Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):507-521. doi:10.7150/ijbs.30575 This issue Cite
Research Paper
Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis
Hao Feng1,2, Xin Zhao2, Qiqiang Guo2, Yanling Feng2, Mengtao Ma2, Wendong Guo2, Xiang Dong2, Chengsi Deng2, Chunlu Li2, Xiaoyu Song2, Shuai Han3 ![]() , Liu Cao2
, Liu Cao2 ![]()
1. Department of Ophthalmology, The First Hospital of China Medical University, Shenyang, Liaoning Province, 110122, China
2. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Liaoning Province, Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China
3. Department of Neurosurgery, The First Hospital of China medical University, Shenyang, Liaoning Province, 110122, China
Received 2018-10-11; Accepted 2018-11-15; Published 2019-1-1
Abstract

Proliferative vitreoretinopathy (PVR) is the most serious fibrous complication that causes vision loss after intraocular surgery, and there is currently no effective treatment in clinical. Autophagy is an important cell biological mechanism in maintaining the homeostasis of tissues and cells, resisting the process of EMT. However, it is still unclear whether autophagy could resist intraocular fibrosis and prevent PVR progression. In this study, we investigated the expression of mesenchymal biomarkers in autophagy deficiency cells and found these proteins were increased. The mesenchymal protein transcription factor Twist can bind to autophagy related protein p62 and promote the degradation of Twist, which reduced the expression of mesenchymal markers. By constructing an EMT model of retinal pigment epithelial (RPE) cells in vitro, we found that autophagy was activated in the EMT process of RPE cells. Moreover, in autophagy deficient RPE cell line via knockdown autophagy related protein 7 (Atg7), the expression of epithelial marker claudin-1 was suppressed and the mesenchymal markers were increased, accompanied by an increase in cell migration and contractility. Importantly, RPE epithelial properties can be maintained by promoting autophagy and effectively reversing TFG-β2-induced RPE fibrosis. These observations reveal that autophagy may be an effective way to treat PVR.
Keywords: Autophagy, Proliferative vitreoretinopathy, Retinal pigment epithelial, EMT, Atg7, Twist
Introduction
Since the importance of retinal tears and detachment in the pathogenesis of rhegmatogenous retinal detachment (RRD) was clarified in 1930 [1], therapeutic interventions of RRD are rapidly developing. Vitrectomy has been implemented and developed continuously and has become the standard for successful treatment of RRD, especially in cases of complex retinal detachment [2]. However, loss of function due to failure after reattachment of the retina, and intraocular intervention given by multiple relapses, is still an important source of morbidity after RRD treatment [3]. The most common cause of retinal detachment after vitreous surgery is proliferative vitreoretinopathy (PVR). Since it was first elaborated so far, there has been no effective clinical progress [4]. Although PVR can occur before surgery, it has a higher incidence of any type of intraocular RRD surgery intervention. PVR accounts for about 75% of the total primary intraocular surgery failure, and the incidence of postoperative RD is 5-10% [5].
The formation of a dense fibrotic contractile membrane on the posterior surface of the vitreous membrane or the detached retinal is the pathological feature of PVR. The retinal distortion and continuous distraction caused by its contraction transforms RRD into traction retinal detachment [6]. In this pathological process, retinal pigment epithelial (RPE) loses epithelial characteristics through an epithelial-mesenchymal transition (EMT), transforms into mesenchymal phenotype, increasing cells migration ability, invasiveness, resistance to apoptosis, and production of extracellular matrix, turning RPE into fibroblast-like cells [7]. From the perspective of the most important cytological features of PVR, many researchers have spent more than 40 years of hard work to explore, but have yet to find effective PVR prevention and treatment methods, which makes us have to pay attention to other possible mechanisms involved in the RD and PVR.
Autophagy is an evolutionarily conserved lysosomal-mediated intracellular degradation process [8]. At the basal level, the primary function of autophagy is to maintain a balance of intracellular proteins and organelles turnover in cells. Under various pathophysiological conditions, autophagy activity can be up-regulated to supply the relevant nutrient or energy requirements within the cell, to cope with development-related intracellular structural remodeling, and to digest intracellular misfolded proteins, redundant or damaged organelles, as well as microorganisms that invade the cells. Even though the morphological features of autophagy have been demonstrated decades ago, the functional role of autophagy in pathological conditions was recognized only because of the recent reports of the molecular regulation mechanisms and functions of autophagy-related genes [9-11]. The significant role of autophagy in human disease has been discovered through studies of mouse models lacking key genes involved in autophagosome formation, including Atg7, Atg5 or Beclin1 [12-14]. Autophagy thus gradually exhibits an important role in pathological conditions and in a variety of disorders such as cancer, neurodegeneration, aging, and heart disease. In the eye, from the anterior cornea to the posterior RPE that provides a protective barrier to the retina, almost all cell types rely on one or more types of autophagy to maintain normal structural and physiological function [15]. Moreover, the expression of autophagy-related proteins in different cells in the eye also sheds light on the importance of autophagy progression in maintaining healthy visual function [16]. In contrast, mutations in related autophagy genes can also directly contribute to the development of ocular diseases. In the meantime, intraocular cell homeostasis also depends on the regulation of the autophagy pathway induced by the interaction of basal and pressure [17].
In retinal RPE cells and photoreceptor cells, autophagy is highly activated, and impairment of autophagy can lead to early degeneration of RPE cells [18, 19]. These characteristics of RPE strongly associate autophagy with retinal degenerative diseases caused by retinal senescent diseases and photodamage. This makes the research of autophagy and retinal diseases focused on degenerative diseases such as age-related macular degeneration (AMD) [20]. Autophagy and EMT could not be linked for a long time in the past because of their independent mechanisms of action leading to alienated associations. However, recent research reveals that these two important processes can be regulated with each other through complex relationships. We speculate that autophagy pathway may play an important role in the pathogenesis of PVR, and the elucidation of this regulatory mechanism will provide new ideas for the prevention and treatment of PVR.
Materials and Methods
Cell lines and reagents
Wild-type (WT) and Atg7-/- mouse embryonic fibroblasts (MEFs) cells were obtained as previously described [13]. In brief, pregnant mice were euthanized to obtain embryos on day 13-15 of embryonation. After removing the limb, head, tail and viscera, the remaining tissues were minced into smaller pieces and digested in 0.25% trypsin/0.01%EDTA (Solarbio, Beijing, China). 10 min later, the mixture was inactivated with fetal bovine serum (FBS; Clark Bioscience, Richmond, VA), centrifuged at 1000 rpm for 5 min, and the cell pellets were resuspended with Dulbecco's modified Eagle's medium (DMEM; Biological Industries, Cromwell, CT) containing 15% FBS and streptomycin (100 μg/ml)/penicillin (100 units/ml) (Genview, Florida, USA). ARPE-19 cells were generously provided by Dr. Mitsuru Arima (Department of Ophthalmology, Kyushu University), and were cultured in DMEM/F12 medium (HyClone, GE lifescience, UK), supplemented with 10% FBS and streptomycin (100 μg/ml)/penicillin (100 units/ml). All cells were cultured in a humidified incubator at 37 °C with 5% (v/v) CO2.
Rabbit anti-Atg7 (A2856), rabbit anti-p62 (p0067), puromycin, chloroquine (CQ) and rapamycin (RAPA) were purchased from Sigma (Sigma-Aldrich, St. Louis, MO). TFG-β2 was purchased from R&D Systems (Minneapolis, MN). HBSS was purchased from Gibco (Thermo Fisher Scientific, Waltham, MA). Rabbit anti-LC3 (#4108), rabbit anti-claudin-1 (#13995), mouse anti-N-cadherin (#14215), rabbit anti-Vimentin (#5741), rabbit anti-mTOR (#2983), rabbit anti-phospho-mTOR (Ser2448) (#5536) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Rabbit anti-αSMA (ab21027) was purchased from Abcam (Cambridge, UK). Mouse anti-Twist (sc-81417) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Mouse anti-c-Myc-tag (A00704) was purchased from Genscript (Piscataway, NJ). Rabbit anti-MMP-2 (10373-2-ap) and rabbit anti-MMP-9 (10375-2-ap) were purchased from Proteintech Group, Inc. (Rosemont, IL, USA). Mouse anti-α-tubulin (AC012), horseradish peroxidase (HRP) goat anti-rabbit IgG (AS014), HRP goat anti-mouse IgG (AS003) were purchased from ABclonal Biotech, Co., Ltd. (Wuhan, China).
Plasmid construction and transfection
GFP-LC3 plasmid was a gift from Prof. Huaqin Wang (Department of Biochemistry and Molecular Biology, China Medical University). Expression of the Myc-Atg7 plasmid was previously described [13]. Myc-Atg7 nontargetable mutant plasmid was generated by mutating three nucleotide base pairs to alternative nucleotides within the shRNA target region without altering the amino acid sequences, constructed by PCR amplification and subcloned into pCMV-myc vector (Clontech, 631604). The plasmid was transfected into cells using Lipofectamine 2000 (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. Cells were harvested 48 h after transfection.
Atg7 shRNA plasmid constructed into lentiviral vector (GV112) was purchased from Genechem (Quebec, Canada) (shAtg7 #1, 5'- CAGCTCTTCCTTACTTCTTAA-3'; shAtg7 #2, 5'-AAGGTTGTGTCTGTCAAATGT-3', shNC, 5'-TTCTCCGAACGTGTCACGT-3'). Lentivirus was produced in HEK-293T cells that were transfected with shRNA-expression vector. Lentiviral supernatant was separately harvested at 24 h, 48 h and 72 h after transfection. After 1500 g centrifugation for 30 min, the lentiviral particles were dissolved in PBS and added into ARPE-19 cells for 24 h infection. Stable cell lines were selected out in 10 μg/mL puromycin for 5 days.
Western blotting analysis
Cells were lysed for 30 min on ice with lysis buffer [50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P‑40, 0.25% Na‑deoxycholate, 1 mM EDTA and protease inhibitor cocktail (B14001, Bimake, Houston, TX, USA)], and the total protein was harvested by centrifugation at 14,000 rpm 4 °C for 20 min. Protein concentration was measured by G250 and 30 μg of cell lysate were used in samples. Samples were loaded on 8 or 15% polyacrylamide gels respectively, and separated by SDS‑PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Merck KGaA, Darmstadt, Germany). The membranes were blocked with 5% nonfat dry milk in 20 mM Tris (pH 7.4), 137 mM NaCl and 0.05% Tween‑20 for 1 h at room temperature. The membranes were probed with the primary antibodies overnight at 4˚C. The secondary antibody with HRP-conjugated were incubated for 1 h at room temperature. All of the PVDF membranes were analyzed by chemiluminescence (Tanon, Tanon Science & Technology Co., Ltd., Shanghai, China).
Co-immunoprecipitation analysis
WT MEF cells were lysed with IP lysis buffer and 1 mg protein was incubated with anti-Twist antibody and protein A/G-Sepharose (Santa Cruz, sc-2003) on a rocking platform overnight at 4°C. The beads were then harvested by centrifugation for 3 min at 600 g at 4°C and the supernatant was removed. The beads were then resuspended in IP lysis buffer and repeatedly inverted on a rocking platform for 3 times (each time for 15 min). The beads were resuspended with loading buffer for Western blot analysis.
Transwell assay
ARPE-19 cells (3 × 104) were suspended in 100 μl serum‑free DMEM/F12 and then added into the upper inserts with 8.0‑μm‑pore polycarbonate membranes and 600 μl DMEM/F12 with 10% FBS was added to the lower chambers (Corning Life Science, Corning, NY, USA). Following incubation with different treatments for 16h, cells on the upper membrane were removed with cotton swabs, while the cells on the other side of the membrane were then fixed with methanol for 5 min, stained with hematoxylin for 15 min at room temperature and counted under a Nikon Eclipse Ni‑U microscope (Nikon Corp., Tokyo, Japan).
Immunocytochemistry assay
ARPE-19 cells were seeded into 24-well plates with a glass coverslip in each well. After different treatments, the cells were rinsed twice with PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, and permeabilized with 0.1% Triton X-100. The cells were blocked with 5% BSA in PBS for 1 h at room temperature and incubated overnight with the primary antibody at 4 °C, followed by incubation with Alexa fluor® 488 goat anti-mouse lgG (H + L) or Alexa fluor® 594 goat anti-rabbit lgG (H + L) (Thermo Fisher Scientific) for 1 h at room temperature, and then analysis using fluorescence microscopy.
Collagen matrix contraction assay
ARPE-19 cells (5 × 105 per well) were resuspended in 2 mg/ml Rat tail type I collagen (c8062, Solarbio, Beijing, China) which was dissolved in 500 μl DMEM/F12 with 6 μl 0.1 M NaOH and seeded in a 24 well plate. After incubated at 37 °C for 1 h polymerization, the cell collagen gels were detached from the bottom of the wells and floated in 1 ml DMEM-F12 containing 10% FBS. The medium was changed every two days. After different treatment for 10 days, the surface area of each matrix was observed, recorded and taken picture by a smartphone.
Statistical analysis
Values are expressed as the mean ± standard deviation of three independent experiments. Statistical significance was analyzed using a Student's t‑test or a one‑way analysis of variance followed by a Tukey's multiple comparisons test using the SPSS 17.0 (SPSS Inc., Chicago, IL, USA) and Prism 5.0 software (GraphPad Software, Inc., LA Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
The alteration of mesenchymal markers' levels correlates with changes of autophagy in MEFs
In mammals, Atg7 was indeed essential for ATG12 conjugation, LC3 modification systems, and autophagosome formation [13, 21]. In order to investigate whether autophagy deficiency influences the expression of mesenchymal related protein, we measured the levels of mesenchymal markers in MEFs of Atg7 deficient mice (Atg7-/-) and their autophagy-sufficient wild type littermates. As shown in Figure 1a, in contrast to WT, the Atg7 knockout MEFs showed that only the LC3-I form was detected. Because that LC3-I is the free mature whereas LC3-II is the lipidated form, it is generally accepted that the increase of LC3-II marks the autophagosome formation. Loss of LC3-II revealed deficient autophagy in Atg7-/- MEFs. We also detected protein p62 level, its increase indicates the inhibition of autophagic flux [22]. Atg7-/- MEFs showed a significant increase of p62 level, which indicated its impaired autophagic function. Accompanied by the disorder of autophagy function, the levels of mesenchymal markers [N-cadherin, Vimentin, alpha smooth muscle actin (αSMA)] were found to have significant increases. It is generally accepted that mutant MEFs separated from genetically manipulated mouse lines are the important tools for revealing the molecular mechanisms and biological behavior of specific gene or function defect [23]. These results revealed that the loss of cellular autophagy function can lead to cell-to-mesenchymal transformation.
Autophagy is involved in the regulation of mesenchymal protein markers expression in MEFs. (a) Immunoblotting with antibodies against N-cadherin, Vimentin, αSMA and autophagy associated markers, Atg7, p62 and LC3 in protein lysates derived from primary MEFs from the indicated genotypes. Quantification of protein expression were shown as the means±S.D. **P<0.01 vs WT. (b) WT MEFs were starved with HBSS for different time points as indicated, lysed, and subjected to immunoblotting with antibodies against N-cadherin, Atg7, p62 and Tubulin used as a loading control. Quantification of protein expression were shown as the means±S.D. **P<0.01 vs 0 h. (c) WT MEFs were starved with serum-free medium for different time points as indicated, lysed, and subjected to immunoblotting with antibodies against N-cadherin, Atg7, p62, LC3, and Tubulin. Quantification of protein expression were shown as the means±S.D. **P<0.01 vs 0 h. (d) WT and Atg7-/- MEFs were or not under serum-starvation for 48 h, lysed, and subjected to immunoblotting with antibodies against N-cadherin, Vimentin, αSMA, and Atg7, p62, LC3, Tubulin used as a loading control. Quantification of protein expression were shown as the means±S.D. **P<0.01 vs Atg7 -/-. ns, not significant.
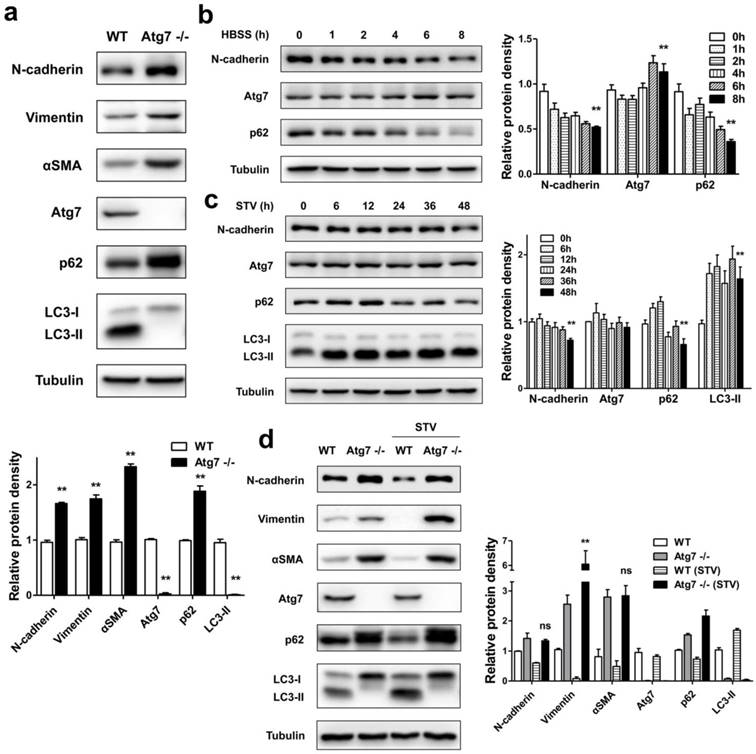
To detect whether the activation of autophagy could influence the expression of mesenchymal markers in MEFs, WT MEFs were treated with either Hank's buffered salt solution (HBSS) or serum-starvation. After nutrient starvation by HBSS, reduced expression of p62 in WT MEFs was observed (Figure 1b). Consistent with the reduced p62 level, there was an N-cadherin degradation followed by HBSS treatment. Similar to the effect of HBSS treatment, long time course of serum-starvation induced both p62 degradation and LC3-II accumulation, accompanied by a reduction of N-cadherin (Figure 1c). These results suggest that promoted autophagy function can delay the mesenchymal process.
Furthermore, we compared changes of mesenchymal markers under serum-starvation condition in autophagy-deficient MEFs and WT MEFs. As shown in Figure 1d, Atg7-/- MEFs displayed a completed block in autophagic flux, both at baseline and in response to starvation with serum-deprived, evidenced by the lack of LC3-II, as well as the accumulation of p62. Meanwhile, in the absence of autophagy function, the promotion of autophagy by serum-starvation did not reduce the expression of mesenchymal markers such as N-cadherin, Vimentin and αSMA compared to WT MEFs, thus further suggesting that lack of autophagy facilitates the mesenchymal process.
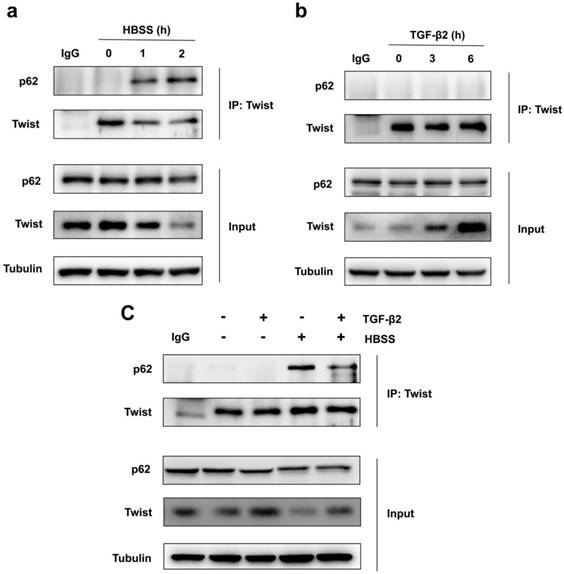
Because Twist acts as the main transcription factor for embryonic morphogenesis, activates mesenchymal markers, regulates cell invasion and metastasis, and promotes EMT [24]. Previous report revealed that p62 regulates cell proliferation and migration through interaction with Twist [25]. Therefore, we speculate that activation of autophagy selectively degrades Twist through p62-mediated selective autophagy, thereby resisting the EMT process of cells. Through co-immunoprecipitation experiments, we found that HBSS-induced starvation makes Twist bind to p62, and this combination does not occur under non-nutrient deprivation conditions (Figure 2a). And the prolongation of starvation time can increase the combination of Twist and p62, indicating that p62-mediated selective autophagy promotes the degradation of Twist. In contrast, TGF-β2 stimulation does not induce the binding of Twist to p62, but blocks the binding of p62 and Twist when autophagy occurs, and up-regulates the expression of Twist (Figure 2b, c), revealing that promoting autophagy may be an effective way to resist TGF-β2-induced EMT.
Autophagy promotes Twist bind to p62. (a) Immunoblotting with antibodies against p62 and Twist following immunoprecipitation using control species matched IgG and anti-Twist antibody in WT MEF cells under HBSS-starvation for indicated hours. (b) Immunoblotting with antibodies against p62 and Twist following immunoprecipitation using control species matched IgG and anti-Twist antibody in WT MEF cells under TGF-β2 (10 ng/ml) treatment for indicated hours. (c) Immunoblotting with antibodies against p62 and Twist following immunoprecipitation using control species matched IgG and anti-Twist antibody in WT MEF cells under 6 h TGF-β2 (10 ng/ml) treatment followed by 1 h HBSS-starvation.
Fibrosis disrupts the homeostasis of RPE cell monolayer accompanied by changes in autophagy
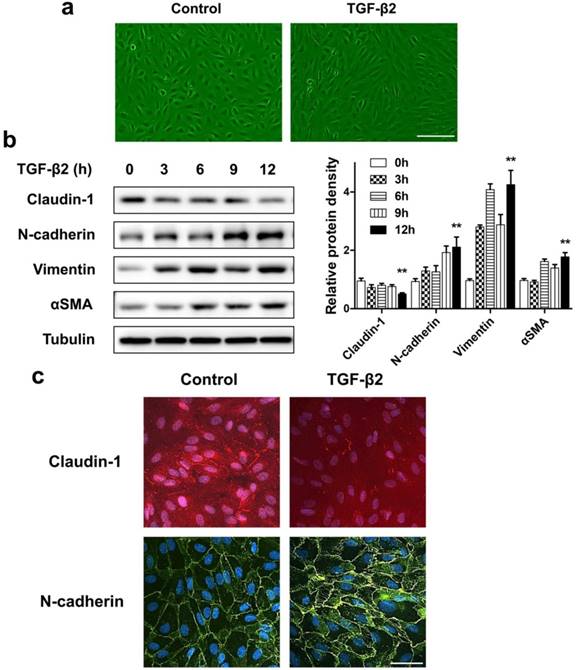
If the destruction of the eye's homeostasis caused by inflammation, infection, or metabolic disease, the final outcome is often fibrosis [26]. The formation of fibrous membranes on the epiretinal surface and the fibrosis contraction are the most direct pathological basis of PVR [27, 28]. In this process, emerging evidence shows that myofibroblast transdifferentiation of RPE cells plays a key role [29-31]. We used TGF-β2, which was detected to upregulate in both the epiretinal membranes and vitreous body in patients enduring PVR [32], to stimulate RPE cell monolayer in vitro to induce myofibroblast transdifferentiation of RPE, resulting in epithelial-mesenchymal transition (EMT) of RPE cells. First, TGF-β2 significantly induced ARPE-19 cells morphological transition into fibroblastoid phenotype (Figure 3a). Western blot analysis showed that, after TGF-β2 stimulation at different time points, the epithelial marker claudin-1 showed a significant downregulation. In contrast, the mesenchymal marker N-cadherin, Vimentin and αSMA showed a significant upregulation (Figure 3b). Similarly, the results of immunocytochemistry also showed that loss of claudin-1 on cell membrane and increased N-cadherin after TGF-β2 treatment (Figure 3c), indicating EMT induction upon TGF-β2 treatment of RPE cells.
ARPE-19 cell monolayer transit into fibroblastoid phenotype under EMT induced by TGF-β2. (a) Images of ARPE-19 cell monolayer either untreated (control) or treated with TGF-β2 (10 ng/ml) for 24 h. scale bar, 100 μm. (b) Western blot analysis of epithelial marker claudin-1 and mesenchymal marker N-cadherin, Vimentin, αSMA, under different time points treatment of TGF-β2 (10 ng/ml). Quantification of protein expression were shown as the means±S.D. **P<0.01 vs 0 h. (c) Immunocytochemistry micrographs showed loss of claudin-1 (red) on cell membrane and increased N-cadherin (green) after TGF-β2 (10 ng/ml) treatment for 24 h. Nuclei were stained with DAPI (blue); scale bar, 50 μm.
Next, we tested whether a functional autophagy is modulated during EMT process in RPE by measuring protein expression and intracellular localization of p62 and LC3. As shown in Figure 4a, under TGF-β2 stimulation p62 significantly decreased from 9 h. Moreover, after 24 h TGF-β2 treatment p62 dots were found marked degradation (Figure 4b, left panels). P62 is able to bind to ubiquitinated cargoes to mediate their selective degradations by autophagy [33]. These results indicate that autophagy was activated during EMT process in RPE. The conversion of endogenous LC3-I to LC3-II was found to start from 3 h after TGF-β2 treatment (Figure 4a). Moreover, TGF-β2 induced stable the GFP-LC3 puncta aggregation, which reveals autophagosome formation (Figure 4b, right panels; Figure 4c).
Autophagy is activated during TGF-β2-induced EMT process in RPE. (a) Immunoblotting analysis of Atg7, p62, and LC3 in ARPE-19 cells treated with TFG-β2 (10 ng/ml) for the indicated hours. Tubulin levels were used as loading control. Quantification of protein expression were shown as the means±S.D. *P<0.05, **P<0.01 vs 0 h. (b) Fluorescence analysis of the localization of p62 or LC3 in ARPE-19 cells expressing either GFP-LC3 (green) or immunofluorescence of p62 (red), respectively, following treatment with TGF-β2 (10 ng/ml) for 24 h. (c) Quantification of the LC3-positive puncta per cell. Data are shown as the means±S.D. n=4, **P<0.01, scale bar, 10 μm. (d) Immunoblotting analysis of p62 and LC3 in ARPE-19 cells treated with TFG-β2 (10 ng/ml; 24 h) either in the presence or absence of the lysosome inhibitor chloroquine (CQ) (30 μM) for the last 12 h of treatment. Tubulin was used for protein loading control. (e) Quantification of the band intensities of p62 and LC3-II obtained by immunoblotting analysis as in panel (d), expressed as the means±S.D. n=3, *P<0.05 **P<0.01.
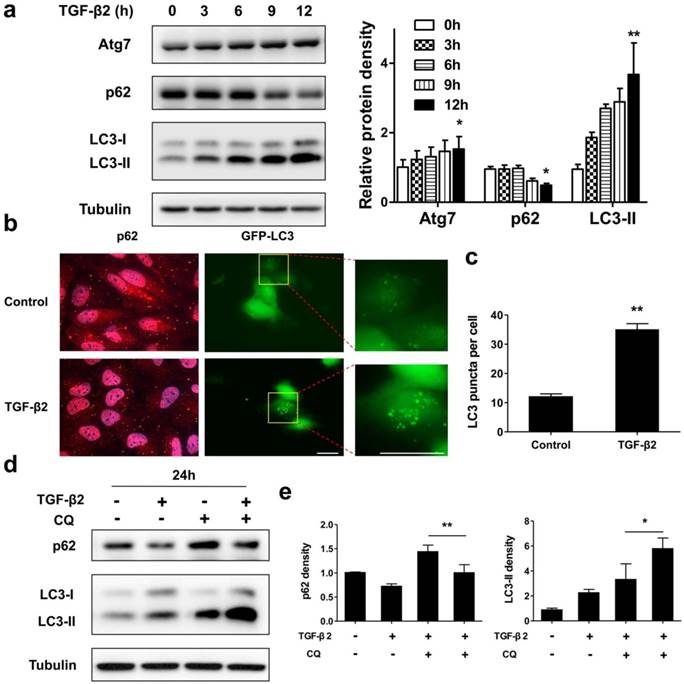
To further determine whether autophagy is activated during EMT, we measured the autophagic flux by analyzing changes in LC3-II and p62 under TGF-β2 stimulation, either in the absence or presence of the lysosome inhibitor chloroquine (CQ), which prevents autophagosomal fusion and degradation. As shown in Figures 4d and e, CQ caused an accumulation of p62 in the absence of TGF-β2, while co-treatment with TFG-β2 downregulated p62 expression. And CQ also caused an accumulation of LC3-II in the absence of TGF-β2, while co-treatment with TFG-β2 aggravated accumulation of LC3-II. Overall, these results demonstrate that the autophagy activity is activated during the TGF-β2-induced EMT process.
Lack of autophagy promotes RPE cells EMT
As during EMT prosses of RPE cells, the autophagy activity was initiated, we asked either autophagy plays a positive or negative role in maintaining RPE epithelial phenotype. By generating stable Atg7 gene KD transfectants of ARPE-19 cells, we detected impaired autophagy in RPE cells. Figure 5a showed increased p62 and decreased LC3-II both in shAtg7 No. 1 and No. 2 cells. At the same time, RPE cells presents a loss of epithelial phenotype, moreover translates to a mesenchymal phenotype. As shown in Figure 5b, both shAtg7 No. 1 and No. 2 cells lost the epithelial marker claudin-1 and upregulated the mesenchymal marker N-cadherin, Vimentin and αSMA. The results of immunocytochemistry also showed that highly expressed N-cadherin is more disorderly and radically arranged in shAtg7 cells (Figure 5c, upper panels). In non-silencing shRNA cells, Vimentin cages showed uniform division, in contrast with those in shAtg7 cells, whose Vimentin showed slender fiber-like changes with closed Vimentin cage (Figure 5c, bottom panels).
Lack of autophagy promotes RPE cells EMT. (a) Western blot analysis of p62 and LC3 in ARPE-19 cells stable knockdown of Atg7. (b) Western blot analysis of EMT markers in ARPE-19 cells stable knockdown of Atg7. Quantification of protein expression were shown as the means±S.D. **P<0.01 vs shNC. (c) Immunocytochemistry staining showed disorder arrangement of N-cadherin (red) and closed Vimentin (green) cage in Atg7-knockdown cells; scale bar, 50 μm. (d) Transwell assay showed enhanced ability of migration in Atg7-knockdown cells. scale bar, 100 μm. (e) Quantification of migration abilities of non-silencing cells and Atg7-knockdown cells in panel (d), expressed as the means±S.D. n=4, **P<0.01. (f) Western blot analysis of MMP-2/9 in Atg7-knockdown cells. Quantification of protein expression were shown as the means±S.D. *P<0.05, **P<0.01 vs shNC. (g) Collagen gel contraction assay in non-silencing cells and Atg7-knockdown cells. (h) Quantification of the percentage of gel contraction by measuring the gel size at the 10 d when compared to the initial size in each group, expressed as the means±S.D. n=4, **P<0.01
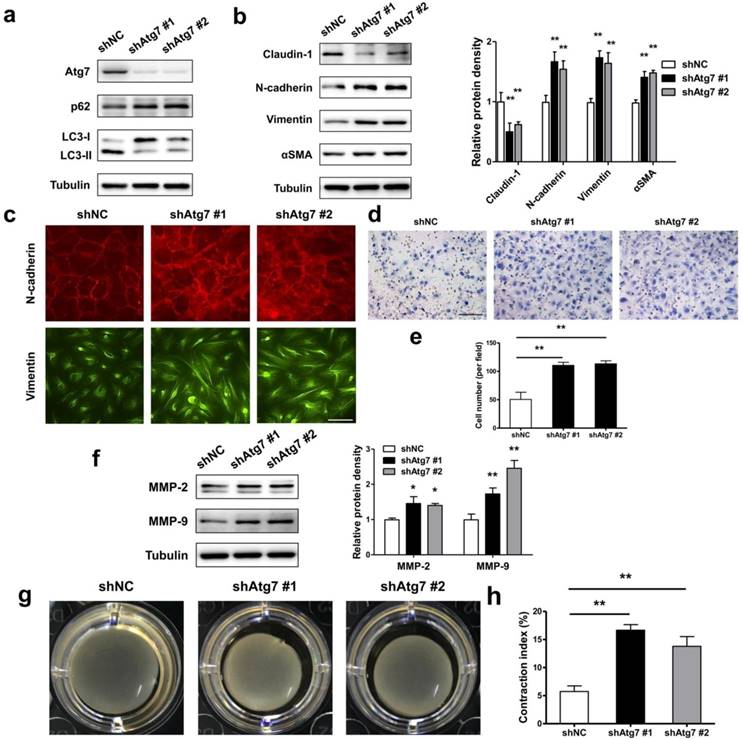
RPE cells undergoing EMT typically lose apicobasal polarity, translate to fibroblastic morphology, and acquire migratory mesenchymal characteristics [34]. By transwell assay, we analyzed the ability of migration in autophagic impaired RPE cells. As shown in Figure 5d and e, shAtg7 cells exhibited enhanced migratory behavior compared to shNC cells. Simultaneously, the expression of matrix metalloproteases MMP-2 and MMP-9, which reflect the promotion of the remodeling of the basement membrane and invasion into surrounding tissues [35], were significantly activated (Figure 5f). By collagen gel contraction assay, we observed a significant reduction in the percentage of the original area occupied by the shAtg7 cells when compared with the shNC cells (Figure 5g and h).
These results demonstrated that dysfunctional autophagy in RPE promotes cell lose epithelial phenotype to translate to a mesenchymal phenotype.
Autophagy is essential to maintain RPE phenotype
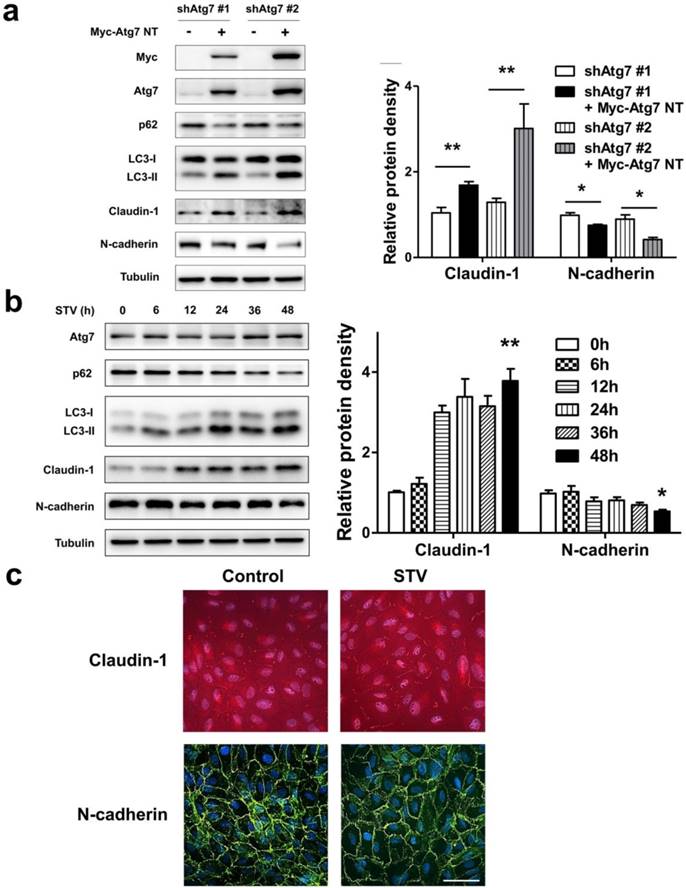
As lack of autophagy promotes RPE lose epithelial phenotype, we speculated autophagy induction could play a positive effect on the maintenance of epithelial phenotype. To address this issue, we overexpressed nontargetable mutant Atg7 in autophagy dysfunction shAtg7 cells to recuse autophagy activity. As we supposed, by overexpressing Atg7, the expression of p62 decreased and the level of LC3-II increased, indicating that autophagy activity was restored (Figure 6a). Meanwhile, the expression of claudin-1 also increased significantly, whereas the expression of N-cadherin reversed (Figure 6a). To further strengthen these findings, we induced autophagy by stimulating RPE cells with serum-starvation. After various time points stimulation, serum-starvation was sufficient for inducing autophagy. As shown in Figure 6b, p62 showed a time-dependent decrease manner, whereas LC3-II significantly increased after starvation. Meanwhile, claudin-1 showed stably increase and N-cadherin decreased after 48 h starvation. Consistent with this, immunocytochemistry analysis showed that exposure to serum-free medium increases claudin-1 expression on cell membrane and weakens N-cadherin expression (Figure 6c). These findings indicate that autophagy is essential to maintain RPE phenotype, autophagy induction enhance RPE cell-cell contact.
Autophagy maintain epithelia phenotype in RPE. (a) Western blot analysis of p62, LC3, claudin-1 and N-cadherin in Atg7-knockdown cells by overexpressing Myc-Atg7 NT (nontargetable mutant). Quantification of protein expression were shown as the means±S.D. *P<0.05, **P<0.01. (b) Western blot analysis of Atg7, p62, LC3, claudin-1 and N-cadherin under different time points of serum-starvation. Quantification of protein expression were shown as the means±S.D. *P<0.05, **P<0.01 vs 0 h. (c) Immunocytochemistry staining showed increased claudin-1 and weaken N-cadherin on cell membrane after 48 h serum-starvation; scale bar, 50 μm.
Autophagy induction by mTOR inhibitor rapamycin reverses TGF-β2-induced RPE fibrosis
To test whether autophagy promotion by pharmacological mTOR inhibition prevents RPE EMT process, we treat ARPE-19 cell monolayer with TGF-β2 combined with rapamycin, which is an activator of autophagy. As shown in Figure 7a, rapamycin blocked TGF-β2 induced phosphorylation of mTOR, enhanced p62 degradation, and increased the accumulation of LC3-II. Moreover, we observed p62 puncta reduction and more GFP-LC3 puncta aggregation compared with those found in control or TGF-β2 stimulation (Figure 7b and c). These data revealed that rapamycin promotes RPE autophagy activity by inhibiting mTOR phosphorylation.
Promoted autophagy by rapamycin reverses TFG-β2-induced EMT in RPE. (a) Western blot analysis of p-mTOR, mTOR, p62 and LC3 in ARPE-19 cells treated with TFG-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM) for 24 h. Quantification of protein expression were shown as the means±S.D. **P<0.01. (b) Fluorescence analysis of the localization of p62 or LC3 in ARPE-19 cells expressing either GFP-LC3 (green) or immunofluorescence of p62 (red), respectively, following treatment with TGF-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM) for 24 h. (c) Quantification of the LC3-positive puncta per cell. Data are shown as the means±S.D. n=4, **P<0.01, scale bar, 10 μm. (d) Western blot analysis of EMT markers in ARPE-19 cells treated with TFG-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM) for 24h. Quantification of protein expression were shown as the means±S.D. *P<0.05, **P<0.01. (e) Immunocytochemistry staining of the localisation of claudin-1 (upper panel, red) and N-cadherin (lower panel, red), Vimentin (lower panel, green) in RPE after treatment with TGF-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM) for 24 h; Nuclei were stained with DAPI (blue); scale bar, 50 μm.
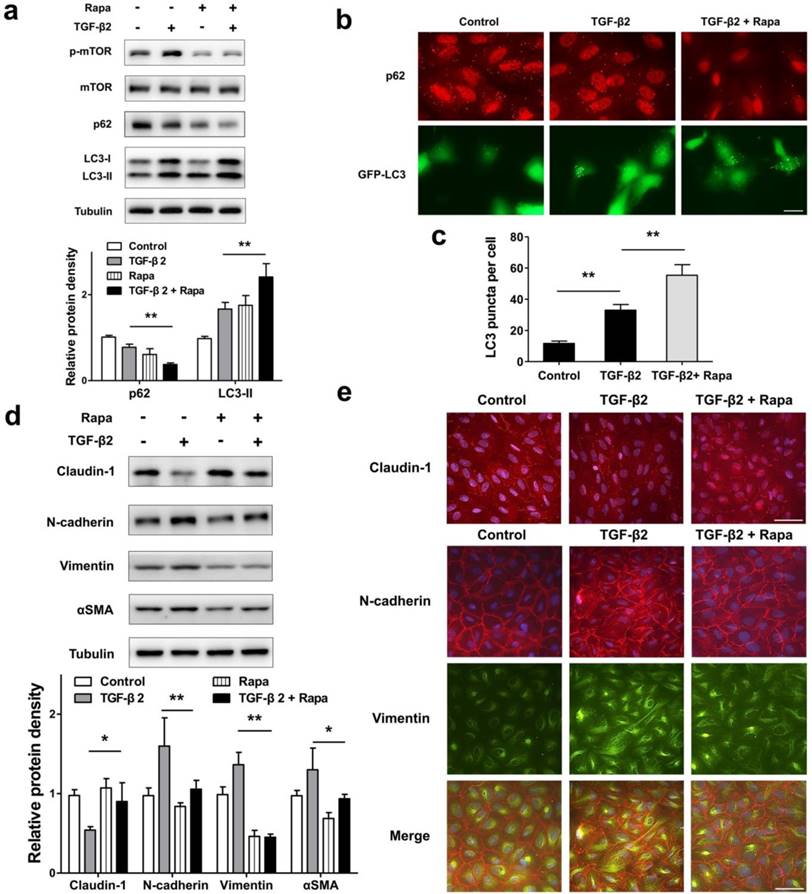
Next, we tested EMT marker levels under TGF-β2 combined with rapamycin. As shown in Figure 7d, rapamycin rescued TGF-β2-induced degradation of claudin-1, and prevent mesenchymal marker increases in N-cadherin, Vimentin, and αSMA. Consistent with this, immunocytochemistry analysis showed that rapamycin rescued loss of claudin-1 on cell membrane, reorganized the distribution of N-cadherin, and released the intensity of Vimentin (Figure 7e). These results indicated that rapamycin protects RPE from EMT stress by elevating autophagy activity.
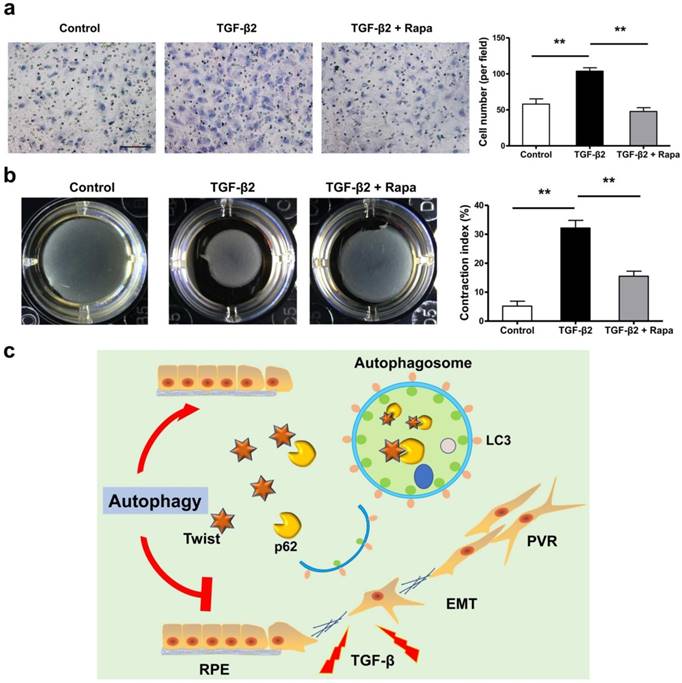
If the EMT of RPE is the initiating factor of PVR, then the subsequent migration of RPE into vitreous and participation in the contraction of the epiretinal membranes are involved in the entire course of PVR [36-40]. We tested the possibility that rapamycin may inhibit RPE cell migration and contraction. Figure 8a and b showed that rapamycin completely suppressed the migration of ARPE-19 cells under stimulation by TGF-β2, and significantly reduced the TGF-β2-induced collagen gel contraction in ARPE-19 cells.
Rapamycin inhibits TFG-β2-induced RPE cell migration and contraction. (a) Transwell assay of ARPE-19 cells treated with TFG-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM) for 16h. The quantification was expressed as the means±S.D. n=4, **P<0.01. Scale bar, 100 μm. (b) Collagen gel contraction assay of ARPE-19 cells treated with TFG-β2 (10 ng/ml) or TFG-β2 combined with rapamycin (1000 nM). Quantification of the percentage of gel contraction by measuring the gel size at the 10 d when compared to the initial size in each group, expressed as the means±S.D. n=4, **P<0.01. (c) Schematic for autophagy preventing the progression of RPE fibrosis and maintaining intracellular environmental homeostasis by p62-mediated Twist degradation.
Discussion
In this study, we observed that loss of autophagic flux may lead to lose epithelial phenotype and promote the development of EMT in RPE cells. Autophagy effectively prevents the progression of ARPE-19 cell fibrosis and maintains intracellular environmental homeostasis. The underlying mechanism is to promote the degradation of Twsit through p62-mediated selective autophagy, thereby increasing the expression of tight junction proteins between cells and inhibiting the progression of EMT (Figure 8c). These data reveal that autophagy function of RPE cells may serve as a novel and previously therapeutic target that has not yet been recognized, limiting the formation of intraocular fibrosis, and providing feasible treatment of PVR which currently have no effective therapy.
EMT is defined as a biological process by which a polarized epithelial cell can detach from the basement membrane or adjacent cells, lose its epithelial cell characteristics, and acquire mesenchymal cell characteristics. This feature includes enhanced migration ability, invasiveness, anti-apoptotic ability, and secretion of extracellular matrix components [41]. In PVR, RPE cells transform into fibroblast-like cells that have the ability to proliferate, migrate, and contract [32]. In this study, we used the cell-cell tight junction protein claudin-1 as a marker of RPE epithelial characteristics. When RPE forms a stable monolayer of epithelium, cell-cell junctions depend on tight junction proteins, claudins. Claudin binds to claudin of adjacent cells, which may only accumulate on adjacent shared lateral membranes of claudin that is identical or compatible [42]. Our study revealed that when EMT occurs in RPE cells, loss of claudin-1 means that the cell-cell tight junction breaks down, loses polarity, and converts to fibroblasts. At the same time, the increase of mesenchymal markers such as N-cadherin, αSMA and Vimentin proves the fibrosis transition of RPE from another aspect (Figure 3b, c). Because RPE originates from N-cadherin-expressed neural tube, RPE is different from other types of epithelial cells and mainly expresses N-cadherin as the main component of adhesive attachment, but not E-cadherin [43]. We also detected uniform and stable expression of N-cadherin in ARPE-19. However, the expression of N-cadherin in RPE was significantly up-regulated in TGF-β2-induced EMT progression, and ICC showed that its arrangement became disordered (Figure 3c). This reveals that excessive expression of N-cadherin may accelerate the detachment of RPE cells. Similarly, previous in vitro studies have shown that the N-cadherin neutralizing antibody can inhibit the invasiveness of RPE cells, indicating that N-cadherin could promote RPE cell migration and invasion [44].
Recent studies show that autophagy and EMT are linked by complex relationships. It has been reported that autophagy can directly regulate the EMT of cancer cells, vascular endothelial cells, hepatocytes, and kidney epithelial cells [45]. In the study of eye diseases, it was also found that autophagy can prevent the RPE cell aging induced by oxidative stress, A2E, and light damage, revealing the protective effect of autophagy in AMD [46-49]. In our study, firstly, we obtained the A7 knock-out MEF cells that cause autophagy defects by breeding Atg7+/- mouse, and detect changes in mesenchymal marker proteins that may be caused by autophagy defects at the initial stage of embryonic development. We found that N-cadherin, Vimentin, and αSMA were significantly up-regulated in autophagy-deficient MEF cells (Figure 1a). This reveals that autophagy is closely related to the expression of mesenchymal proteins. The autophagy promoted by HBSS or serum-free starvation significantly down-regulate the expression of mesenchymal markers in WT MEF, but in autophagy-deficient MEF, this phenomenon is reversed, indicating that autophagy activity has an inevitable regulatory relationship with these proteins (Figure 1b, c and d). In exploring its underlying mechanism, we found that promoting autophagy can induce Twist binding to p62, suggesting that p62-mediated selective autophagy is an important factor affecting the stability of Twist (Figure 2a). Base on the above results, we revealed that autophagy plays a role in inhibiting the process of cell fibrosis, which provides a basis for the subsequent description of the role of autophagy in fibrotic diseases.
Because the pathological features and terminal phenotype of PVR are intraocular fibrosis [50], autophagy plays a crucial role in maintaining the homeostasis of RPE cells, maintaining normal function and resisting environmental stress [20]. Therefore, we speculate that autophagy in RPE protects against changes in the intraocular environment, resists EMT stress, and maintains the epithelial cell characteristics of RPE. By stimulating the RPE cell monolayer with TGF-β2, we found that the autophagy activity of RPE cells was activated (Figure 4a). This reveals that autophagy is involved in the process of RPE cells coping with EMT stress. In order to further clarify the role of autophagy behavior, we constructed an autophagy defect ARPE-19 cell line by stable silencing Atg7. We found that similar characteristics to Atg7-/- MEF cells, autophagy-deficient RPE cells spontaneously showed high expression of mesenchymal cell characteristic proteins: N-cadherin, Vimentin, αSMA. Most cells with high expression of N-cadherin exhibit fibroblast morphology and strongly express Vimentin, an intermediate filament protein, which is often used as a marker for EMT in RPE cells [51]. At the same time, these cells also strongly express αSMA, a marker of myofibroblasts, and MMP-2/9, extracellular matrix proteins often used as markers for fibrosis. α-SMA-positive myofibroblasts are thought to play a role in wound contraction and fibrosis in various organs, including PVR [51]. These results suggest that autophagy is indispensable in maintaining the characteristics of RPE cells. Through transwell and collagen gel contraction assays, we also found that autophagy-deficient RPE cells obtained stronger migration ability and contractile ability (Figure 5d and g), indicating that the function of RPE cells changed with the loss of autophagy and was more prone to fibrosis. Previous studies showed that RPE lacking Atg7 does have an increase in intracellular stress, which is manifested in cell hypertrophy and increased undigested debris in the cell [52]. The insufficiency of intracellular stress caused by insufficient autophagy make the RPE layer to lose integrity. In our study, it was observed that due to the occurrence of EMT in RPE monolayers, the tight junctions between cells were lost, and the integrity of RPE was destroyed. It could be speculated that autophagy may be critical to maintaining the basic health of RPE and protecting cell survival under stress. By over-expressing Atg7 in stable silencing Atg7 RPE cells, we found that the loss of the tight junction protein claudin-1 and the up-regulation of N-cadherin were reversed (Figure 5a), suggesting that the recovery of autophagy in the cell, accompanied by the recovery of RPE epithelial properties. When the RPE monolayer under serum-free starvation, the up-regulation of tight junction protein claudin-1 and the ordered arrangement of n-cadherin were also observed (Figure 6b and c), revealing that promoting autophagy of RPE may be a target for maintaining the morphology and function of RPE.
Based on the above inference, we chose to use the mTOR inhibitor rapamycin as a drug that may reverse the induction of RPE fibrosis in vitro. The AKT/mTOR pathway is a classical pathway that represses autophagy. mTOR acts as an important protein kinase that regulates cellular functions such as cell growth, protein synthesis and transcription. mTOR acts as a negative regulator by inhibiting the formation of phagosomes and blocks the earliest stages of autophagy, thus leading to a decrease in autophagosome formation and accumulation of LC3-II. By inhibiting mTOR with rapamycin, autophagy can be pharmacologically induced [8]. Previous research has shown that, rapamycin promoted the autophagy induced by A2E in RPE, and protected RPE against cell death caused by A2E toxicity. This revealed that promoting autophagy could protect RPE cells against A2E-induced AMD [49]. Similarly, the decrease in RPE activity caused by chronic H2O2 damage is also accompanied by the dysfunction of autophagy, while giving rapamycin to promote cell autophagy can resist RPE oxidative stress and increase cell survival [46]. In this study, we induced EMT stress in RPE by TGF-β2, observed an increase in autophagy, and combined with the characteristics of autophagy in maintaining RPE epithelial phenotype, the autophagy response caused by this EMT stress is likely a RPE strategy to deal with EMT stress. By co-processing with RPE monolayer TGF-β2 and rapamycin, we found that promoting autophagy can significantly prevent the degradation of tight junction protein claudin-1, down-regulate the expression of N-cadherin, Vimentin, α-SMA (Figure 7d). By immunofluorescence assay, the release of Vimentin in the cells was observed to relieve the stress of cell fibrosis (Figure 7e). Furthermore, the contraction process of the preretinal membrane during the formation of PVR was simulated by collagen gel contraction assay in vitro. It was observed that autophagy promotion by rapamycin could significantly inhibit the contraction of RPE collagen gel induced by TGF-β2 (Figure 8b), revealing the possible protective effect of autophagy in the pathogenesis of PVR.
Collectively, these findings indicate that autophagy is the key pathway to maintain the normal epithelial morphology of RPE. Promoting autophagy can protect RPE against EMT stress, maintain normal cell tight junction structure, and reverse fibrosis (Figure 8c). Our study provides a more complete understanding of the relationship between autophagy and intraocular RPE fibrotic disease, and a new perspective for autophagy as a therapeutic target for PVR. Further clinically relevant therapeutic experiments targeting autophagy still need to be performed to provide guidance for clinical treatment.
Acknowledgements
This work was supported by National Key R&D Program of China (2016YFC1302400). The Ministry of education innovation team development plan to Liu Cao (IRT_17R107&IRT13101). Key project of the National Natural Science Foundation to Liu Cao (81130042, 31171323, 81770001, 2015225003) National Science Foundation of China to Xiaoyu Song (31300963, LFWK201725), Hao Feng (81800839), Qiqiang Guo (81502400), Yi Guan (81502447) and Fei Yi (81502414). Liaoning education department general project to Hongde Xu (LK201627).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gonin J. The treatment of detached retina by searing the retinal tears. Archives of Ophthalmology. 1930;4:621-5
2. D'Amico DJ. Clinical practice. Primary retinal detachment. The New England journal of medicine. 2008;359:2346-54
3. Pastor JC, Rojas J, Pastor-Idoate S, Di Lauro S, Gonzalez-Buendia L, Delgado-Tirado S. Proliferative vitreoretinopathy: A new concept of disease pathogenesis and practical consequences. Progress in retinal and eye research. 2016;51:125-55
4. Pennock S, Haddock LJ, Eliott D, Mukai S, Kazlauskas A. Is neutralizing vitreal growth factors a viable strategy to prevent proliferative vitreoretinopathy? Progress in retinal and eye research. 2014;40:16-34
5. Pastor JC, de la Rua ER, Martin F. Proliferative vitreoretinopathy: risk factors and pathobiology. Progress in retinal and eye research. 2002;21:127-44
6. Machemer R, Aaberg TM, Freeman HM, Irvine AR, Lean JS, Michels RM. An updated classification of retinal detachment with proliferative vitreoretinopathy. American journal of ophthalmology. 1991;112:159-65
7. Rouberol F, Chiquet C. [Proliferative vitreoretinopathy: pathophysiology and clinical diagnosis]. Journal francais d'ophtalmologie. 2014;37:557-65
8. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
9. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682-95
10. Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA. et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nature medicine. 2013;19:1281-7
11. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814-22
12. Lee IH, Kawai Y, Fergusson MM, Rovira, II, Bishop AJ, Motoyama N. et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225-8
13. Feng Y, Liu J, Guo W, Guan Y, Xu H, Guo Q. et al. Atg7 inhibits Warburg effect by suppressing PKM2 phosphorylation resulting reduced epithelial-mesenchymal transition. International journal of biological sciences. 2018;14:775-83
14. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell death and differentiation. 2011;18:571-80
15. Kurz T, Karlsson M, Brunk UT, Nilsson SE, Frennesson C. ARPE-19 retinal pigment epithelial cells are highly resistant to oxidative stress and exercise strict control over their lysosomal redox-active iron. Autophagy. 2009;5:494-501
16. Frost LS, Mitchell CH, Boesze-Battaglia K. Autophagy in the eye: implications for ocular cell health. Exp Eye Res. 2014;124:56-66
17. Boya P, Esteban-Martinez L, Serrano-Puebla A, Gomez-Sintes R, Villarejo-Zori B. Autophagy in the eye: Development, degeneration, and aging. Progress in retinal and eye research. 2016;55:206-45
18. Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:562-4
19. Besirli CG, Chinskey ND, Zheng QD, Zacks DN. Autophagy activation in the injured photoreceptor inhibits fas-mediated apoptosis. Investigative ophthalmology & visual science. 2011;52:4193-9
20. Kaarniranta K. Autophagy-hot topic in AMD. Acta ophthalmologica. 2010;88:387-8
21. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of cell biology. 2005;169:425-34
22. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26
23. Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Current protocols in molecular biology. 2005 Chapter 28: Unit 28 1
24. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
25. Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S. et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9241-6
26. Friedlander M. Fibrosis and diseases of the eye. The Journal of clinical investigation. 2007;117:576-86
27. Charteris DG. Proliferative vitreoretinopathy: pathobiology, surgical management, and adjunctive treatment. The British journal of ophthalmology. 1995;79:953-60
28. Tamiya S, Kaplan HJ. Role of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Exp Eye Res. 2016;142:26-31
29. Xiao W, Chen X, Liu X, Luo L, Ye S, Liu Y. Trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and epithelial-mesenchymal transition in retinal pigment epithelium cells. Journal of cellular and molecular medicine. 2014;18:646-55
30. Strauss O. The retinal pigment epithelium in visual function. Physiological reviews. 2005;85:845-81
31. Campochiaro PA. Cytokine production by retinal pigmented epithelial cells. International review of cytology. 1993;146:75-82
32. Winkler J, Hoerauf H. TGF-ss and RPE-derived cells in taut subretinal strands from patients with proliferative vitreoretinopathy. European journal of ophthalmology. 2011;21:422-6
33. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279-96
34. Tamiya S, Liu L, Kaplan HJ. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investigative ophthalmology & visual science. 2010;51:2755-63
35. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell. 2012;22:725-36
36. Jin M, He S, Worpel V, Ryan SJ, Hinton DR. Promotion of adhesion and migration of RPE cells to provisional extracellular matrices by TNF-alpha. Investigative ophthalmology & visual science. 2000;41:4324-32
37. Schiro JA, Chan BM, Roswit WT, Kassner PD, Pentland AP, Hemler ME. et al. Integrin alpha 2 beta 1 (VLA-2) mediates reorganization and contraction of collagen matrices by human cells. Cell. 1991;67:403-10
38. Hiscott P, Hagan S, Heathcote L, Sheridan CM, Groenewald CP, Grierson I. et al. Pathobiology of epiretinal and subretinal membranes: possible roles for the matricellular proteins thrombospondin 1 and osteonectin (SPARC). Eye. 2002;16:393-403
39. Yang CH, Liu CZ, Huang TF, Yang CM, Lui KR, Chen MS. et al. Inhibition of RPE cell-mediated matrix adhesion and collagen gel contraction by crovidisin, a collagen-binding snake venom protein. Current eye research. 1997;16:1119-26
40. Leiderman YI, Miller JW. Proliferative vitreoretinopathy: pathobiology and therapeutic targets. Seminars in ophthalmology. 2009;24:62-9
41. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178-96
42. Tsukita S, Furuse M. Pores in the wall: claudins constitute tight junction strands containing aqueous pores. The Journal of cell biology. 2000;149:13-6
43. Burke JM, Hong J. Fate of E-cadherin in early RPE cultures: transient accumulation of truncated peptides at nonjunctional sites. Investigative ophthalmology & visual science. 2006;47:3635-43
44. Van Aken EH, De Wever O, Van Hoorde L, Bruyneel E, De Laey JJ, Mareel MM. Invasion of retinal pigment epithelial cells: N-cadherin, hepatocyte growth factor, and focal adhesion kinase. Investigative ophthalmology & visual science. 2003;44:463-72
45. Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell death & disease. 2016;7:e2520
46. Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D. et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989-2005
47. Klettner A, Kauppinen A, Blasiak J, Roider J, Salminen A, Kaarniranta K. Cellular and molecular mechanisms of age-related macular degeneration: from impaired autophagy to neovascularization. The international journal of biochemistry & cell biology. 2013;45:1457-67
48. Baek A, Yoon S, Kim J, Baek YM, Park H, Lim D. et al. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy. 2017;13:248-63
49. Zhang J, Bai Y, Huang L, Qi Y, Zhang Q, Li S. et al. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: implications for age-related macular degeneration. Cell death & disease. 2015;6:e1972
50. Shu DY, Lovicu FJ. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Progress in retinal and eye research. 2017;60:44-65
51. Casaroli Marano RP, Vilaro S. The role of fibronectin, laminin, vitronectin and their receptors on cellular adhesion in proliferative vitreoretinopathy. Investigative ophthalmology & visual science. 1994;35:2791-803
52. Perusek L, Sahu B, Parmar T, Maeno H, Arai E, Le YZ. et al. Di-retinoid-pyridinium-ethanolamine (A2E) Accumulation and the Maintenance of the Visual Cycle Are Independent of Atg7-mediated Autophagy in the Retinal Pigmented Epithelium. The Journal of biological chemistry. 2015;290:29035-44
Author contact
![]() Corresponding authors: Liu Cao, Institute of Translational Medicine, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang, Liaoning Province, 110122, China. Tel: +86 024 31939630, Fax: +86 024 31939630, E-mail: lcaoedu.cn; Shuai Han, Department of Neurosurgery, The First Hospital of China medical University, Shenyang, Liaoning Province, 110122, China. Tel: +86 13840569777, E-mail: hanshuai537197com.
Corresponding authors: Liu Cao, Institute of Translational Medicine, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang, Liaoning Province, 110122, China. Tel: +86 024 31939630, Fax: +86 024 31939630, E-mail: lcaoedu.cn; Shuai Han, Department of Neurosurgery, The First Hospital of China medical University, Shenyang, Liaoning Province, 110122, China. Tel: +86 13840569777, E-mail: hanshuai537197com.