Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):522-532. doi:10.7150/ijbs.30572 This issue Cite
Research Paper
Combined Androgen receptor blockade overcomes the resistance of breast cancer cells to palbociclib
Wenfei Ji1#, Yaqin Shi2#, Xin Wang2, Weiwei He1, Lin Tang2, Shengwang Tian4, Hua Jiang5, Yongqian Shu3 ![]() , Xiaoxiang Guan1,2,3
, Xiaoxiang Guan1,2,3 ![]()
1. Department of Medical Oncology, Jinling Clinical College of Nanjing Medical University, Nanjing 210002, China.
2. Department of Medical Oncology, Jinling Hospital, Medical School of Nanjing University, Nanjing 210002, China.
3. Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China.
4. Department of Oncology, JinTan People's Hospital, Jintan 213200, China.
5. Department of Oncology, The Affiliated Changzhou No.2 People's Hospital with Nanjing Medical University, Changzhou 213003, Jiangsu, China.
#These authors contributed equally to this work.
Received 2018-10-11; Accepted 2018-11-28; Published 2019-1-1
Abstract

The dysregulation of cyclin D -Cyclin-dependent kinase 4/6 (CDK4/6)-Rb axis has been implicated in breast cancer progression and the selective CDK4/6 inhibitors have shown effective activity in advanced breast cancer, especially in tumors driven by the estrogen receptor (ER). However, resistance to these small molecular inhibitors has become an inevitable clinical issue after their initial use. Here, we investigated the potential mechanism of resistance by establishing a CDK4/6 inhibitor palbociclib-resistant breast cancer cell line (MCF-7pR). After prolonged exposure to palbociclib, we detected the loss of the ER signaling and an increase in androgen receptor (AR). Moreover, we demonstrated more localization of AR in the cell nucleus of MCF-7pR compared to the parental cell (MCF-7). We also reported that AR could promote the progression of the cell cycle. Blockade of AR signaling could reduce the level of the relative G1-S cyclins, abolish Rb phosphorylation and inhibit the activation of transcriptional programs in S phase. Furthermore, dual inhibition of AR and CDK4/6 could reverse the resistance of palbociclib both in vitro and in vivo. In sum, our studies provide evidence that AR activation promotes cell cycle progression and cell proliferation in CDK4/6 inhibitor resistance, and identify AR inhibition as a putative novel therapeutic strategy to treat CDK4/6 inhibitor resistance in cancer.
Keywords: drug resistance, breast cancer, palbociclib, enzalutamide, cell cycle
Introduction
Breast cancer is the most common malignancy, and is the second cause of cancer death among females worldwide, accounting for an estimated 266120 new cases and 40,920 deaths in 2018 [1]. For hormone receptor (HR)-positive breast cancer patients, which account for approximately 60%-70% of all cases, hormonal monotherapy including selective ER modulator (SERM) or selective estrogen receptor down-regulator (SERD), aromatase inhibitor (AI), or even their combination as doublets have contributed to the decline in breast cancer mortality [2-4]. Unfortunately, disease progression occurs inevitably after endocrine treatment in patients with metastatic breast cancer [5]. CDK4/6 inhibitors suppress cell proliferation by repressing the CDK4/6-dependent cell cycle in HR-positive tumors which is driven by ER related transcriptional signaling [6]. Palbociclib is an oral and highly selective CDK4/6 inhibitor which is effective in inducing cell cycle arrest and cell mitosis. Palbociclib has been approved by the Food and Drug Administration (FDA) and European Medicine Agency (EMA) for the treatment of patients with HR-positive locally advanced or metastatic breast cancer [7]. Despite of the promising clinical response, the development of drug resistance is common in breast cancer patients in the metastatic setting and the mechanisms of the resistance are poorly understood. Based on the known function of CDK4/6 dependent cell cycle signaling, the loss of the retinoblastoma tumor suppressor protein (RB), the amplification of Cyclin D1and CDK6, and the activation of Cyclin E-CDK2 are potential mechanisms leading to the occurrence of drug resistance[8, 9]. Furthermore, it has also been reported that the mechanism of resistance is independent of the alteration or activation of PI3K/AKT/mTOR pathway [10, 11]. Herein, identifying mechanisms involved in the resistance to palbociclib may improve the efficacy and prevent therapeutic resistance.
In this study, we established the specific HR-positive cell line which was exposed to the CDK4/6 inhibitor palbociclib chronically. Eventually, the cancer cells acquired the resistance to palbociclib and lost the expression of ER signaling. Additionally, acquired resistance to palbociclib was associated with the activation of AR which was expressed in more than 70% breast cancer [12] and participates in the tumorigenesis and the development of cancers in a ligand-dependent or a ligand-independent manner [13, 14]. Furthermore, enzalutamide, as a novel and selective AR inhibitor [15], could sensitize breast cancer to palbociclib and reverses the palbociclib resistance. In conclusion, this article illustrates the involvement of AR signaling activation in the resistance of palbociclib and nominates AR inhibitor enzalutamide as a new treatment strategy for advanced cancers upon CDK 4/6 inhibitor resistance to maximize its clinical effectiveness.
Materials and methods
Cell culture and resistant cell line establishment
The MCF-7 was purchased in 2017 to 2018 from the Chinese Academy of Science Committee Type Culture Collection Cell Bank (Shanghai, China). MCF-7 cells were cultured in DMEM (Dulbecco's modified Eagle's medium) high glucose supplemented with 10% fetal bovine serum (FBS) and 100 units/ml of penicillin and 100 units /ml of streptomycin. The MCF-7 cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C.
MCF-7 cells were initially cultured in medium containing 1 µM palbociclib (MedChem Express, Shanghai, China), and then cells were subcultured every 2-3 weeks in medium with increased concentrations of palbociclib (a 25% increase each time). Finally, the obtained cells grew exponentially in the presence of 30 µM palbociclib and were named as MCF-7pR.
Cell Viability assay and agents
In cell viability assay, 4000 cells per well were planted in 96-well plates and cells were exposed to different concentrations of agents in the following day. After 48-hour treatment, cell survival was assessed with the Cell Counting kit-8 in according to the recommended guideline (KeyGEN Biotech, Nanjing, China). Combination index (CI) values were calculated using CompuSyn software (ComboSyn, Inc., NJ, USA).
Palbociclib, Ribociclib, Abemaciclib, enzalutamide, Fulvestrant and Dihydrotestosterone (DHT) were obtained from MedChem Express, and palbociclib was diluted in sterile water, DHT was dissolved in ethanol and others above were diluted in DMSO.
Cell cycle analyses
Cells were incubated with culture medium in 6-well plates for 24 hours and then cells were treated with the indicated drugs. For the purpose of cell cycle analysis, cells were washed with ice-cold PBS and then fixed with 70% ethanol overnight at -20 °C. The fixed cells were rehydrated in PBS for 10 minutes and subjected to PI/RNase staining. The cell cycle in each specific sample were evaluated by the flow cytometer (FACS Calibur, BD Biosciences, USA) following the manufacture's instruction.
Colony formation assay
Cells were diluted and replaced in six-well plates at 500 cells per well. After incubating for 24 hours, cells were treated with indicated drugs for 24 hours. After the drug administration, cells were washed with PBS and were incubated in fresh medium for 14 days. Finally, cells were fixed with 100% methanol and stained with 0.5% crystal violet. Colonies containing over 50 cells were counted as survivors.
Western blot and Co-Immunoprecipitation assay
Cells were washed three times with ice-cold PBS and lysed in RIPA buffer containing protease and phosphatase inhibitor cocktails. Protein concentrations were determined with BCA kit (All kits from KeyGEN biotech, Nanjing, China). Subsequent steps were performed as recently described [16].
In co-immunoprecipitation assay, cells were lysed with NETN buffer (20mM Tris-HCl, pH 8.0, 100mM NaCl, 1mM EDTA, 0.5% Nonidet P-40) containing 50mM b-glycerophosphate, 10mM NaF and 1mgml-1 each of pepstatin A and aprotinin. The assay was performed as previously described [17].
The specific antibodies used in western blot were anti-MDR1 (ab170904), anti-Rb (ab181616), anti-E2F1 (ab179445), anti-cyclin A2 (ab181591), anti-cyclin E2 (ab226388), anti-PR (ab131486), anti-cyclin D1 (ab326977), anti-cyclin E1 (ab213142), anti-CDK 2 (ab101682) from Abcam and anti-pRb (S795) (#9301), anti-ER (#13258), anti-AR (#5153), anti-ki67 (#9449), GAPDH (#2118), mouse IgG (#7076), rabbit IgG (#7074) from Cell Signaling Technologies.
Real-time polymerase chain reaction (real-time RT-PCR) analysis
Total RNA was extracted from cultured cells using Trizol Reagent (Invitrogen, CA, USA) following the manufacturer's instructions. For qPCR analyses of ESR1, PGR, XBP1, TFF1 and AR, the cDNA was synthesized via a PrimeScriptTM RT Master Mix Kit (Takara, China), followed by PCR using Powers SYBR Green PCR Master Mix (Life Technology, USA), according to the manufacturer's instructions. GAPDH was used as internal control. Primer sequences (forward and reverse, respectively) were as follows: ESR1 F: 5'-TATGTGTCCAGCCACCAACC-3', R: 5'-CTTCGTAGCATTTGCGGAGC-3'; PGR F: 5'-GACTGAGCTGAAGGCAAAGG-3', R: 5'-AACTTCAGGCAAGGTGTCCG-3'; XBP1 F: 5'-CTGAGTCCGCAGCAGGTG-3', R: 5'-GTCCAGAATGCCCAACAGGA-3'; TFF1 F: 5'-GTGGTTTTCCTGGTGTCACG-3', R: 5'-AGGATAGAAGCACCAGGGGA-3'; AR R: 5'-GGGCGAAGTAGAGCATCCT-3', F: 5'-GACGACCAGATGGCTGTCATT3'; GAPDH F: 5'-CATCTTCTTTTGCGTCGCCA-3', R: 5'-TTAAAAGCAGCCCTGGTGACC-3'. Real-time PCR analysis was performed on an ABI 7300 Sequence Detection System (Applied Biosystems, CA, USA).
Immunofluorescence and agents
The cells were plated on eight-well chamber slides (Lab-Tek Products, Illinois, USA) and washed with PBS. Then, the cells were fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.1% TritonX-100 for 10 minutes and incubated for one hour at 37°C with anti-AR (#5153) antibody which was used at the manufacturer's recommended dilution. Cells were then washed with PBS and incubated for 30 min at 37°C with mouse anti-rabbit IgG conjugated with FITC (Invitrogen; 1:200). Subsequently, nuclei were counterstained with 4', 6-diamidino-2-phenylindole (DAPI; Sigma) for 10 min. Finally, cells were mounted with mounting solution (DAKO, Glostrup, Denmark) and examined under a LSM510 confocal microscope (Carl Zeiss, Gottingen, Germany).
Transfection
AR shRNA (shAR) and scramble shRNA control (NC) were first cloned into the pGpU6/GFP/Neo vector purchased from GenePharma (Shanghai, China), and then transfected into cells according to the manufacturer's protocol. The shRNA sequences were as follows: shAR-1, 5'-TTGCCAGAGAGCTGCATCA-3', shAR-2, 5'-CTGGCGATCCTTCACCAAT-3', shAR-3, 5'-CACCAATGTCAACTCCAGGAT-3', and shNC, 5'-AGTGCACGTGCATGTCCTA-3'.
Xenograft Experiments
MCF-7pR xenograft was established as previously described [18].When tumor volume reached 100 mm3 (±50), mice were randomized to four treatment groups (six animals in each group). The mice were treated with vehicle (orally), or palbociclib at a dose of 100 mg/kg twice a week by oral gavage (p.o.), or enzalutamide at a dose of 50 mg/kg five times a week (p.o.), or both palbociclib and enzalutamide. Tumors were measured once every three days and tumor volume were calculated as (width × length2)/2. Animal studies were performed in accordance with institutional guidelines.
Immunohistochemistry
Following being deparaffinized in a series of xylenes and ethanols, slides were rehydrated and subjected to antigen retrieval using citrate buffer (BioGenex, USA). Slides Sections were incubated overnight with primary antibodies against Ki67, and AR antibodies. Immunostained sections were scanned using a microscope. The percentages of positive cells were scored according to the previous researches [19].
Statistical Analysis
All statistical tests were conducted with GraphPad Prism version 6.0. Data were analyzed using a Student's t-test. Data are presented as mean ± SD of three independent experiments unless stated otherwise. A P value of < 0.05 was considered statistically significant. *P < 0.05, or **P < 0.01 or ***P < 0.001.
Results
Establishment of CDK4/6 inhibitor-resistant cells
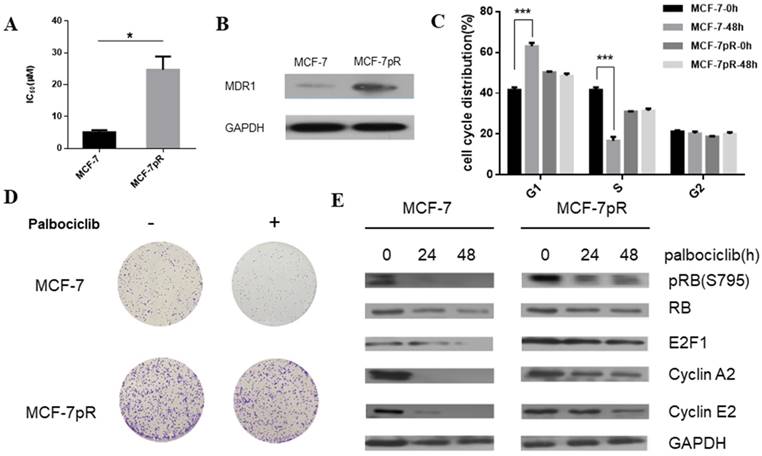
To generate palbociclib-resistant clones from MCF-7 cells, we exposed MCF-7 cells to palbociclib continuously. The palbociclib resistant cells (MCF-7pR) were maintained in growth medium with increasing doses of palbociclib for over 1 year. The MCF-7pR cells were repeatedly confirmed as insensitive to palbociclib with a 5-fold increase in IC50 compared to the parental MCF-7 cells. The mean IC50 of the palbociclib resistant cells (MCF-7pR) and parental cells (MCF-7) were detected as 24.72 µM and 5.077 µM respectively (Figure 1A). To ascertain if the palbociclib resistant cells are resistant to other CDK4/6 inhibitors, we tested the growth response of the resistant cells and the parental cells to other novel and selective CDK4/6 inhibitors Ribociclib and Abemaciclib. In both cases, MCF-7pR cells had significantly higher IC50. (Supplementary Figure 1). These findings might indicate that MCF-7pR cells were resistant to broad CDK4/6 inhibitors. Increased multidrug resistance protein 1 (MDR1) expression was detected in MCF-7pR cells compared to the parental MCF7 cells, confirming the successful generation of palbociclib-resistant cells (Figure 1B). We further investigate if there are any differences in cell cycle distribution between the parental MCF-7 cells and the resistant cells. Data from flow cytometry assay showed that the proportion of cells in the S phase was markedly decreased by the treatment with 4uM palbociclib for 48 hours, whereas the proportion of cells in the S phase was not decreased in MCF-7pR cells (Figure 1C). Moreover, we detected the increased colony formation in MCF-7pR compared to the parental MCF-7 cells. The exposure to palbociclib resulted in a significant reduction of colony formation in parental MCF-7; however, there is no decrease of colony formation in MCF-7pR cells (Figure 1D). To further verify that palbociclib resistance was associated with the alteration of the cyclin D1-CDK4/6-Rb axis, we examined the level of Rb phosphorylation after the exposure to palbociclib in 0 hours, 24 hours and 48 hours respectively. The results demonstrated that the treatment of 4 µM palbociclib for 24 hours reduced the level of phosphor-Rb (pRb) in MCF-7 cells while the exposure to palbociclib for 48 hours did not significantly inhibit the phosphorylation of Rb in MCF-7pR cells. Consistent with the change of the Rb phosphorylation, the expression of E2F1and E2F1 target genes (cyclinA2 and cyclin E2) in parental MCF-7 cells was inhibited by palbociclib significantly but not in the MCF-7pR (Figure 1E). In conclusion, palbociclib-resistant breast cancer cells showed higher IC50 of palbociclib, increased expression of MDR1, improved colony-forming capacity than parental cells. Moreover, acquired resistance could impair the inhibitory effect of palbociclib on the G1-S phase transition.
Generation of CDK4/6 inhibitor-resistant cells. A. Relative cell viability of MCF-7 and MCF-7pR cells after being treated with indicated concentrations of palbociclib for 48 hours was analyzed by cell viability assay. IC50 of the indicated drug in MCF-7 and MCF-7pR cells were shown. *P < 0.05. B. Western blot determined the MDR-1 expression level in MCF-7 and MCF-7pR cells. C. MCF-7 and MCF-7pR cells were treated with or without palbociclib (4 µM) for 48 hours and measured by a flow cytometer. Results were reported as mean percent cell cycle distribution with standard errors. *P < 0.05, or **P < 0.01 or ***P < 0.001. D. Respective images of colony-formation results in MCF-7 and MCF-7pR with the treatment of palbociclib or with no treatment. E. MCF-7 and MCF-7pR cells were treated with 4 µM palbociclib and collected at indicated times. Immunoblots were performed with the indicated antibodies.
The dysregulation of hormone receptors in CDK4/6 inhibitor-resistant cells
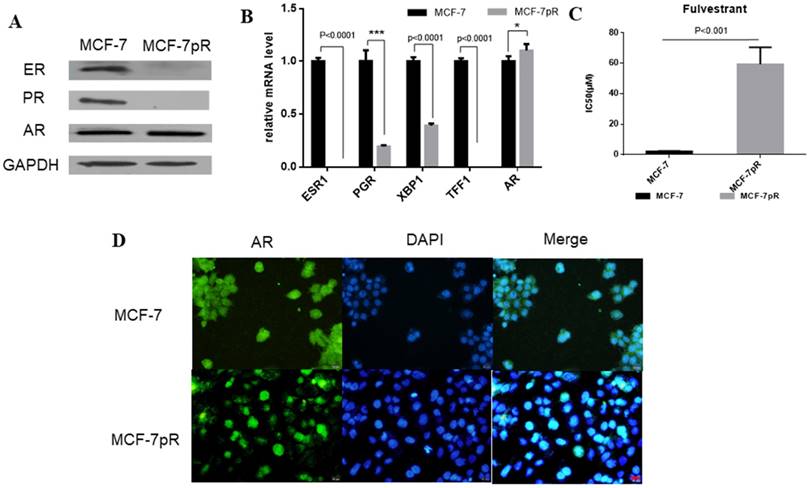
In breast cancer cells, the activation of ER can continuously enhance the expression of cyclin D1 through directly binding to the promoter, which encodes the cyclin D1 isoform [20]. Furthermore, E2F, which is involved in cyclin D1-CDK4/6-Rb axis, can regulate the expression of ER [21, 22]. We speculated whether ER signaling was altered in MCF-7pR cells where the cyclin D1-CDK4/6-Rb axis is dysregulated. Compared with the parental MCF-7 cells, ER and progesterone receptor (PR) expression were reduced obviously in the mRNA and protein level in the resistant cells (Figure 2A and B). Moreover, XBP1 and TFF1, as the ER-regulated genes, were also inhibited in palbociclib-resistant cells (Figure 2B). Considering that ER plays a crucial role in the proliferation and progression in ER-positive breast cancer [23], we further investigated whether the loss of ER signaling could induce cell proliferation in the resistant clones independent of ER. We examined the effects of fulvestrant in MCF-7pR and the parental MCF-7 cells via cell viability assay. The results showed that the IC50 value was higher in MCF-7pR cells than that in MCF-7 cells (Figure 2C). Collectively, these analyses indicate that cells resistant to CDK4/6 inhibitors might also be insensitive to endocrine therapy. In addition to ER and PR, AR is another steroid-hormone activated transcription factor belonging to the nuclear receptor superfamily [24]. There also exists a dynamic interplay between AR and ER signaling in breast cancer, which established a specific transcriptional network. Recently, several studies have demonstrated that the ratio of nuclear AR to ER might critically influence tumor biology and response to endocrine therapy. Besides, overexpression of AR might induce the resistance to endocrine therapy [25-28]. Therefore, we next sought to determine the expression of AR in resistant cells. The elevated expression of AR was observed in mRNA and protein level of MCF-7pR compared to the parental cells (Figure 2A and B). Furthermore, the immunostaining showed more localizations of AR in the cell nucleus of resistant cells compared to that in the parental MCF-7 cells (Figure 2D). These observations suggested that AR might act as a mediator in the proliferation of resistant cells and participate in the resistance to the CDK4/6 inhibitors in absence of ER.
The dysregulation of hormone receptors in CDK4/6 inhibitor-resistant cells. A. Western blot results showed the expression of ER, PR and AR in MCF-7 and MCF-7pR cells. B. mRNA from MCF-7 and MVF-7pR cells were analyzed by the real-time RT-PCR for levels of ESR1, AR and ER-regulated genes PGR, XBP1, TFF1. Data were from triplicate experiments and the reported results were shown as the mean ± SD, *P < 0.05, or **P < 0.01 or ***P < 0.001. C. Relative cell viability of MCF-7 and MCF-7pR cells after treated with indicated concentrations of fulvestrant for 48 hours was analyzed by cell viability assay. IC50 of fulvestrant in MCF-7 and MCF-7pR cells were shown. *P < 0.05, or **P < 0.01 or ***P < 0.001. D. Fluorescence microscopy analysis of the location of AR by IF. The green signal represents the staining of AR protein, and the blue signal represents the nuclear DNA staining by DAPI. Scale bar: 25 µm.
The regulation of AR in the cell cycle of resistant cells
We demonstrated that the non-aromatizable androgen 5-α-dihydrotestosterone (DHT) could inhibit the proliferation of MCF-7. Contrary to the inhibitory effect of DHT in parental cells, the DHT treatment enhanced the cell proliferation of MCF-7pR cells at different time points (24, 48 or 72 hours) (Figure 3A). Furthermore, since the different G1/S transition was observed in the resistant MCF-7pR cells compared with the parental MCF-7 cells, we inquired whether different regulation of cyclin D1, which is a key rate-limiting event in mitogenic signaling leading to S-phase entry, exists between the resistant clones and parental cells. As shown in Figure 3B, MCF-7 cells exhibited a decrease in the level of cyclin D1 following the 48 hours of DHT treatment while AR activated by the DHT increased the cyclin D1 expression in MCF-7pR (Figure 3B). To examine whether AR associates with cyclin D1 in the resistant cells, we performed the co-immunoprecipitation experiments. The MCF-7pR cells were treated with DHT agents. Cell lysates were subjected to immunoprecipitation assay with anti-AR or anti-cyclin D1 antibody. These results provided the evidence of the interaction between the AR and cyclin D1 proteins (Figure 3C). Taken together, our findings suggest that AR can bind to cyclin D1 and regulate the expression of cyclin D1 positively in MCF-7pR cells. AR-mediated cyclin D1 expression may be involved in the resistance to CDK4/6 inhibitors.
The regulation of AR in the cell cycle of resistant cells. A. Having been treated with 100 nM DHT for 24, 48 or 72 hours, Cell viability of MCF-7 and MCF-7pR was measured by the Cell Counting kit-8 assay. *P < 0.05, or **P < 0.01 or ***P < 0.001. B. The expression of AR and CCND1 was detected by Western blot following the treatment of vehicle or 100 nM DHT for 48 hours in MCF-7 and MCF-7pR cells. C. The lysates of MCF-7pR treated with vehicle or 100 nM DHT were subjected to immunoprecipitation with AR antibodies. The immunoprecipitates were then blotted with indicated antibodies. D. AR mRNA levels were determined by real-time PCR in MCF-7pR cells following transfection with shAR or shNC for 48 hours. Data shown were from triplicate experiments and reported as the mean ± SD, ***P < 0.001. E. The expression of AR was detected by Western blot following transfection with shAR or shNC for 48 hours. *P < 0.05, or **P < 0.01 or ***P < 0.001. F. MCF-7pR cells were treated with DHT and/or transfected with shAR for 48 hours and measured by a flow cytometer. Results are reported as mean percent cell cycle distribution with standard errors. G. The protein expression of AR, CCND1, CDK2 and CCNE1 was determined by Western blot in MCF-7pR following treatment DHT and/or transfection with AR for 48 hours.
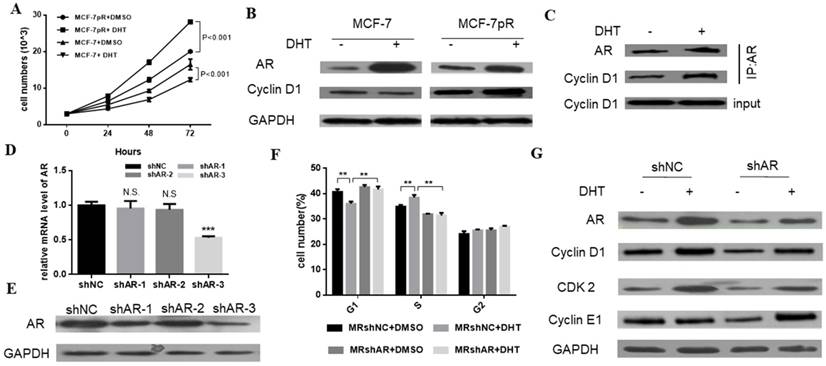
To further validate the role of AR in the regulation of cell cycle, AR short hairpin RNA (shAR) was transfected into the MCF-7pR cells. Silencing of AR was confirmed in the mRNA and protein level (Figure 3C and D). ShAR-3 was the most transfection‐efficient and was chosen for additional study. Data from cell cycle analysis showed that DHT treatment resulted in a decrease of cells in G0/G1 phase and a concurrent decrease in the S phase in MCF-7pR cells. Addition of shRNA effectively reversed the induction effect of DHT in G1/S transition (Figure 3E). Recent in vitro studies in ER-positive breast cancer cells have shown that the hyperactivity of cyclin E-CDK2 axis is highly associated with palbociclib resistance. Chronic exposure to palbociclib could resulted in increased expression of CDK2 and cyclin E [8-10]. We next investigated whether AR could regulate CDK2 and CCNE1 expression in resistant cells. Data from western blot experiments demonstrated that AR activation due to DHT treatment elevated the expression of the CDK2 and CCNE1 in MCF-7pR cells. Besides, silencing of AR inhibited the expression of these two genes (Figure 3F). Consequently, AR is involved in the regulation of cell cycle progression in the resistant MCF-7pR cells and might contribute to explain partial molecular basis of palbociclib resistance in the resistant cells.
AR blockade restores the sensitivity to palbociclib
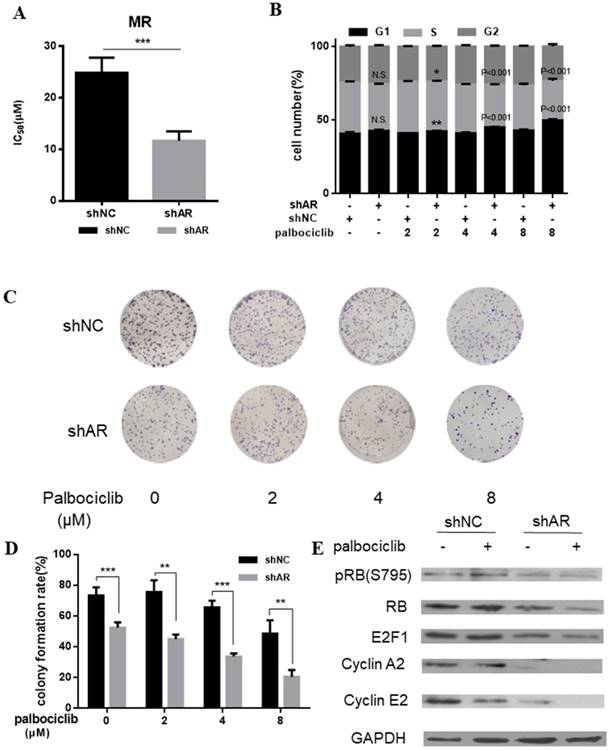
Based on the above evidence that activation of AR signaling was possibly associated with mediating cell cycle progression, we sought to determine whether inhibition of AR could increase the sensitivity of MCF-7pR cells to palbociclib. Results from cell viability assays revealed the decreased IC50 of palbociclib in MCF-7pR cells transfected with shAR compared with the control group, which suggesting that silencing of AR could sensitize MCF-7pR cells to palbociclib (Figure 4A). Moreover, by performing the cell-cycle analysis, we confirmed that the combination of AR blockade and palbociclib resulted in substantially increased G1-S phase transition arrest. It was noted that shAR significantly potentiated the effects of palbociclib in inducing cell cycle arrest in MCF-7pR cells relative to control shRNA (Figure 4B). In addition, the increasing concentration of palbociclib at 2 to 8 µM decreased the clonogenicity in MCF-7pR cells transfected with AR shRNA (Figure 4C and D). Furthermore, palbociclib treatment led to decreased expression of several G1-restriction point markers including RB, pRb, E2F1, cyclin A2 and cyclin E2 in cells transfected with shAR relative to control (Figure 4E). Conclusively, our data suggested that AR inhibition could impair the G1-S phase transition and inhibit cell proliferation in palbociclib-resistant cells.
AR blockade restored the sensitivity to palbociclib. A. Relative cell viability of MCF-7pR cells transfected with shAR or shNC were treated with indicated concentrations of palbociclib for 48 hours and then evaluated with the Cell Counting kit-8. IC50 are shown, ***P < 0.001. B. MCF-7pR cells were treated with indicated concentrations of palbociclib and transfected with shAR or shNC. Cell cycle distribution was determined by flow cytometry. The percentage of cells in each phase of the cell cycle (G1, S and G2) is indicated. C&D. MCF-7pR cells were transfected with shAR or shNC and treated with indicated concentrations of palbociclib for 48 hours, and the cell proliferation was determined by clone formation assay. Data represented the mean of the three independent experiments, **P < 0.01 or ***P < 0.001. E. MCF-7pR cells were treated with vehicle or palbociclib and/or transfected with shAR, collected after 48 hours and the immunoblotted with the indicated antibodies.
Enzalutamide resensitizes palbociclib-resistant cells to palbociclib
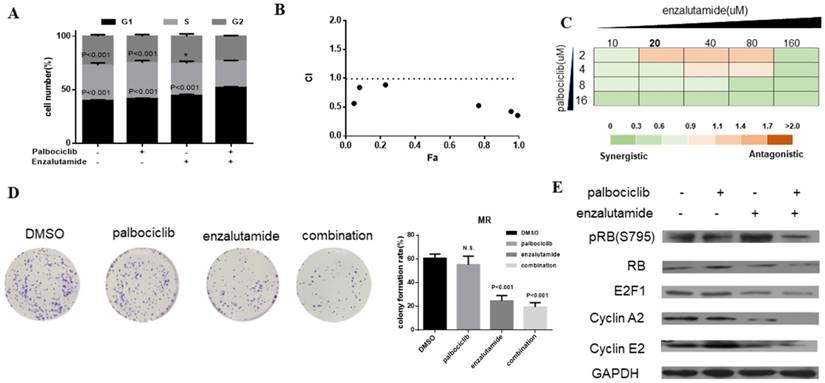
Enzalutamide is an AR antagonist and has been approved by FDA for the treatment of patients with metastatic prostate cancer [29, 30]. Next, we investigate whether treatment of CDK4/6 inhibitor in combination with AR antagonist could resensitize MCF-7pR cells to CDK4/6 inhibitor. The combination induced the suppression of S phase entry significantly in MCF-7pR cells (Figure 5A). To further study the synergistic effect of enzalutamide and palbociclib, we used a fixed radio model of combination therapy in MCF-7pR cells. We utilized the combination radio (1:1) and then calculated the combination index (CI). The combination index vs. fraction affected plot (CI vs. Fa plot) showed that all the CI values in the resistant cells were below 1, which indicating that the combination of these two drugs had a significant synergistic effect (Figure 5B). We also treated cell with increasing concentrations of palbociclib and enzalutamide, and MCF-7pR cells exhibited additive to synergistic effects (Figure 5C). To further evaluate the long-term effects of the combination treatment, we examined the effects of palbociclib, enzalutamide or the combination in colony-formation assays. For MCF-7pR cells, single palbociclib administration failed to inhibit the colony formation but the same concentration of palbociclib significantly reduced the colony formation when combined with enzalutamide (Figure 5D). In line with the cell-cycle analysis, the combination therapy also suppressed the expression of the pRB and related G1-S cyclins (Figure 5E). Thus, treatment with enzalutamide and palbociclib can overcome the acquire resistance to palbociclib in palbociclib-resistant breast cancer cells.
Enzalutamide resensitizes palbociclib-resistant cells to palbociclib. A. MCF-7pR cells were treated for 48 hours with 20 µM of enzalutamide, 4 µM of palbociclib or both. Flow cytometry was used to identify the distribution of cell cycle compared to the cells with no treatment. B. MCF-7pR cells were treated with the combination of palbociclib and enzalutamide. The dose radio of palbociclib and enzalutamide was 1:1. Cell viability was investigated by Cell Counting kit-8 assay. CI vs. Fa plot (combination index vs. fraction affected) for cell viability data are shown. The CI values were calculated by Compusyn software. CI values below 1 are considered to have a synergistic interaction. Data represented the mean of the three independent experiments. C. MCF-7pR cells were treated with 10-160 µM enzalutamide combined with a constant dose of palbociclib (2, 4, 8, 16 µM) for 48 hours. CI values calculated by Compusyn software are shown. D. MCF-7pR cells were treated with palbociclib and enzalutamide alone or in combination for 48 hours and cell proliferation was determined by clone formation assay. Data represents the mean of the three independent experiments, ***P < 0.001. E. MCF-7pR cells were treated with 20 µM of enzalutamide with or without 4 µM of palbociclib for 48 hours. After treatment of drugs, western blot was performed using anti-pRb (s795), anti-Rb, anti-E2F1, anti-cyclin A2 and anti-cyclin E2.
The synergistic effect of enzalutamide and palbociclib on palbociclib-resistant cells in vivo
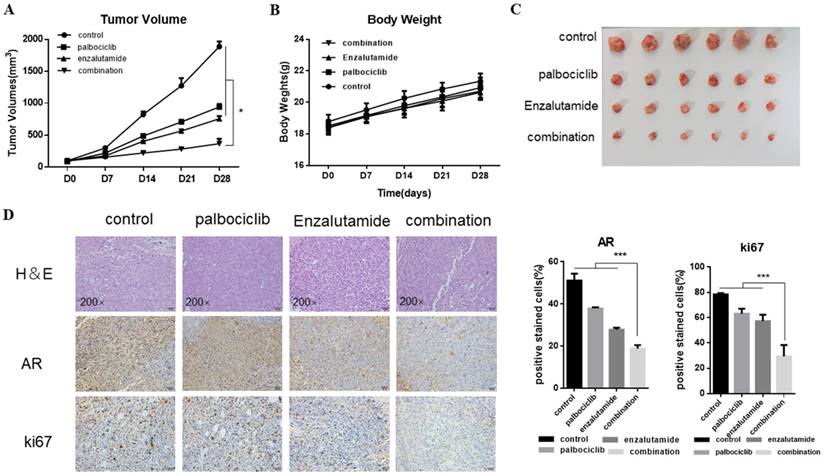
Next, we explored the combination of palbociclib and enzalutamide in vivo using the MCF-7pR xenograft model. MCF-7pR cells were injected into nude mice. When tumors reached an average of 100mm3, treatment of enzalutamide, palbociclib, or the combination of these two were administrated. Mice were treated with vehicle, 100 mg/kg of palbociclib twice a week orally, 50 mg/kg of enzalutamide 5 times a week orally, or a combination. The combination of enzalutamide and palbociclib reduced significantly tumor growth compared to single drug treatments (Figure 6A and C). Additionally, the single drug or combination treatment did not cause body weight loss apparently (Figure 6B). The dual treatment was superior to any of the single treatments and resulted in significant shrinkage as evidenced by decreased levels of Ki67 (Figure 6D).
The synergistic effect of enzalutamide and palbociclib on palbociclib-resistant cells in vivo. Mice bearing MCF-7pR xenografts with a tumor volume of 100±50mm3 (6 for each group) were dosed with palbociclib (100mg/kg/d, p.o.) twice a week or enzalutamide (50 mg/kg/d, p.o.) five times a week or both. A. Tumor volumes were evaluated once a week and calculated by the formula:(S2 × L) /2. S: short diameter; L: long diameter, *P < 0.05. B. Body weight data for MCF-7pR xenograft mouse. C-D. Tumors from mice treated with each of the treatment arms were extracted and stained with routine H&E staining (200×). The xenograft tumors were subjected to immunohistochemical analysis of treated tumors for Ki67 and AR. Expression ratio of ki67 and AR in each of the treatment arms are shown, ***P < 0.001.
Discussion
CDK4/6 inhibitors have shown clinical benefit in advanced ER-positive breast cancer; however, these agents are unlikely to provide a durable cure, and development of resistance is anticipated. Herein, we illustrate the molecular mechanisms of resistance to CDK4/6 inhibitors to explore combined treatment of CDK4/6 inhibitors with other targeted agents to shed light on optimal therapy in ER-positive breast cancer.
To identify the underlying mechanism of palbociclib resistance, we exposed the HR-positive sensitive cell line to the CDK4/6 inhibitor palbociclib. Long-term exposure of MCF-7 cell line to the drug led to the loss of ER signaling and the activated AR signaling. We speculate that one potential effector of the change may be downstream E2F family which is more persistently liberated from Rb and act as a repressor of ER expression [21, 22]. Data herein suggest that cell cycle progression is independent of ER signaling occurring and resistance to palbociclib may also induce an acquired insensitivity to ER antagonists. Additionally, it has been reported that Rb interacts with AR in an androgen-independent manner and acts as a coactivator for AR. Besides, CCNE1 can also function as an AR coactivator, and contribute to AR activation [31]. Thus, in resistant cells, dysregulated cell cycle progression may induce the abnormal change of HR expression. Consistent with the speculation, induction of the AR pathway was observed in CDK4/6 inhibitor resistance, determined by the elevated expression and more nuclear localizations of AR.
AR is a member of nuclear receptors and is expressed in a significant subset (60-90%) of both ER-positive and ER-negative BCs [32, 33]. AR expression has been shown to be associated with a better prognosis, particularly in the luminal breast cancer [34]. Conversely, loss of AR expression induced the transformation from in situ to invasive basal subtype of high-grade ductal BC [35]. Previous studies have shown that DHT-activated AR inhibits MCF-7 cells proliferation by targeting the G1/S transition of the cell cycle [36]. Contrary to the role of repressor in cell cycle regulation [36], AR activation in the resistant cells may participate in the promoting ongoing cell-cycle entry. AR can bind to and activate cyclin D1 which is in line with the data in recent studies[11, 37] and CDK4/6 exert the kinase function and phosphorylate RB partly by binding cyclin D1. Therefore, one speculation is that the elevated AR signaling bypasses palbociclib by cyclin D1 which can induce a cyclin D1/CDK2 complex to develop early adaption to palbociclib [10]. Previous studies have demonstrated that the activated CDK2-CCNE1 complex induces palbociclib resistance. Actually, AR can improve the expression of CDK2 and CCNE1 in our resistant cell models. Conclusively, these data show that AR participates in the cell cycle progression of resistant cells to palbociclib.
Enzalutamide is a second-generation AR antagonist and has been utilized for the treatment of patients with metastatic castration-resistant or chemotherapy-resistant prostate cancer. Enzalutamide has already entered clinical trials in BC patients (NCT02689427, NCT01889238 and NCT02457910) [38]. Strikingly, inhibition of AR can resensitize our resistant cells to palbociclib. Enzalutamide, as an AR antagonist, yields the synergistic growth inhibitory effect on resistant cells when given with palbociclib. Considering the synergistic growth inhibitory effect of enzalutamide and palbociclib on the palbociclib-resistant cells in vitro and in vivo, the clinical application of enzalutamide could be considered in the context of palbociclib resistance. Moreover, mechanistic investigation identified that AR activation improved the expression of CCNE1 in our resistant cell models. This finding assigns useful predictive relevance to CCNE1, which provides the ability to identify CDK4/6 inhibitor resistant tumors with CCNE1 overexpression and more prompted to the combination of CDK4/6 inhibitors and AR inhibitor enzalutamide.
However, this study also has some limitations. Only one palbociclib-resistant cell line was used and there might be other proteins or mutants that could participate in the resistance to palbociclib. Furthermore, the acquired mutation of RB1 is common in palbociclib-resistant breast cancers, which may lead to the resistance to palbociclib in a Rb-independent mechanism. AR in our palbociclib-resistant cells mediates palbociclib resistance in a Rb-dependent manner and therapeutic effect of combination between palbociclib and enzalutamide appears to be uncertain in the lack of Rb. Therefore, it is necessary to assess the effect of the combination treatment in palbociclib-resistant cells with genomic and functional RB loss.
In conclusion, we first identified the activated AR played an important role in palbociclib resistance in breast cancer and illustrate the synergy between AR inhibitor enzalutamide and palbociclib, which might be a promising strategy for the palbociclib-resistant breast cancer.
Supplementary Material
Supplementary figure.
Acknowledgements
This research was supported by a Foundation for Clinical Medicine Science and Technology Special Project of the Jiangsu Province, China (No. BL2014071) (to XG), and National Natural Science Foundation of China (No. 81773102, No. 81470357).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233-47
3. Lee CI, Goodwin A, Wilcken N. Fulvestrant for hormone-sensitive metastatic breast cancer. Cochrane Database Syst Rev. 2017;1:CD011093
4. Mehta RS, Barlow WE, Albain KS, Vandenberg TA, Dakhil SR, Tirumali NR. et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med. 2012;367:435-44
5. Yeo B, Turner NC, Jones A. An update on the medical management of breast cancer. BMJ. 2014;348:g3608
6. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417-30
7. Kwapisz D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: palbociclib, ribociclib, and abemaciclib. Breast Cancer Res Treat. 2017;166:41-54
8. Garrido-Castro AC SGS. CDK4/6 Inhibition in Breast Cancer: Mechanisms of Response and Treatment Failure. Curr Breast Cancer Rep. 2017:26-33
9. Gu G, Dustin D, Fuqua SA. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol. 2016;31:97-103
10. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I. et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016;76:2301-13
11. Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE. et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017;77:2488-99
12. McNamara KM, Yoda T, Takagi K, Miki Y, Suzuki T, Sasano H. Androgen receptor in triple negative breast cancer. J Steroid Biochem Mol Biol. 2013;133:66-76
13. Mangelsdorf DJ TC, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P ER. The nuclear receptor superfamily: the second decade. cell. 1995;83:835-9
14. Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ. et al. The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012;14:R27
15. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V. et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787-90
16. Zhang W, Luo J, Yang F, Wang Y, Yin Y, Strom A. et al. BRCA1 inhibits AR-mediated proliferation of breast cancer cells through the activation of SIRT1. Sci Rep. 2016;6:22034
17. Li Y, Luo K, Yin Y, Wu C, Deng M, Li L. et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nature communications. 2017;8:15752
18. Zhu A, Li Y, Song W, Xu Y, Yang F, Zhang W. et al. Antiproliferative Effect of Androgen Receptor Inhibition in Mesenchymal Stem-Like Triple-Negative Breast Cancer. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;38:1003-14
19. Shen Y, Yang F, Zhang W, Song W, Liu Y, Guan X. The Androgen Receptor Promotes Cellular Proliferation by Suppression of G-Protein Coupled Estrogen Receptor Signaling in Triple-Negative Breast Cancer. Cell Physiol Biochem. 2017;43:2047-61
20. VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin Cancer Res. 2015;21:2905-10
21. Umemura S, Shirane M, Takekoshi S, Kusakabe T, Itoh J, Egashira N. et al. Overexpression of E2F-5 correlates with a pathological basal phenotype and a worse clinical outcome. British Journal of Cancer. 2009;100:764-71
22. Macaluso M, Cinti C, Russo G, Russo A, Giordano A. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene. 2003;22:3511-7
23. Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC. et al. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc Natl Acad Sci U S A. 2014;111:1933-8
24. Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev. 1995;16:271-321
25. Panet-Raymond VG, B.; Beitel LK, Pinsky L, Trifiro M.A. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000;167:139-50
26. Cochrane DR BS, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gómez F, Medicherla S, Alfaro IE, McCullagh E, Jedlicka P, Torkko KC, Thor AD, Elias AD, Protter AA, Richer JK. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Research. 2014;16:R7
27. Need EF, Selth LA, Harris TJ, Birrell SN, Tilley WD, Buchanan G. Research resource: interplay between the genomic and transcriptional networks of androgen receptor and estrogen receptor alpha in luminal breast cancer cells. Mol Endocrinol. 2012;26:1941-52
28. De Amicis F, Thirugnansampanthan J, Cui Y, Selever J, Beyer A, Parra I. et al. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat. 2010;121:1-11
29. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. New England Journal of Medicine. 2014; 371: 1755-6.
30. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-97
31. Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001
32. Riva C, Dainese E, Caprara G, Rocca PC, Massarelli G, Tot T. et al. Immunohistochemical study of androgen receptors in breast carcinoma. Evidence of their frequent expression in lobular carcinoma. Virchows Arch. 2005;447:695-700
33. Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW. et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin Cancer Res. 2018
34. Aleskandarany MA, Abduljabbar R, Ashankyty I, Elmouna A, Jerjees D, Ali S. et al. Prognostic significance of androgen receptor expression in invasive breast cancer: transcriptomic and protein expression analysis. Breast Cancer Res Treat. 2016;159:215-27
35. Hanley K, Wang J, Bourne P, Yang Q, Gao AC, Lyman G. et al. Lack of expression of androgen receptor may play a critical role in transformation from in situ to invasive basal subtype of high-grade ductal carcinoma of the breast. Hum Pathol. 2008;39:386-92
36. Lanzino M, Sisci D, Morelli C, Garofalo C, Catalano S, Casaburi I. et al. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells-identification of a novel androgen response element. Nucleic Acids Res. 2010;38:5351-65
37. C Yang ZL, T Bhatt, M Dickler, D Giri, M Scaltriti, J Baselga, N Rosen, S Chandarlapaty. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2016
38. Liu CY, Lau KY, Hsu CC, Chen JL, Lee CH, Huang TT. et al. Combination of palbociclib with enzalutamide shows in vitro activity in RB proficient and androgen receptor positive triple negative breast cancer cells. PLoS One. 2017;12:e0189007
Author contact
![]() Corresponding authors: Yongqian Shu and Xiaoxiang Guan, Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China. E-mail: shuyongqianorg.cn (Yongqian Shu), xguanedu.cn (Xiaoxiang Guan).
Corresponding authors: Yongqian Shu and Xiaoxiang Guan, Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China. E-mail: shuyongqianorg.cn (Yongqian Shu), xguanedu.cn (Xiaoxiang Guan).