Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(1):148-157. doi:10.7150/ijbs.28874 This issue Cite
Research Paper
Sulforaphane Attenuates Angiotensin II-Induced Vascular Smooth Muscle Cell Migration via Suppression of NOX4/ROS/Nrf2 Signaling
Min Zhang, Yingjie Xu, Zhaohui Qiu ![]() , Li Jiang
, Li Jiang ![]()
Division of Cardiology, TongRen Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200336, China
Received 2018-7-30; Accepted 2018-9-28; Published 2019-1-1
Abstract

Angiotensin II (Ang II) is involved in the pathogenic progress of cardiovascular diseases via the promotion of abnormal proliferation and migration of human vascular smooth muscle cells (HVSMCs). Sulforaphane (SFN) exerts potent anti-inflammatory effects both in vitro and in vivo. In the present study, we aimed to investigate the effects of SFN on Ang II-induced abnormal migration of HVSMCs as well as the underlying mechanisms of those effects. The results showed that Ang II-induced HVSMC proliferation and migration were inhibited by treatment with SFN. SFN also exhibited anti-inflammatory activity, as indicated by its reduction of monocyte adhesion to HVSMCs via the reduction of ICAM1 and VCAM1 levels. Moreover, SFN reduced the Ang II-induced upregulation of HVSMC migration; this effect was inhibited by pretreatment with inhibitors of NADPH oxidase and ROS or transfection with siNOX4. In addition, SFN reversed the Ang II-induced upregulation of HVSMC migration via elevation of Nrf2 activation and expression. Taken together, the results indicate that SFN reverses Ang II-induced HVSMC migration through suppression of the NOX4/ROS/Nrf2 pathway. Thus, SFN is a potential agent to reverse the pathological changes involved in various cardiovascular diseases.
Keywords: sulforaphane, angiotensin II, human vascular smooth muscle cells, nuclear respiratory factor 2, reactive oxygen species
Introduction
Human vascular smooth muscle cells (HVSMCs) are critical for maintaining vascular homeostasis, mainly by manipulating contraction and relaxation. Abnormal HVSMC proliferation and migration into the intima, processes that are predominantly triggered by injured endothelial cells (ECs), are key stages for the initiation and development of hypertension and atherosclerosis (AS) [1]. However, the molecular mechanism of HVSMC proliferation and migration after vascular injury is not completely known.
Angiotensin ΙΙ (Ang ΙΙ) induces the pathogenesis of several cardiovascular diseases, such as cardiomyopathy[2], coronary artery disease[3], AS [4] and vascular injury[5]. Numerous interventions have been used to downregulate the renin-angiotensin system (RAS) system or to directly block Ang II expression for the treatment of cardiovascular disease [6, 7]. Ang II is also known to enhance vascular remodeling through NADPH oxidase-derived ROS generation and inflammatory responses in HVSMCs; this remodeling then induces an abnormal proliferative capacity and the migration of HVSMCs [8, 9]. Then, these abnormal activities induce formation of neointima, accelerate vascular damage and cause AS. Moreover, in vivo studies have revealed that endogenous Ang II is associated with plaque vulnerability. Thus, it is well known that prevention of Ang ΙΙ-induced HVSMC hyperactivation is a potential therapeutic intervention for vascular diseases.
The potent antioxidant compound sulforaphane (SFN) suppresses platelet-derived growth factor (PDGF)-induced VSMC proliferation and neointima formation through a p53 signaling-independent increase in Nrf2 expression [10-12]. SFN suppresses leptin-stimulated VSMC proliferation via p70S6K/ S6 signaling [10]. Furthermore, SFN is regarded as a promoter for the activation of nuclear factor erythroid 2-related factor 2 (Nrf2)[13]. However, the effect of SFN on Ang ΙΙ-induced HVSMC proliferation and migration has not yet been fully investigated.
Nrf2 plays a key role in VSMC cellular proliferation and neointima formation[14]. An investigation has determined that enhanced Nrf2 activity has an antiproliferative effect, thus contributing to the induction of Nrf2 target genes, including HO-1 and NQO1 [15]. This research also showed that the abnormal VSMCs migration decreases via inducing Nrf2 activity[15]. Further, Nrf2 depletion induces VSMC migration and neointimal hyperplasia [16].Nevertheless, the role of Nrf2 in HVSMC proliferation and migration and neointima formation remains unknown. The kelch-like ECH-associated protein 1 (Keap1) is the key negative regulator of Nrf2, and SFN activated Nrf2 combination with antioxidant response element (ARE), which has a key role in the protection of cells under oxidative stress. It is also unclear whether SFN-induced Nrf2 activation contributes to inhibition of Ang ΙΙ-induced HVSMC migration.
Nox4 is a key source of ROS, which has an important role in the cell damage. Under oxidative stress, Nrf2 disassociates from KEAP1 and moves into the nucleus. Nox4 is regarded to be regulated by Nrf2 in various models in vitro and in vivo. These data suggest that Nox4 may be associated with Nrf2 in Ang II-stimulated HVSMCs. However, few studies have investigated the connection between Nox4 and Nrf2 in Ang II-stimulated HVSMCs. The present study aims to assess the effects of SFN on Ang II-induced HVSMC migration and to explore the potential underlying mechanisms.
Results
SFN ameliorates Ang II-stimulated HVSMC proliferation
To determine effective treatment concentrations, the cytotoxic effects of SFN were investigated by MTT colorimetric assay. Data on the percentage of viable cells after treatment revealed that various concentrations of SFN (1-10 μM) were not cytotoxic toward HVSMCs after 24 h of exposure (Figure 1A). Further, HVSMC viability was increased by Ang II, whereas SFN inhibited Ang II-induced increases in HVSMC viability (p< 0.05, Figure 1B).
The preventive effects of SFN on Ang II-induced increases in HVSMC viability. (A) Cell viability was measured in Ang II-stimulated HVSMCs using an MTT assay. (B) Cell viability was measured for 24 h after Ang II stimulation in the presence or absence of SFN. (C) LDH release was measured for 24 h after Ang II stimulation in the presence or absence of SFN. (D) Cell proliferation was measured for 24 h after Ang II stimulation in the presence or absence of SFN by [3H] thymidine incorporation assay. These data were obtained from at least three independent experiments; *P < 0.05, #P < 0.05, ##P < 0.01 (n = 3).
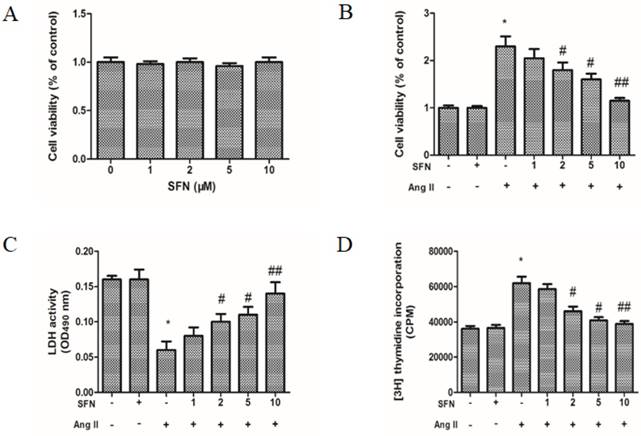
To test whether SFN could inhibit Ang II-stimulated HVSMC proliferation, LDH assays and [3H] thymidine incorporation assays were also performed. The data showed that Ang II caused a decrease in LDH release (Figure 1C) and an increase in HVSMC proliferation (Figure 1D). However, SFN significantly inhibited the Ang II-mediated effects on LDH release and proliferation in a dose-dependent manner in HVSMCs (Figure 1C and D). LDH assays and [3H] thymidine incorporation assays revealed similar inhibitory effects of SFN on Ang II-induced HVSMC proliferation.
SFN ameliorates Ang II-induced migration of HVSMCs
To determine the effects of SFN on migration, a transwell migration chamber assay and a wound healing assay were used. As shown in Figure 2A and B, Ang II increased the migratory ability of HVSMCs, whereas SFN reversed the Ang II-induced HVSMC migration in a dose-dependent manner.
The preventive effects of SFN on Ang II-stimulated HVSMC migration. (A) Cell migration was evaluated by Boyden chamber assay after Ang II stimulation in the presence or absence of SFN for 12 h. (B) Quantification of cell migration was performed with a Boyden chamber assay. (C) Cell migration was evaluated by wound healing assay after Ang II stimulation in the presence or absence of SFN for 24 h. (D) Quantification of cell migration was performed with a wound healing assay. These data were obtained from at least three independent experiments; *P < 0.05 (n = 3), #P < 0.05, ##P < 0.01 (n = 3).
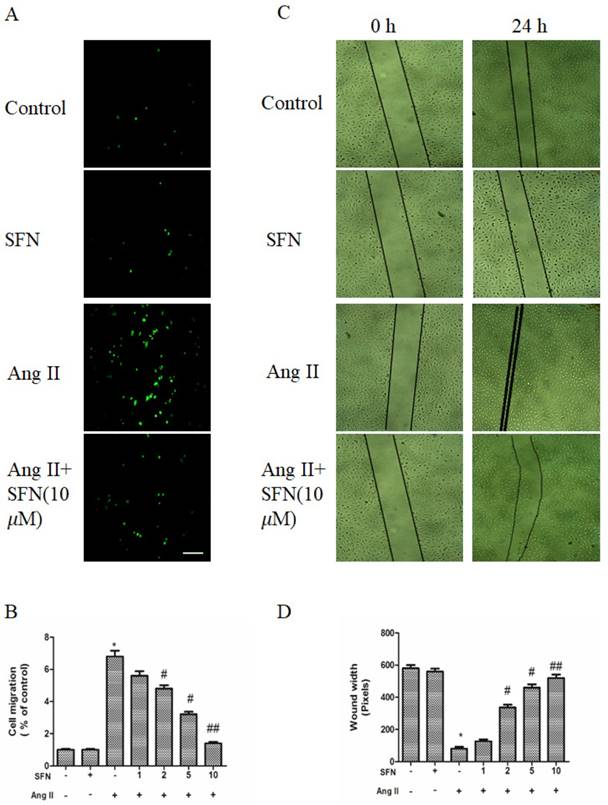
The wound healing assay also revealed a consistent tendency of SFN to reduce Ang II-stimulated HVSMC migration (Figure 2C and D).
SFN ameliorates monocyte adhesion to HVSMCs by reducing the expression of adhesion molecules
HVSMCs directly interact with monocytes under conditions of inflammation, including during neointima formation and AS, so we determined whether SFN could inhibit monocyte attachment to Ang II-stimulated HVSMCs. The results indicated that Ang II promoted monocyte binding to HVSMCs, whereas SFN attenuated this response (Figure 3A and B). In addition, both ICAM1 and VCAM1 are important for elevating the adherence of monocytes, so we investigated their levels in HVSMCs. Ang II increased the levels of ICAM1 and VCAM1, whereas SFN attenuated ICAM1 and VCAM1 levels (Figure 3C).
The preventive effects of SFN (10 μM) on Ang II (100 ng/mL) -stimulated inflammatory responses in HVSMCs for 24 h. (A) RAW264.7 cells were stained to visualize monocyte adhesion to HVSMCs. The red color indicates PKH-labeled monocytes. (B) Relative monocyte adhesion was estimated and analyzed by relative fluorescence intensity. (C) Protein levels of ICAM1 and VCAM1 were analyzed by western blot assay. (D) NF-κB activity was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. (E) The level of VEGF was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. (F) The level of PlGF was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. (G) The level of IL-6 was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. These data were obtained from at least three independent experiments; *P < 0.05 (n = 3), #P < 0.05,**P < 0.01 (n = 3).
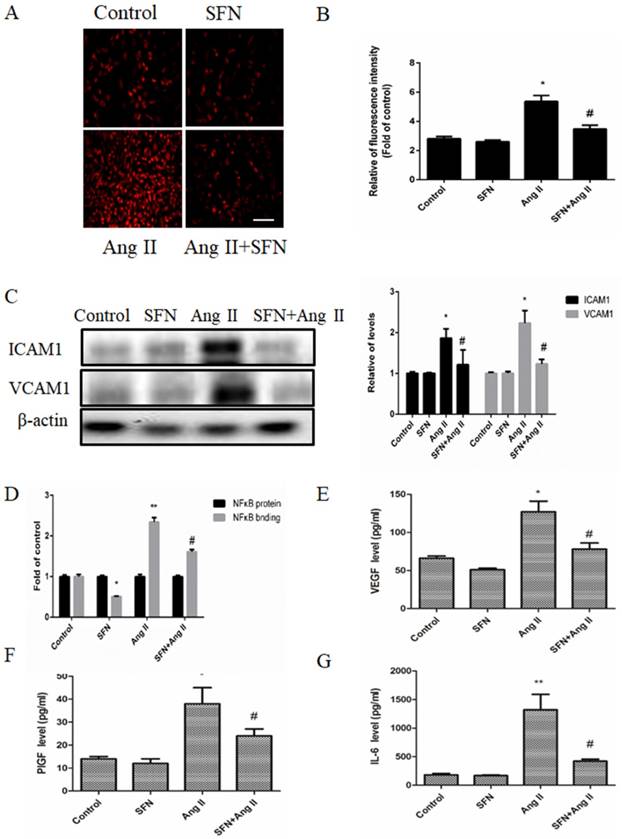
NF-κB is a key transcription factor that regulates ICAM1 and VCAM1 expression in many types of cells. Therefore, we determined whether SFN could suppress Ang II-mediated NF-κB activation in HVSMCs. Our data showed that Ang II caused an increase in NF-κB activation. However, SFN significantly decreased the Ang II-induced elevations in NF-κB activation (Figure 3D). Therefore, the data suggest that SFN inhibits Ang II-induced inflammatory responses via inhibition of NF-κB activation in HVSMCs.
To determine whether SFN could affect inflammatory cytokine expression in HVSMCs, the levels of VEGF, PlGF and IL-6 were assessed by ELISA. Ang II caused increases in VEGF (Figure 3E), PlGF (Figure 3F) and IL-6 (Figure 3G) expression compared to the expression in the control group. However, SFN reduced the Ang II-induced elevations in VEGF (Figure 3E), PlGF (Figure 3F) and IL-6 (Figure 3G) expression in HVSMCs. Taken together, the results suggest that SFN inhibits Ang II-induced inflammatory responses in HVSMCs.
SFN inhibits Ang II-induced migration of HVSMCs via reducing ROS generation and NADPH oxidase activity
DCF-DA fluorescence assay was used to determine total intracellular ROS levels, and the intensity of MitoSOX Red fluorescence was used to determine the mtROS levels. SFN decreased intracellular ROS accumulation compared with the that of the Ang II group (Figure 4A); moreover, SFN had a similar effect on mtROS accumulation in HVSMCs (Figure 4B). Furthermore, we found that SFN reversed Ang II-induced NADPH oxidase activity (Figure 4C) in HVSMCs.
SFN (10 μM) inhibits Ang II (100 ng/mL) -induced migration of HVSMCs via reducing ROS generation and NADPH oxidase activity. (A) Intracellular ROS generation was evaluated by DCF-DA fluorescence assay after Ang II stimulation in the presence or absence of SFN. (B) Cellular mtROS levels were measured by MitoSOX Red fluorescence after Ang II stimulation in the presence or absence of SFN. (C) Cellular NADPH oxidase activity was evaluated after Ang II stimulation in the presence or absence of SFN. (D) Cell migration was evaluated by Boyden chamber assay after Ang II stimulation in the presence or absence of NAC, DPI or APO. (E) The expression of Nox4 in HVSMCs was evaluated by western blot assay after Ang II stimulation in the presence or absence of SFN. (F) Quantification of ROS generation was performed with a DCF-DA fluorescence assay after Ang II stimulation in the presence or absence of SFN and siNOX4. These data were obtained from at least three independent experiments; *P < 0.05, **P < 0.01, #P < 0.05, # #P < 0.01 (n = 3).
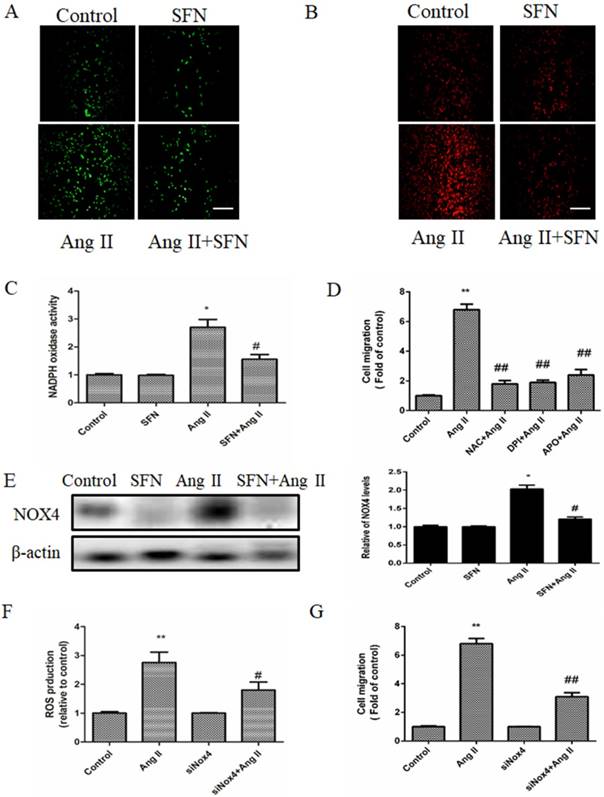
Pretreatment with inhibitors of ROS (NAC) or NADPH oxidase (DPI or APO) suppressed the Ang II-induced HVSMC migration (Figure 4D). On the other hand, SFN reduced the expression of NADPH oxidase subunit NOX4 in Ang II-treated HVSMCs (Figure 4E). To investigate whether Nox4 was involved in the Ang II-induced stimulation of HVSMC migration, siNOX4 was used. The results showed that transfection with siNOX4 inhibited Ang II-induced ROS production (Figure 4F) and HVSMC migration (Figure 4G). Taken together, these results suggest that SFN decreases Ang II-induced HVSMCs migration via inhibition of NADPH oxidase activity and ROS generation.
SFN amelioration of the Ang II-induced migration of HVSMC requires Nrf2 activation
To gain insight into the mechanism by which SFN reversed HVSMC migration in response to Ang II treatment, we investigated Nrf2 activity and expression by ELISA and western blot. Compared to the Ang II group, the SFN treatment group had elevated Nrf2 activity and expression levels (Figure 5A and B).
SFN (10 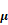 M) amelioration of the Ang II (100 ng/mL) -induced migration of HVSMCs requires Nrf2 activation. (A) The activity of Nrf2 was measured in HVSMCs after Ang II stimulation in the presence or absence of SFN. (B) The expression of Nrf2 was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. (C) The location of Nrf2 in HVSMCs was evaluated after Ang II stimulation in the presence or absence of SFN. (D) Western blot assay confirmed the expression of Nrf2 in transfected HVSMCs. (E) Cell migration was evaluated by Boyden chamber assay after Ang II stimulation in the presence or absence of SFN and siNrf2. These data were obtained from at least three independent experiments; *P < 0.05, **P < 0.01, #P < 0.05, # #P < 0.01, $$ P < 0.01 (n = 3).
M) amelioration of the Ang II (100 ng/mL) -induced migration of HVSMCs requires Nrf2 activation. (A) The activity of Nrf2 was measured in HVSMCs after Ang II stimulation in the presence or absence of SFN. (B) The expression of Nrf2 was evaluated in HVSMCs after Ang II stimulation in the presence or absence of SFN. (C) The location of Nrf2 in HVSMCs was evaluated after Ang II stimulation in the presence or absence of SFN. (D) Western blot assay confirmed the expression of Nrf2 in transfected HVSMCs. (E) Cell migration was evaluated by Boyden chamber assay after Ang II stimulation in the presence or absence of SFN and siNrf2. These data were obtained from at least three independent experiments; *P < 0.05, **P < 0.01, #P < 0.05, # #P < 0.01, $$ P < 0.01 (n = 3).
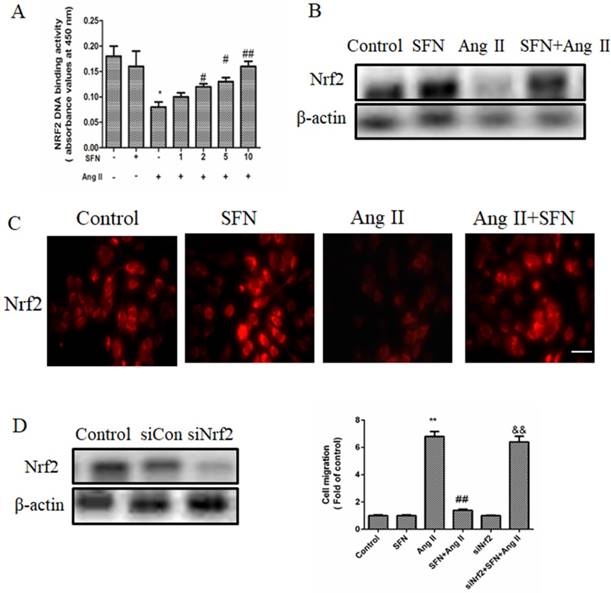
In the control group, Nrf2 was located in both the cytoplasm and the nucleus. Ang II reduced the cytoplasmic and nuclear localization of Nrf2, whereas SFN increased Nrf2 expression in both the cytoplasm and the nucleus compared the expression in the Ang II group (Figure 5C). These results suggested that SFN reverses Ang II-mediated reductions in Nrf2 activity and expression.
Next, we investigated the effect of Nrf2 on Ang II-induced migration in HVSMCs. Depletion of Nrf2 by siRNA caused an approximately 70 % reduction in expression (Figure 5D) that was mediated by a promotion of HVSMC migration (Figure 5E). Taken together, these data demonstrated that Nrf2 is required for SFN to reverse the Ang II-induced migration of HVSMCs.
Discussion
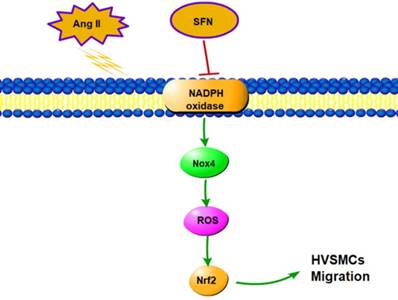
In this study, we demonstrated that SFN attenuates the Ang II-induced abnormal migration of HVSMCs through suppression of the NOX4/ ROS/Nrf2 pathway (Fig. 6). Therefore, administration of SFN may serve as a potential therapeutic strategy for cardiovascular diseases.
Schematic diagram showing the role of SFN in Ang II-mediated signaling pathways in HVSMCs.
Ang II has been identified as a key vasoactive peptide that has important roles in the pathogenesis of vascular diseases, such as in growth and migration of VSMCs [17-19]. Recently, increasing numbers of studies have investigated the effects of Ang II (and the molecular mechanisms of those effects) on processes involved in blood vessel injury, including induction of the cAMP/PKA signaling pathway [20], the ERK1/2 pathway[21], the NF-κB signaling pathway [22] and the NADPH oxidase subunit signaling pathway[23]. Therefore, more detailed elucidation of the mechanisms underlying Ang II-induced inflammatory responses and oxidative stress in the cardiovascular system can contribute to the development of new therapeutic strategies. Our results showed that Ang II induced HVSMC proliferation and migration. In addition, Ang II increased monocyte adhesion to HVSMCs, ICAM1 and VCAM1 expression and inflammatory cytokine expression. Furthermore, we measured intracellular ROS and mtROS generation in HVSMCs, as Ang II is an activator of NADPH oxidase pathway-mediated ROS generation in many types of cells, including VSMCs. Ang II increased intracellular ROS and mtROS generation, NADPH oxidase activation and NOX4 expression in HVSMCs.
SFN reduced the proliferation and migration of HVSMCs subjected to Ang II. SFN also decreased monocyte adhesion to HVSMCs, which may have been mediated by a reduction in ICAM1 and VCAM1 expression, inflammatory cytokine expression and NF-κB activity. Overall, we confirmed that Ang II promotes increased levels of adhesion molecules and elevated monocyte adhesion and that SFN reduces this inflammatory activity.
In addition, we measured intracellular ROS and mtROS levels in vitro. In HVSMCs, SFN showed a protective effect against Ang II-induced increases in intracellular ROS, mtROS, NADPH oxidase activity and NOX4 expression in HVSMCs. The use of inhibitors of ROS (NAC) and NADPH oxidase (DPI or APO) or siNOX4 decreased the HVSMC migration induced by Ang II, implying that SFN reversed the Ang II-induced ROS generation and NADPH oxidase activation that were responsible for HVSMC migration.
Nrf2 is widely recognized as a potential modulator of cell proliferation and migration [24-26]. SFN modulates cellular homeostasis via Nrf2 activation [27]. Our results showed that SFN triggers a stimulation of Nrf2 activity and expression in Ang II-treated HVSMCs. When HVSMCs were transfected with Nrf2 siRNA, the SFN-mediated attenuation of the Ang II-stimulated HVSMC migration was diminished. We also found SFN obviously decreases Ang II-induced the expression of ICAM-1 and VCAM-1 in HVSMCs. SFN attenuates Ang II-induced HVSMCs inflammatory cytokine expression. SFN protect HVSMCs against Ang II-induced injury by both Nrf2-dependent and independent pathways.
Conclusions
In summary, we have presented evidence that SFN attenuates Ang II-promoted HVSMC migration via reducing NOX4 /ROS/Nrf2 signaling and suppressing the NF-κB pathway. These findings reveal the molecular mechanism underlying the increase in HVSMC migration induced by Ang II, which may aid in the development of new therapies that can be used to prevent cardiovascular diseases.
Materials and methods
Cell culture and treatments
Human aortic VSMCs were purchased from ATCC (Manassas, VA, USA) and maintained in vascular cell basal medium with smooth muscle growth supplement (SMGS) and 1% penicillin and streptomycin in a 5 % CO2 atmosphere. HVSMCs from passages 3-6 were used. In all experiments, HVSMCs were exposed to vascular cell basal medium containing 0.05% FBS (Gibco) for 24 h.
SFN and Ang ΙΙ were purchased from Sigma-Aldrich (St. Louis, MO, USA). SFN stock solution was prepared in sterile double-distilled water.
MTT Assay
Cell viability was measured by using an MTT colorimetric assay[28]. Cells (2×105 cells/well) were seeded in 96-well plates, and the cells were treated with various concentrations of SFN (0-10 μM) in the presence of Ang ΙΙ (100 ng/mL) for 24 h. After treatment, the cells were incubated with 400 μL of MTT (Sigma-Aldrich, St. Louis, MO, USA) reagent at 37 °C for 2 h, and the absorbance was evaluated at 540 nm by a microplate reader (Thermo Scientific, CA, USA).
For the LDH assay, cell lysate was collected with 1% Triton-100, and LDH activity was measured as previously described [28].
For the [3H] thymidine incorporation assay, the cells were labeled with [3H] thymidine at a concentration of 1 μCi/ml for 24 h. The cultures were then placed on ice to terminate the reaction, and the cells were washed with PBS with trichloroacetic acid (10%). Acid-insoluble [3H] thymidine was released through cell lysis, mixed with scintillation cocktail (3 ml), and evaluated by a liquid scintillation counter (PerkinElmer, Waltham, MA).
Cell migration assay
Cell migration was evaluated by a modified Boyden chamber assay (Corning, NY, pore diameter 8.0 μm) as previously described[29]. First, 2×104 cells were added to the upper chamber. Then, vascular cell basal medium (500 μL) containing 10 % FBS was added to the lower chamber. After 12 h, the migrated cells were stained with calcein AM. Migrated cells were evaluated with a fluorescence microscope (Nikon, Minato, Tokyo, Japan).
Wound healing assay
Cells migration was also measured by a wound healing assay[30]. Cells (5 × 105/well) were seeded in 12-well plates overnight. After cells reached 90% confluence, the scratches were made with scratched with a 200 μl plastic pipette tip. The cells were washed with PBS, the cells were incubated in free-serum medium and the wound region was imaged at 0 and 24 h with a microscope (Nikon, Minato, To'kyo, Japan) and evaluated by using ImageJ software.
Monocyte adherence assay
Cells (2 × 104) were seeded in a 4-well chamber slide (Nunc® Lab-Tek® II Chamber Slide™ System). Then, RAW264.7 cells (2 ×105) were stained with PKH26 (Sigma-Aldrich) following the manufacturer's instructions. After incubation for 30 min at 37 °C, nonadherent cells were removed, and the adherent cells were fixed with 4% formalin. Images were obtained by using a fluorescence microscope (Nikon, Minato, Tokyo, Japan).
Measurement of cellular and mitochondrial reactive oxygen species (ROS)
CM-H2DCFDA (Thermo Scientific) and MitoSOX™ Red (Invitrogen) were used to detect total cellular ROS and mitochondrial ROS formation as previous described [31]. The cells were treated with SFN (10 μM) 24 h followed by incubation with Ang ΙΙ (100 ng/mL) for 24 h. Cells were stained with CM-H2DCFDA (10 μM) or MitoSOX (2 μM) for 10 min at 37 °C in the dark. After the incubation, cells were washed with PBS, fixed in 4% paraformaldehyde and evaluated with a fluorescence microscope (Nikon, Minato, Tokyo, Japan; CM-H2DCFDA excitation/emission: 492/527 nm and MitoSOX™ Red excitation/emission: 510/580 nm).
Measurement of NADPH oxidase activity
The activity of NADPH oxidase was evaluated by lucigenin chemiluminescence assay as described previously [32]. The absorbance was detected by a microplate reader (Thermo Scientific, CA, USA).
In vitro small interfering RNA assay
Human scrambled, Nrf2 and Nox4 siRNAs were obtained from Life Technologies. Transfections with the various siRNAs (100 nM) were performed with Lipofectamine RNAiMAX reagent following the manufacturer's instructions.
Nuclear and cytoplasmic protein extraction
Nuclear and cytoplasmic protein fractions were obtained with a nuclear and cytoplasmic extraction kit according to the manufacturer's instructions (Thermo Scientific, Rockford, IL). Briefly, cells were centrifuged at 500 × g for 5 min. Then, ice-cold cytoplasmic extraction reagents (CER I and CER II) with protease/phosphatase inhibitor cocktail were added to the cells. After centrifugation at 16000 × g for 5 min, the nuclear proteins were obtained with the nuclear extraction reagent (NER) with a protease/phosphatase inhibitor cocktail.
Western blotting
Cells were lysed with ice-cold RIPA lysis buffer with protease inhibitor. The protein concentration was measured with a BCA kit (Beyotime Institute of Biotechnology, China). The proteins were separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in 5% nonfat milk at room temperature for 1 h and then incubated with the diluted primary antibody at 4°C overnight. The primary anti-Nrf2 antibody was obtained from abcam (ab62352, Abcam, UK). In addition, the membranes were incubated with the corresponding secondary antibodies for 1 h at room temperature. The immunoreactive bands were detected with ECL reagent (Thermo Scientific, Rockford, IL).
Measurement of Nrf2 activity
Nrf2 activity was measured by using a TransAM Nrf2 assay kit (Active Motif, Carlsbad, CA) as previously described [28]. The cell nuclear extracts (5 μg) were added to a 96-well plate that had been precoated with an oligonucleotide with a binding site for Nrf2. The absorbance was detected at 450 nm by a microplate reader.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde and immersed in 0.1% TritonX-100 for 30 min at room temperature. The cells were blocked with 10% BSA in PBS for 1 h and incubated with the primary anti-Nrf2 antibody (1:200 dilution) at 4 °C overnight; then, the cells were incubated in Alexa Fluor 488-conjugated goat anti-rabbit IgG for 1 h at room temperature. The images were analyzed with a Zeiss LSM 710 confocal scanning laser microscope (Carl Zeiss Microscopy GmbH, Jena, Germany).
Measurement of cytokines by ELISA
To measure vascular endothelial growth factor (VEGF), placenta growth factor (PlGF) and IL-6 expression, ELISA kits for proinflammatory cytokines including VEGF, PlGF and IL-6 were used according to the manufacturer's instructions.
NF-κB p65 transcription factor assay
Cell nuclear fractions were isolated, and NF-κB activity was detected by using a NF-κB p65 Transcription Factor Assay Kit (ab133112, Abcam, UK) according to the manufacturer's instructions.
Statistical Analysis
Each experiment was repeated three times. Data are expressed as the means ± SD from at least three independent experiments. Comparisons of more than two groups were completed by one-way ANOVA followed by a Bonferroni test using GraphPad Prism software version 2.01 (GraphPad Software, La Jolla, CA, USA). P <0.05 was considered significant.
Acknowledgements
This study was supported by grants (Nos. 81401870) from the Natural Science Foundation of China (NSFC), the research fund of Shanghai Tongren Hospital, Shanghai Jiao Tong University School of Medicine (TRYJ201614) and Sixth people's hospital of Shanghai medical group projects.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen D, Xia M, Hayford C. et al. Expression of Human Tissue Factor Pathway Inhibitor on Vascular Smooth Muscle Cells Inhibits Secretion of Macrophage Migration Inhibitory Factor and Attenuates Atherosclerosis in ApoE-/- Mice. Circulation. 2015;131(15):1350-60
2. Ma H, Kong J, Wang Y L. et al. Angiotensin-converting enzyme 2 overexpression protects against doxorubicin-induced cardiomyopathy by multiple mechanisms in rats. Oncotarget. 2017;8(15):24548-24563
3. Sen A, Shin-Ichiro M, Yuhei S. et al. Depressor and Anti-Inflammatory Effects of Angiotensin II Receptor Blockers in Metabolic and/or Hypertensive Patients With Coronary Artery Disease: A Randomized, Prospective Study (DIAMOND Study). Journal of Clinical Medicine Research. 2016;8(10):743-748
4. Yano H, Hibi K, Nozawa N. et al. Effects of valsartan, an angiotensin II receptor blocker, on coronary atherosclerosis in patients with acute myocardial infarction who receive an angiotensin-converting enzyme inhibitor. Japanese Circulation Journal. 2012;76(6):1442-1451
5. Seara F D A C, Oliveira D F D, Silva D L S G D. et al. Administration of anabolic steroid during adolescence induces long-term cardiac hypertrophy and increases susceptibility to ischemia/reperfusion injury in adult Wistar rats. Journal of Steroid Biochemistry & Molecular Biology. 2017:171
6. Gao X M, Tsai A, Al-Sharea A. et al. Inhibition of the Renin-Angiotensin System Post Myocardial Infarction Prevents Inflammation-Associated Acute Cardiac Rupture. Cardiovascular Drugs & Therapy. 2017;31(2):1-12
7. Elkhatali S, Maayah Z H, El-Sherbeni A A. et al. Inhibition of Mid-chain HETEs Protects Against Angiotensin II-induced Cardiac Hypertrophy. J Cardiovasc Pharmacol. 2017;70(1):16-24
8. Zhang F, Ren X, Zhao M. et al. Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Scientific Reports. 2016;6:34621
9. Cui R, Tieu B, Recinos A. et al. RhoA mediates angiotensin II-induced phospho-Ser536 nuclear factor kappaB/RelA subunit exchange on the interleukin-6 promoter in VSMCs. Circulation Research. 2006;99(7):723
10. Shawky N M, Pichavaram P, Shehatou G S G. et al. Sulforaphane improves dysregulated metabolic profile and inhibits leptin-induced VSMC proliferation: implications toward suppression of neointima formation after arterial injury in western diet-fed obese mice. Journal of Nutritional Biochemistry. 2016;32:73-84
11. Shawky N M, Segar L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacological Research. 2017;119:251-264
12. Yoo SH, Lim Y, Kim SJ. et al. Sulforaphane inhibits PDGF-induced proliferation of rat aortic vascular smooth muscle cell by up-regulation of p53 leading to G1/S cell cycle arrest. Vascul Pharmacol. 2013;59(1-2):44-51
13. Gan N, Wu Y C, Brunet M. et al. Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. Journal of Biological Chemistry. 2010;285(46):35528-35536
14. Ashino T, Yamamoto M, Numazawa S. Nrf2/Keap1 system regulates vascular smooth muscle cell apoptosis for vascular homeostasis: role in neointimal formation after vascular injury. Sci Rep. 2016;6:26291
15. Choi S H, Park S, Oh C J. et al. Dipeptidyl peptidase-4 inhibition by gemigliptin prevents abnormal vascular remodeling via NF-E2-related factor 2 activation. Vascular Pharmacology. 2015;73:11-19
16. Ashino T, Yamamoto M, Yoshida T. et al. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler Thromb Vasc Biol. 2013;33(4):760-U273
17. Abbas M, Jesel L, Auger C. et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation. 2017;135(3):280-296
18. Herbert K E, Mistry Y, Hastings R. et al. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circulation Research. 2008;102(2):201
19. Browatzki M, Larsen D, Pfeiffer C A H. et al. Angiotensin II Stimulates Matrix Metalloproteinase Secretion in Human Vascular Smooth Muscle Cells via Nuclear Factor-κB and Activator Protein 1 in a Redox-Sensitive Manner. Journal of Vascular Research. 2005;42(5):415-423
20. Crajoinas R O, Polidoro J Z, Cp C D M. et al. Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. American Journal of Physiology Cell Physiology. 2016;311(5):C768-C776
21. Liu G C, Oudit G Y, Fang F. et al. Angiotensin-(1-7)-induced activation of ERK1/2 is cAMP/protein kinase A-dependent in glomerular mesangial cells. American Journal of Physiology - Renal Physiology. 2012;302(6F):784-790
22. Chen J, Peters A, Papke C L. et al. Loss of Smooth Muscle α-Actin Leads to NF-κB-Dependent Increased Sensitivity to Angiotensin II in Smooth Muscle Cells and Aortic Enlargement. Circulation Research. 2017;120(12):1903
23. Wei Y; Sowers JR; Nistala R; Gong H; Uptergrove GM; Clark SE; Morris EM; Szary N; Manrique C; Stump CS. Angiotensin II-induced NADPH Oxidase Activation Impairs Insulin Signaling in Skeletal Muscle Cells. Journal of Biological Chemistry. 2006;281(46):35137-35146
24. Florczyk U, Jazwa A, Maleszewska M. et al. Nrf2 regulates angiogenesis: effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid Redox Signal. 2014;20(11):1693-1708
25. Bai Q, Yang X, Lyu Z. et al. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behavioural Brain Research. 2017;321:79-86
26. Dai X, Yan X, Zeng J. et al. Elevating CXCR7 Improves Angiogenic Function of EPCs via Akt/GSK-3β/Fyn-Mediated Nrf2 Activation in Diabetic Limb Ischemia. Circulation Research. 2017;120(5):e7
27. Russo M, Spagnuolo C, Russo G L. et al. Nrf2 targeting by sulforaphane: a potential therapy for cancer treatment[J]. Critical Reviews in Food Science & Nutrition. 2016
28. Zhang M, Pan H, Xu Y. et al. Allicin Decreases Lipopolysaccharide-Induced Oxidative Stress and Inflammation in Human Umbilical Vein Endothelial Cells through Suppression of Mitochondrial Dysfunction and Activation of Nrf2. Cellular Physiology & Biochemistry. 2017;41(6):2255-2267
29. Wang L, Yang L, Lu Y, Chen Y, Liu T, Peng Y. et al. Osthole Induces Cell Cycle Arrest and Inhibits Migration and Invasion via PTEN/Akt Pathways in Osteosarcoma. Cellular Physiology & Biochemistry. 2016;38(6):2173-2182
30. Chen Y, Peng Y, Xu Z. et al. LncROR Promotes Bladder Cancer Cell Proliferation, Migration, and Epithelial-Mesenchymal Transition. Cellular Physiology & Biochemistry. 2017;41(6):2399-2410
31. Yang F, Yan G, Li Y. et al. Astragalus Polysaccharide Attenuated Iron Overload-Induced Dysfunction of Mesenchymal Stem Cells via Suppressing Mitochondrial ROS. Cellular Physiology & Biochemistry. 2016;39(4):1369-1379
32. Zhou S, Yu D, Ning S. et al. Augmented Rac1 Expression and Activity are Associated with Oxidative Stress and Decline of u03b2 Cell Function in Obesity. Cellular Physiology & Biochemistry. 2015;35(6):2135-2148
Author contact
![]() Corresponding author: Zhaohui Qiu, Li Jiang, Tel: (+86) 021 62909911; E-mail: qzh3503com, jianglicom, Address: Division of Cardiology, TongRen Hospital, Shanghai Jiao Tong University School of Medicine, 1111 Xianxia Road, Shanghai 200336, China
Corresponding author: Zhaohui Qiu, Li Jiang, Tel: (+86) 021 62909911; E-mail: qzh3503com, jianglicom, Address: Division of Cardiology, TongRen Hospital, Shanghai Jiao Tong University School of Medicine, 1111 Xianxia Road, Shanghai 200336, China