wortmannin, resulting in the inhibition of cell viability and increase of cytochrome c release. SNAP induced phosphorylation of Akt and Bad and then increased the interactions between 14-3-3β and p-Bad. These data suggest that the NO suppresses H2O2-induced SK-N-MC cell apoptosis by suppressing apoptosis signal mediating the interaction between 14‐3‐3β and Bad phosphorylation via PKG/PI3K/Akt.
Keywords: Nitric oxide, hydrogen peroxide, PKG, PI3K, Akt
Introduction
Hydrogen peroxide (H2O2) has been known as a signaling molecule in various biological processes including cell proliferation, differentiation, migration, autophagy, and apoptosis [1-4]. These molecules can decompose into a hydroxyl radical through Fenton reaction or Haber-Weiss reaction [5]. Hydroxyl radicals can react with and damage vital cellular components, especially the mitochondria. At last cells or tissues induces cell death, or accelerates aging and cancer [6, 7].
Nitric oxide (NO) has been known to be produced by the conversion of L-arginine to L-citrulline, a reaction catalyzed by one of three NO synthases (NOS): neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) [8, 9]. NO has been shown as a tissue-protection through its physiologic regulation of vascular tone, inhibition of platelet aggregation, attenuation of leukocyte adherence to the endothelium, scavenging of oxygen-derived free radicals, maintenance of normal vascular permeability, inhibition of smooth muscle proliferation, immune defenses, and stimulation of endothelial cell regeneration [9]. Furthermore, NO is to be a mediator/protector of ischemia and reperfusion tissue-mediated injury [9] and neuroprotector in neurodegenerative diseases including Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) [8].
In PC12 cells, NO protects against serum withdrawal-cell death by inhibiting the activation of caspase through cGMP production and activation of protein kinase G (PKG) [10]. Also, this molecule suppresses 6-hydroxydopamine (6-OHDA)-induced apoptosis by activating PKG via phosphatidylinositol-3 kinase (PI3K)/Akt-dependent Bad phosphorylation [11]. In the primary neurons, NO-activated survival signaling involves nerve growth factor (NGF)-like effects, including enhanced phosphorylation of TrkA and activation of PI3K/Akt and MAPK pathways [12]. Andoh et al. [13] reported that the cytoprotective effect of NO in human neuroblastoma SH-SY5Y cells appears to proceed via the PKG-mediated pathway and the expression of thioredoxin and thioredoxin peroxidase-1. Ciani et al. [14] showed that NO protection in serum starved-SK-N-BE neuroblastoma cell apoptosis is associated with cAMP-response element-binding protein (CREB) activation. The SK-N-MC cells are a human neuroblastoma cell line. This cell line was established in 1971 from the supraorbital metastatic site in a 14-year-old Caucasian female with an Askin's tumor related to Ewing's sarcoma. The SK-N-MC cells show little or no dopamine-beta-hydroxylase activity but increase choline acetyltransferase activity compared to other neuroblastoma cell lines such as the SK-N-SH, SK-N-BE, and SH-SY5Y. Therefore, in the present study, the NO suppresses H2O2-induced human neuroblastoma SK-N-MC cell apoptosis by suppressing apoptosis signal through activating PKG/PI3K/Akt and result in the increase of the interaction between 14‐3‐3β and Bad phosphorylation.
Results
The protection of NO against H2O2-induced apoptosis in SK-N-MC cells
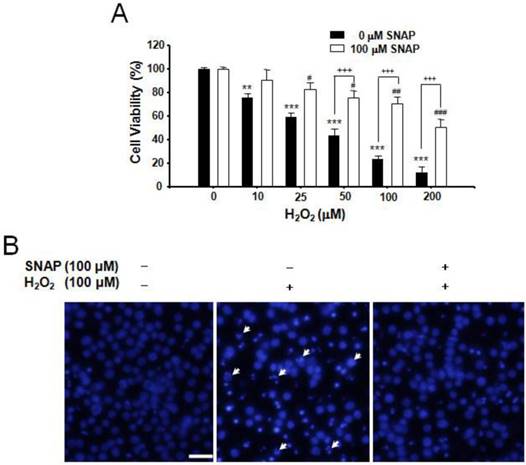
We investigated the protection of NO against H2O2-induced apoptosis in human neuroblastoma SK-N-MC cells. H2O2 treatment (10-200 μM) induced cell death in a dose-dependent manner and pretreatment of cells with 100 μM SNAP, an NO donor, attenuated the occurrence of H2O2-induced cell death; viabilities of cells treated with SNAP were about 90, 83, 75, 70, and 52%, respectively, of the control value (100%) (Figure 1A). DAPI staining showed H2O2-induced nuclear fragmentation and NO treatment suppressed it (Figure 1B).
Protective effect of SNAP against the H2O2-induced death of SK-N-MC cells. The SK-N-MC cells were cultured in DMEM supplemented with 10% heat-inactivated FBS at 37°C in 5% CO2, 95% air in a humidified cell incubator. Cells were treated with H2O2 for 24 hr, and SNAP was added 10 hr prior to H2O2 treatment. Cell viability assay was performed by Cell Counting Kit-8 (A). DAPI staining was analyzed by fluorescence microscopy (B). Values were represented as mean ± SD from three independent experiments. Scale bar, 50 μm. **P < 0.01, ***P < 0.01 versus no treatment of SNAP. #p < 0.01, ##p < 0.01, ###p < 0.001 versus 100 μM SNAP treatment. +++p < 0.001, no treatment of 100 μM SNAP versus 100 μM SNAP treatment.
The effect of SNAP and inhibitors of caspase on SK-N-MC cell apoptosis
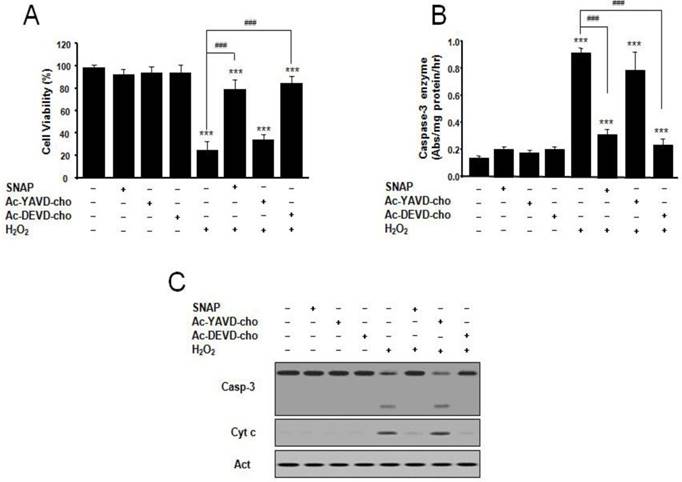
We examined the effect of SNAP and inhibitors of caspase on SK-N-MC cell apoptosis and caspase 3-like activity. The caspase-3-like protease inhibitor Ac-DEVD-cho prevented H2O2-induced cell death, whereas the caspase-1-like protease inhibitor Ac-YVAD-cho did not (Figure 2A). The protective effect of Ac-DEVD-cho was comparable with the effect of SNAP (100 μM) treatment. Caspase-3-like enzyme activity was measured in the cytosol from H2O2-treated cells by a colorimetric assay using substrate-specific tetrapeptides (Figure 2B). Caspase-3-like enzyme activity in H2O2-treated cell extracts was about 6-fold higher than that of untreated control cells. Caspase-3-like enzyme activity was increased by the addition of Ac-YVAD-cho but decreased by Ac-DEVD-cho or SNAP (Figure 2B). In these conditions, Western blot analyses of caspase-3 activation and mitochondrial cytochrome c release were performed (Figure 2C). Caspase-3 was activated under H2O2 treatment condition, and the activation was suppressed by addition of SNAP and Ac-DEVD-cho, respectively, but not by the addition of Ac-YVAD-cho (Figure 2C). These results indicate that the NO on H2O2-treated SK-N-MC cells may inhibit by an inhibition of caspase 3-like enzyme activation and/or activity.
Protective effect of SNAP and caspase inhibitors on the survival of SK-N-MC cells. The SK-N-MC cells were cultured in DMEM supplemented with 10% heat-inactivated FBS at 37°C in 5% CO2, 95% air in a humidified cell incubator. Cells were treated with H2O2 for 24 hr, and SNAP was added 10 hr prior to H2O2 treatment. Cells were treated with SNAP (100 μM), Ac-YVAD-cho (200 μM), or Ac-DEVD-cho (200 μM) in media. At 24 hr, cell viability was determined by Cell Counting Kit-8 (A). SK-N-MC cells were collected, washed with ice-cold PBS, and lysed in 100 mM HEPES buffer, pH 7.4, containing protease inhibitors. Caspase-3 enzyme activity was determined by measuring the absorbance of the fraction at 405 nm, with 200 μM Ac-DEVD-pNA as a substrate for caspase-3 enzyme (B). Western blot analyses of caspase-3 activation and cytochrome c (C) were determined as described in Materials and methods. Values were represented as mean ± SD from three independent experiments. ***p < 0.01 versus nontreatment of SNAP. ###p < 0.001, 100 μM H2O2 treatment versus 100 μM SNAP/H2O2 or Ac-DEVD-cho/H2O2 treatment.
The role of cGMP in NO-mediated protection
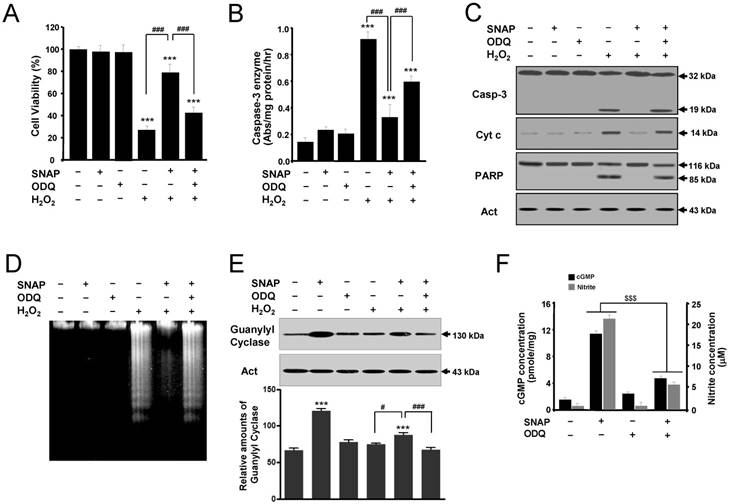
To determine the role of cGMP in NO-mediated protection, the effects of SNAP on SK-N-MC cells were examined in the presence of an inhibitor of soluble guanylyl cyclase ODQ (Figure 3). ODQ did not affect the cell viability of cells but inhibited the protective effect of SNAP on H2O2-treated cells (Figure 3A). Also, treatment with ODQ prevented the capacity of SNAP to decrease caspase-3-like enzyme activity (Figure 3B) and to identify caspase-3 activation and mitochondrial cytochrome c release by Western blot (Figure 3C). One of the established substrates for a caspase-3 enzyme in cells is PARP, which is cleaved from 116 kDa intact protein into 85 and 31 kDa fragments during apoptosis [10]. The cleaved product (85 kDa) of PARP was identified in H2O2-treated cells (Figure 3C). PARP cleavage was almost completely inhibited by SNAP treatment, and this inhibition was primarily reversed by the addition of ODQ (Figure 3C). DNA fragmentation was identified in H2O2-treated cells (Figure 3D). SNAP-mediated guanylyl cyclase increase was inhibited by ODQ (Figure 3E). These results showed that SNAP prevented DNA fragmentation, and ODQ significantly blocked this effect of SNAP. SNAP treatment increased the concentration of cGMP production and ODQ inhibited the cGMP level in response to NO induction (Figure 3F). Together, these findings indicate that a major effector for NO-mediated inhibition of SK-N-MC cell apoptosis is cGMP, which inhibits the activation of the caspase-3 enzyme.
A guanylyl cyclase inhibitor reverses the protective effect of SNAP on SK-N-MC cells. The SK-N-MC cells were cultured in DMEM supplemented with 10% heat-inactivated FBS at 37°C in 5% CO2, 95% air in a humidified cell incubator. Cells were treated with H2O2 for 24 hr, and SNAP was added 10 hr prior to H2O2 treatment. Cells were treated with SNAP (100 μM) or/and ODQ (40 μM) in media. At 24 hr, cell viability (A), caspase-3 enzyme activity (B), Western blot analyses (C) of the caspase-3 enzyme, cytochrome c, and PARP fragmentation, and DNA fragmentation (D) were determined as described in Materials and Methods. Western blot analyses of guanylyl cyclase and the relative amounts of guanylyl cyclase (E) were determined as described in Materials and Methods. cGMP levels were measured in whole cell lysate with enzyme immunoassay kits and nitrite concentrations were determined from the Griess reagent (F). Values were represented as mean ± SD from three independent experiments. ***p < 0.01 versus no treatment of SNAP. #p < 0.05, ###p < 0.001, H2O2 + SNAP treatment versus H2O2 alone or H2O2 + 100 μM SNAP + ODQ treatment. $$$p < 0.001, SNAP treatment versus 100 μM SNAP + ODQ treatment.
The cGMP-mediated protection through protein kinase G activation
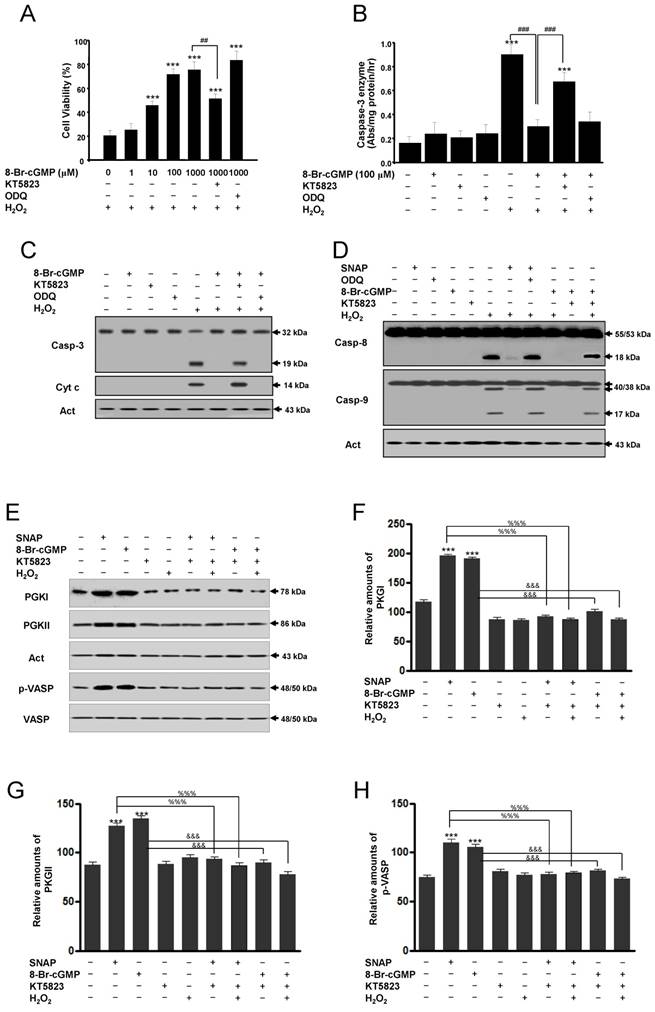
Actions of cGMP are implicated in the activation of cGMP-dependent protein kinase or protein kinase G (PKG) [11-13]. Thus, we used the PKG inhibitor KT5823 to determine whether the protective actions of cGMP were mediated by the activation of PKG. The cell-permeable cGMP analog 8-Br-cGMP protected SK-N-MC cells from H2O2-induced apoptosis in a concentration-dependent manner. This protection was significantly inhibited by the addition of KT5823 but not by ODQ (Figure 4A). In this condition, treatment with 8-Br-cGMP significantly inhibited caspase-3-like enzyme activity in H2O2-treated cells, and this inhibition was reversed partly by KT5823 but not ODQ (Figure 4B). Western blot analyses of caspase-3 activation and cytochrome c release were consistent with this condition (Figure 4C). Also, Western blot analyses of caspase-8 and -9 enzymes activation were identified. Caspase-8 and -9 activations by Western blot were increased in treatment with SNAP under ODQ and 8-Br-cGMP by the addition of KT5823 (Figure 4D). To identify inhibition of the PKG activity by KT5823, PKGI, PKGII, and PKG-specific vasodilator-stimulated phosphoprotein (VASP) phosphorylation (Ser239) were analyzed (Figure 4E-H). Levels of PKGI, PKGII, and p-VASP proteins were increased by SNAP and 8-Br-cGMP and suppressed by KT5823 treatment. These results indicate that the effects of cGMP on the activation of caspase-3, -8 and -9 enzymes are mediated via the activation of PKG.
A protein kinase G inhibitor reverses the cGMP-dependent protection of H2O2-induced apoptosis of SK-N-MC cells. Cells were cultured in media containing different concentrations of 8-Br-cGMP (A) or 100 μM 8-Br-cGMP (B) with or without KT5823 (180 nM) or ODQ (40 μM). Cell viability was measured by Cell Counting Kit-8 (A). Caspase-3 enzyme activity was determined by a colorimetric assay using Ac-DEVD-pNA after incubation of lysate for 30 min (B). Western blot analyses of caspase-3 enzyme and cytochrome c (C), caspase-8 and caspase-9 enzymes (D), PKGI (E), PKGII (E), and p-VASP (E) and the relative amounts of PKGI (F), PKGII (G), and p-VASP (F) were determined as described in Materials and Methods. Values were represented as mean ± SD from three independent experiments. ***p < 0.01, versus non-treatment. ##p < 0.01, ###p < 0.001, 8-Br-cGMP + H2O2 treatment versus H2O2 alone or 8-Br-cGMP + KT5823 + H2O2 treatment. %%%p < 0.001, SNAP treatment versus SNAP + KT5823 or SNAP + KT5823 + H2O2 treatment. &&&p < 0.001, 8-Br-cGMP treatment versus 8-Br-cGMP + KT5823 or -Br-cGMP + KT5823 + H2O2 treatment.
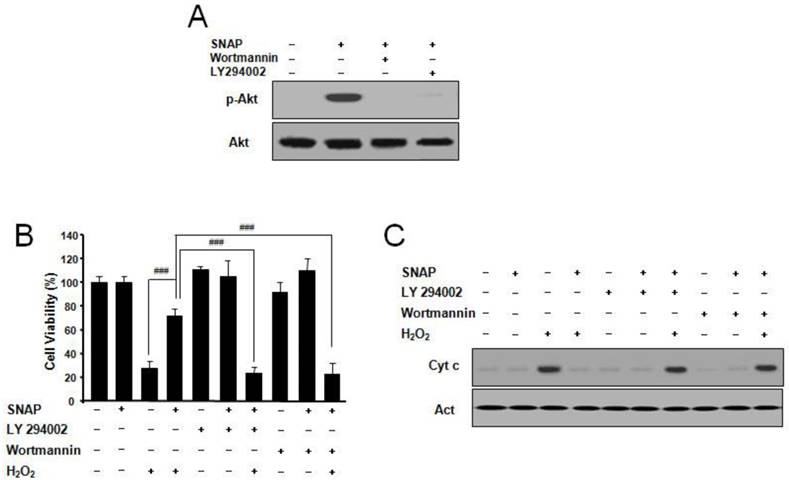
SNAP activates PI3K-dependent Akt activation
PI3K/Akt pathway suppresses apoptosis and promotes cell survival [11]. To explore whether NO may suppress apoptosis via the PI3K/Akt pathway, we examined the effects of NO on Akt activation. SNAP treatment resulted in the p-Akt increase and the PI3K inhibitor LY294002 or Wortmannin markedly abolished p-Akt activation by SNAP (Figure 5A). LY294002 or Wortmannin did not affect the cell viability of cells but inhibited the protective effect of SNAP on H2O2-treated cells (Figure 5B). Cytochrome c by Western blot was increased in treatment with SNAP under LY294002 or Wortmannin (Figure 5C). These results indicate that the PI3K-dependent Akt activation may play an important role in the anti-apoptotic effects of NO in SK-N-MC cells.
SNAP increases PI3K-dependent Akt activation and prevents H2O2-induced SK-N-MC cell apoptosis. Cells were treated with H2O2 (100 μM) and/or SNAP (100 μM) in the presence or absence of LY294002 (10 μM) or Wortmannin (20 nM) for 1 hr. Western blot analyses of phospho-Akt (Ser473) (A) and cytochrome c (C) were determined as described in Materials and Methods. Cell viability was measured by Cell Counting Kit-8 (B). Values were represented as mean ± SD from three independent experiments. ###P < 0.001, SNAP + H2O2 treatment versus H2O2 alone, SNAP + LY294002 + H2O2, or SNAP + Wortmannin + H2O2 treatment.
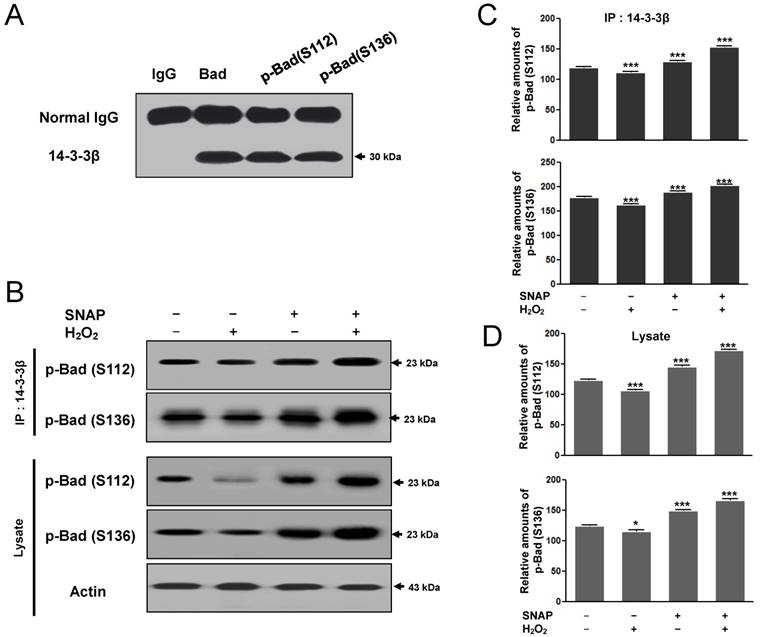
The binding of the 14-3-3β and p-Bad to prevent H2O2-induced SK-N-MC cell apoptosis
Phozsphorylation of Bad (Ser 112, 136) may result in the binding of the neuronal cell type protein 14-3-3 and alter the subcellular distribution of Bad to block pro-apoptotic death by Bad [15]. As the association between 14-3-3β and p-Bad inhibits the apoptotic pathway, we examined whether SNAP would affect the protein-protein interaction between 14-3-3β and p-Bad. First, the interaction between p-Bad (Ser 112, 136) and 14-3-3β was identified by immunoprecipitation. Ser 112 and Ser136 of p-Bad antibodies were carried out immunoprecipitation and detected with 14-3-3β after SDS-PAGE (Figure 6A). 14-3-3β binding to p-Bad (Ser 112, 136) was increased in the SNAP-treated SK-N-MC cells (Figure 6B, C) and p-Bad was increased in cell lysate (Figure 6B, D); thus, the anti-apoptotic effect of SNAP is closely associated with the phosphorylation of Akt and Bad in H2O2-induced apoptosis. This finding suggests that NO inhibits H2O2-induced apoptosis via an increased interaction between 14-3-3β and p-Bad.
The binding of the 14-3-3β and p-Bad to prevent H2O2-induced SK-N-MC cell apoptosis. Ser 112 and Ser136 of p-Bad antibodies were carried out immunoprecipitation and Western blot analysis was detected with 14-3-3β after SDS-PAGE (A). 14-3-3β was carried out immunoprecipitation and Western blot analysis was detected with Ser 112 and Ser136 of p-Bad antibodies after SDS-PAGE (B). The image intensities and optical densities of Western blots were quantified using ImageJ software and Prism Graph Pad v4.0 (C). These experiments were determined as described in Materials and Methods.
Discussion
Treatment of exogenous NO as NO donors (sodium nitroprusside, SNP; S-nitroso-N-acetylpenicillamine, SNAP; or diethylenetriamine/NO adduct, DETA/NO) has been studied to protect against the cell death in serum-deprived or 6-hydrodopamine-induced PC12 cells [10, 11] and in nerve growth factor-deprived sympathetic neurons [16]. These neuroprotective effects occurred only when the NO donors added at lower concentrations (below 100 μM), which may generate locally submicromolar concentration range from NO donors. NO at this concentration may activate sGC, bind to the heme of sGC and triggers formation of cGMP from GTP (17, 18). Therefore, NO elevate intracellular levels of cGMP, but it probably could not efficiently combine with superoxide anion to form the toxic peroxynitrite anion. At higher concentrations (above 100 μM) of these NO donors, NO may form peroxynitrite anion and cause toxic effects in some types of cells. Nevertheless, these studies including the present study suggest that the cGMP pathway in neurons may be an important protective mechanism [10, 11, 16-18].
The present study demonstrated that sGC inhibitor ODQ and a PKG inhibitor KT5823 were essential to mediated with cGMP/PKG-dependent apoptotic cell death in SK-N-MC cells. ODQ inhibited sGC-induced cGMP production and KT5823 prevented PKG-dependent effect (10, 11) (Figure 7). Furthermore, treatment of the cGMP analog 8‐Br‐cGMP as a cell‐permeable PKG activator prevented the apoptosis. Thus, the NO-stimulated cGMP/PKG-dependent pathway appears to play an important role in preventing activation of a proapoptotic pathway and results in promoting SK-N-MC cell survival or proliferation. cGMP/PKG pathway involves regulation of apoptosis and survival/proliferation. Antiapoptotic effect shows in different types of cells: neural cells including cortical neurons [19], PC12 cells, motor neurons, neurons of dorsal root ganglia, hippocampus and sympathetic nerves [10, 11, 20, 21], cerebellar granule neurons [22-25], and avian retinal neurons [26]; non-neuronal cells such as bone marrow stromal cells [27] and immortalized uterine epithelial cells [28]. Especially, Bonthius et al. [22-24] reported that the activation of the NO/cGMP/PKG pathway in cerebellar granule neurons inhibits alcohol-induced cell death. Yoneyama et al. [29] demonstrated for the first time the NO/cGMP/PKG signaling-mediated enhancement of cell proliferation in the neural stem/progenitor cells derived from the embryonic mouse hippocampus. The other studies have demonstrated that cGMP/PKG contains the proapoptotic actions of NO in vascular smooth muscle cells [30], cardiac myocytes [31, 32], a pancreatic beta cells such as HIT-T15 and RINm5F [33, 34], vascular endothelial cells [35], colon tumor cells [36], primary midbrain cultures [37], SH-SY5Y human neuroblastoma cells [38, 39], and ischemia/reperfusion injury model [40, 41]. Therefore, the cGMP/PKG pathway appears to contribute to NO-induced cell protection and toxicity.
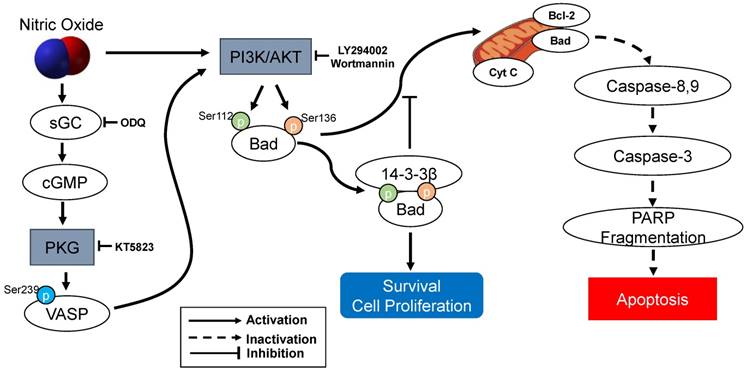
The protection of NO against H2O2-induced apoptosis in SK-N-MC cells via PKG/PI3K/Akt pathway. sGC, soluble guanylyl cyclase; PKG, cGMP-dependent protein kinase or protein kinase G; PI3K, phosphatidylinositol 3-kinase; Cyt c, cytochrome c; VASP, vasodilator-stimulated phosphoprotein; PARP, poly(ADP-ribose) polymerase.
Although there is little solid evidence for the direct or indirect links between the PI3K/Akt pathway and cGMP/PKG, Kawasaki et al. [42] indicate that exogenous NO promotes cell migration and angiogenesis through cGMP/PKG-dependent activation of the PI3K/Akt pathway in endothelial cells. This study may support that NO-induced activation of cGMP/PKG produces activation of the PI3K/Akt pathway as proliferation/survival signaling in our present experiment with SH-SY5Y cells, in the neural stem/progenitor cells [29], and in indirectly cerebellar granule neurons [22]. Especially, He et al. [43] clarify that the PI3K/Akt pathway contributes to the action of astragaloside IV, a major active ingredient of the traditional Chinese herb Radix Astragali, by activating NO generation leading to activation of the cGMP/PKG signaling in H9c2 cardiac cells.
As an important component of mitochondria-mediated apoptosis, the Bad is normally maintained in the cytosol in an active form, p-Bad and dimerized with 14-3-3 protein [15, 44-48]. These studies indicate that the association of Bad and 14-3-3 protein regulates cellular processes such as cell proliferation and survival. When apoptotic signals are triggered, Bad could be dephosphorylated through its release from 14-3-3 protein and interacts with some proapoptotic proteins on the outer membrane of mitochondria, initiating the opening of mitochondrial permeability transition pores and subsequent releasing cytochrome c into the cytosol [15, 45, 46]. In the present research, the protection of NO against H2O2-induced apoptosis in SK-N-MC cells mediated by cGMP through PKG activation and then activated PI3K-dependent Akt activation via an increased interaction between 14-3-3β and p-Bad. A flow Chart for the entire mechanistic pathway was shown in Figure 7.
Materials and Methods
Cell culture
The SK-N-MC cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GibcoBRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, GibcoBRL) at 37°C in 5% CO2, 95% air in a humidified cell incubator. Cells were treated with H2O2 (Sigma-Aldrich, St. Louis, MO, USA) for 24 hr, and S-nitroso-N-acetylpenicillamine (SNAP, Sigma-Aldrich) was added 10 hr prior to H2O2 treatment. Cells were treated with Ac-YVAD-cho and Ac-DEVD-cho (Alexis corporation, San Diego, CA, USA); 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, Santa Cruz Biotechnology, Santa Cruz, CA, USA); 8-Br-cGMP, KT5823, LY294002, and Wortmannin (Calbiochem, San Diego, CA, USA) in media.
Cell viability assay
Cell survival was quantified using a Cell Counting Kit-8 (Dojindo, Tokyo, Japan). SK-N-MC cells were cultured in 96-well plates (Corning Inc., Corning, NY, USA) at a density of 5 × 103 cells per well. The cells were cultured in the presence or absence SNAP. After 24 hr, the cells were washed and treated with the Cell Counting Kit-8 reagents, and then the plate was incubated in the dark for 4 hr, after which absorbance at 450 nm was measured using a plate reader (Molecular Device, Sunnyvale, CA, USA). Percent viability was calculated as the absorbance of the melatonin-treated sample/control absorbance × 100.
Nuclear staining
Cells were fixed in 4% paraformaldehyde buffered with 0.1 M phosphate (pH 7.3) for 30 min and then washed with phosphate buffered saline (PBS). The cells were permeabilized with 0.3% Triton X-100 for 20 min, washed with PBS, and then stained with 4,6-diamidino-2-phenylindole (DAPI, Santa Cruz, USA) for 10 min. Cells were observed by fluorescence microscopy (OlympusIX71, Japan) at a peak excitation wavelength of 340 nm.
Caspase-3 enzyme activity assay
Cell pellets were washed with ice-cold PBS and resuspended in 100 mM HEPES buffer, pH 7.4, containing protease inhibitors (5 mg/mL aprotinin, 5 mg/mL pepstatin, 10 mg/mL leupeptin and 0.5 mM phenylmethylsulfonyl fluoride). The cells were lysed via freeze-thawing, which was repeated three times, and the cytosolic fraction was obtained by centrifugation at 100,000 × g for 1 hr at 4°C. Activities of caspase-3 enzymes were determined by measuring the absorbance of the fraction at 405 nm, with 200 μM Ac-DEVD-pNA (Alexis Corporation) as a substrate for a caspase-3 enzyme.
DNA fragmentation analysis
Cell pellets were resuspended in 750 μL of lysis buffer (20 mM Tris-HCl, 10 mM EDTA, and 0.5% Triton X-100, pH 8.0) and left on ice for 45 min with occasional shaking. DNA was extracted with phenol/chloroform and precipitated with alcohol. The precipitate was dried and resuspended in 100 μL of 20 mM Tris-HCl, pH 8.0. After degradation of RNA with RNase (0.1 mg/mL) at 37°C for 1 hr, samples (15 μL) were electrophoresed on a 1.2% agarose gel in 450 mm Tris borate-EDTA buffer (TBE, pH 8.0) and photographed under UV light.
Western blot analysis
Cells were harvested, washed two times with ice-cold PBS, and then resuspended in 20 mM Tris-HCl buffer (pH 7.4) containing a protease inhibitor mixture (0.1 mM phenylmethylsulfonyl fluoride, 5 µg/mL aprotinin, 5 µg/mL pepstatin A, 1 µg/mL chymostatin) and phosphatase inhibitors (5 mM Na3VO4, 5 mM NaF). Whole cell lysates were prepared with a Dounce homogenizer using 20 strokes, followed by centrifugation of the lysates at 13,000 × g for 20 min at 4°C. Protein concentration was determined using the BCA assay (Sigma, St Louis, CA, USA). Proteins (40 µg) were separated by 12% SDS-PAGE, and then transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were hybridized with antibodies specific for caspase-3, -8, -9 (Cell Signaling Technology, Beverly, MA, USA), cytochrome C (Santa Cruz Biotechnology), PARP (Santa Cruz Biotechnology), guanylyl cyclase (Santa Cruz Biotechnology), p-VASP (Ser239) (Cell Signaling Technology), VASP (Cell Signaling Technology), PKGI (Cell Signaling Technology), PKGII (Santa Cruz Biotechnology), p-Akt and Akt (Cell Signaling Technology), p-Bad (Ser112, Ser136) and Bad (Cell Signaling Technology), and Actin (Assay Designs, Michigan, USA). Immunoreactive proteins were visualized by exposure of the membrane to X-ray film. The films were scanned and the band intensities and optical densities were quantified using ImageJ software (version 1.52, Wayne Rasband, NIH), after correction by background subtraction, and then normalized.
Intracellular cGMP measurement
cGMP levels were measured with enzyme immunoassay kits (Cayman Chemical, Ann Arbor, MI) and were expressed as picomoles of cGMP generated per milligram total protein of the cells.
Measurement of NO Production
Culture supernatant (100 μL) from each group was mixed with 100 μL of the Griess reagent (mixture in 1:1 ratio of 0.1% N-1-napthylethylenediamine dihydrochloride in water and 1% sulphanilamide in 5% phosphoric acid) and then incubated for 20 min. Absorbance at 540 nm was measured using a plate reader. Nitrite concentrations were determined from a standard curve prepared with known concentrations of NaNO2.
Immunoprecipitation
Immunoprecipitation was performed using an indirect technique involving magnetic Dynabead separation (Dynal ASA, Oslo, Norway) at 4°C. The SK-N-MC cells were homogenized by passing the sample through a syringe fitted with a 20-gauge needle eight times. The resulting homogenate was centrifuged at 10,000 × g at 4°C for 15 min. The protein concentration of the supernatant was determined with the BCA assay; 50 μL (∼3.4 × 107 beads) sheep anti-rabbit immunoglobulin G (IgG) M-280 Dynabeads (Dynal Biotech) were incubated at 4°C for 2-3 hr with gentle shaking on a bi-directional mixer with anti-14-3-3β (Santa Cruz Biotechnology). The supernatant was removed and the beads were washed thrice in 500 μL PBS with 0.1% protease-free BSA for 10 min. The beads were collected on a magnet, transferred to a 500 μL PBS mixture with protease inhibition for each cell lysate (500 μg protein), and were incubated in a bi-directional mixer overnight at 4°C. Following this step, the supernatant was removed, the beads were washed thrice in 500 μL PBS; 30 μL of sample loading buffer was added, and the mixture was boiled for 5 min. The samples were separated by 12% SDS-PAGE and electroblotted onto PVDF membranes. Subsequently, each membrane was hybridized with anti-14-3-3β (Santa Cruz Biotechnology), anti-p-Bad (Ser136) and anti-p-Bad (Ser112) (Cell Signaling Technology). The protein bands were viewed by exposure to x-ray film.
Statistical analysis
Significant differences were determined by ANOVA, followed by Tukey's test for multiple comparisons. The analysis was performed with Prism Graph Pad v4.0 (Graph Pad Software Inc., San Diego, CA, USA). Values are expressed as means ± SD. A P value of < 0.05 was considered statistically significant.
Abbreviations
NO: Nitric oxide; SNAP: S-nitroso-N-acetylpenicillamine; DAPI: 4',6-diamidino-2-phenylindole; ODQ: 1H-(1,2,4)-oxadiazole[4,3-a]quinoxalon-1-one; PARP: poly(ADP-ribose) polymerase; 8-Br-cGMP: 8-bromo-cGMP; PKG: cGMP-dependent protein kinase G; PI3K: phosphatidylinositol 3-kinase; NOS: NO syntheses; nNOS: neuronal NOS; eNOS: endothelial NOS; iNOS: inducible NOS; PD: Parkinson's disease; AD: Alzheimer's disease; HD: Huntinton's disease; ALS: amyotrophic lateral sclerosis; 6-OHDA: 6-hydroxydopamine; PBS: phosphate buffered saline; TBE: Tris borate-EDTA buffer; PVDF: polyvinylidene difluoride; SNP: sodium nitroprusside; DETA/NO: diethylenetriamine/NO adduct.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant of Korean government (MEST) (2017R1A2B2005031).
Author Contributions
Yeong-Min Yoo and Eui-Bae Jeung designed the experiments, analyzed the data, and wrote the paper. Article drafting: Eui-Man Jung and Changhwan Ahn had final approval of the version to be submitted. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26:1-14
2. Veal E, Day A. Hydrogen peroxide as a signaling molecule. Antioxidants & Redox Signaling. 2011;15:147-51
3. Hu J, Cui W, Ding W, Gu Y, Wang Z, Fan W. Globular Adiponectin attenuated H2O2-induced apoptosis in rat chondrocytes by inducing autophagy through the AMPK/ mTOR pathway. Cellular Physiology and Biochemistry. 2017;43:367-82
4. Guo X, Chen Y, Hong T, Chen X, Duan Y, Li C. et al. Induced pluripotent stem cell-derived conditional medium promotes Leydig cell anti-apoptosis and proliferation via autophagy and Wnt/β-catenin pathway. Journal of Cellular and Molecular Medicine. 2018;22:3614-26
5. Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology. 2000;149:43-50
6. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nature Reviews Molecular Cell Biology. 2007;8:722-8
7. López-Lázaro M. Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer Letters. 2007;252:1-8
8. Mohanakumar KP, Thomas B, Sharma SM, Muralikrishnan D, Chowdhury R, Chiueh CC. Nitric oxide: an antioxidant and neuroprotector. Annals of the New York Academy of Sciences. 2002;962:389-401
9. Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric oxide mechanism of protection in ischemia and reperfusion injury. Journal of Investigative Surgery. 2009;22:46-55
10. Kim YM, Chung HT, Kim SS, Han JA, Yoo YM, Kim KM. et al. Nitric oxide protects PC12 cells from serum deprivation-induced apoptosis by cGMP-dependent inhibition of caspase signaling. Journal of Neuroscience. 1999;19:6740-7
11. Ha KS, Kim KM, Kwon YG, Bai SK, Nam WD, Yoo YM. et al. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB Journal. 2003;17:1036-47
12. Culmsee C, Gerling N, Landshamer S, Rickerts B, Duchstein HJ, Umezawa K. et al. Nitric oxide donors induce neurotrophin-like survival signaling and protect neurons against apoptosis. Molecular Pharmacology. 2005;68:1006-17
13. Andoh T, Chiueh CC, Chock PB. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. Journal of Biological Chemistry. 2003;278:885-90
14. Ciani E, Guidi S, Della Valle G, Perini G, Bartesaghi R, Contestabile A. Nitric oxide protects neuroblastoma cells from apoptosis induced by serum deprivation through cAMP-response element-binding protein (CREB) activation. Journal of Biological Chemistry. 2002;277:49896-902
15. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231-41
16. Farinelli SE, Park DS, Greene LA. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. Journal of Neuroscience. 1996;16:2325-34
17. Zhao Y, Brandish PE, Ballou DP, Marletta MA. A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14753-58
18. Bellamy TC, Wood J, Garthwaite J. On the activation of soluble guanylyl cyclase by nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:507-10
19. Fernández-Tomé P, Lizasoain I, Leza JC, Lorenzo P, Moro MA. Neuroprotective effects of DETA-NONOate, a nitric oxide donor, on hydrogen peroxide-induced neurotoxicity in cortical neurones. Neuropharmacology. 1999;38:1307-15
20. Fiscus RR. Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neurosignals. 2002;11:175-90
21. Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacological Reviews. 2010;62:525-63
22. Bonthius DJ, Karacay B, Dai D, Hutton A, Pantazis NJ. The NO-cGMP-PKG pathway plays an essential role in the acquisition of ethanol resistance by cerebellar granule neurons. Neurotoxicology and Teratology. 2004;26:47-57
23. Bonthius DJ, Bonthius NE, Li S, Karacay B. The protective effect of neuronal nitric oxide synthase (nNOS) against alcohol toxicity depends upon the NO-cGMP-PKG pathway and NF-kappaB. Neurotoxicology. 2008;29:1080-91
24. Bonthius DJ, Luong T, Bonthius NE, Hostager BS, Karacay B. Nitric oxide utilizes NF-kappaB to signal its neuroprotective effect against alcohol toxicity. Neuropharmacology. 2009;56:716-31
25. Kouzoukas DE, Bhalla RC, Pantazis NJ. Activation of cyclic GMP-dependent protein kinase blocks alcohol-mediated cell death and calcium disruption in cerebellar granule neurons. Neuroscience Letters. 2018;676:108-112
26. Mejía-García TA, Paes-de-Carvalho R. Nitric oxide regulates cell survival in purified cultures of avian retinal neurons: involvement of multiple transduction pathways. Journal of Neurochemistry. 2007;100:382-94
27. Wong JC, Fiscus RR. Essential roles of the nitric oxide (NO)/cGMP/protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. Journal of Cellular Biochemistry. 2011;112:829-39
28. Chan SL, Fiscus RR. Guanylyl cyclase inhibitors NS2028 and ODQ and protein kinase G (PKG) inhibitor KT5823 trigger apoptotic DNA fragmentation in immortalized uterine epithelial cells: anti-apoptotic effects of basal cGMP/PKG. Molecular Human Reproduction. 2003;9:775-83
29. Yoneyama M, Kawada K, Shiba T, Ogita K. Endogenous nitric oxide generation linked to ryanodine receptors activates cyclic GMP/protein kinase G pathway for cell proliferation of neural stem/progenitor cells derived from embryonic hippocampus. Journal of Pharmacological Sciences. 2011;115:182-95
30. Pollman MJ, Yamada T, Horiuchi M, Gibbons GH. Vasoactive substances regulate vascular smooth muscle cell apoptosis. Countervailing influences of nitric oxide and angiotensin II. Circulation Research. 1996;79:748-56
31. Wollert KC, Fiedler B, Gambaryan S, Smolenski A, Heineke J, Butt E. et al. Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension. 2002;39:87-92
32. Wu CF, Bishopric NH, Pratt RE. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. Journal of Biological Chemistry. 1997;272:14860-6
33. Loweth AC, Williams GT, Scarpello JH, Morgan NG. Evidence for the involvement of cGMP and protein kinase G in nitric oxideinduced apoptosis in the pancreatic B-cell line, HIT-T15. FEBS Letters. 1997;400:285-8
34. Tejedo JR, Ramírez R, Cahuana GM, Rincón P, Sobrino F, Bedoya FJ. Evidence for involvement of c-Src in the anti-apoptotic action of nitric oxide in serum-deprived RINm5F cells. Cellular Signalling. 2001;13:809-17
35. Suenobu N, Shichiri M, Iwashina M, Marumo F, Hirata Y. Natriuretic peptides and nitric oxide induce endothelial apoptosis via a cGMP-dependent mechanism. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19:140-6
36. Liu L, Li H, Underwood T, Lloyd M, David M, Sperl G. et al. Cyclic GMP-dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. Journal of Pharmacology and Experimental Therapeutics. 2001;299:583-92
37. Canals S, Casarejos MJ, de Bernardo S, Rodríguez-Martín E, Mena MA. Glutathione depletion switches nitric oxide neurotrophic effects to cell death in midbrain cultures: implications for Parkinson's disease. Journal of Neurochemistry. 2001;79:1183-95
38. Canzoniero LM, Adornetto A, Secondo A, Magi S, Dell'aversano C. et al. Involvement of the nitric oxide/protein kinase G pathway in polychlorinated biphenyl-induced cell death in SH-SY 5Y neuroblastoma cells. Journal of Neuroscience Research. 2006;84:692-7
39. Nashida T, Takuma K, Fukuda S, Kawasaki T, Takahashi T, Baba A. et al. The specific Na+/Ca2+ exchange inhibitor SEA0400 prevents nitric oxide-induced cytotoxicity in SH-SY5Y cells. Neurochemistry International. 2011;59:51-8
40. Kuo KK, Wu BN, Chiu EY, Tseng CJ, Yeh JL, Liu CP. et al. NO donor KMUP-1 improves hepatic ischemia-reperfusion and hypoxic cell injury by inhibiting oxidative stress and pro-inflammatory signaling. International Journal of Immunopathology and Pharmacology. 2013;26:93-106
41. Salloum FN, Das A, Samidurai A, Hoke NN, Chau VQ, Ockaili RA. et al. Cinaciguat, a novel activator of soluble guanylate cyclase, protects against ischemia/reperfusion injury: role of hydrogen sulfide. American Journal of Physiology Heart and Circulatory Physiology. 2012;302:H1347-54
42. Kawasaki K, Smith RS. Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Molecular and Cellular Biology. 2003;23:5726-37
43. He Y, Xi J, Zheng H, Zhang Y, Jin Y, Xu Z. Astragaloside IV inhibits oxidative stress-induced mitochondrial permeability transition pore opening by inactivating GSK-3β via nitric oxide in H9c2 cardiac cells. Oxidative Medicine and Cellular Longevity. 2012;2012:935738
44. Chen XQ, Fung YW, Yu AC. Association of 14-3-3gamma and phosphorylated bad attenuates injury in ischemic astrocytes. Journal of Cerebral Blood Flow & Metabolism. 2005;25:338-47
45. Fan J, Xu G, Nagel DJ, Hua Z, Zhang N, Yin G. A model of ischemia and reperfusion increases JNK activity, inhibits the association of BAD and 14-3-3, and induces apoptosis of rabbit spinal neurocytes. Neuroscience Letters. 2010;473:196-201
46. Fan J, Zhang N, Yin G, Zhang Z, Cheng G, Qian W. et al. Edaravone protects cortical neurons from apoptosis by inhibiting the translocation of BAX and Increasing the interaction between 14-3-3 and p-BAD. International Journal of Neuroscience. 2012;122:665-674
47. Fong WH, Tsai HD, Chen YC, Wu JS, Lin TN. Anti-apoptotic actions of PPAR-gamma against ischemic stroke. Molecular Neurobiology. 2010;41:180-6
48. Oh JE, Jang DH, Kim H, Kang HK, Chung CP, Park WH, Min BM. alpha3beta1 integrin promotes cell survival via multiple interactions between 14-3-3 isoforms and proapoptotic proteins. Experimental Cell Research. 2009;315:3187-200
Author contact
![]() Corresponding author: Eui-Bae Jeung, PhD. Laboratory of Veterinary Biochemistry and Molecular Biology, College of Veterinary Medicine, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: ebjeungac.kr
Corresponding author: Eui-Bae Jeung, PhD. Laboratory of Veterinary Biochemistry and Molecular Biology, College of Veterinary Medicine, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: ebjeungac.kr