Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):714-725. doi:10.7150/ijbs.31425 This issue Cite
Research Paper
Differential Expression Patterns of Glycolytic Enzymes and Mitochondria-Dependent Apoptosis in PCOS Patients with Endometrial Hyperplasia, an Early Hallmark of Endometrial Cancer, In Vivo and the Impact of Metformin In Vitro
Tao Wang1,2*, Jiao Zhang3*, Min Hu4,5,6, Yuehui Zhang2,4, Peng Cui4, Xin Li4,7,8, Juan Li4,5, Edvin Vestin1, Mats Brännström9, Linus R Shao4 ![]() , Håkan Billig4
, Håkan Billig4
1. The School of Basic Medical Science, Heilongjiang University of Chinese Medicine, 150040 Harbin, China
2. Department of Obstetrics and Gynecology, Key Laboratory and Unit of Infertility in Chinese Medicine, First Affiliated Hospital, Heilongjiang University of Chinese Medicine, 150040 Harbin, China
3. Department of Acupuncture and Moxibustion, Second Affiliated Hospital, Heilongjiang University of Chinese Medicine, 150040 Harbin, China
4. Department of Physiology/Endocrinology, Institute of Neuroscience and Physiology, The Sahlgrenska Academy, University of Gothenburg, 40530 Gothenburg, Sweden
5. Department of Traditional Chinese Medicine, The First Affiliated Hospital of Guangzhou Medical University, 510120 Guangzhou, China
6. Institute of Integrated Traditional Chinese Medicine and Western Medicine, Guangzhou Medical University, 510120 Guangzhou, China
7. Department of Gynecology, Obstetrics and Gynecology Hospital of Fudan University, 200011 Shanghai, China
8. Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, 200011 Shanghai, China
9. Department of Obstetrics and Gynecology, Sahlgrenska University Hospital, Sahlgrenska Academy, University of Gothenburg, 41345 Gothenburg, Sweden
*These authors should be regarded as joint first authors
Received 2018-11-12; Accepted 2018-12-23; Published 2019-1-24
Abstract

The underlying mechanisms of polycystic ovarian syndrome (PCOS)-induced endometrial dysfunction are not fully understood, and although accumulating evidence shows that the use of metformin has beneficial effects in PCOS patients, the precise regulatory mechanisms of metformin on endometrial function under PCOS conditions have only been partially explored. To address these clinical challenges, this study aimed to assess the protein expression patterns of glycolytic enzymes, estrogen receptor (ER), and androgen receptor (AR) along with differences in mitochondria-dependent apoptosis in PCOS patients with and without endometrial hyperplasia in vivo and to investigate the effects of metformin in PCOS patients with endometrial hyperplasia in vitro. Here, we showed that compared to non-PCOS patients and PCOS patients without hyperplasia, the endometria from PCOS patients with hyperplasia had a distinct protein expression pattern of glycolytic enzymes, including pyruvate kinase isozyme M2 isoform (PKM2) and pyruvate dehydrogenase (PDH), and mitochondrial transcription factor A (TFAM). In PCOS patients with endometrial hyperplasia, increased glandular epithelial cell secretion and infiltrated stromal cells in the glands were associated with decreased PDH immunoreactivity in the epithelial cells. Using endometrial tissues from PCOS patients with hyperplasia, we found that in response to metformin treatment in vitro, hexokinase 2 (HK2) expression was decreased, whereas phosphofructokinase (PFK), PKM2, and lactate dehydrogenase A (LDHA) expression was increased compared to controls. Although there was no change in PDH expression, metformin treatment increased the expression of TFAM and cleaved caspase-3. Moreover, our in vivo study showed that while endometrial ERβ expression was no different between non-PCOS and PCOS patients regardless of whether or not hyperplasia was present, ERα and AR protein expression was gradually increased in women with PCOS following the onset of endometrial hyperplasia. Our in vitro study showed that treatment with metformin inhibited ERα expression without affecting ERβ expression. Our findings suggest that decreased glycolysis and increased mitochondrial activity might contribute to the onset of ERα-dependent endometrial hyperplasia and that metformin might directly reverse impaired glycolysis and normalize mitochondrial function in PCOS patients with endometrial hyperplasia.
Keywords: glycolysis, mitochondria, apoptosis, endometrial hyperplasia, metformin, PCOS
Introduction
Polycystic ovary syndrome (PCOS) describes a spectrum of reproductive and metabolic diseases characterized by hyperandrogenism and insulin resistance [1, 2]. Although PCOS affects 4%-21% of all adolescent and reproductive-aged women globally depending on the criteria used to define PCOS and on the ethnicity of the group being studied [3], the underlying cellular and molecular mechanisms of the pathogenesis of this syndrome are far from being completely understood. Furthermore, despite the impairment of endometrial dysfunction that causes endometrial hyperplasia and carcinoma, implantation failure, recurrent pregnancy loss, and premature delivery in PCOS patients [4, 5], an established cause-and-effect relationship between PCOS-related hyperandrogenism and insulin resistance and endometrial dysfunction-induced infertility is still lacking.
The human endometrium is an ovarian steroid hormone-responsive tissue composed of luminal and glandular epithelial cells, stroma with stromal fibroblastic cells, immunocompetent cells, and blood vessels [6]. It has been shown that 17β-estradiol (E2) drives epithelial cell proliferation whereas progesterone inhibits E2-stimulated epithetlial cell proliferation [7], and thus aberrant hormone levels might induce endometrial cell dysfunction. It is widely accepted that prolonged estrogen excess or lack of progesterone results in endometrial overgrowth and atypical endometrial hyperplasia [8, 9], which is the precursor of endometrial carcinoma [10, 11], and the majority of endometrial cancers are estrogen-dependent endometrial carcinomas [8, 9]. Due to chronic anovulation, PCOS patients experience sustained and persistent estrogen stimulation but minimal or completely absent progesterone stimulation [11], and PCOS patients with endometrial hyperplasia have a four-fold greater risk of developing endometrial carcinoma than non-PCOS controls [12]. Moreover, PCOS is a hyperandrogenic state that results in increased bioavailability of unopposed estrogens due to the increased peripheral conversion of endogenous androgens such as testosterone and androstenedione into estrogen [13]. In addition to changes in endogenous steroid hormone metabolism in PCOS patients, insulin resistance and obesity can also contribute to the development of endometrial cancer [13-15]. Although the metabolic tissues such as skeletal muscle, adipose tissue, and liver are considered to be the major tissues that contribute to whole-body insulin resistance [16], we and others have previously shown that the onset of endometrial insulin resistance also occurs in PCOS patients and PCOS-like animals [17-19]. Thus, abnormal steroid hormone responsiveness accompanied by hyperandrogenism as well as both peripheral and tissue-specific insulin resistance are all likely to be responses to pathophysiological alterations in the PCOS patient's endometrium.
Glucose is a primary source of energy and is used to maintain the cellular energy balance through the process of aerobic glycolysis and mitochondrial oxidative phosphorylation [20], and studies have indicated that abnormal glycolysis and mitochondrial function are involved in the progression of metabolic diseases, including PCOS. For example, gene expression analysis of endometrial and skeletal muscle tissues has shown that the expression of key genes involved in cytosolic glycolysis and mitochondrial oxidative metabolism is lower in PCOS patients compared to non-PCOS controls [21, 22]. Moreover, a clinical study showed that the levels of glycolysis- mediated and mitochondria-dependent metabolic products are decreased in the follicular fluid in PCOS patients compared to non-PCOS controls [23]. It is well known that the insulin-insulin receptor signaling pathway contributes to the regulation of glycolysis and mitochondrial function [24]. Compared to physiological conditions, some PCOS patients exhibit the onset and progression of insulin resistance and dysregulation of glucose homeostasis by reduced insulin action in multiple tissues [1, 2]. Furthermore, abnormal regulation of glycolytic enzyme activities is found in women with endometrial cancer [25]. Based on these findings, we hypothesized that alterations of glycolytic enzyme levels and mitochondrial function are different between non-endometrial hyperplasia and endometrial hyperplasia in PCOS patients. However, to the best of our knowledge, there are no available reports or studies demonstrating the expression of glycolytic enzymes or differences in mitochondria-dependent apoptosis in PCOS patients with and without endometrial hyperplasia either in vivo or in vitro.
PCOS patients with early-stage endometrial cancer are often of reproductive age, and they usually wish to retain their potential fertility. It is thus imperative to develop new and effective non-surgical and conservative treatments for these patients [26, 27]. Several clinical studies have demonstrated that treatment with metformin (N,N-dimethylbiguanide), an oral biguanide insulin-sensitizing drug [28], decreases the incidence, progression, and mortality of endometrial cancer [29, 30], and we and others have reported that metformin is sufficient to reverse atypical endometrial hyperplasia and carcinoma in some PCOS patients in addition to improving hyperandrogenism and insulin resistance and preserving their fertility [26, 31-33]. Although metformin is a promising drug for treating PCOS patients with endometrial hyperplasia and carcinoma [34, 35], the molecular and functional metformin-induced alterations in the uterus under PCOS conditions have only been studied to a limited extent [36].
In this study, we analyzed the expression pattern of key glycolytic enzymes, mitochondria- related molecules, and potential molecular markers (estrogen receptor (ER) alpha and beta and androgen receptor (AR)) in the endometrial tissues obtained from non-PCOS patients and from PCOS patients with and without endometrial hyperplasia. Using Western blot and immunohistochemical and immunofluorescent analyses, we measured protein changes in glycolytic enzymes, mitochondrial transcription factor A (TFAM), both ER subtypes, and AR in the endometrium to determine the possible role of glycolysis and mitochondrial function under PCOS conditions in vivo. In addition, endometrial tissues from PCOS patients with endometrial hyperplasia were used for studying the effects of metformin on estrogen-dependent endometrial hyperproliferation in vitro.
Materials and Methods
Ethical approval of human blood and tissue use
Experiments involving human samples were reviewed and approved by the institutional ethical review committee of the Obstetrics and Gynecology Hospital of Fudan University (OGHFU 2013-23). Appropriate informed consent was obtained from all patients who were attending the Obstetrics and Gynecology Hospital of Fudan University in Shanghai, China.
Reagents and antibodies
Metformin, 3,3-diaminobenzidine tetrahydrochloride (DAB), and anti-mouse IgG horseradish peroxidase (HRP)-conjugated goat (A2304) and anti-rabbit IgG HRP-conjugated goat (A0545) secondary antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 594-conjugated goat polyclonal anti-mouse IgG was purchased from Invitrogen (Sollentuna, Sweden). The avidin-biotinylated-peroxidase complex detection system (ABC kit) and DAB-Ni were purchased from Vector Laboratories Inc. (Burlingame, CA). The primary antibodies used for Western blot analysis and immunostaining in the present study, along with their species, molecular weight, dilutions, and sources are listed in Supplemental Table 1.
Patient selection and endometrial tissue collection
Reproductive-aged women with PCOS (n = 10) and without PCOS (n = 3) were recruited into the study. All PCOS patients were diagnosed by the presence of any two of three cardinal features such as clinical/biochemical hyperandrogenism, polycystic ovarian morphology, and chronic anovulation according to the Rotterdam criteria provided by the American Society for Reproductive Medicine and the European Society for Human Reproduction and Embryology [37]. Based on our previous study [26], body weight (BW), body mass index (BMI), follicle- stimulating hormone (FSH), luteinizing hormone (LH), total testosterone, glucose and insulin area under curve (AUC), and homeostasis model assessment of insulin resistance (HOMA-IR) score were determined before endometrial tissue collection. The oral glucose tolerance test (OGTT), insulin tolerance test, and HOMA-IR were performed as previously described [26, 38]. Neither non-PCOS nor PCOS subjects were exposed to any insulin sensitizing, hormonal, or steroidal therapies within three months prior to tissue sampling [26, 39]. All endometrial tissues were obtained during the proliferative phase of the menstrual cycle by endometrial curettage of the bisected uteri obtained after hysterectomy for benign gynecological indications. Each endometrial sample was diagnosed and staged by routine pathology analysis based on standard histological criteria [40]. All endometrial samples were fixed in 10% neutral formalin solution for 24 h at 4ºC and embedded in paraffin for histochemical analysis or snap frozen in liquid nitrogen for subsequent protein analyses in a non-blinded manner.
Endometrial tissue culture and treatment
The culture conditions and dose and time for metformin treatment were as described previously [41, 42].
Western blot analysis
The protein extractions for human endometrial tissue/cells were performed as previously described [42, 43], and a detailed explanation of the Western blot analysis protocol has been published in our previous studies [39, 42].
Histology and immunostaining
Immunohistochemistry and immunofluorescence were performed according to previously described methods [18, 39]. Background settings were adjusted based on the examination of negative control specimens, and different controls for nonspecific staining have been described [39].
Statistics
Results are presented as means ± SEM. Statistical analyses were performed using SPSS version 24.0 statistical software for Windows (SPSS Inc., Chicago, IL). The normal distribution of the data was tested with the Shapiro-Wilk test. Differences between groups were analyzed by one-way ANOVA or two-way ANOVA followed by Tukey's post-hoc test for normally distributed data or the Kruskal-Wallis test for skewed data. A p-value less than 0.05 was considered statistically significant, and statistical significance is shown as *p < 0.05, **p < 0.01, or ***p < 0.001.
Results
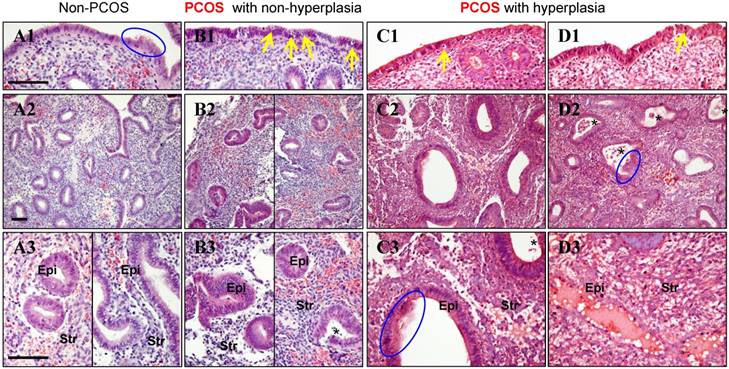
In non-PCOS patients without hyperplasia, general histological evaluation of the endometrial sections stained with H&E showed that in the proliferative phase luminal epithelial cells exhibited secretory activity (Fig. 1A1) and the glands had pseudostratified columnar cells surrounded by dense stroma cells (Fig. 1A2). Based on the endometrial pathology analysis, we divided all PCOS patients into those without endometrial hyperplasia (Fig. 1B1-B3) and those with simple (Fig. 1C1-C3) and complex (Fig. 1D1-D3) hyperplasia. Both simple and complex endometrial hyperplasia had an increased risk of developing an endometrioid-type endometrial carcinoma [8, 9], and histological analysis showed that secretory epithelial cells were also detected in PCOS patients with simple and complex hyperplasia (Fig. 1C3 and D2). Although different numbers of apoptotic bodies were found between PCOS patients without hyperplasia (Fig. 1B1) and those with hyperplasia (Fig. 1C1-D1), more apoptotic bodies were seen increased in the luminal epithelial cells in all PCOS patients compared to non-PCOS patients (Fig. 1A1-D1). Compared to non-PCOS patients, PCOS patients without hyperplasia (Fig. 1B3) and PCOS patients with hyperplasia (Fig. 1C3 and D2) showed increased infiltration of stromal cells into the endometrial gland, and these cells were greater in number in those with hyperplasia. In addition, while the endometrial sections contained histologically normal glands with no evidence of glandular dilatation in PCOS patients without hyperplasia (Fig. 1B2 and B3), we observed that in PCOS patients with simple and complex hyperplasia the glands exhibited multiple glandular dilatations (Fig. 1C2, C3 and D2) filled with large amounts of secretory fluid (Fig. 1D3).
(A1-D3) Representative images of histological changes in endometrial tissues collected from non-PCOS patients (A1-A3), PCOS patients without hyperplasia (B1-B3), PCOS patients with simple hyperplasia (C1-C3), and PCOS patients with complex hyperplasia (D1-D3). The endometrial tissue sections were stained with hematoxylin and eosin. Enhanced magnifications are shown in A3-D3. The blue rings indicate the secretory epithelial cells, the yellow arrows indicate the apoptotic bodies, and the black stars indicate infiltrated stromal cells in the endometrial gland. Epi, epithelial cells; Str, stromal cells. Scale bars (100 μm) are indicated in the photomicrographs.
The clinical and biochemical features of this study cohort are presented in Table 1. All patients had similar age, BW, BMI, and serum FSH levels, and all PCOS patients regardless of hyperplasia had higher serum LH and total testosterone levels as well as higher LH:FSH ratios compared to non-PCOS patients. Although there was no significant alteration in fasting serum glucose level between non-PCOS and PCOS patients, PCOS patients had higher stimulated serum glucose levels at 30 minutes in the OGTT. Further, stimulated serum glucose levels at 30 and 180 minutes in the OGTT were higher in PCOS patients with hyperplasia than PCOS and non-PCOS patients without hyperplasia. Although no significant difference in fasting serum insulin level between non-PCOS and PCOS patients without hyperplasia was noted, significantly higher stimulated serum insulin at 60 minutes and insulin AUC were observed in PCOS patients with hyperplasia.
Main clinical and biochemical profiles in non-PCOS controls and PCOS patients with and without hyperplasia (only the proliferation phase).
| Characteristic | Non-PCOS (n = 3) | PCOS | |
|---|---|---|---|
| Non-hyperplasia (n = 3) | Hyperplasia (n = 7) | ||
| Age (years) | 30.0 ± 3.6 | 29.3 ± 0.9 | 30.3 ± 1.5 |
| BW (kg) | 67.23 ± 1.47 | 62.67 ± 6.17 | 67.43 ± 2.78 |
| BMI (kg/m2) | 26.08 ± 0.64 | 24.76 ± 2.79 | 25.53 ± 1.27 |
| FSH (IU/l) | 5.51 ± 0.62 | 7.30 ± 0.86 | 6.43 ± 0.78 |
| LH (IU/l) | 4.61 ± 0.73 | 17.75 ± 5.56 a | 13.04 ± 2.10 b |
| LH/FSH | 0.83 ± 0.04 | 2.33 ± 0.52 a | 2.10 ± 0.34 b |
| Total T (nmol/l) | 0.20 ± 0.05 | 0.53 ± 0.06 b | 0.69 ± 0.03 b |
| OGTT | |||
| Glucose (mmol/l) | |||
| Glucose 0 min | 4.33 ± 0.57 | 4.83 ± 0.13 | 5.19 ± 0.26 |
| Glucose 30 min | 8.80 ± 0.44 | 7.40 ± 0.30 b | 9.01 ± 0.38 c |
| Glucose 60 min | 10.43 ± 1.33 | 8.67 ± 0.76 | 11.87 ± 0.97 |
| Glucose 120 min | 8.03 ± 0.41 | 6.97 ± 0.26 | 10.39 ± 1.48 |
| Glucose 180 min | 3.97 ± 0.26 | 6.23 ± 0.37 | 6.29 ± 0.71 b |
| Glucose AUC | 15.71 ± 0.89 | 14.28 ± 0.59 | 18.50 ± 1.43 |
| Insulin (mIU/l) | |||
| Insulin 0 min | 12.06 ± 4.14 | 10.31 ± 2.08 | 15.83 ± 3.96 |
| Insulin 30 min | 54.62 ± 14.65 | 79.01 ± 28.09 | 71.04 ± 13.12 |
| Insulin 60 min | 60.07 ± 5.18 | 59.19 ± 5.11 | 99.88 ± 9.56 b |
| Insulin 120 min | 41.76 ± 3.68 | 61.93 ± 15.86 | 142.65 ± 43.67 |
| Insulin 180 min | 21.60 ± 6.09 | 54.38 ± 23.20 | 76.87 ± 12.78 |
| Insulin AUC | 86.65 ± 12.88 | 116.24 ± 28.43 | 179.96 ± 27.96 b |
| HOMA-IR | 2.29 ± 0.90 | 2.24 ± 0.51 | 3.86 ± 1.08 |
BW, body weight; BMI, body mass index; FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, testosterone; OGTT, oral glucose tolerance test; AUC, area under the curve, (AUC = 0.5 × [BG0 + BG30] / 2 + 0.5 × [BG30 + BG60] / 2 + 0.5 × [BG60 + BG120] / 2 + 0.5 × [BG120 + BG180] / 2); HOMA-IR, homeostasis model assessment of insulin resistance, (HOMA-IR = fasting blood glucose (mmol/l) × fasting serum insulin (mIU/ml) / 22.5).
Values are means ± SEM. The multiple comparisons between data were performed using one-way ANOVA followed by the Bonferroni post hoc test for normally distributed data or the Kruskal-Wallis test followed by the Mann-Whitney U-test for skewed data. A p-value less than 0.05 was considered statistically significant.
a p = 0.05 versus non-PCOS patients. b p < 0.05 versus non-PCOS patients. c p < 0.05 versus PCOS patients with non-hyperplasia.
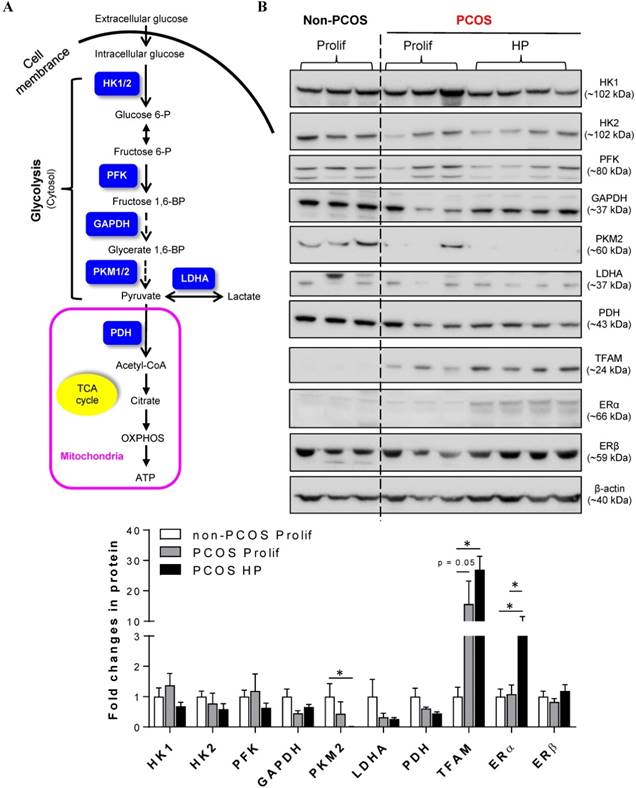
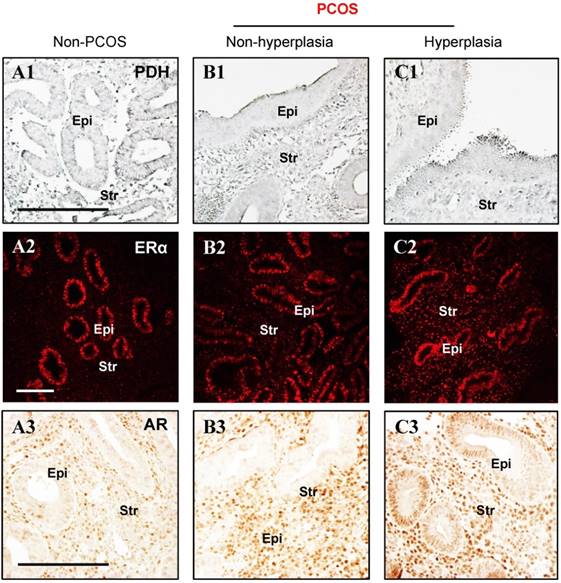
Because cellular glycolysis is an enzymatic metabolic process that depends on the relative expression levels and activities of multiple enzymes [20] (Fig. 2A), we profiled the expression of hexokinase (HK) 1, HK2, phosphofructokinase (PFK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase isozyme M2 isoform (PKM2, an alternative splicing isoform of pyruvate kinase), and lactate dehydrogenase A (LDHA) in the endometrium by Western blot analysis. Quantitative data indicated that PKM2 was decreased in PCOS patients with hyperplasia compared to non-PCOS patients (Fig. 2B). PDH is a key enzyme for pyruvate decarboxylation in the mitochondria and links mitochondrial oxidative phosphorylation to the glycolytic metabolic pathway [44]. Although our Western blot analysis failed to detect a significant difference in endometrial PDH protein abundance between non-PCOS and PCOS patients (Fig. 2B), immunohistochemical analysis showed that the levels of PDH immunoreactivity were decreased in the epithelial cells in PCOS patients regardless of whether or not hyperplasia was present (Fig. 3B1 and C1) compared to non-PCOS patients (Fig. 3A1). At the same time, we observed that endometrial mitochondrial transcription factor A (TFAM) protein was increased in PCOS patients regardless of whether or not hyperplasia was present compared to non-PCOS patients (Fig. 2B). ERβ protein expression was no different between non-PCOS and PCOS patients (Fig. 2B), while ERα protein abundance (Fig. 2B) and immunoreactivity (Fig. 3C2) were highest in PCOS patients with hyperplasia, which was in line with previous studies [39, 45, 46]. Moreover, elevated ERα protein expression (Fig. 2B) was associated with high levels of cell proliferative factors (vimentin and Ki-67) in PCOS patients with endometrium hyperplasia [39, 47]. This indicates that activation of estrogen-ERα signaling is related to sustained endometrial proliferation in PCOS patients with endometrium hyperplasia. Also in line with previous studies [39, 46], the levels of AR immunoreactivity were increased in the stromal cells in PCOS patients regardless of whether or not hyperplasia was present (Fig. 3B3 and C3) compared to AR expression in the stromal cells in non-PCOS patients (Fig. 3A3). Of note, we found strong and heterogeneous positive nuclear staining for AR in the epithelial cells in PCOS patients with hyperplasia (Fig. 3C3).
(A) Schematic illustration of the glycolytic metabolic pathway and mitochondria-mediated energy metabolism, including key enzymes that have been identified in human endometria (labeled in blue). HK, hexokinases; PFK, phosphofructokinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PKM2, pyruvate kinase M2 isoforms; LDHA, lactate dehydrogenase A; PDH, pyruvate dehydrogenase; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid. (B) Comparison of the expression patterns of multiple glycolytic enzymes, mitochondria-related proteins, and estrogen receptor (ER) subtypes in the endometria between non-PCOS patients and PCOS patients with non-hyperplasia and hyperplasia. Endometrial biopsy samples were collected, and tissue lysates were directly immunoblotted with antibodies against HK1, HK2, PFK, GAPDH, PKM2, LDHA, PDH, TFAM, ERα, and ERβ as indicated. β-actin and gels stained with Coomassie blue (data not shown) served as loading controls. Prolif., the proliferative phase; HP, endometrial hyperplasia.
(A1-C3) Immunohistochemical and immunofluorescent staining of PDH (A1-C1), ERα (A2-C2), and AR (A3-C3) in the endometrial tissue sections from non-PCOS (n = 3) and PCOS patients with non-hyperplasia (n = 3) and hyperplasia (n = 4). The images are representative of those observed in numerous sections from multiple uterine tissues. Epi, epithelial cells; Str, stromal cells. Scale bars (100 μm) are indicated in the photomicrographs.
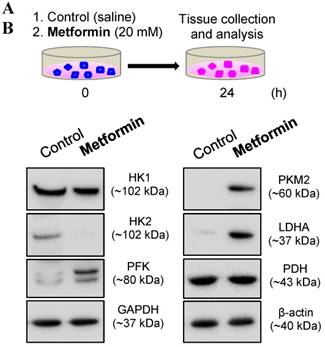
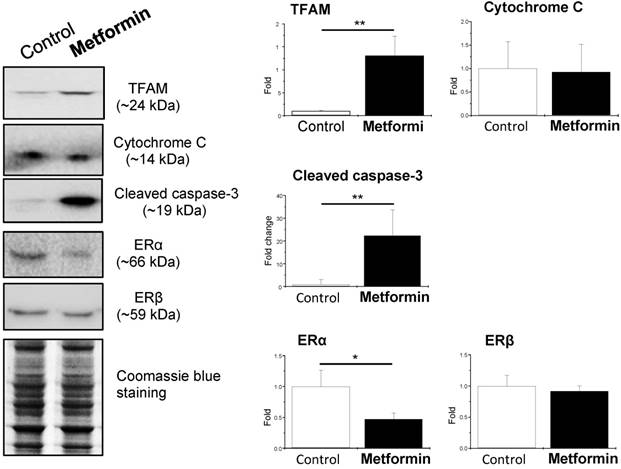
Because in vivo and in vitro pre-clinical and clinical studies have shown that metformin reverses PCOS-induced inhibition of glucose transporter (Glut) isoforms [17, 42] and reverses PCOS-induced enhancement of ERα mRNA expression in the uterus [42, 47], it is possible that metformin can directly regulate endometrial glycolysis and mitochondrial function in PCOS patients with hyperplasia. To address this question, we cultured endometrial hyperplasia tissues in the presence of 20 mM metformin. The results from the Western blot analysis showed decreased HK2 expression and increased PFK, PKM2, LDHA, TFAM, and cleaved caspase-3 expression (Fig. 4) in parallel with decreased ERα expression (Fig. 5) in the endometrial hyperplasia tissues after 24 h of metformin treatment.
(A) Schematic of the in vitro experimental procedure. (B) Effects of metformin on the protein levels of the major glycolytic enzymes in cultured human endometrial tissues. Endometrial biopsy samples from PCOS patients with endometrial hyperplasia were treated with saline or 20 mM metformin for 24 h. Endometrial tissue lysates were directly immunoblotted with antibodies against HK1, HK2, PFK, GAPDH, PKM2, LDHA, and PDH as indicated. β-actin and gels stained with Coomassie blue (data not shown) served as loading controls. Representative images (n = 2 patients/group) from two independent experiments with identical results are shown.
Effects of metformin on mitochondria-related protein levels and nuclear estrogen receptor subtype expression in cultured human endometrial tissues. Endometrial biopsy samples from PCOS patients with endometrial hyperplasia were treated with saline or 20 mM metformin for 24 h. Endometrial tissue lysates were directly immunoblotted with antibodies against TFAM, cytochrome C, cleaved caspase-3, ERα, and ERβ as indicated. Representative images (n = 3-4 patients/group) from three independent experiments are shown. The gels stained with Coomassie blue served as loading controls.
Discussion
PCOS is commonly considered to be a significant risk factor for the development and progression of estrogen-dependent endometrial cancer [36]. While endometrial hyperplasia predisposes for and coexists with endometrial cancer [48], a number of molecules such as estrogen and ER are important pathogenic factors that are involved in the onset and progression of endometrial hyperplasia [8]. Nowadays attention has started to focus on the role of metabolic abnormalities in the development of endometrial hyperplasia [9, 13], but to our knowledge the present study was the first to compare the expression of key glycolytic enzymes and mitochondria-related molecules between PCOS patients with endometrial hyperplasia and PCOS patients without endometrial hyperplasia.
It is well known that aerobic glycolysis- mediated catabolization of glucose to generate ATP is required for cellular proliferation under both physiological and pathological conditions [20], and previous reports have indicated that key glycolytic enzyme mRNAs and enzymatic activities can be detected in human endometrial tissues [21, 25]. While decreased levels of glycolytic enzyme mRNAs are found in the endometrium collected from PCOS patients compared to non-PCOS controls [21], our previous findings also showed that the expression of several glycolytic enzymes such as PKM1/2 is significantly decreased in the PCOS-like rat uterus [18]. Consistent with these previous reports, our Western blotting data showed that the amount of PKM2 protein was decreased in PCOS patients, particularly in those with endometrial hyperplasia, compared to non-PCOS controls. Because Gluts are not only the rate-limiting step of glycolysis, but also the first step of glucose metabolism [49], a number of Glut isoforms, including Glut4, are aberrantly expressed in the endometrium in PCOS patients and PCOS-like rats [18, 41, 42]. Thus, it appears that decreased glycolysis is a consequence of abnormal regulation of Gluts and impaired glucose uptake and utilization in the endometrium under PCOS conditions. Preclinical and clinical evidence suggests that tissue hemostasis, cell proliferation, differentiation, cell death, and remodeling are critical to normal endometrial function. The endometrium is one of main pathologically targeted tissues in PCOS patients, and the development of endometrial hyperplasia is mediated by excessive exposure to estrogen [8, 9]. However, E2 is a master regulator of endometrial cell proliferation [7] and has been shown to upregulate PKM2 protein expression in association with increased glucose metabolism in the rat uterus in vivo [50] and in human endometrial stromal cells in vitro [51]. While our data indicate that the levels of estrogen-regulated ERα protein are positively associated with endometrial hyperplasia in PCOS patients, it remains unclear to what degree such differences in the regulation of glycolysis reflect endometrial tissue responses to endogenous estrogenic stimulation or represent variations in different endometrial cell types or even cell compartments in these patients. Due to the increased endometrial AR expression ([39] and this study) and impaired insulin/PI3K/Akt signaling pathway in PCOS patients and PCOS-like rats [18, 19], we speculate that the decreased glycolysis seen in PCOS patients with endometrial hyperplasia might be accounted for by other etiological factors such as hyperandrogenism and endometrial insulin resistance.
Under both physiological and pathological conditions, mitochondrial function is essential for metabolic reprogramming and cellular energy production via glycolysis and the tricarboxylic acid cycle and plays a fundamental role in cell proliferation [52]. Previous clinical studies have reported that PCOS patients often exhibit increased reactive oxygen species (ROS) production in their ovarian granulosa cells [53] and leukocytes [54, 55]. While intracellular ROS are generated mainly in the mitochondria [56, 57] and inhibit PKM2 activity [58], our observations suggest that decreased metabolic glycolysis is a reflection of increased mitochondria- induced oxidative response in the endometrium in PCOS patients. TFAM functions as a transcription factor to protect tissues and cells from oxidative stress [59], and we have shown here that expression of endometrial TFAM protein is increased in PCOS patients. Both in vivo and in vitro preclinical studies have shown that PKM2 is critical for the growth of different human tumors [60], and the specific cell deletion of the PKM2 protein resulted in increased tumor cell proliferation in a mouse model of breast cancer [61]. Thus, abnormal regulation of endometrial PKM2 protein expression in PCOS patients suggests an important role for PKM2 in the onset and progression of endometrial hyperplasia. Our findings also suggest that upregulation of TFAM protein might lead, at least in part, to the mitochondria compensating for the PKM2 loss and repairing ROS-induced endometrial cell damage in order to maintain the endometrial cell proliferation in PCOS patients during the onset and progression of endometrial hyperplasia. However, the detailed mechanism of the regulation of endometrial cell survival and proliferation through PKM2 and its downstream targets in PCOS patients with endometrial hyperplasia remains to be further investigated.
Clinical studies have reported that apoptosis is rarely detected in the glandular epithelium during the proliferative phase or at the beginning of the secretory phase of the menstrual cycle [62]. Despite the lack of quantification, we show that the number of apoptotic bodies is increased in the epithelial cells in PCOS patients without hyperplasia compared to non-PCOS controls and PCOS patients with hyperplasia. While the development and progression of endometrial hyperplasia is strongly influenced by elevated estrogen levels [8], which are negatively associated with endometrial cell apoptosis [62], androgens act as anti-proliferative factors to regulate endometrial function [63]. Although PCOS patients exhibit hyperandrogenism regardless of whether they have endometrial hyperplasia or not, our data show that epithelial AR expression is not significantly increased in PCOS patients without endometrial hyperplasia compared to PCOS patients with endometrial hyperplasia. Because endometrial ER expression is paralleled with endometrial AR expression under physiological conditions [26], one might speculate that impaired coordinated regulation of ER and AR signaling pathways is involved in inducing epithelial cell apoptosis in PCOS patients without endometrial hyperplasia.
In addition to hyperandrogenism and insulin resistance [1, 2], impaired endometrial progesterone- progesterone receptor (PR) signaling is often seen in PCOS patients [7, 46] and PCOS-like animals [17, 47]. It has been reported that PR expression is decreased in PCOS patients with endometrial hyperplasia compared to those without endometrial hyperplasia [26], which might explain the loss of progesterone responsiveness seen in PCOS patients with endometrial hyperplasia. Interestingly, selective PR modulators such as ulipristal acetate play beneficial roles in endometrial function by up-regulation of both endometrial PR and AR expression in patients with heavy menstrual bleeding [64]. While PR and AR are both members of the steroid receptor superfamily and regulate the transcription of target genes by either forming structural homodimers themselves or by heterometrically binding to each other [65], it will be interesting to explore whether ulipristal acetate modulates endometrial PR expression and activity and counterbalances hyperandrogenism-induced AR activation and thus limits endometrial hyperplasia in PCOS patients.
Several clinical studies have previously shown that treatment of some PCOS patients with metformin improves endometrial receptivity and enhances endometrial vascularity and blood flow [66, 67]. While metformin has been shown to inhibit gluconeogenesis by modulation of AMP-activated protein kinase activity [36], metformin has also been reported to correct defective glycolytic processes and impaired mitochondrial respiration in metabolic tissues in vivo [68, 69]. Thus, as expected, our in vitro studies showed that metformin reverses abnormal alterations in glycolytic enzyme expression and triggers mitochondria-mediated caspase-3-dependent apoptosis in PCOS patients with endometrial hyperplasia. These results indicate that metformin reverses impaired uterine metabolism by inducing glycolysis and mitochondria-mediated cell death in PCOS patients with endometrial hyperplasia. The indirect (systemic) and direct (local) beneficial effects of metformin on uterine function in PCOS patients have been proposed [33, 36]. There is convincing evidence that organic cation transporters (OCTs) and multidrug and toxin extrusion proteins (MATEs) contribute to the cellular uptake and the excretion of metformin [70], and because human and rat endometrial cells express different OCT and MATE proteins [36] and because in vitro treatment with metformin regulates OCT protein expression in PCOS patients with endometrial hyperplasia [42], our findings suggest that metformin has direct effects on glycolytic enzyme and mitochondria-related protein expression.
Overweight/obesity-induced insulin resistance as a critical contributor to the pathophysiology of PCOS [1, 2] and plays important roles in altered glucose homeostasis and mitochondrial dysfunction in multiple tissues in vivo [13]. In the current study, there was no difference in BMI between non-PCOS and PCOS patients (Table 1). Because several clinical studies indicate that greater insulin resistance is independent of higher BMI in various phenotypes of PCOS [3], this suggests that our PCOS patients appear to more accurately represent the lean PCOS phenotype.
The weaknesses of our study include the small sample size and the different protein expression originating from endometrial tissues but not separated into epithelial and stromal cells. The diversity of PCOS etiologies (hyperandrogenism and insulin resistance) and main hormone-responding endometrial cells types (epithelial and stromal cells) poses challenges for elucidating how aberrant glycolysis and mitochondrial dysfunction contribute to the development of endometrial hyperplasia in PCOS patients. Our study also raises the question of how the effect of metformin at the molecular level might inhibit the development of atypical endometrial hyperplasia and endometrial carcinoma in PCOS patients and how our findings affect therapeutic guidelines for PCOS patients with and without endometrial hyperplasia. Thus, in addition to studies featuring PCOS-like animal models to confirm the current findings, our data reinforce the need for a large clinical trial using multiple institutions/centers with different populations of PCOS patients with various phenotypes.
In summary, this study provides in vivo and in vitro evidence showing that abnormal expression of glycolytic enzymes and mitochondria-related molecules in PCOS patients with endometrial hyperplasia can be directly corrected by metformin treatment. The identification of the molecular mechanisms behind the initiation and development of endometrial hyperplasia under the conditions of PCOS will provide an opportunity for developing new preventative treatments for those patients at risk of developing endometrial cancer. Therefore, it will be important in future studies to determine the precise regulatory mechanisms and consequences of impaired glycolysis and mitochondrial dysfunction in the endometrium of PCOS patients.
Supplementary Material
Supplementary table.
Acknowledgements
We thank all of our patients for their voluntary participation in this study. This work was supported by the Swedish Medical Research Council (grant number 10380), the Swedish federal government under the LUA/ALF agreement (grant number ALFGBG-147791), the Jane and Dan Olsson's Foundation, and the Adlerbert Research Foundation to HB and LRS as well as the National Natural Science Foundation of China (grant number 81774136), the Project of Young Innovation Talents in Heilongjiang Provincial University (grant number UNPYSCT-2015121), the Project of Innovation Talents (Young Reserve Talents) in Harbin City (grant number 2015RAQYJ089), the Scientific Research Foundation of Heilongjiang Province for Returned Chinese Scholars, the Scientific Research Foundation for Postdoctoral Researchers of Heilong Jiang Province, the Project of Science Foundation by Heilongjiang University of Chinese Medicine, and the Project of Excellent Innovation Talents by Heilongjiang University of Chinese Medicine to YZ. The Guangzhou Medical University High-level University Construction Talents Fund (grant number B185006010046) supported MH. We also acknowledge the Centre for Cellular Imaging at the University of Gothenburg and the National Microscopy Infrastructure (VR-RFI 2016-00968) for providing assistance in microscopy.
Author Contributions
LRS conceptualized the experiments, supervised the study, and provided key research direction; YW, JZ, MH, YZ, PC, XL, JL, EV, and LRS collected the endometrial biopsies, performed the experiments, and analyzed the data; LRS wrote, edited, and revised the manuscript; and MB, LRS, and HB provided scientific oversight and guidance. All authors read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rosenfield RL, Ehrmann DA. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr Rev. 2016;37:467-520
2. Azziz R, Carmina E, Chen Z. et al. Polycystic ovary syndrome. Nat Rev Dis Primers. 2016;2:16057
3. Lizneva D, Suturina L, Walker W. et al. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril. 2016;106:6-15
4. Goodarzi MO, Dumesic DA, Chazenbalk G. et al. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219-31
5. Cooney LG, Dokras A. Beyond fertility: polycystic ovary syndrome and long-term health. Fertil Steril. 2018;110:794-809
6. Critchley HO, Saunders PT. Hormone receptor dynamics in a receptive human endometrium. Reprod Sci. 2009;16:191-9
7. Li X, Feng Y, Lin JF. et al. Endometrial progesterone resistance and PCOS. J Biomed Sci. 2014;21:2
8. Sanderson PA, Critchley HO, Williams AR. et al. New concepts for an old problem: the diagnosis of endometrial hyperplasia. Hum Reprod Update. 2017;23:232-54
9. Chandra V, Kim JJ, Benbrook DM. et al. Therapeutic options for management of endometrial hyperplasia. J Gynecol Oncol. 2016;27:e8
10. Chittenden BG, Fullerton G, Maheshwari A. et al. Polycystic ovary syndrome and the risk of gynaecological cancer: a systematic review. Reprod Biomed Online. 2009;19:398-405
11. Hardiman P, Pillay OC, Atiomo W. Polycystic ovary syndrome and endometrial carcinoma. Lancet. 2003;361:1810-2
12. Fearnley EJ, Marquart L, Spurdle AB. et al. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an Australian case-control study. Cancer Causes Control. 2010;21:2303-8
13. Li X, Shao R. PCOS and obesity: insulin resistance might be a common etiology for the development of type I endometrial carcinoma. Am J Cancer Res. 2014;4:73-9
14. Burzawa JK, Schmeler KM, Soliman PT. et al. Prospective evaluation of insulin resistance among endometrial cancer patients. Am J Obstet Gynecol. 2011;204:355 e1-7
15. Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer epidemiology, biomarkers & prevention: a publication of the Am Asso Cancer Res. 2002;11:1531-43
16. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23:804-14
17. Zhang Y, Hu M, Meng F. et al. Metformin Ameliorates Uterine Defects in a Rat Model of Polycystic Ovary Syndrome. EBioMedicine. 2017;18:157-70
18. Zhang Y, Sun X, Sun X. et al. Molecular characterization of insulin resistance and glycolytic metabolism in the rat uterus. Sci Rep. 2016;6:30679
19. Qi J, Wang W, Zhu Q. et al. Local Cortisol Elevation Contributes to Endometrial Insulin Resistance in Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2018;103:2457-67
20. Burns JS, Manda G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int J Mol Sci. 2017:18
21. Kim JY, Song H, Kim H. et al. Transcriptional profiling with a pathway-oriented analysis identifies dysregulated molecular phenotypes in the endometrium of patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1416-26
22. Skov V, Glintborg D, Knudsen S. et al. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes. 2007;56:2349-55
23. Zhao H, Zhao Y, Li T. et al. Metabolism alteration in follicular niche: The nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Rad Biol Med. 2015;86:295-307
24. Wu C, Khan SA, Lange AJ. Regulation of glycolysis-role of insulin. Exp Gerontol. 2005;40:894-9
25. Marshall MJ, Goldberg DM, Neal FE. et al. Enzymes of glucose metabolism in carcinoma of the cervix and endometrium of the human uterus. Br J Cancer. 1978;37:990-1001
26. Li X, Guo JR, Lin JF. et al. Combination of Diane-35 and metformin to treat early endometrial carcinoma in PCOS women with insulin resistance. J Cancer. 2014;5:173-81
27. Tong XM, Lin XN, Jiang HF. et al. Fertility-preserving treatment and pregnancy outcomes in the early stage of endometrial carcinoma. Chin Med J. 2013;126:2965-71
28. Nestler JE. Metformin for the treatment of the polycystic ovary syndrome. N Engl J Med. 2008;358:47-54
29. Nevadunsky NS, Van Arsdale A, Strickler HD. et al. Metformin use and endometrial cancer survival. Gynecol Oncol. 2014;132:236-40
30. Ko EM, Walter P, Jackson A, Clark L. et al. Metformin is associated with improved survival in endometrial cancer. Gynecol Oncol. 2014;132:438-42
31. Shen ZQ, Zhu HT, Lin JF. Reverse of progestin-resistant atypical endometrial hyperplasia by metformin and oral contraceptives. Obstet Gynecol. 2008;112:465-7
32. Session DR, Kalli KR, Tummon IS. et al. Treatment of atypical endometrial hyperplasia with an insulin-sensitizing agent. Gynecol Endocrinol. 2003;17:405-7
33. Shao R, Li X, Billig H. Promising clinical practices of metformin in women with PCOS and early-stage endometrial cancer. BBA Clin. 2014;2:7-9
34. Palomba S, Falbo A, Zullo F, Orio F Jr. Evidence-based and potential benefits of metformin in the polycystic ovary syndrome: a comprehensive review. Endocr Rev. 2009;30:1-50
35. Sivalingam VN, Myers J, Nicholas S. et al. Metformin in reproductive health, pregnancy and gynaecological cancer: established and emerging indications. Hum Reprod Update. 2014;20:853-68
36. Shao R, Li X, Feng Y. et al. Direct effects of metformin in the endometrium: a hypothetical mechanism for the treatment of women with PCOS and endometrial carcinoma. J Exp Clin Cancer Res. 2014;33:41
37. Rotterdam-ESHRE/ASRM-Sponsored-PCOS-Consensus-Workshop-Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod. 2004;19:41-7
38. Legro RS, Zaino RJ, Demers LM. et al. The effects of metformin and rosiglitazone, alone and in combination, on the ovary and endometrium in polycystic ovary syndrome. Am J Obstet Gynecol. 2007;196:402 e1-10; discussion e10-1
39. Li X, Pishdari B, Cui P. et al. Regulation of androgen receptor expression alters AMPK phosphorylation in the endometrium: in vivo and in vitro studies in women with polycystic ovary syndrome. Int J Bio Sci. 2015;11:1376-89
40. Noyes RW, Hertig AT, Rock J. Dating the endometrial biopsy. Am J Obstet Gynecol. 1975;122:262-3
41. Cui P, Li X, Wang X. et al. Lack of cyclical fluctuations of endometrial GLUT4 expression in women with polycystic ovary syndrome: Evidence for direct regulation of GLUT4 by steroid hormones. BBA Clin. 2015;4:85-91
42. Li X, Cui P, Jiang HY. et al. Reversing the reduced level of endometrial GLUT4 expression in polycystic ovary syndrome: a mechanistic study of metformin action. Am J Transl Res. 2015;7:574-86
43. Shao R, Wang X, Weijdegard B. et al. Coordinate regulation of heterogeneous nuclear ribonucleoprotein dynamics by steroid hormones in the human Fallopian tube and endometrium in vivo and in vitro. Am J Physiol Endocrinol Metab. 2012;302:E1269-82
44. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71:2577-604
45. Piltonen TT. Polycystic ovary syndrome: Endometrial markers. Best Prac Res Clin Obstet Gynaecol. 2016;37:66-79
46. Hu M, Li J, Zhang Y. et al. Endometrial progesterone receptor isoforms in women with polycystic ovary syndrome. Am J Transl Res. 2018;10:2696-705
47. Hu M, Zhang Y, Feng J. et al. Uterine progesterone signaling is a target for metformin therapy in PCOS-like rats. J Endocrinol. 2018;237:123-37
48. Shao R. Progesterone receptor isoforms A and B: new insights into the mechanism of progesterone resistance for the treatment of endometrial carcinoma. Ecancermedicalscience. 2013;7:381
49. Frolova AI, Moley KH. Glucose transporters in the uterus: an analysis of tissue distribution and proposed physiological roles. Reproduction. 2011;142:211-20
50. Singhal RL, Valadares JR. Estrogenic regulation of uterine pyruvate kinase. Am J Physiol. 1970;218:321-7
51. Salama SA, Mohammad MA, Diaz-Arrastia CR. et al. Estradiol-17beta upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-alpha and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J Clin Endocrinol Metab. 2014;99:3790-9
52. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann Rev Cell Dev Biol. 2011;27:441-64
53. Lai Q, Xiang W, Li Q. et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Front Med. 2017 In press
54. Banuls C, Rovira-Llopis S, Martinez de Maranon A. et al. Metabolic syndrome enhances endoplasmic reticulum, oxidative stress and leukocyte-endothelium interactions in PCOS. Metabolism. 2017;71:153-62
55. Victor VM, Rocha M, Banuls C. et al. Induction of oxidative stress and human leukocyte/endothelial cell interactions in polycystic ovary syndrome patients with insulin resistance. J Clin Endocrinol Metab. 2011;96:3115-22
56. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M. et al. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304-12
57. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158-67
58. Anastasiou D, Poulogiannis G, Asara JM. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278-83
59. Uchiumi T, Kang D. The role of TFAM-associated proteins in mitochondrial RNA metabolism. Biochim Biophys Acta. 2012;1820:565-70
60. Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721-30
61. Israelsen WJ, Dayton TL, Davidson SM. et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397-409
62. Harada T, Kaponis A, Iwabe T. et al. Apoptosis in human endometrium and endometriosis. Hum Reprod Update. 2004;10:29-38
63. Miller N, Bedard YC, Cooter NB. et al. Histological changes in the genital tract in transsexual women following androgen therapy. Histopathology. 1986;10:661-9
64. Whitaker LH, Murray AA, Matthews R. et al. Selective progesterone receptor modulator (SPRM) ulipristal acetate (UPA) and its effects on the human endometrium. Hum Reprod. 2017;32:531-43
65. Helsen C, Claessens F. Looking at nuclear receptors from a new angle. Mol Cell Endocrinol. 2014;382:97-106
66. Jakubowicz DJ, Seppala M, Jakubowicz S. et al. Insulin reduction with metformin increases luteal phase serum glycodelin and insulin-like growth factor-binding protein 1 concentrations and enhances uterine vascularity and blood flow in the polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:1126-33
67. Palomba S, Russo T, Orio F Jr. et al. Uterine effects of metformin administration in anovulatory women with polycystic ovary syndrome. Hum Reprod. 2006;21:457-65
68. Da Silva D, Ausina P, Alencar EM. et al. Metformin reverses hexokinase and phosphofructokinase downregulation and intracellular distribution in the heart of diabetic mice. IUBMB life. 2012;64:766-74
69. Da Silva D, Zancan P, Coelho WS. et al. Metformin reverses hexokinase and 6-phosphofructo-1-kinase inhibition in skeletal muscle, liver and adipose tissues from streptozotocin-induced diabetic mouse. Arch Biochem Biophys. 2010;496:53-60
70. Nies AT, Koepsell H, Damme K. et al. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011:105-67
Author contact
![]() Corresponding author: Linus R Shao, M.D., Ph.D., Tel: +46 31 7863408; Fax: +46 31 7863512; E-mail: linus.r.shaogu.se
Corresponding author: Linus R Shao, M.D., Ph.D., Tel: +46 31 7863408; Fax: +46 31 7863512; E-mail: linus.r.shaogu.se