Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):453-463. doi:10.7150/ijbs.26703 This issue Cite
Research Paper
Changes in Chemokines and Chemokine Receptors Expression in a Mouse Model of Alzheimer's Disease
Adrián Jorda1, Omar Cauli2, Jose M. Santonja3, Martin Aldasoro1, Constanza Aldasoro1, Elena Obrador1, Jose Ma Vila1, Ma Dolores Mauricio1, Antonio Iradi1, Sol Guerra-Ojeda1, Patricia Marchio1, Soraya L. Valles1 ![]()
1. Department of Physiology, School of Medicine, University of Valencia. Spain
2. Faculty of Surgery and Chiropody, University of Valencia. Spain
3. Clinic Hospital of Valencia. Spain
Received 2018-4-16; Accepted 2018-11-21; Published 2019-1-1
Abstract

The amyloid precursor protein plus presenilin-1 (APP/PS1) mice are a frequently-used model for Alzheimer's disease studies (AD). However, the data relevant to which proteins are involved in inflammatory mechanism are not sufficiently well-studied using the AD mouse model. Using behavioral studies, quantitative RT-PCR and Western-blot techniques, significant findings were determined by the expression of proteins involved in inflammation comparing APP/PS1 and Wild type mice. Increased GFAP expression could be associated with the elevation in number of reactive astrocytes. IL-3 is involved in inflammation and ABDF1 intervenes normally in the transport across cell membranes and both were found up-regulated in APP/PS1 mice compared to Wild type mice. Furthermore, CCR5 expression was decreased and both CCL3 and CCL4 chemokines were highly expressed indicating a possible gliosis and probably an increase in chemotaxis from lymphocytes and T cell generation. We also noted for the first time, a CCR8 increase expression with diminution of its CCL1 chemokine, both normally involved in protection from bacterial infection and demyelination. Control of inflammatory proteins will be the next step in understanding the progression of AD and also in determining the mechanisms that can develop in this disease.
Keywords: Alzheimer's disease, chemokine receptors, chemotaxis, inflammation, behavior
Introduction
Alzheimer's disease (AD) is a common multifactorial neurodegenerative disorder that occurs mainly with aging. The neuropathological hallmarks of AD are amyloid plaques, intraneuronal tangles, and activation of glial cells [1-4]. Glial swelling and astrogliosis are a characteristic reaction of astrocytes to inflammation, oxidative stress and trauma, leading to the secretion of several potentially toxic products, including inflammatory agents [1] and oxidative stress mediators [5]. Data from our laboratory have shown the possible pathway to increase mitochondriogenesis and anti-inflammatory proteins to recover from amyloid beta (Aβ) action in primary culture neural cells [6]. Aβ deposition can result in brain damage but whether different chemokines in brain could participate in the Aβ-induced brain damage and neurodegeneration is unclear. Our study evaluates first, if changes in young and old-mice behavior occur. Second, whether changes in chemokine expressions, which act in inflammatory processes with chemotaxis and infiltration across blood- brain barrier (BBB), take place in transgenic APP/PS1 mice aged 7 months and 22-24 months, as we demonstrated previously in cells in primary culture [6-9]. Chemokine signaling has been implicated in the pathogenesis of diabetic neuropathy but the involvement of the chemokine CC motif ligand 1 (CCL1) and chemokine CC motif receptor 8 (CCR8) expressions remain unknown in AD. Different authors, have demonstrated the involvement of chemokines in APP/PS1 mice. Zhu et al. [10] have showed an increase in CCL4 mRNA and protein expression in the brains of APP/PS1 mice vs. controls. This data is consistent with a potential inflammatory contribution to the neurodegenerative cascade in this model [10]. Also, high CCL4 expression has been found in multiple tissues affected by chronic inflammatory disease including AD [11]. In AD patients and in AD animal models, included APP/PS1, the chemokine CXCL10 is found in high concentrations, suggesting a pathogenic role for this chemokine and its receptor, CXCR3 [12]. Furthermore, plaque burden and Aβ levels were strongly reduced in CXCR3-deficient APP/PS1 mice and CXCR3 antagonist increased microglia Aβ phagocytosis [12]. In other chemokines, such as CCL11 was increase observed in APP/PS1 double transgenic mice [10]. CCL11, which is predominantly expressed by the activated microglia, was observed in the cerebrospinal fluid of APP/PS1 double transgenic mice compared with wild-type (WT) mice. Also, the deletion of CCR3 in APP/PS1 mice significantly reduced the phosphorylation of CDK5 [13]. The goal of this study was to examine, for the first time, the protein expression of CCL1 chemokine and its receptor CCR8, and also expression of the chemokines CCL3, CCL4, CCL5 and their receptor CCR5 in APP/PS1 mice, implicated in brain damage. Our findings may lead to more specific control of inflammatory changes that occur in AD brain.
Material and Methods
Animals
APP/PS1 mice (B6C3-Tg) and wild type littermates were used. APP/PS1 mice express a chimeric mouse/human APP 695 cDNA containing the Swedish (KM670/671 NL) mutation co-integrated with the human presenilin 1 (PS1) gene harboring the DE9 mutation. Two groups were assayed, APP/PS1 and Wild type mice and both of them were fed ad libitum on standard diet (Letica, Barcelona, Spain). Mice were kept on a 12-h light/12-h dark cycle with the room temperature maintained at 22ºC. All animal procedures were carried out in accordance with the European legislation on the use and care of laboratory animals (CEE 86/609). Experimental research on mice was performed with the approval of the ethics committee on animal research of the University of Valencia (Spain).
Behavior studies
APP/PS1 and Wild type mice (N=25, 6 months) were used to examine motor function and memory and learning functions using active avoidance and object recognition tests and motor coordination (rotarod test). The tests were performed at different ages (at 7, 21 and 24 months).
Active avoidance
The active avoidance task is designed to test the ability of the mice to avoid an aversive event by first learning to perform a specific behavior in response to a stimulus cue. The test was performed in one single day and consisted of 50 trials per animal, as previously described [14].
Object recognition test
This test exploits the tendency of mice to preferentially explore novel elements of their environment or the same element located in different position. Thus, when a mouse is presented with both a novel and recently presented familiar object, it will spend significantly more time exploring the novel object (object recognition memory). When a mouse is presented with two identical known objects but one of them is located in a different position it will spend significantly more time exploring the object in the new location (object location memory test). The familiar objects were presented in a previous training session 1 h before the test. The percentage of time exploring the non-familiar object in the training session over total exploration time (exploration time of the familiar plus the non-familiar objects) was represented. The object recognition or location index was calculated as follows: time of exploration of novel object (or the same object in a new location-time of exploration of familiar object + time of exploration of novel object) [15].
Balance beam test
This task is used to assess sensorimotor function and balance in rodents.
In this test, the ability of mice to pass through a narrow beam to reach a dark box is evaluated. To force mice to pass through the beam, a white light illuminates the beginning of the beam. The wooden beam (1 x 100 cm) was elevated to a height of 1 m above the floor. The time required to cross to the escape box at the other end of beam and the number of forelimb and hind-limb paw slips were recorded. A paw slip was defined as any paw coming off the top of the beam or any limb use on the side of the beam. The day of the test, four trials were performed before recording the results. The goal was to familiarize the mice to the beam and to train them to the presence of the dark box at the end of the beam. The narrow beam and the dark box were cleaned after each trial with ethanol. The latency between trials.
Grip strength
Grip strength is measured non-invasively by taking advantage of the mouse's instinctive tendency to grab as they are gently pulled backward. This method can be used to test for limb grip using a horizontal bar with small diameter 0.1 cm which is attached to a force transducer. The advantages are that the grip strength is reproducible and can be measured repeatedly in rodents to detect progressive motor deficits, value of therapy and genetic modifications [16]. Mean latency to fall was determined with a maximum cut-off at 60 s.
Biochemical studies
APP/PS1 and Wild type mice (N=10, 6 months) were used. All the biochemical studies were done in 7 month-old mice.
Real-time polymerase chain reaction analyses
Cortical brain samples were collected from each mouse into an RNAlater solution (Ambion, Austin, TX, USA), an RNA stabilization reagent, following the manufacturer's instructions. Total RNA was extracted with Tripure isolation reagent (Roche Molecular Biochemical, Basel, Switzerland) and concentration and integrity were assessed in RNA 6000 Nano Labchips using Agilent 2100 Bio-analyzer (Agilent Technologies, Foster City, CA, USA). Ready-to-use primers and probes from the assay-on-demand service of Applied Biosystems were used for the quantification of selected target gene: CCL1 (Mm99999220_Mh), CCL3 (Mm00441259_g1), CCL4 (Mm00443111_m1), CCL5 (Mm01302428_m1), CCR5 (Mm01963251_s1), CCR8 (Mm99999115_s1), ABCF1 (Mm01275245_m1), IL-3 (Mm00439631_m1), and endogenous reference gene β-actin (Mm00607939_s1). RNA samples were reverse-transcribed using random hexamers and MultiScribe reverse transcriptase (Applied Biosystems). After complementary DNA synthesis, real-time polymerase chain reaction (RT-PCR) was carried out using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Samples were run in triplicate, and expression changes were generated by calculating 2-ΔΔCt.
Western-blot analysis
Protein extracts from cortex brain were mixed with equal volumes of SDS buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 0.5% (v/v) 2-mercaptoethanol, 1% bromophenol blue and 19% glycerol) and then boiled for 5 min. Protein concentration was determined using a modified Lowry method [17]. Proteins were separated by SDS-PAGE gels and transferred to nitrocellulose membranes using standard techniques. Membranes were blocked with 5% dried milk in TBS containing 0.05% Tween-20 and then incubated with the corresponding antibodies following manufacturer's recommendations. The blots were washed three times with a washing buffer (phosphate-buffered saline, 0.2% Tween 20) for 15 min each and then incubated for 1 h with a secondary horseradish peroxidase-linked anti-rabbit or anti-mouse IgG antibody (Cell Signaling Technologies, Barcelona, Spain). As above, the blots were washed three times and developed using the enhanced chemiluminescence (ECL) procedure as specified by the manufacturer (Pharmacia biotechnology, San Francisco, CA, USA). Auto-radiographic signals were assessed using a Bio-Rad scanning densitometer. Antibodies: Monoclonal anti-glial fibrillary acidic protein (GFAP) and anti-microtubule associated protein 2 (MAP-2) from Santa Cruz Biotechnology. Monoclonal anti-CCR5 antibody and anti-CCR8 antibody from Abcam Biotechnology. All other reagents are analytical or culture grade purity.
Data analysis and statistics
All values are expressed as means ± SD. The differences between transgenic (APP/PS1) and Wild type (WT) mice were determined with unpaired Student's t-test. All statistical analyses were performed using the Graph-Pad Prism software (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was accepted at p values < 0.05.
Results
General motor, muscular impairment and coordination
The ability of mice to pass through a narrow beam showed the difference of activity between APP/PS1 and Wild type mice using the behavior activity of the beam-walking test. Behaviorally, APP/PS1 mice at 7 and 22-24 months did not display general motor and muscular impairment as demonstrated by unaffected exploratory cage activity, grip strength and toe hold ability to cross a beam in the beam-walking test (Figure 1 A-C).
Behavior assays. A: Exploration of two identical object (sec in 5 minutes), with no significantly differences between wild type (WT) and transgenic (TG) mice. B: Grip strength (time in 60 seconds). C: Time to cross the beam (sec). D: number of foot faults, ***p<0.01 vs. wild type mice.
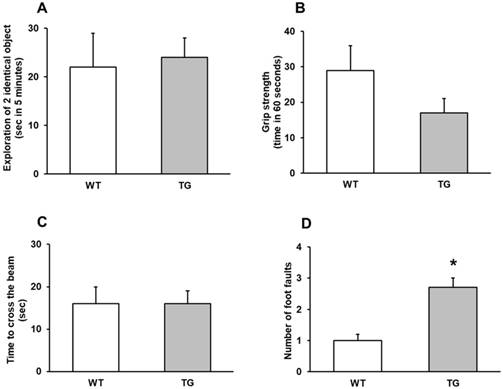
By contrast, a significant number of foot faults in the beam-walking test was observed in APP/PS1 mice 22-24 months compared to Wild type mice (p<0.001) (Figure 1D).
Recall spatial information and memory test
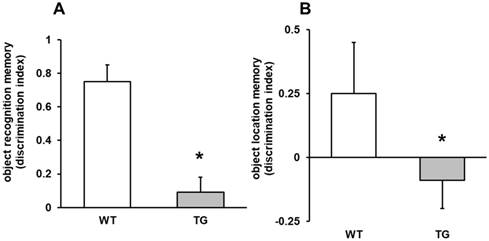
The ability of mice to acquire and recall spatial information was assessed by active avoidance task and by object recognition test. In the memory test, APP/PS1 mice displayed memory impairment on both object recognition (p<0.001) and object location memory (p<0.05) compared to Wild type mice (Figures 2 A-B). Memory changes were not detected in 7 month mice.
Object location memory and object recognition memory. A: object recognition memory (discrimination index), ***p<0.01 vs. wild type mice. B: object location memory (discrimination index), *p<0.05 vs. WT mice.
Expression of GFAP and MAP-2 on the APP/PS1 mice
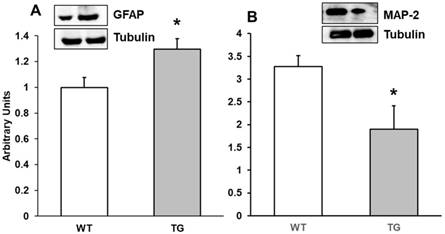
Alterations in motor, memory and object recognition may be related to brain inflammation, demyelination and cell infiltration in APP/PS1 mice. APP/PS1 mice show increase in Aβ generation and accumulation and activation of astrocytes and microglia is critical in this process. For this reason, we determined by Western-blot both GFAP and MAP-2 expression. GFAP is an astrocyte protein marker and MAP-2 a marker of microtubule assembly in neurons. Our results showed that expression of GFAP was higher in APP/PS1 cortex at 7 months compared with Wild type mice (Figure 3A), which could indicate an increase in the number of astrocytes in APP/PS1 compared to Wild type mice. Conversely, we noted a decrease in MAP-2 expression on APP/PS1 compared with Wild type mice (Figure 3B).
Protein expression of A: GFAP and B: MAP-2 in cortex of APP/PS1 and WT mice. A representative immunoblot is shown in the panel. Data are mean ± SD of four independent experiments. *p<0.05 vs. WT.
Expression of CCL1 chemokine and its CCR8 receptor in brain cortex
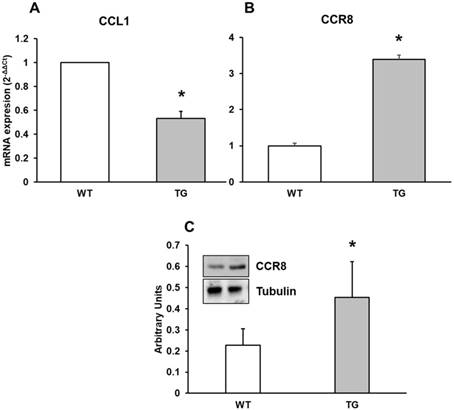
In the present study, we determined mRNA expression of CCL1 chemokine by real-time RT-PCR from APP/PS1 and Wild type cortex in mice. Also we assessed CCR8 protein expression by RT-PCR and Western-blot techniques in all mice. CCL1 expression was higher in Wild type compared with APP/PS1 mice (Figure 4A) and CCR8 showed higher expression in APP/PS1 compared to Wild type mice (Figure 4B and 4C), demonstrating different expression values between CCR8 and its chemokine in cortex of APP/PS1 and Wild type mice. This expression was inversely correlated with the expression of CCL1 in APP/PS1 mice.
mRNA expression of A: CCL1 and B: CCR8 in cortex of APP/PS1 and WT mice. C: Protein expression of CCR8 by Western-blot. A representative immunoblot is shown in the panel. Data are mean ± SD of four independent experiments. *p<0.05 vs. WT.
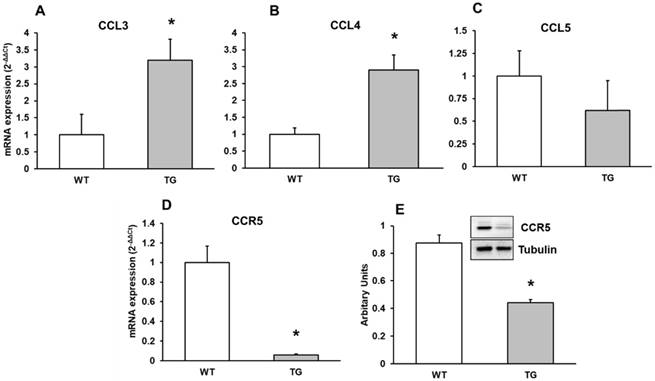
Expression of CCL3, CCL4, CCL5 chemokines and the CCR5 receptor
Here we detected a higher CCL3 and CCL4 expression without a different expression in CCL5 chemokine in APP/PS1 to Wild type mice (Figure 5A, 5B, 5C). We therefore performed real-time RT-PCR and Western-blot methods expression of CCR5 in mouse brain cortex. CCR5 expression decreased in transgenic compared to Wild type mice (Figure 5D and 5E).
mRNA expression of A: CCL3, B: CCL4 and C: CCL5 and D: CCR5 by Western-blot in cortex of APP/PS1 and WT mice. A representative immunoblot is shown in the panel. Data are mean ± SD of four independent experiments. *p<0.05 vs. WT.
IL-3 mRNA expression in APP/PS1 transgenic compared to Wild type mice
IL-3 regulates the proliferation, survival and differentiation of hematopoietic cells and has been detected in the CNS, but its physiological role in neural cells is poorly understood in AD. IL-3 prevents neuronal death induced by Aβ and has been detected in higher expression in AD, demonstrating that IL-3 could play a neuroprotective role in AD. We showed here an increase in IL-3 expression in APP/PS1 compared to Wild type mice (Figure 6A).
mRNA expression of A: IL-3 and B: ABCF1 in cortex of APP/PS1 and WT mice. Data are mean ± SD of four independent experiments. *p<0.05 vs. WT.
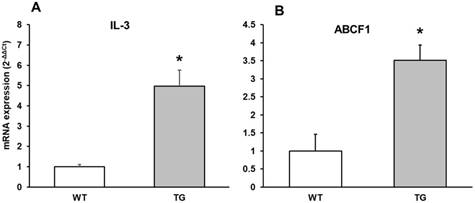
ABCF1 mRNA expression in APP/PS1 compared to Wild type mice
ABCF1 gene expression was determined. The ATP binding cassette (ABC) super-family of proteins are present in brain and harness the energy from the hydrolysis of ATP in order to transport substrates across cellular membranes and power cellular machinery. ABCF1 can import and export molecules across the cell and was initially identified as a protein that was up-regulated in synoviocytes. Also ABCF1 is important in translation initiation. Here, we found that its expression is higher in APP/PS1 compared with Wild type mice, and increase 3 times respect to Wild type mice (Figure 6B).
Discussion
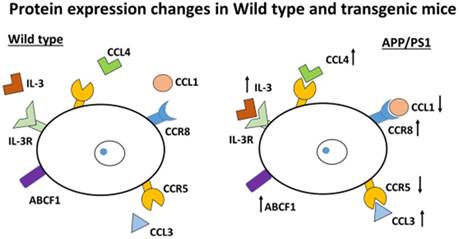
This study indicates that changes in chemokines and chemokine receptors are important in the involvement of inflammation in AD and one of the possible causes for the development of this disorder. This finding suggests BBB damage and chemotaxis occur with inflammatory consequences in the brain. In addition, the molecules movement across cell membranes could be possibly higher in transgenic mice compared to Wild type mice which suggests that compensation of energy loss occurred in transgenic mice compared with Wild type mice as evidenced by the results obtained with the changes in ABCF1 protein. Demyelination occurs due to an increase in CCR8, as different authors have suggested, and as we have confirmed here (Figure 7).
Inflammatory protein changes in APP/PS1 mice. The schema shows the up-regulation or down-regulation of different proteins. Pro-inflammatory mediators, such as CCL3 and CCL4, are increased. By contrast, CCR5 is down-regulated. The protein ABCF1 (a member of the superfamily of ATP-binding cassette (ABC) transporters) is increased. IL-3 (involved in the immune system) is highly expressed. Also CCR8 is increased (important for the migration of various cell types into the inflammatory sites).
Our previous results demonstrated that Aβ peptide causes oxidative stress in neurons [8] and in astrocytes in primary culture [9]. Others have demonstrated that secretion of reactive oxygen species (ROS) and reactive nitrogen species (RNS) by inflammatory cells is a major mechanism for attacking opsonized targets while activated glial cells can produce large amounts of ROS/RNS by various mechanisms [10]. The APP/PS1 mice is well known model of AD and the behavior deficits are not detected until an elderly age in mice. Our present results indicate that the alterations in the cortex of 7 month APP/PS1 mice take place before deficits in behavior appear. In the present study we show that transgenic mice (APP/PS1) (22-24-months) display severe motor incoordination and amnesic impairment on both object recognition memory and object location memory. On the contrary, behavior experiments with 7 month mice did not show changes between transgenic and Wild type mice in this study. The present study aimed to evaluate also whether a deregulation of the expression of certain chemokines involved in demyelination, phagocytosis and chemotaxis, occurs in the APP/PS1 mice. The measurements were made in the cortex of 7 month APP-PS1 mice because this is the second brain region most affected when memory is damaged. Our data shows a decrease in CCL1 expression and an increased expression of its receptor CCR8 and also an upregulation of CCL3 and CCL4 chemokine expressions with a reduced expression of their receptor CCR5 in APP/PS1 mice compared to Wild type mice. CCR8 encodes a member of the beta chemokine receptor family and its ligand is CCL1 [18]. Here, we detected for the first time, changes in expression of CCR8 and its chemokine, CCL1 in APP/PS1. CCR8 expression was increased in transgenic mice, indicating a rise in receptor expression to supply downregulation of its chemokine CCL1. CCL1 attracts immune cells by interacting with the cell surface chemokine receptor CCR8 [19] and contributed to the development of neuropathic pain [20]. CCR8 expression is controlled by skin-specific factors derived from epidermal keratinocytes and not by resident dendritic cells. It is also expressed in neurons, microglia and astrocytes [21] and has been reported to accumulate microglial cells in the CNS of APP/PS1 [21]. This is related to the action of promoting chemotaxis of mononuclear cells and phagocytosis [22]. Trebst et al., suggested that CCR8 is not expressed in Alzheimer's type macrophages surrounding senile plaques [23]. Trebst et al. data include tissue from AD humans, whereas our results were done in 7 month APP/PS1 mice (corresponding to nearly 30 years old in human). At this age in humans, Aβ1-42 plaques are detected and probably eliminated. On the other hand, there is no elimination of those plaques in elderly AD patients. Perhaps expression of CCR8 disappears when AD has developed. The combination of 1α,25-dihydroxyvitamin D3 and prostaglandin E2 (PGE2) results in a high CCR8 expression, demonstrating the importance of tissue environments in maintaining cellular immune surveillance networks within distinct healthy tissues [24]. This receptor is expressed on primary cultured microglia and astrocytes and CCL1 mRNA is more robustly expressed in neurons than in glia [21]. This data indicates that CCL1 works as a mediator between neuron and microglia in the CNS. Thus, a decrease in CCL1 expression, such as we observed, could indicate a reduction in communication between neurons and glia and also a decrease in neuron density in APP/PS1 mice as we show by a reduction of MAP-2 expression.
Inflammation occurs in Alzheimer disease with a production of cytokines and chemokines inducing progression of the inflammatory cascade [25, 26]. Lipopolysaccharide stimulation of primary microglial cultures is reported to diminish the levels of CCL1 in a diabetic neuropathy mouse model [27]. Our results support the hypothesis that CCL1 has an important role as a mediator in neuro-immune interactions and that CCL1/CCR8 cross-talk contributes to the development of inflammation in AD. Another key function of CCL1 is the role in microglial phagocytosis eliminating neurotoxic molecules, cellular debris or microbes [23]. A decrease in CCL1 expression could explain the decline in elimination of Aβ by phagocytosis detected in AD [28]. In Taiep rats, an upregulation in CCR8 mRNA and protein were detected at 1 month old, explaining the accumulation of microglial cells in the CNS due to the action of promoting chemotaxis and phagocytosis [29].
The volume of the human brain has important variation especially in relation to age. Interleukin-3 (IL-3) is reported to be strongly associated with brain volume changes [30]. Moreover, IL-3 receptor alpha is expressed in neural progenitors and neurons, and promotes proliferation and survival of neural progenitors with novel roles of IL-3 in regulating brain development [30]. Here we show that IL-3 is highly expressed in APP/PS1 mice indicating possible changes in brain volume and confirming previous findings [30]. Furthermore, correlation between IL-3 and CCR8 has been detected in transcriptional analysis of multiple sclerosis brain lesions demonstrating a complex pattern of cytokine expression in this disease [31]. Also, overexpression of CCR5 and IL-3 has been detected in severe tuberculosis [32].
CCR5 is expressed in neurons associated with memory and in monocytes, macrophages, astrocytes, and microglia as well as in epithelium, endothelium, vascular smooth muscle, and fibroblasts [33]. The pro-inflammatory cytokines CCL3, CCL4 and CCL5 are its ligands, involved in the effector responses [34]. Here we detected a downregulation of CCR5 in APP/PS1 mice with upregulation of CCL3 and CCL4 chemokines and without changes in CCL5 expression. The role of CCR5 has been mainly studied in the context of HIV (human immunodeficiency virus) infection [35]. In 1996, different laboratory groups indicated that CCR5 is a major co-receptor for the entry of HIV into target cells [36-38]. But the receptor is also expressed in the CNS [39-41] and in addition, to the modulation of immune response. CCR5 can influence neuronal survival and has been suggested to be involved in neuroprotective mechanisms [42]. It is important to note that the cell death in the brains of CCR5-/- mice are significantly higher than in the brains of CCR5+/+ mice [43] and also an increase of reactive astrocytes exist in these mice, suggesting that absence of CCR5 leads to the activation of astrocytes. Furthermore, CCR5 is implicated in neuronal growth and differentiation during development and is related to striatal dopamine release [44]. Deletion of CCR5 could be implicated in a deficient development and maturation of dopaminergic neurons and prevents macrophage infiltration and demyelination [45, 46]. Moreover, Hwang and collaborators have indicated an increase in Aβ deposits and impaired memory when CCR5 declines and supports cell death that occurs in AD patients [47]. In the brain of CCR5 knockout mice, LPS injection significantly increases astrogliosis, β-secretase expression and Aβ deposition compared with CCR5 Wild type mice. Furthermore, deficiency of CCR5 results in activation of astrocytes and Aβ deposits [47].
We here detected an increase in chemokines CCL3 and CCL4 expression in cortex of 7-month-old APP/PS1 mice. There could be two possibilities for this finding. First, by the increment of the chemokines CCL3 and CCL4 that have as consequence the decrement in CCR5 expression, or contrarily, by decrease in CCR5 expression that produces an increase in CCL3 and CCL4 chemokine expressions. According to our data we cannot deduce what is the sequence of events that would lead to that process in APP/PS1 mice. Studies by Man and collaborators [48] show that AD patients had a higher CCL3 level compared to healthy controls in peripheral T lymphocytes. A study in 2017, demonstrated that an increase in CCL3 expression produced accumulation of glial cells, monocytes and lymphocytes in a hypoxia mouse model and probably contributes to the pathogenesis of AD [49]. Furthermore, CCL3 is upregulated in the CNS during Alzheimer's disease [50, 51] and is located in microglia, astrocytes and perivascular macrophages [52, 53]. CCL3 is a member of β-chemokine subfamily and is involved in the recruitment and activation of polymorph-nuclear leukocytes. Accumulated T cells lead to the increased levels of pro-inflammatory cytokines and cause chronic inflammation, which enhance neurotoxicity and impair microglial function [54]. Cell infiltration in the brain might also contribute to the cognitive impairment of tau pathology. Other authors found that hippocampal T cells might modulate microglial and/or astrocytic activation status and lead to detrimental impact on synaptic plasticity [55]. Furthermore, CCL3 is constitutively expressed by the nigral dopaminergic neurons and modulates neurotransmission by liberating transmitters across ion-channel gating and also long-term potentiation [56]. Others have indicated that active transport of CCL3 across the BBB in vitro at the luminal side occurs independently of the chemokine receptors CCR1 and CCR5. Moreover, leukocyte transporter was decreased in inflammatory conditions [57], suggesting additional pathways to increase immune cell transport across BBB. One of those pathways, we hypothesize could be ABCE1. CCL4 mRNA and protein are upregulated in APP/PS1 mice compared with Wild type and correlates with Aβ increased levels [13]. These authors also demonstrated a relationship among astrocytes, Aβ and CCL4 location in the brain of APP/PS1 transgenic mice. Astrocytes are the source of Aβ1-42 because they produce an overexpression of β-secretase (BACE1), an enzyme that cleaves amyloid precursor protein (APP) to produce Aβ [58] which accelerates Aβ1-42 deposition and can possibly result in impaired memory function [59]. These results correlate well with the data published by Zhu and collaborators where a clean-up of amyloid toxicity occurs inside the brain of APP/PS1 mice [13]. Furthermore the production of CCL4 is controlled by peripheral blood mononuclear cells in AD patients [60]. Song and collaborators demonstrate an increase in deposition and soluble Aβ1-42 in the brain of TL4RM (Toll-like receptor 4 mutation), which was associated with decreased expression levels of CCL4 and CCL3. This action increases cognitive functions in the hippocampus of these mice. Consequently, microglia is activated via TLR4 signaling to reduce Aβ deposits and preserve cognitive functions from Aβ-mediated neurotoxicity [61].
Our results also demonstrated a higher mRNA expression of ABCF1 in APP/PS1 compared to Wild type mice. ABCF1 is a member of the super-family ABC that can be divided in three categories, importers, exporters and DNA repair and translation [62]. ABCF1 lack trans-membrane domain so it falls into the third category [63]. Its function could be interacting in the initiation of translation through an interaction with eukaryotic translation initiation factor 2. Abcf1-/- mice are embryonic lethal and Abcf1+/- mice are fertile and develop normally in adults. This protein was highly correlated with active proliferation and differentiation of cell types [64]. Furthermore, it is expressed in many tissues, including brain, where it is particularly expressed in pyramidal cells, cerebellar Purkinje cells and in hippocampus. On the contrary, it is lower expressed in the pyramidal tracts and the thalamus [64]. Recently investigators have reported that ABCF1 is a critical mediator of N6-methyladenosine (m6A) translation promoter under both stress and physiological conditions [65], supporting the role of ABCF1 in illnesses such as AD.
Conclusions
Our results show that changes in chemokines and chemokine receptors expression could explain the induction of chronic inflammation and events such as demyelination, phagocytosis and chemotaxis which are likely involved in AD. Pro-inflammatory mediators, such as CCL3 and CCL4, are increased and by contrast, CCR5 is down-regulated indicating probably an increase in astrogliosis and number of astrocytes. In addition, we detected an increased expression by induction of IL-3 protein. The ABCF1 increase detected here reinforces the idea of further investigation concerning the import and/or export of molecules between brain cells that may be affected in AD. Since chemokines can cross BBB in this mouse model of AD, disturbed normal physiology is expected. Chemotaxis with a possible accumulation of microglia and demyelination could be produced by CCR8 (Figure 7). These findings suggest that no appropriate crosstalk seems to occur between brain cells.
Proper control of inflammatory chemokines, chemokine receptors and proteins helping molecules that cross cell membranes could be a potential next step in Alzheimer's disease treatment.
Acknowledgements
We thank Mrs. Pilar Ribera for its help in the laboratory.
Funding
Not applicable. The APP/PS1 mice were provided by Dr. Juan Gambini, School of Medicine. All other material was obtained from each researcher.
Authors' contributions
SLV conceived of and designed the study, collected data, interpreted the data, and drafted the manuscript. AI performed statistical analysis and interpreted the data. AJ and MPR did the majority of the experiments, interpreted the data and collect data. OC did behavior experiments and interpreted the data. JMS interpreted the data from western-blot. JCC did ABCF1 experiments and interpreted the data. PM, CA, MA, MDM, EB, JMV, MP, SGO collected data and critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal procedures were carried out in accordance with the European legislation on the use and care of laboratory animals (CEE 86/609). Experimental research on mice was performed with the approval of the ethics committee on animal research of the University of Valencia (Spain) and all participants provided written informed consent. All procedures were performed in accordance with the 1964 Helsinki declaration and its later amendments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Giunta B, Fernandez F, Nikolic WV, Obrego E, Rrapo E, Town T, Tan J. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation. 2008;5:51-65
2. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741-66
3. Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417-20
4. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nature Medicine. 2006;12:1005-15
5. Perry G, Cash AD, Smith M. Alzheimer disease and oxidative stress. J Biomed Biotechnol. 2002;2:120-23
6. Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, Iradi A, Obrador E, Ortega A, Mauricio MD, Vila JM, Valles SL. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int J Med Sci. 2015;12:48-56
7. Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, Iradi A, Obrador E, Mauricio MD, Vila JM, Marchio P, Valles SL. WIN 55,212-2, agonist of cannabinoid receptors, prevents amyloid β1-42 effects on astrocytes in primary culture. PLoS One. 2015;10:e0122843. doi: 10.1371/journal.pone.0122843
8. Valles SL, Borrás C, Gambini J, Furriol J, Ortega A, Sastre J, Pallardo FV, Viña J. Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging cell. 2008:7 112-18
9. Valles SL, Dolz-Gaiton P, Gambini J, Borras C, Lloret A, Pallardo FV, Viña J. Estradiol or genistein prevent Alzheimer's disease-associated inflammation correlating with an increase PPAR gamma expression in cultured astrocytes. Brain Res. 2010;1312:138-44
10. Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci. 2007;64:2202-2210
11. Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. 1998;153:31-37
12. Krauthausen M, Kummer MP, Zimmermann J, Reyes-Irisarri E, Terwel D, Bulic B, Heneka MT, Müller M. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer's disease model. J Clin Invest. 2015;125:365-78
13. Zhu M, Allard JS, Zhang Y, Perez E, Spangle EL, Becker KG, Rapp PR. Age-related brain expression and regulation of the chemokine CCL4/MIP-1β in APP/PS1 double-transgenic mice. J Neuropathol Exp Neurol. 2014;73:362-74
14. Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;1:47-59
15. Aguilar MA, Miñarro J, Felipo V. Chronic moderate hyperammonemia impairs active and passive avoidance behavior and conditional discrimination learning in rats. Exp Neurol. 2000;161:704-13
16. Connolly JF. Applying cognitive research in the twenty-first century: event-related potentials in assessment. Brain Cogn. 2000;42:99-101
17. Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346-56
18. Garlisi CG, Xiao H, Tian F, Hedrick JA, Billah MM, Egan RW, Umland SP. The assignment of chemokine-chemokine receptor pairs: TARC and MIP-1 beta are not ligands for human CC-chemokine receptor 8. Eur J Immunol. 1999;29:3210-15
19. Oshio T, Kawashima R, Kawamura YI, Hagiwara T, Mizutani N, Okad T, Otsubo T, Inagaki-Ohara K, Matsukawa A, Haga T. Chemokine receptor CCR8 is required for lipopolysaccharide triggered cytokine production in mouse peritoneal macrophages. PLoS One. 2014;9:e94445
20. Akimoto N, Honda K, Uta D, Beppu K, Ushijima Y, Matsuzaki Y, Nakashima S, Kido MA, Imoto K, Takano Y, Noda M. CCL-1 in the spinal cord contributes to neuropathic pain induced by nerve injury. Cell Death Dis. 2013;20:4 e679
21. Akimoto N, Ifuku M, Mori Y, Noda M. Effects of chemokine (C-C motif) ligand 1 on microglial function. Biochem Biophys Res Commun. 2013;436:455-61
22. Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030-38
23. Trebst C, Staugaitis SM, Kivisäkk P, Mahad D, Cathcart MK, Tuck B, Wei T, Rani MR, Horu R, Aldape KD. CC chemokine receptor 8 in the central nervous system is associated with phagocytic macrophages. Am J Pathol. 2003;162:427-38
24. McCully ML, Collins PJ, Hughes TR, Thomas CP, Billen J, O'Donnell VB, Moser B. Skin Metabolites Define a New Paradigm in the Localization of Skin Tropic Memory T Cells. J Immunol. 2015;195:96-104
25. Gorelick PB. Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann N Y Acad Sci. 2010;1207:155-62
26. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161-67
27. Zychowska M, Rojewska E, Piotrowska A, Kreiner G, Nalepa I, Mika J. Spinal CCL1/CCR8 signaling interplay as a potential therapeutic target - Evidence from a mouse diabetic neuropathy model. Int Immunopharmacol. 2017;52:261-71
28. Takata K, Amamiya T, Mizoguchi H, Kawanishi S, Kuroda E, Kitamura R, Ito A, Saito Y, Tawa M, Nagasawa T. et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Aβ accumulation through suppression of neuronal γ-secretase activity and promotion of microglial amyloid-β phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer's disease. Neurobiol Aging. 2018;62:197-209
29. Soto-Rodriguez G, Gonzalez-Barrios JA, Martinez-Fong D, Blanco-Alvarez VM, Eguibar JR, Ugarte A, Martinez-Perez F, Brambila E, Peña LM, Pazos-Salazar NG. et al. Analysis of chemokines and receptors expression profile in the myelin mutant taiep rat. Oxid Med Cell Longev. 2015:397310 doi: 10.1155/2015/397310
30. Luo XJ, Li M, Huang L, Nho K, Deng M, Chen Q, Weinberger DR, Arias Vasquez A, Rijpkema M, Mattay VS, Saykin AJ, Shen L. et al. The Interleukin 3 Gene (IL3) Contributes to Human Brain Volume Variation by Regulating Proliferation and Survival of Neural Progenitors. PLoS One. 2012;11(30):doi.org /10.1371/journal.pone.0050375
31. Baranzini SE, Elfstrom C, Chang SY, Butunoi C, Murray R, Higuchi R, Oksenberg JR. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. J Immunol. 2000;165:6576-6582
32. Qiu L, Huang D, Chen CY, Wang R, Shen L, Shen Y, Hunt R, Estep J, Haynes BF, Jacobs WR Jr. et al. Severe tuberculosis induces unbalanced up-regulation of gene networks and overexpression of IL-22, MIP-1alpha, CCL27, IP-10, CCR4, CCR5, CXCR3, PD1, PDL2, IL-3, IFN-beta, TIM1, and TLR2 but low antigen-specific cellular responses. J Infect Dis. 2008;198:1514-9
33. Corbeau P, Reynes J. CCR5 antagonism in HIV infection: ways, effects, and side effects. Aids. 2009;23:1931-43
34. Trifilo MJ, Bergmann CC, Kuziel WA, Lane TE. CC chemokine ligand 3 (CCL3) regulates CD8(+)-T-cell effector function and migration following viral infection. J Virol. 2003;77:4004-14
35. Venuti A, Pastori C, Lopalco L. The Role of Natural Antibodies to CC Chemokine Receptor 5 in HIV Infection. Front Immunol. 2017;8:1358
36. Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955-58
37. Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W. et al. The betachemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135-48
38. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM. et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661-6
39. Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 2001;22:147-84
40. Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res. Rev. 2005;48:16-42
41. Rostene W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895-903
42. Sorce S, Myburgh R, Krause KH. The chemokine receptor CCR5 in the central nervous system. Prog Neurobiol. 2011;93:297-311
43. Kaul M, Ma Q, Medders KE, Desai M, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death and Differentiation. 2007;14:296-305
44. Park MH, Lee YK, Lee YH, Kim YB, Yun YW, Nam SY, Hwang SJ, Han SB, Kim SU, Hong JT. Chemokines released from astrocytes promote chemokine receptor 5-mediated neuronal cell differentiation. Exp Cell Res. 2009;315:2715-26
45. Choi DY, Lee MK, Hong JT. Lack of CCR5 modifies glial phenotypes and population of the nigral dopaminergic neurons, but not MPTP-induced dopaminergic neurodegeneration. Neurobiology of Disease. 2013;49:159-68
46. Glass WG, Liu MT, Kuziel WA, Lane TE. Reduced macrophage infiltration and demyelination in mice lacking the chemokine receptor CCR5 following infection with a neurotropic coronavirus. Virology. 2001;288:8-17
47. Hwang CJ, Park MH, Hwang JY, Kim JH, Yun NY, Oh SY, Song JK, Seo HO, Kim YB, Hwang DY. et al. CCR5 deficiency accelerates lipopolysaccharide-induced astrogliosis, amyloid-beta deposit and impaired memory function. Oncotarget. 2016;7:11984-99
48. Man SM, Ma YR, Shang DS, Zhao WD, Li B, Guo DW, Fang WG, Zhu L, Chen YH. Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging. 2007;28:485-96
49. Zhang F, Zhong R, Li S, Fu Z, Cheng C, Cai H, Le W. Acute Hypoxia Induced an Imbalanced M1/M2 Activation of Microglia through NF-κB Signaling in Alzheimer's Disease Mice and Wild-Type Littermates. Front Aging Neurosci. 2017;9:282
50. Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM Brachova L, Yan SD, Walker DG, Shen Y. et al. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001;35:72-9
51. Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging. 2001;22:957-66
52. Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F. Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. Journal of Neuropathology & Experimental Neurology. 2005;64:706-15
53. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proceedings of the National Academy of Sciences. 1999;96:6873-6878
54. Mietelska-Porowska A, Wojda UT. Lymphocytes and Inflammatory Mediators in the Interplay between Brain and Blood in Alzheimer's disease: Potential Pools of New Biomarkers. J Immunol Res. 2017:4626540 doi: 10.1155/2017/4626540
55. Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, Dong Y, Legeron FP, Leboucher A, Burnouf S. et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140:184-200
56. Dong J, Xiong H. Human immunodeficiency virus type 1 gp120 inhibits long-term potentiation via chemokine receptor CXCR4 in rat hippocampal slices. J Neurosci Res. 2006;15:489-496
57. De Laere M, Sousa C, Meena M, Buckinx R, Timmermans JP, Berneman Z, Cools N. Increased Transendothelial Transport of CCL3 Is Insufficient to Drive Immune Cell Transmigration through the Blood-Brain Barrier under Inflammatory Conditions In Vitro. Mediators Inflamm. 2017 doi: 10.1155/2017/6752756
58. Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR. Alzheimer's disease beta-secretase BACE1 is not a neuron-specific enzyme. Journal of Neurochemistry. 2005;92:226-34
59. Lee YK, Kwak DH, Oh KW, Nam SY, Lee BJ, Yun YW, Kim YB, Han SB, Hong JT. CCR5 deficiency induces astrocyte activation, Abeta deposit and impaired memory function. Neurobiol Learn Mem. 2009;92:356-63
60. Verite J, Janet T, Julian A, Chassaing D, Page G, Paccalin M. Peripheral Blood Mononuclear Cells of Alzheimer's Disease Patients Control CCL4 and CXCL10 Levels in a Human Blood Brain Barrier Model. Curr Alzheimer Res. 2017;14:1215-28
61. Song M, Ji J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J Neuroinflammation. 2011;9:8 92
62. Jones PM, O'Mara ML, George AM. ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem Sci. 2009;34:520-31
63. Richard M, Drouin R, Beaulieu AD. ABC50, a novel human ATP-binding cassette protein found in tumor necrosis factor-alpha-stimulated synoviocytes. Genomics. 1998;53:137-45
64. Wilcox SM, Arora H, Munro L, Xin J, Fenninger F, Johnson LA, Pfeifer CG, Choi KB, Hou J, Hoodless P. et al. The role of the innate immune response regulatory gene ABCF1 in mammalian embryogenesis and development. PLoS One. 2017:19 12(5): e0175918. doi: 10.1371/journal.pone.0175918
65. Coots RA, Liu XM, Mao Y, Dong L, Zhou J, Wan J, Zhang X, Qian SB. m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol Cell. 2017;68:504-14
Author contact
![]() Corresponding author: Dr. Soraya L. Valles. Department of Physiology. School of Medicine, University of Valencia. Email: lilian.valleses. Phone: 0034-963983813
Corresponding author: Dr. Soraya L. Valles. Department of Physiology. School of Medicine, University of Valencia. Email: lilian.valleses. Phone: 0034-963983813