Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(1):138-147. doi:10.7150/ijbs.28873 This issue Cite
Research Paper
Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats
Yuchen Shi, Shaoping Wang, Hongyu Peng, Yuan Lv, Wenzheng Li, Shujuan Cheng, Jinghua Liu ![]()
Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, and Beijing Institute of Heart, Lung and Blood Vessel Diseases, Beijing, China
Received 2018-7-30; Accepted 2018-10-30; Published 2019-1-6
Abstract

Fibroblast growth factor 21 (FGF21), a hormone with multiple metabolic properties, has proven to be pleiotropic biological effects and may play pivotal role in numerous cardiovascular and metabolic diseases in the future. Vascular calcification (VC) is a concomitant pathological process of various cardiovascular and metabolic diseases. However, the effects of FGF21 on VC remain unclear. Therefore, in this research, we aimed to explore the roles and mechanisms of FGF21 in VC induced by vitamin D3 plus nicotine (VDN) treatment rats. After 28 days VDN treatment, the calcium overload was confirmed by blood pressure, ultrasound imaging, calcium content, ALP activity and aortic pathological characteristics. In terms of FGF21, exogenous FGF21 can ameliorate the elevation of blood pressure, aortic calcification and related injury in VC rats. To investigate the mechanisms of FGF21 on VC, the endoplasmic reticulum stress (ERS) mediated apoptosis pathways were tested. As a method to detect apoptosis, the increased positive TUNEL staining cells were alleviated by FGF21 treatment. Furthermore, exogenous FGF21 can suppress the increased ERS chaperone, GRP78, in the calcified aortas. In the three pathways of ERS mediated apoptosis, we found CHOP pathway and caspase-12 pathway were involved in the treatment of FGF21, but not p-JNK/JNK pathway. Our study proved for the first time that FGF21 can inhibit the progress of VC by alleviating ERS mediated apoptosis in rats. FGF21 might be a new target for preventing and treating VC.
Keywords: fibroblast growth factor 21, vascular calcification, endoplasmic reticulum stress, apoptosis
Introduction
Vascular calcification (VC) is a concomitant pathological process of various cardiovascular diseases[1]. The deposition of calcium-phosphorus contributed to the structural damage, fibrosis proliferation and the formation of calcium nodules in the middle layer of the artery, which can result in the thickening of arterial walls and the reducing of vascular compliance[2]. In contrary to the traditional view, VC is now regarded as an automatic, regulatable and revisable procedure controlled by multiple factors[3]. Particularly, previous researchers have proven that apoptosis of vascular smooth muscle cells (VSMCs) exist extensively in calcified aorta[4]. In addition, several pathological studies have found that apoptotic bodies released from VSMCs can initiate VC[5]. Thus, inhibiting apoptosis of VSMCs may alleviate VC.
Endoplasmic reticulum stress (ERS), a key reason to mediate apoptosis, has a significant effect on various metabolic disorders related diseases such as diabetes[6], atherosclerosis[7] and chronic kidney disease[8], all of which are related to VC. Furthermore, in the procedure of VC, calcium overloaded in VSMCs may disrupt the endoplasmic reticulum homeostasis and result in the unfolded protein response, which can induce the ERS and apoptosis[9]. Stated thus, to restrain ERS mediated apoptosis may attenuate the development of VC.
Recently, many studies have revealed that various biological factors such as cardiovascular peptides[10], cytokines and hormone[11] have key regulatory roles in the progression of VC. FGF21 is an endocrine hormone with multiple biological properties[12, 13]. Unlike other paracrine/autocrine FGFs, FGF21 is released from metabolic organs/ tissues into blood and mediates its pleiotropic effects as an extracellular protein by activating its receptor FGFR1 and its cofactor β-Klotho[14, 15]. Increasing evidences point out that FGF21 plays a protective role on many cardiovascular events or metabolism associated diseases such as coronary atherosclerotic heart disease[16, 17], obesity[18, 19] and diabetes[20, 21]. Furthermore, our previous study has proven that FGF21 could ameliorate atherosclerosis via inhibiting ERS mediated apoptosis[22]. However, the biological functions of FGF21 in VC by modulating ERS mediated apoptosis are unknown yet. The thorough investigation into the mechanisms of the ameliorative effect of FGF21 on VC may contribute to the transformation from bench to bedside and be useful to discover a new strategy for treating VC.
Vitamin D3 plus nicotine (VDN) induced rat model, first established by Niederhoffer et al, is a classical rat model of VC[23]. Therefore, we used the VDN rat model and administrated FGF21 to investigate the mechanisms of FGF21 on VC. Our results first reveal the evidences that FGF21 can protect VDN induced VC by inhibiting the ERS mediated apoptosis in vivo.
Materials and methods
Materials and reagents
FGF21 was purchased from Phoenix Pharmaceuticals (Belmont, CA, USA). Alzet Mini-Osmotic Pumps (model 2004) were purchased from Durect Corp. (Cupertino, CA, USA). Vitamin D3, nicotine, Alizarin-red S and Hoechst 33342 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Calcium assay kit was purchased from Zhongsheng Biosino Bio-technology and Science INC (Beijing, China). Alkaline phosphatase (ALP) kit was purchased from Jiancheng Biological Corp. (Nanjing, Jiangsu, China). Antibodies against GRP78, CHOP and caspase-12 were purchased from Abcam (Cambridge, UK). Antibodies against p-JNK and JNK were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against GAPDH and all secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The kit for terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was purchased from Roche Applied Science (Indianapolis, IN, USA). All other reagents were of analytical grade.
Rats model of VC and experimental groups
Male Sprague-Dawley rats (180-200g) were purchased from the Laboratory Animal Center of Peking University (Beijing, China). All animal care and experimental protocols were performed in according with the Animal Management Rule of the Ministry of Health (Document No. 55, 2001), People's Republic of China. Rats were randomly divided into three groups (n=13): (1) control group (Con group), (2) VDN group and (3) FGF21 treatment VDN group (FGF21 group). The method of establishing VDN model was described as previously[23]. Briefly, the VDN and FGF21 group rats were administrated with nicotine (25 mg/kg in 5 ml peanut oil) intragastrically and injected intramuscularly with vitamin D3 (3×105 IU/kg) at 9 a.m. on day 1. And the nicotine was administrated again at 5 p.m. on the same day. The control group rats received the saline injection and peanut oil oral gavage only. For FGF21 treatment, all rats were anesthetized by 1.5% pentobarbital sodium (40 mg/kg), then an osmotic mini pump was implanted subcutaneously. The FGF21 group pumps were infused with FGF21 (70 μg/kg/d), together with the infusion of saline for control and VDN group [24].
Ultrasound imaging
Before the examination, the rats were anesthetized by 1.5% pentobarbital sodium (40 mg/kg). Then the thoracoabdominal hair was shaved carefully, and the ultrasound transmission gel was smeared on the naked skin. The ultrasonographic images were obtained by the imaging system (Vevo770TM Visual Sonics Inc., Toronto, Canada) in B mode. All rats survived in the imaging process.
Blood pressure measurement and specimens collection
At the end of experiment, rats were anesthetized with ethylcarbamate (10 mg/kg, i.p.). Intubate a polyethylene catheter (PE-50) filled with heparin through the right carotid artery for 30 min, then the rat aortic blood pressure was measured by a pressure transducer of PowerLab instrument (model BL-420F) (TaiMeng, Chengdu, Sichuan, China). Finally, the blood samples were drawn and mixed with 500 KIU/ml aprotinin and 1 mg/ml ethylene diamine tetraacetic acid-Na2. Plasma was obtained by blood samples centrifuging at 3000 g for 15 min at 4℃ and stored at -70℃. The intact aorta was removed to 4℃ PBS and all samples of tissues were stored at -70℃ until use[24].
Hematoxylin and eosin (HE) staining
After fixed in formalin overnight, the aortic tissue was embedded in paraffin and sectioned for HE staining. Briefly, the slides were placed in the hematoxylin staining solution first, and then were washed and treated with 1% acidic alcohol. Afterwards, the slides were stained by eosin. Finally, the slides were washed, dehydrated and treated by xylene before microscopy. The aortic slides were evaluated by use of Image Pro Plus 6.0 system.
Alizarin-red S staining
After dehydrated and rinsed rapidly in distilled water, the aortic slides were placed in the Alizarin-red S staining solution for 30 min at room temperature. When a red-orange color appeared, removed all unbound stain from the slides, then the slides were photographed by use of a microscope, Olympus BX 50 (Olympus Optical, Tokyo, Japan).
Calcium content in aorta
The calcium content in aortic tissue was extracted and measured by a calcium assay kit according to the manufacturer instructions. Briefly, 10 mg of aortas were separated and dried at 55℃, and the dry weights were recorded. Then the dried tissue was nitrified by nitric acid. After removing the nitric acid by heating, the calcium was dissolved in the solution containing 27 NM KCl and 27 μM LaCl3. Finally, the calcium content of the aorta was measured and standardized by the aortic tissues dry weight.
ALP activity in aorta
The ALP activity in aortic tissue was measured by an ALP assay kit followed its instructions. Briefly, the aortic tissue homogenate was mixed with a reaction mixture (stock substration solution: alkaline buffer =1:1) at a ratio of 1:1, respectively. Then the mixtures were incubated at 37℃ for 15 min. After incubation, 1.5 ml of the developer was added to each mixture, and the absorbance of each well at 520 nm was recorded. The activity of ALP in each specimen was calculated from the standard curve. Besides, for protein quantification, the aortic tissue was homogenized in ice-cold buffer (20 mmol/L MgCl2, 20 mmol/L HEPES and 0.2% NP-40, pH 7.4) and centrifuged at 8000 g at 4℃ for 10 min. The protein content in supernatant was determined by the Bradford method [25]. One unit of ALP activity was defined as 1 g protein producing 1 mg phenol for 15 min.
Western blot analysis
The rat tissues were homogenized with a lysis buffer which contains 0.1 mol/L NaCl, 500 KIU/mL aprotinin, 0.01 mol/L Tris-HCl (pH 7.5) and 1 mmol/L ethylene diamine tetraacetic acid (EDTA). Equal amounts of protein sample were loaded at 10% or 12% SDS gels and then transferred to a nitrocellulose membrane. Afterwards, incubated the membrane with 5% non-fat dry milk for 1 h to block the non-specific proteins. Then the membranes were incubated with primary antibodies, anti-GAPDH (1:1000), anti-GRP78 (1:1000), anti-CHOP (1:200), anti-p-JNK (1:1000), anti-JNK (1:1000) and anti-Caspases-12 (1:500) overnight at 4℃, and with secondary antibody (HRP-conjugated anti-rabbit or anti-goat IgG) at room temperature for 1 h. The protein expression was analyzed by NIH image software and normalized to that of GAPDH. All experiments were repeated at least 3 times.
TUNEL staining
Apoptosis of aortas was evaluated by TUNEL staining according to the TUNEL TMR red kit. Briefly, the ascending aorta was separated and fixed in formalin overnight, then embedded in paraffin and cut at 40 μM intervals along the aortic tissue. Then the sections were permeabilized and stained with TMR Red. The nuclei were counterstained with Hoechst 33342. Finally, the fluorescence signal was monitored under a fluorescence inverted microscope (Leica Imaging Systems, Cambridge, UK).
Statistical analysis
The data were calculated as the mean ± SD form at least three independent experiments and analyzed by a one-way ANOVA. The difference was accepted at P < 0.05. A P < 0.05 was considered statistically significant, and a P < 0.01 was considered very statistically significant.
Results
FGF21 improved the general characteristics of VDN rats
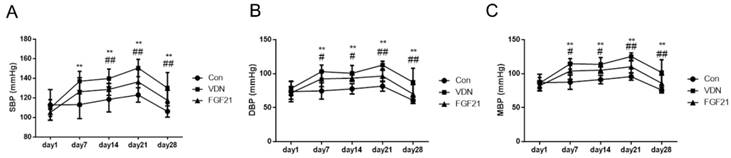
First, the effects of FGF21 on the blood pressure of VC rats were evaluated. Under VDN treatment, as compared to the normal group, the systolic blood pressure (SBP), diastolic blood pressure (DBP) and mean blood pressure (MBP) of model group began to increase at the day 7 by 21.0% (P< 0.01), 38.1% (P< 0.01) and 30.9% (P< 0.01), respectively. At the day 21, the blood pressures got to the top with 22.3% (P< 0.01), 38.2% (P< 0.01) and 31.1% (P< 0.01), respectively. At the end of our study, SBP, DBP and MBP of model group were dramatically increased by 22.5% (P< 0.01), 43.9% (P< 0.01) and 33.9% (P< 0.01), respectively. And compared with the model group, FGF21-treated group shows the mitigation of VDN-induced hemodynamic disorders at the beginning of day 14 by 7.9% (P< 0.01), 7.2% (P< 0.05) and 7.4% (P< 0.05), respectively. And at the end of the study, the decreasing rate of SBP, DBP and MBP were 9.7% (P< 0.01), 19.9% (P< 0.01) and 15.5% (P< 0.01) compared to the VDN group, respectively (Figure 1A, B and C).
General hemodynamic tendency of rats in vitamin D3 plus nicotine (VDN) model. (A) Hemodynamic function of systolic blood pressure (SBP) tendency in vascular calcification (VC) rats. (B) Diastolic blood pressure (DBP) tendency in VDN induced VC rats. (C) Mean blood pressure (MBP) tendency in VDN induced VC rats (n=11, respectively). **P < 0.01 compared with the normal control group. #P < 0.05 compared with the VDN group. ##P < 0.01 compared with the VDN group.
FGF21 restored VDN-induced pathological injury of the aorta in VC rats
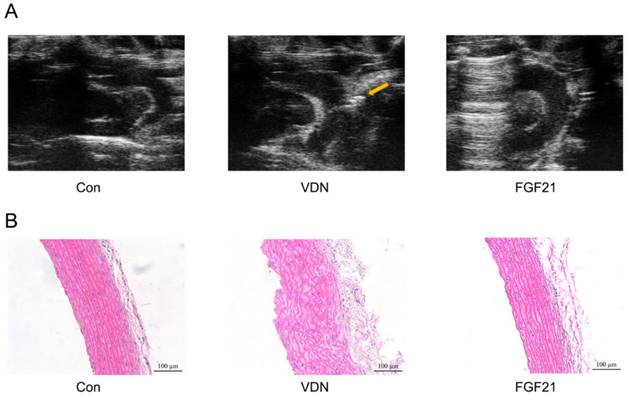
Next, the influence of FGF21 on the structure of calcified aortas was identified. As shown in Figure 2A, the ultrasonography showed the formation of calcified plaques and aorta wall thicken distinctly, resulting in serious VC by VDN treatment. In terms of the microscopic pathological changes, the histopathologic examination also showed disordered elastic fibers and thickened vessel walls (Figure 2B) in the aortas. And compared with the VDN group, FGF21 treatment significantly restored the pathological disorder caused by VDN through relieving the irregular proliferation of fibrous tissue and formation of calcified nodules in the middle layer of the aorta (Figure 2A and B).
Morphological study of calcified aortas by echocardiogram and Hematoxylin and eosin (HE) staining. (A) Images show short-axis views of ascending aorta, including the brachiocephalic artery branch, aortic arch and proximal descending aorta. Arrow: Calcified plaque. (B) HE staining of disordered elastic fibers in aortas of VC rats (magnification 200×).
FGF21 ameliorated VC in VDN induced rats
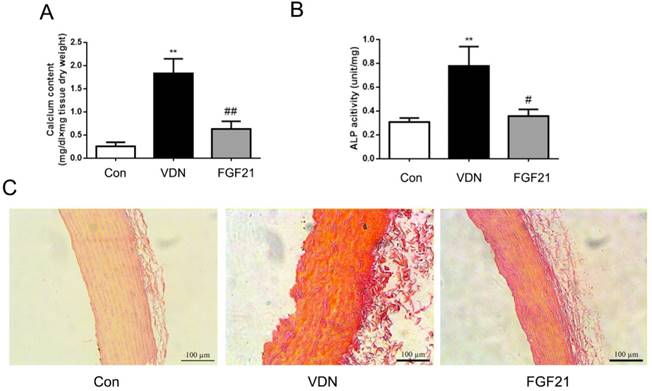
The roles of FGF21 in the mineralization of aorta were further investigated. Compared with the normal group, the model group showed stronger calcium salt deposition in aortas that the aortic calcium content was significantly increased by 7.03-fold (P <0.01, Figure 3A). Meanwhile, the ALP activity, a key inducer of mineralization in tissue calcification, was also increased by 2.51-fold in aortas (P < 0.01, Figure 3B). In addition, the Alizarin-red S staining also showed widespread calcification in the middle layer of the aorta (Figure 3C). As compared with the VC alone group, FGF21 group showed lower calcium deposition by Alizarin-red S staining (Figure 3C) as well as markedly decreased the aortic calcium content, together with aortic ALP activity by 65.77% (P <0.01) and 54.18% (P <0.05) respectively (Figure 3A and B).
FGF21 ameliorated VDN-induced VC in rats. (A) Effect of exogenous FGF21 on the aortic calcium content in VC rats (n=7). (B) Effect of exogenous FGF21 on aortic ALP activity in VC rats (n=7). (C) Alizarin-red S staining of vascular calcium deposition in aortas of VC rats (magnification 200×). **P < 0.01 compared with the control group. #P < 0.05 compared with the VDN group. ##P < 0.01 compared with the VDN group.
FGF21 inhibited apoptosis in the VSMCs
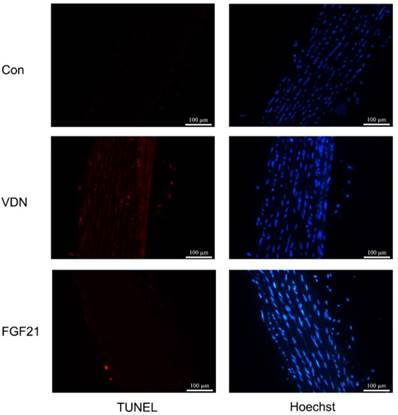
Since apoptosis of VSMCs has been proven to be a major cause of VC, the aortic tissues were drawn for TUNEL staining to test whether FGF21 shows effects on the apoptosis of VSMCs. Compared to the control group, the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells of aortic tissues could be observed dispersedly in the VDN group, which represented an extensive apoptosis in VSMCs. Whereas, compared to the model group, FGF21 treatment for 28 days significantly decreased the amount of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells in the aortic media (Figure 4). This result indicated that the effects of FGF21 on VC may be resulted from its inhibiting effect on apoptosis.
The apoptosis level of vascular smooth muscle cells (VSMCs) in rat aortas. Representative immunofluorescence staining of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL, TMR Red) in calcified aortas. Blue indicates VSMCs nuclei by Hoechst staining (magnification 200×).
FGF21 down-regulated the ERS chaperones in calcified aortas
There have been studies proving that the apoptosis of VSMC in VC is caused by ERS. To assess whether FGF21 has function in the ERS pathways, we further detected the expression of GRP78, a main ERS chaperone in calcified aortas. Western blot analysis showed that the GRP78 level was significantly elevated by 108.8% (P < 0.01) in the model group as compared with controls (Figure 5A). And with the treatment of FGF21, the GRP78 protein level was down-regulated by 31.9% (P < 0.05, Figure 5A) which indicates that FGF21 may inhibit apoptosis by down-regulating the ERS pathways.
The levels of endoplasmic reticulum stress (ERS) marker and ERS mediated apoptosis pathways expression in rat aortas. (A) Western blot analysis of GRP78 in VC rats. Western blot analysis of CHOP (B), p-JNK/JNK (C) and caspase-12 (D) in VC rats. GAPDH was a control for protein loading. Results are from one representative experiment of three, and densitometric analysis of autoradiograms is shown as a ratio to GAPDH. **P < 0.01 compared with the control group. #P < 0.05 compared with the VDN group.
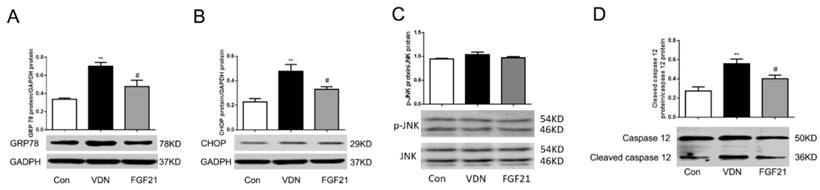
FGF21 decreased the ERS mediated apoptosis pathways in VDN induced VC rats
As is well known, there are three major signaling pathways in the ERS, and CHOP, JNK and caspase-12 are three cardinal biomolecules in the branches of ERS mediated apoptosis. As the results showed above, ERS and apoptosis joined the VDN induced VC and FGF21 could restrain the ERS and apoptosis respectively. However, which signaling pathway can FGF21 play a role in is unclear. Therefore, we examined these three key biomarkers respectively by Western blot analysis furtherly. As shown in Figure 5B and 5D, in VDN group, the CHOP and cleaved caspase-12 significantly increased by 109.3% (P< 0.01) and 103.1% (P< 0.01), respectively, compared with the controls. Moreover, FGF21 significantly suppressed the CHOP and cleaved caspase-12 activation by 31.0% (P< 0.05) and 27.9% (P< 0.05), respectively. However, the level of JNK phosphorylation seemed not involved in the ERS caused by VDN (Figure 5C). These results demonstrated that FGF21 attenuated VC through suppressing ERS mediated apoptosis by inhibiting the activation of CHOP and caspase-12 pathways, but not by p-JNK/JNK.
Discussion
FGF21, a peculiar member of the FGFs superfamily, has multiple metabolic effects and anticipated therapeutic applications on various diseases such as atherosclerosis[26], diabetes[27], and chronic kidney disease[28]. In our study, FGF21 treatment can ameliorate the elevation of blood pressure induced by VC in clinical manifestation. Going deep into the pathology, exogenous FGF21 can alleviate the histological disorder, calcium content and ALP activity of the aortas. For the mechanism research, FGF21 can reduce the apoptosis mediated by ERS in the aortic tissue of VDN rats. These results suggest that FGF21 may mitigate VC by inhibiting ERS mediated apoptosis. Thus, FGF21 may be a new target to prevent and alleviate VC.
VC is a crucial risk factor of many cardiovascular diseases[29]. The increased vessel wall stiffness and vascular wall thicken resulted from VC leads to increased pulse pressure and pulse wave velocity, which subsequently brings on various cardio-cerebrovascular diseases[2]. Several mechanisms have been proven involved in the progress of VC, including apoptosis, loss of inhibition and osteogenic differentiation[30].
In our study, a rat model of VC was established using the VDN method, to research the effects of FGF21 on VC and reveal the mechanism behind it. VDN induced rat model, first established by Niederhoffer et al in 1997[23], is a classical rat model of VC which is stable and dependable, widely used to investigate VC. Hypervitaminosis D increases the deposition of calcium on the elastic fibers of arteries. And nicotine amplifies these effects of Vitamin D in arteries. VDN-induced calcium overload on the arteries has similarities with the calcification in human athero- and arteriosclerosis[31]. In our VDN model, the calcium content and ALP activity, key factors and markers for active osteochondrogenesis, were dramatically higher than that of the normal group. HE staining and Alizarin-red S staining observed that the aortic elastic plate of the VDN group was significantly thicker and more disordered, with breakage of the elastic fibers and widespread calcification compared with the control group. The increased SBP, MBP and DBP we found in VDN rats might result from the increased arterial stiffness, which consisted with the alterations in human hemodynamic characteristics of calcified arteries. All these data agree with those of prior researches[23] which indicated that our construction of the rat VC model was successful and stable.
As a special member of FGFs, FGF21 has a major role in maintaining cardiovascular homeostasis[12, 32]. In our research, compared with the model group, rats treated with FGF21 showed alleviated aortic calcification induced by VDN, as shown by decreased SBP, DBP and MBP, ameliorated aortic calcium content and ALP activity, and regular elastic fibers and less calcification in the vessel wall. These results suggested that FGF21 can inhibit VC in rats.
Apoptosis is considered as a forerunner to vascular barrier destruction, which is a critical mechanism serving to VC[33]. During the procedure of VC, both apoptotic bodies induced by exposing to calcium and phosphate deposition and matrix vesicles derived from VSMCs of vessels may lead to concentrate the local calcium-phosphate, which will provide an appropriate microenvironment for nucleation[34]. As well-known, the transformation of VSMCs phenotype into osteoblast-like phenotype is involved in the pathogenesis of VC. Apoptosis is involved in the process of VSMCs phenotypic transformation[35]. Apoptosis occurred prior to the onset of calcification and that VSMC “blebs” or apoptotic bodies could induce calcium concentration in a crystallized form. Apoptotic bodies derived from VSMCs have similarities with matrix vesicles. Therefore, apoptosis is a key initiating process for VC and that, once an original nucleation site is formed, calcium-phosphate crystal growth can occur[36]. Based on this theory, attenuating the osteoblast-like phenotypic transition of VSMCs through inhibition of apoptosis might be an effective treatment for VC. In addition, accumulating data suggests that apoptosis mediated by ERS is close related to VC because the calcium accumulation in the cytoplasm may damage endoplasmic reticulum homeostasis[37]. Endoplasmic reticulum is the primary site for Ca2+ storage. Any stimulus that interferes with calcium homeostasis may lead to endoplasmic reticulum function disturbance, accumulating unfolded or misfolded protein in the endoplasmic reticulum, namely ERS[38]. Therefore, inhibition of ERS induced VSMC apoptosis may alleviate the development of VC.
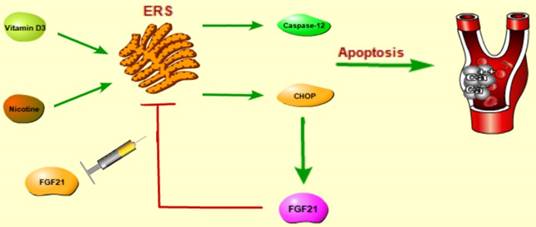
Schematic of pathways for FGF21 signal transduction. FGF21 attenuated VC through suppressing ERS mediated apoptosis by inhibiting the activation of CHOP and caspase-12 pathways, but not by p-JNK/JNK.
Consistent with previous research[39], in the aorta of VDN challenged rats, positive TUNEL staining confirmed the apoptotic VSMCs in the aortas. Furthermore, the Western blot analysis showed a significant increase of ERS markers, GRP78, in the VDN model group. GRP78, a defined biomolecular chaperone, is a critical regulator for the unfolded protein response network by binding to endoplasmic reticulum sensors and inhibiting their activation[40]. The up-regulation of the ERS landmark and the increased positive apoptotic cells suggested an ERS mediated apoptosis response during VC.
The initiation of ERS mediated apoptosis may trigger by three signaling pathways: CHOP, caspase-12 and JNK pathways[41]. Therefore, we researched the possible signaling pathways to ERS mediated apoptosis in VC furtherly. CHOP, one of the ERS response hallmarks, can directly regulate the expression of numerous pro-apoptotic proteins, which leads to enhanced oxidative damage and thus apoptosis[42]. In our VC rats, CHOP was up-regulated in the aortas, which demonstrated that CHOP pathway mediated apoptosis procedure was closely involved in VC. Caspases, a superfamily of cysteine proteases, are major mediators of programmed cell death, namely apoptosis. Among these superfamily, caspase-12 is activated only by ERS[43]. When activated to the cleaved form, caspase-12 can activate caspase-9 directly, which in turn activates caspase-3, and finally leads to apoptosis[43]. In our research, caspase-12 was cleaved, namely being activated, which suggested that caspase-12 pathway mediated apoptosis was also involved in VC. Besides, the JNK pathway mediates another apoptotic pathway[44]. However, in our VDN model, there were no changes in protein levels of p-JNK, which means that the p-JNK/JNK pathway may not be the major pathway of apoptosis in VC rats.
For FGF21 treatment, we found FGF21 administration can decrease TUNEL-positive cells in the aortas as well as alleviate the increased expression of ERS marker GRP78 in the calcified aortas. In the three pathways to ERS, the expression of CHOP in calcified aorta was significantly down-regulated under FGF21 treatment. Also, FGF21 can inhibit the caspase-12 activation but not the p-JNK/JNK. Therefore, FGF21 may reduce apoptosis via inhibiting ERS in VC. These results are in accordance with recent studies that FGF21 can inhibit ERS induced apoptosis of VSMC in atherosclerotic mice[22].
As the possible mechanism of the vascular protective and anti-ERS effect of FGF21, some reports demonstrate that FGF21 is a downstream factor of ERS which can exert beneficial feedback effects through counteracting ERS[45]. However, because FGF21 was originally found in the liver and highly secreted by the adipose tissue, most researches focused on the effects of FGF21 on the hepatocyte, adipocyte and even cardiomyocyte[46] that the effects of FGF21 on the artery has been neglected. Actually, the existence of FGF21, β-Klotho and FGFR1 in the cardiac microvascular endothelial cells and VSMCs suggests that FGF21 may be a key target in the physiological and pathophysiological aspects of artery[47].
For the pathogenesis of liver disease study, ERS stimulated by triglycerides can directly increase FGF21 expression via a PERK-ATF4-CHOP dependent mechanism[48]. When the ERS occurs, one of ERS specific pathway, containing CHOP, is activated, and consequently expression of endogenous FGF21 is upregulated[49]. Furthermore, intraperitoneal injection of tunicamycin, an endoplasmic reticulum stressor, to normal mice can also induce hepatic FGF21 expression directly[50]. The same results are also confirmed in the adipocyte[45] and H9C2 cardiomyocytes[51]. Especially for the CHOP, some studies have found CHOP can induce the expression of FGF21 by activating transcription via promoter elements and enhancing FGF21 mRNA stability in ERS[49]. And in our previous study, we have confirmed that the expression of endogenous FGF21 were increased in the development of VC[52]. Taken together, the CHOP pathway in ERS may play a crucial role in ERS induced FGF21 elevation in the related diseases. These findings may further explain the correlation between ERS and increased FGF21 expression. As a result, the elevation of FGF21 can feedback control the ERS responses and attenuate the development of the ERS related diseases. The negative feedback mechanisms can prevent an excessive activation during the ERS and they can trigger cellular recovery. For instance, Zarei et al. revealed that FGF21 may protect against hepatic steatosis by alleviating ERS[53]. Other researchers reported that the administration of FGF21 could reverse the suppression of adiponectin expression, counteracts obesity and alleviates insulin resistance via inhibiting ERS in mice[45, 54]. In particular for the cardiovascular diseases, FGF21 could reduce the hypoxia‑induced apoptosis of human pulmonary arterial endothelial cells and prevent endothelial dysfunction via the inhibition of ERS[55]. In addition, FGF21 overexpression can protect against ERS induced cardiomyocyte injury via increasing cell viability and reducing cell apoptosis[51]. FGF21 can also protect against ischemia cerebral injury via decreasing ERS[56]. In addition, our team first reported that FGF21 can protect against atherosclerosis at least in part by its inhibitory effects on ERS mediated apoptosis[22]. These researches clearly demonstrate that FGF21 administration or overexpression can protect tissues and organs against the adverse effects induced by ERS. In our study, administration of FGF21 in turn ameliorated the ERS responses of VC by suppressing CHOP and caspase-12 pathways. These imply that the ERS-FGF21 axis creates a negative feedback loop and our results show that FGF21 inhibits two of the three ERS induction downstream effectors in VC. Therefore, FGF21 appears to be a regulatory factor in the proteostasis network and not merely an endpoint regulator in the negative feedback loop. ERS is thought to be caused not only by the excess misfolded or unfolded proteins in the endoplasmic reticulum but also by pathogenic conditions, such as atherosclerosis[57] and diabetes[58]. In addition, treatment with chemical chaperones that reduce ERS can improve several metabolic diseases, including VC and obesity[59]. Based on our data, together with these previous findings, we propose that FGF21 may function as a critical regulator in the proteostasis network to maintain protein homeostasis by protecting against ERS not only in the VC but also in other metabolic disorders caused by ERS.
There were also several limitations in our research, which still need to be explored in the future. Firstly, we have been yet regretting that we did not confirm our in vivo study in vitro. Actually, it is worth to have a further discussion that in vitro study we plan next. In addition, our team had just published a paper recently which matched our in vivo study in vitro[60]. Involvement of RANK/RANKL/OPG/TRAIL system in VC has been researched widely, but it is still full of controversy. In our previous study, FGF21 may inhibit VSMCs calcification through OPG/RANKL system by P38 and PI3K/AKT pathways in vitro. Secondly, in this study, we used the echocardiography to evaluate the calcification condition, but in the clinical study, intravascular ultrasound (IVUS) is more common to investigate the vascular lumen morphology and calcification condition. However, the rat aorta was too tiny that the IVUS cannot pass. Subsequently, we plan to use the IVUS in a VC pig model to evaluate the VC continuously and dynamically.
In conclusion, FGF21 is likely an endogenous cardiovascular protective peptide. Using a rat model of VC, we propose that FGF21 can significantly alleviate the progression of VC in vivo. These data, to our knowledge, provide the first evidence that in the aorta of VDN challenged VC rat, the expression of apoptotic cells and pro-apoptosis protein levels of ERS pathway: CHOP and cleaved caspase-12, were decreased by FGF21. Thus, FGF21 is a potent cardiovascular protective peptide which can inhibit VC at least in part by alleviating ERS mediated apoptosis in VSMCs. This study provides new avenues and targets for prevention and treatment of VC.
Abbreviations
FGF21: fibroblast growth factor 21; VDN: vitamin D3 plus nicotine; VC: vascular calcification; FGFR1: fibroblast growth factor receptor 1; ALP: alkaline phosphatase; VSMC: Vascular smooth muscle cell; ERS: endoplasmic reticulum stress; GRP 78: glucose-regulated protein 78; JNK: c-JUN NH2-terminal kinase; CHOP: C/EBP homologous protein.
Acknowledgements
National Natural Science Fund of China (No. 81570388); Beijing Natural Science Foundation (No. 7142048); and the Major State Basic Research Development Program of China (973 Program, No. 2015CB554404) supported this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kobayashi S. Cardiovascular Events in Hemodialysis Patients: Challenging against Vascular Calcification. Ann Vasc Dis. 2017;10:1-7
2. Lacolley P, Regnault V, Segers P, Laurent S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol Rev. 2017;97:1555-617
3. Kwon D, Kim Y, Kook H. New Aspects of Vascular Calcification: Histone Deacetylases and Beyond. J Korean Med Sci. 2017;32:1738-48
4. Trion A, van der Laarse A. Vascular smooth muscle cells and calcification in atherosclerosis. Am Heart J. 2004;147:808-14
5. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724-36
6. Ariyasu D, Yoshida H, Hasegawa Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. Int J Mol Sci. 2017:18
7. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839-50
8. Cybulsky A. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13:681-96
9. Hetz C, Papa F. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69:169-81
10. Han X, Wang L, Diao Z, Liu W. Apelin: A novel inhibitor of vascular calcification in chronic kidney disease. Atherosclerosis. 2016;244:1-8
11. Zhang K, Zhang Y, Feng W, Chen R, Chen J, Touyz R. et al. Interleukin-18 Enhances Vascular Calcification and Osteogenic Differentiation of Vascular Smooth Muscle Cells Through TRPM7 Activation. Arterioscler Thromb Vasc Biol. 2017;37:1933-43
12. Domouzoglou E, Naka K, Vlahos A, Papafaklis M, Michalis L, Tsatsoulis A. et al. Fibroblast growth factors in cardiovascular disease: The emerging role of FGF21. Am J Physiol Heart Circ Physiol. 2015;309:H1029-38
13. Luo Y, Lu W, Li X. Unraveling Endocrine FGF Signaling Complex to Combat Metabolic Diseases. Trends Biochem Sci. 2018;43:563-6
14. Luo Y, Ye S, Chen X, Gong F, Lu W, Li X. Rush to the fire: FGF21 extinguishes metabolic stress, metaflammation and tissue damage. Cytokine Growth Factor Rev. 2017;38:59-65
15. Yang C, Jin C, Li X, Wang F, McKeehan W, Luo Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PloS one. 2012;7:e33870
16. Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y. et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation. 2015;131:1861-71
17. Jin L, Lin Z, Xu A. Fibroblast Growth Factor 21 Protects against Atherosclerosis via Fine-Tuning the Multiorgan Crosstalk. Diabetes Metab J. 2016;40:22-31
18. Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Jaiwongkam T, Wang X, Liang G. et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. Biomed Pharmacother. 2018;97:1663-72
19. Wang Q, Yuan J, Yu Z, Lin L, Jiang Y, Cao Z. et al. FGF21 Attenuates High-Fat Diet-Induced Cognitive Impairment via Metabolic Regulation and Anti-inflammation of Obese Mice. Mol Neurobiol. 2018;55:4702-17
20. Kharitonenkov A, DiMarchi R. Fibroblast growth factor 21 night watch: advances and uncertainties in the field. J Intern Med. 2017;281:233-46
21. Lin Z, Tian H, Lam K, Lin S, Hoo R, Konishi M. et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779-89
22. Wu X, Qi Y, Chang J, Lu W, Zhang J, Wang S. et al. Possible role of fibroblast growth factor 21 on atherosclerosis via amelioration of endoplasmic reticulum stress-mediated apoptosis in apoE(-/-) mice. Heart Vessels. 2015;30:657-68
23. Niederhoffer N, Bobryshev Y, Lartaud-Idjouadiene I, Giummelly P, Atkinson J. Aortic calcification produced by vitamin D3 plus nicotine. J Vasc Res. 1997;34:386-98
24. Zhou Y, Zhang J, Cai Y, Teng X, Duan X, Song J. et al. Insulin resistance induces medial artery calcification in fructose-fed rats. Exp Biol Med (Maywood). 2012;237:50-7
25. Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54
26. Kokkinos J, Tang S, Rye K, Ong K. The role of fibroblast growth factor 21 in atherosclerosis. Atherosclerosis. 2017;257:259-65
27. Staiger H, Keuper M, Berti L, Hrabe de Angelis M, Häring H. Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr Rev. 2017;38:468-88
28. Shi Y, Liu J. [Study on the kidney impairment and expressions of FGF21 from a rat model of vascular calcification]. Zhonghua Yi Xue Za Zhi. 2016;96:3741-4
29. Rochette L, Meloux A, Rigal E, Zeller M, Cottin Y, Vergely C. The role of osteoprotegerin in the crosstalk between vessels and bone: Its potential utility as a marker of cardiometabolic diseases. Pharmacol Ther. 2018;182:115-32
30. Ter Braake A, Shanahan C, de Baaij J. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arterioscler Thromb Vasc Biol. 2017;37:1431-45
31. Cai Y, Xu M, Teng X, Zhou Y, Chen L, Zhu Y. et al. Intermedin inhibits vascular calcification by increasing the level of matrix gamma-carboxyglutamic acid protein. Cardiovasc Res. 2010;85:864-73
32. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J. et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab. 2018;27:1323-37.e5
33. Clarke M, Littlewood T, Figg N, Maguire J, Davenport A, Goddard M. et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529-38
34. Duan X, Zhou Y, Teng X, Tang C, Qi Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun. 2009;387:694-9
35. Ma W, Sun X, Wang Y, Zhu Y, Han X, Liu N. Restoring mitochondrial biogenesis with metformin attenuates β-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol Cell Endocrinol. 2018
36. Proudfoot D, Skepper J, Hegyi L, Farzaneh-Far A, Shanahan C, Weissberg P. The role of apoptosis in the initiation of vascular calcification. Z Kardiol. 2001;90(Suppl 3):43-6
37. Lu Y, Bian Y, Wang Y, Bai R, Wang J, Xiao C. Globular adiponectin reduces vascular calcification via inhibition of ER-stress-mediated smooth muscle cell apoptosis. Int J Clin Exp Pathol. 2015;8:2545-54
38. Yang R, Teng X, Li H, Xue H, Guo Q, Xiao L. et al. Hydrogen Sulfide Improves Vascular Calcification in Rats by Inhibiting Endoplasmic Reticulum Stress. Oxid Med Cell Longev. 2016;2016:9095242
39. Li H, Teng X, Yang R, Guo Q, Xue H, Xiao L. et al. Hydrogen Sulfide Facilitates the Impaired Sensitivity of Carotid Sinus Baroreflex in Rats with Vascular Calcification. Front Pharmacol. 2017;8:629
40. Guzel E, Arlier S, Guzeloglu-Kayisli O, Tabak M, Ekiz T, Semerci N. et al. Endoplasmic Reticulum Stress and Homeostasis in Reproductive Physiology and Pathology. Int J Mol Sci. 2017:18
41. Dong Y, Fernandes C, Liu Y, Wu Y, Wu H, Brophy M. et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diab Vasc Dis Res. 2017;14:14-23
42. Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai). 2014;46:629-40
43. García de la Cadena S, Massieu L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis. 2016;21:763-77
44. Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640-52
45. Guo Q, Xu L, Liu J, Li H, Sun H, Wu S. et al. Fibroblast growth factor 21 reverses suppression of adiponectin expression via inhibiting endoplasmic reticulum stress in adipose tissue of obese mice. Exp Biol Med (Maywood). 2017;242:441-7
46. Wu F, Wang B, Zhang S, Shi L, Wang Y, Xiong R. et al. FGF21 ameliorates diabetic cardiomyopathy by activating the AMPK-paraoxonase 1 signaling axis in mice. Clin Sci. 2017;131:1877-93
47. Fon Tacer K, Bookout A, Ding X, Kurosu H, John G, Wang L. et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol. 2010;24:2050-64
48. Liu W, Huang S, Shi K, Zhao C, Chen L, Braddock M. et al. The role of fibroblast growth factor 21 in the pathogenesis of liver disease: a novel predictor and therapeutic target. Expert Opin Ther Targets. 2014;18:1305-13
49. Wan X, Lu X, Xiao Y, Lin Y, Zhu H, Ding T. et al. ATF4- and CHOP-dependent induction of FGF21 through endoplasmic reticulum stress. Biomed Res Int. 2014;2014:807874
50. Maruyama R, Shimizu M, Hashidume T, Inoue J, Itoh N, Sato R. FGF21 Alleviates Hepatic Endoplasmic Reticulum Stress under Physiological Conditions. J Nutr Sci Vitaminol. 2018;64:200-8
51. Liang P, Zhong L, Gong L, Wang J, Zhu Y, Liu W. et al. Fibroblast growth factor 21 protects rat cardiomyocytes from endoplasmic reticulum stress by promoting the fibroblast growth factor receptor 1-extracellular signal-regulated kinase 1/2 signaling pathway. Int J Mol Med. 2017;40:1477-85
52. Shi Y, Lu W, Hou Y, Fu K, Gan F, Liu J. Fibroblast growth factor 21 ameliorates vascular calcification by inhibiting osteogenic transition in vitamin D3 plus nicotine-treated rats. Biochem Biophys Res Commun. 2018;495:2448-55
53. Zarei M, Barroso E, Palomer X, Dai J, Rada P, Quesada-López T. et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol Metab. 2018;8:117-31
54. Ye M, Lu W, Wang X, Wang C, Abbruzzese J, Liang G. et al. FGF21-FGFR1 Coordinates Phospholipid Homeostasis, Lipid Droplet Function, and ER Stress in Obesity. Endocrinology. 2016;157:4754-69
55. Chen A, Liu J, Zhu J, Wang X, Xu Z, Cui Z. et al. FGF21 attenuates hypoxia-induced dysfunction and apoptosis in HPAECs through alleviating endoplasmic reticulum stress. Int J Mol Med. 2018;42:1684-94
56. Yang X, Hui Q, Yu B, Huang Z, Zhou P, Wang P. et al. Design and Evaluation of Lyophilized Fibroblast Growth Factor 21 and Its Protection against Ischemia Cerebral Injury. Bioconjug Chem. 2018;29:287-95
57. Yingchun H, Yahong M, Jiangping W, Xiaokui H, Xiaohong Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018;17:220
58. Pang X, Zhang Y, Shi X, Li D, Han J. ERp44 depletion exacerbates ER stress and aggravates diabetic nephropathy in db/db mice. Biochem Biophys Res Commun. 2018
59. Kim K, Ahn N, Jung S. Comparison of endoplasmic reticulum stress and mitochondrial biogenesis responses after 12 weeks of treadmill running and ladder climbing exercises in the cardiac muscle of middle-aged obese rats. Braz J Med Biol Res. 2018;51:e7508
60. Cao F, Liu X, Cao X, Wang S, Fu K, Zhao Y. et al. Fibroblast growth factor 21 plays an inhibitory role in vascular calcification in vitro through OPG/RANKL system. Biochem Biophys Res Commun. 2017;491:578-86
Author contact
![]() Corresponding author: Prof. Jinghua Liu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, and Beijing Institute of Heart, Lung and Blood Vessel Diseases, 2 Anzhen Road, Chaoyang District, Beijing, 100029, China. Email: liujinghuasina.com; Fax: + (86) 010 64456998
Corresponding author: Prof. Jinghua Liu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, and Beijing Institute of Heart, Lung and Blood Vessel Diseases, 2 Anzhen Road, Chaoyang District, Beijing, 100029, China. Email: liujinghuasina.com; Fax: + (86) 010 64456998