Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Mechanism of autophagy
3. Ovarian follicular...
4. Autophagy in ovarian...
4. Perspective
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):726-737. doi:10.7150/ijbs.30369 This issue Cite
Review
Autophagy in Ovarian Follicular Development and Atresia
Jiawei Zhou1,2, Xianwen Peng1,2, Shuqi Mei1,2 ![]()
1. Institute of Animal Science and Veterinary Medicine, Hubei Academy of Agricultural Sciences, Wuhan 430064, China
2. Hubei Key Lab for Animal Embryo Engineering and Molecular Breeding, Wuhan 430064, China
Received 2018-10-2; Accepted 2018-12-15; Published 2019-1-29
Abstract

Autophagy is a mechanism that exists in all eukaryotes under a variety of physiological and pathological conditions. In the mammalian ovaries, less than 1% of follicles ovulate, whereas the remaining 99% undergo follicular atresia. Autophagy and apoptosis have been previously found to be involved in the regulation of both primordial follicular development as well as atresia. The relationship between autophagy, follicular development, and atresia have been summarized in this review with the aim to obtain a more comprehensive understanding of the role played by autophagy in follicular development and atresia.
Keywords: autophagy, follicular development, atresia, granulosa cells
1. Introduction
Follicular development and atresia begins in the fetus. Ovarian follicles are structures generated in the ovaries and have two major functions, the production of hormones and oocytes. Antral follicles regulate these functions via the inner wall of granulosa cells (GCs) that rest on a distinct basal lamina. Ovarian follicles are made up of oocytes surrounded by pre-granulosa cells or GCs. Because of the specialized extracellular matrix, the epithelial layer is separated from the connective tissue. As a consequence, this matrix affects proliferation and differentiation of GCs [1,2]. Ovarian follicles can either generate oocytes or they can breakdown by a process known as follicular atresia. Although the endocrine system, including the hypothalamus, pituitary gland, and ovaries, plays an important role in follicular development and atresia, an increasing number of pathways and cellular processes such as autophagy have also been found to be involved in this regulation. Autophagy is critical for the natural disassembly of unnecessary or dysfunctional components of cells. Cytoplasmic components, such as proteins, lipids, nucleotides, and organelles, such as mitochondria and peroxisomes, are degraded and recycled through this process [3]. Autophagy can also deliver foreign entities, such as bacteria and viruses, to the lysosome for degradation and also regulates inflammatory responses [4]. This cellular degradation mechanism allows for the recycling of cellular components and organelles, thus maintaining cell metabolism [5]. Therefore, autophagy is essential for normal cellular function and plays a wide variety of physiological and pathophysiological roles. Compared to other cellular processes, autophagy displays unique functions in ovarian follicular development and atresia. The role of autophagy in ovarian follicular development and atresia has recently gained a lot of attention due to an increasing amount of evidence suggesting a relationship between autophagy and the above reproductive events [6]. In this review, we first discuss the mechanisms underlying autophagy, ovarian follicular development, and atresia, and then discuss the relationship between these three processes. This review provides a comprehensive overview of the involvement of autophagy in the regulation of the female reproductive system, specifically in terms of follicular development and atresia.
2. Mechanism of autophagy
2.1 Defining autophagy
Autophagy, meaning “self-devouring”, denotes the mechanism by which cells disassemble unnecessary or dysfunctional cellular components. There are several different types of autophagy, including macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy is the classic autophagy mechanism wherein targeted cytoplasmic constituents are isolated from the rest of the cell within a double-membraned vesicle known as an autophagosome [8]. The autophagosome then fuses with lysosomes and the contents are eventually degraded and recycled (Figure 1).
Overview of autophagy. Schematic depicting the main autophagic pathways. Briefly, a series of factors including nutrient depletion and growth factor deprivation induce autophagy. The induction of autophagy is mediated by the ULK1, VPS34, and ATG12-ATG5-ATG16 complexes. The formation mechanisms are described in the text.
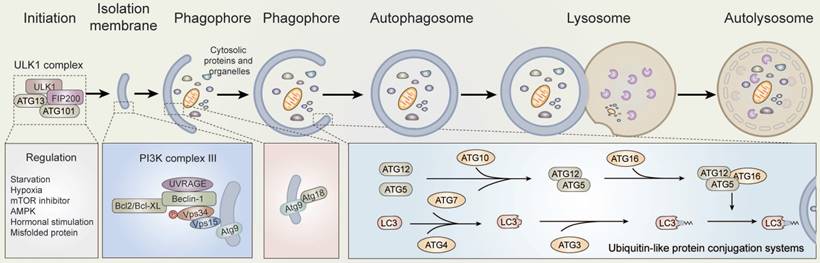
According to previous researches, autophagy is executed by autophagy-related genes [9-11]. Notably, the activity of the mechanistic target of rapamycin, mammalian target of rapamycin (mTOR), is maintained by amino acids and serum starvation in mammals. Starvation consequently induces low energy conditions or glucose deprivation, which results in the up-regulation of 5'-AMP activated protein kinase (AMPK) activity [12,13]. The early stages of autophagy are induced by the dephosphorylation and activation of ULK kinases and proteins, as part of a complex containing the ATG5, ATG12, ATG16, focal adhesion kinase (FAK), and family-interacting protein of 200kD (FIP2000), among other proteins [14]. Subsequently, with the phosphorylation of ULK and up-regulation of AMPK, the mammalian homologue of ATG6 (Beclin-1), a protein complex containing the proteins Vps15 (p150), ATG14L, and the class III phosphoinositide 3-kinase (PI3K) Vps34, is activated [15-17]. Thus, the ULK and Beclin-1 complexes are re-localized to the phagophore site through their activation, which generates autophagosomes and contributes to the subsequent activation of downstream autophagy components [13,14,18].
Once activated, VPS34 generates phosphatidylinositol 3-phosphate (Ptdlns(3)P) on the surface of the phagophore to provide a docking point for Ptdlns(3)P binding proteins. Next, with the help of ATG7 and ATG10, ubiquitin-like protein ATG5 and ATG12 undergo covalent binding. ATG5-ATG12 conjugates then interact non-covalently with ATG16L1 to form an E3-like complex [19]. The E3-like complex binds and activates ATG3 to the lipid phosphatidylethanolamine (PE) on the surface of autophagosomes through the covalent binding of ATG7 with ATG8, inducing the formation of LC3-II, and enables the docking of specific cargos and adaptor proteins such as Sequestosome-1/p62 [20-22], NBR1, and NDP52. As a result, multiple LC3-positive autophagosomes are transported along microtubules to the lysosomes, where autophagosomes fuse to lysosomes through the actions of multiple proteins, including soluble NSF attachment protein receptors (SANREs) [23,24]. In the meantime, the contents of autolysosomes are degraded and released from the vesicles [13,25].
2.2 Function of autophagy
Several functions have been attributed to autophagy. For example, in programmed cell death, autophagy is physiologically very distinct from other cellular processes, such as apoptosis and necrosis, the former which involves the active and programmed process of cellular death via plasma membrane blebbing but with no modifications to cytoplasmic organelles, while the latter involves passive cell death induced by environmental perturbations resulting in plasma membrane rupture, the loss of intracellular content, and the subsequent release of inflammatory signals [26].
To respond to the physiological and pathophysiological stresses of nutrient starvation, cells have developed a variety of strategies and cellular metabolic/catabolic pathways, among which autophagy is a prominent mechanism. In disease, autophagy has always been described as a double-edged sword, which promotes adaptive response to stress for cellular survival on the one hand, but also promotes cell death and morbidity on the other hand [27]. Even under extreme starvation conditions, autophagy has been shown to be a mechanism of energy production. Through the breakdown of cellular components, autophagy promotes cell survival by maintaining energy levels [6].
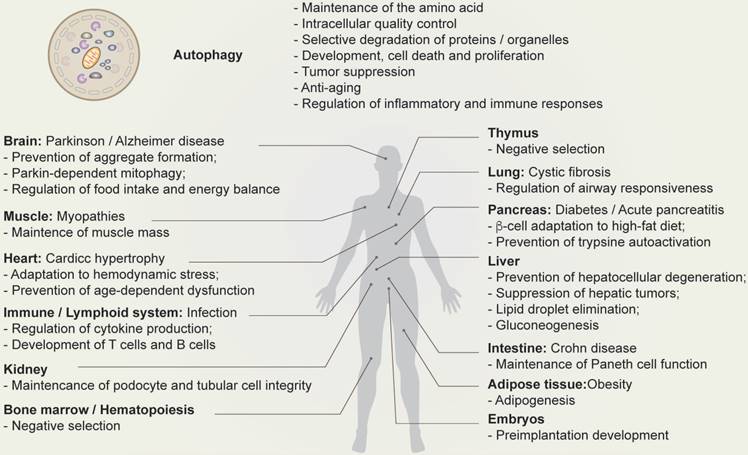
Multiple lines of evidence suggest that autophagy has a number of different functions, including protection from stress, energy regulation, immune regulation, differentiation, proliferation, cell death, and the regulation of reproduction (Figure 2) [7,28,29].
Physiological function of autophagy. Schematic depicting the function of autophagy (top) and autophagy-related human diseases, including organ-specific and systemic problems (bottom).
2.3 Regulation of autophagy
The signal pathways of autophagy are stimulated and maintained by several factors (Figure 3). In addition to the induction of amino acids and glucose, the stimulation of IGF1R (insulin-like growth factor receptors) is one of the most crucial pathways for regulating the mTOR pathway when bound to insulin-like growth factor. Through the signal transduction of effectors such as IRS1 and IRS2 (insulin receptor substrates), AKT phosphorylation is activated and inhibits TSC1/TSC2 (tuberous sclerosis protein 1/2) complex activity, which performs as a negative regulator of mTOR pathway through Rheb eventually.
Regulation of autophagy. Nutrients, growth factors, hormones, and stress signals may regulate the induction of autophagy. Several signaling pathways are involved in the regulation of autophagy, within which the AMPK and mTOR pathways perform critical functions. mTOR inhibits autophagy while AMPK induces autophagy. Similarly, proteins such as p53, JNK, ERK, and their relevant pathways are involved in the regulation of autophagy. In addition, as indicated, the modifications of histones and transcriptional factors such as HIF-1 participate in the regulation process. Please refer to the text for more details.
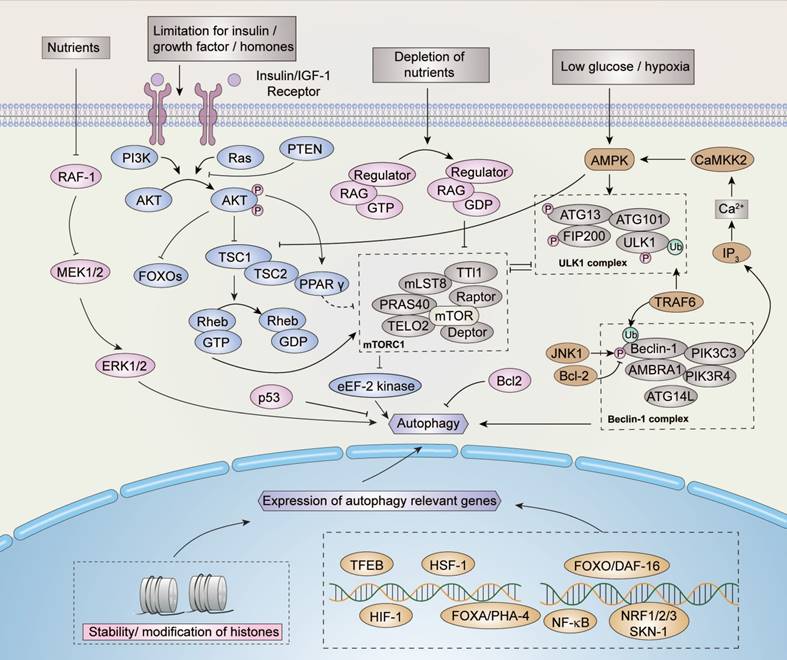
mTOR activity is always inhibited under starvation conditions. With the clustering of mTOR complex 1 (mTORC1, an autophagy regulator) around the microtubule organizing center (MTOC), lysosomes fuse with autophagosome to form autophagosome-lysosomes [30]. Aside from the conditions described above, mTORC1 is regulated by GTPases under certain conditions, which influences lysosomal localization [31].
Autophagy is induced when the ULK/ATG complex is phosphorylated by activated AMPK. However, the ULK/ATG complex is inhibited by the TSC1/TSC2 and mTORC1 complexes, such that autophagy is blocked by their activation [32,33]. The deprivation of nutrients can negatively regulate the expression of proto-oncogene serine/threonine protein kinase (RAF-1), which reversely up-regulates the level of extracellular signal-regulated kinases 1/2 (ERK1/2), eventually inducing autophagy [34]. Another pathway involved in starvation-induced autophagy is maintained by the inhibition of apoptosis-related protein B cell lymphoma 2 (Bcl-2). Under starvation conditions, the phosphorylation of Bcl-2 is activated by c-Jun N-terminal kinase-1 (JNK-1), which in turn disrupts the stability of Bcl-2 and thus induces autophagy [13,35].
Several cellular factors, including DAP kinase-containing TIR-domain-containing adapter-inducing interferon (TRIF), activated by death signals, as well as myeloid differentiation primary response gene 88 (MyD88) pathogens and BCL/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), activated by hypoxia, can enhance autophagy by reducing or interfering with the interaction of the Beclin-1/Bcl-2 complex [36-39]. Autophagy can also be up-regulated by the stress-induced eIF2 eukaryotic translation initiation factor 2 kinase signaling pathway, however, the mechanism of eIF2α kinase signaling remains to be elucidated.
Besides cytosolic regulation, nuclear regulation of autophagy also occurs under oxidative and genotoxic stress conditions. Under oxidative stress conditions, JNK-1 is induced and activates forkhead box (FOXO) proteins, such as FOXOA [40], and several other molecules, including transcription factor EB (TFEB), heat shock factor 1 (HSF-1), and hypoxia-inducible factors 1 (HIF-1) to bind to DNA and promote the expression of autophagy-related genes. Moreover, AKT activated by IRS1/2 inhibits FOXO and down-regulates the expression of autophagy-related genes [13,41-44]. The tumor protein p53 is maintained by genotoxic stress and regulates autophagy through the mTOR pathway [12,45]. p53 is a context-dependent regulator, and only cytoplasmic p53 is capable of inhibiting autophagy [45]. Sirtuin-1 also maintains autophagy inside the nucleus by countering the acetylation effect of trifunctional enzyme subunit beta, also known as acetyl-CoA acyltransferase (KAT8), to maintain the stability of histones, such as Histone H4 (H4K), thereby inducing the expression of autophagy-relevant genes [46-48]. Spermidine can also counter the effect of acetyl-CoA to maintain the stability of certain histones and induce autophagy [49]. The nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) defends cells against damage and environmental stress by repressing autophagy and is inhibited by sirtuin-1 [50]. In addition to the acetylation of histones, several other histone methylation marks, such as H3K9 and methyltransferase euchromatic histone-lysine N-methyltransferase 2, also known as G9a (EHMT2), repress autophagy through demethylation [51].
3. Ovarian follicular development and atresia
3.1 Follicular development
Mammalian oocytes maturation is reached in the follicles (Figure 4) [52]. Ovarian development initiates in the embryo, during which several cellular events take place, such as massive colonization of the ovary [53], the generation of the primordial germ cells (PGCs), the migration of the primordial germ cells into the genital ridge, the colonization of the gonads by the PGCs, gonadal sex differentiation, mitosis, and apoptosis of the germ-cells, among others [2]. After PGC differentiation, oocyte development initiates in females fetuses. Firstly, proliferating PGC migrate towards the nascent genital ridges, where they differentiate into oogonia, which are enveloped by primary follicles accompanied by a monolayer of follicular epithelial/ GCs. Thereafter, oogonia enter meiosis to become primary oocytes, within which the primary follicles become secondary follicles accompanied by stratified GCs without antrum [54]. Subsequently, secondary follicles develop into tertiary follicles, with a follicular antrum, which are characterized by a full layer of proliferative cuboidal GCs [55]. GCs continue to proliferate throughout follicular development, and the theca layer develops to provide the follicle with an independent blood supply [56]. With regards to the antral, depending on the species, a cavity forms when follicles reach a diameter between 200-500 μm [57]. This becomes filled with follicular fluid which acts as a source of oxygen, buffering molecules, carbohydrates, amino acids, growth factors, hormones, and other molecules [58]. Oocytes then undergo ovulation, growth, and maturation, stockpiling mRNAs, proteins, metabolic substrates, and organelles during post-natal life. During this process, maternal stores are tasked with supporting fertilization, unpacking the paternal genome, karyogamy, and early cleavage division of the zygote until the initiation of embryo genome activation (EGA).
Development and atresia of follicles in mammalian ovaries. Schematic depicting follicle development and atresia in mammalian ovaries. Primary follicles (with a monolayer of follicular epithelial/granulosa cells) transform into secondary follicles (with stratified granulosa cells but no antrum) and subsequently develop into tertiary follicles (with a follicular antrum). During follicular growth and development, most follicles undergo a degenerative process known as atresia. As such, very few follicles eventually develop into ovum by the process of ovulation.
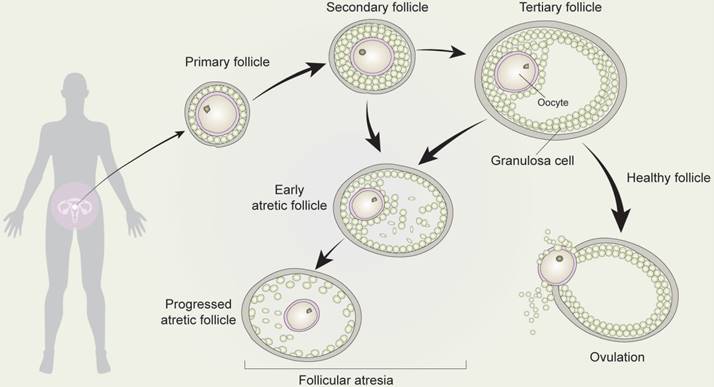
As primary oocytes become enveloped by a single layer of flattened pre-granulosa cells and a basement membrane, they are transformed into primary follicles. However, during follicular growth and development, most follicles undergo a degenerative process known as atresia, such that the ovarian population of primordial follicles is continuously depleted [54]. Secondary or tertiary follicles are highly likely to become early atretic follicles, which undergo follicular atresia.
3.2 Oocytogenesis
The oocyte undergoes a 100-300-fold increase in volume within the growing follicle [59] and is completed around the time the antrum is formed. In addition, the oocyte is supported by an increasing number of surrounding GC [59]. During this process, the ooplasm of the growing oocyte continues to accumulate glycogen granules, lipid droplets [60], proteins, and mRNA for later developmental stages. The high synthetic activity of oocytes leads to the structural changes and redistribution of the endoplasmic reticulum and Golgi complexes. With the increasing number of vesicles and ribosomes, cortical granules form and the zona pellucida begins to export glycoproteins [57,61]. Glutathione (GSH), which protects the oocyte from oxidative stress, accumulates during both oocyte growth and maturation. Mitochondria replicate from about 200 per oogonia to 6000 per oocyte in the primordial follicles during oogenesis, eventually reaching 300000-400000 in the mature human oocyte [62]. This rise in the number of mitochondria is thought to compensate for their apparent lack of maturity and activity, minimizing the oxidative stress by low mitochondrial activity in individuals [63]. Mitochondrial replication then ceases shortly and resumes only after implantation [62,63]. During oogenesis, the expansion of the mitochondrial population is vital for preimplantation development as it is essential for sustaining the numbers and function of mitochondria of the preimplantation embryo.
In the latter stage, the pre-ovulatory surge of gonadotropins maintains the selection and final growth of the follicles destined for ovulation. In the meantime, the surge of gonadotropins triggers the expansion of the cumulus-oocyte complex, gap junction loss, and ultimately the maturation and ovulation of oocytes [64]. Oocyte maturation involves both cytoplasmic and nuclear maturation, which must be completed in order for the gamete to acquire developmental competence [65]. The are several steps involved in the process of nuclear maturation, including nuclear changes relevant to the resumption of meiosis I followed by progression to metaphase II (MII) and extrusion of the first polar body, before arresting at MII [66]. Sperm chromatin decondensation, pronuclei formation, and the support of the first embryonic divisions are also involved in maturation [67,68]. Moreover, cytoplasmic maturation during oocyte growth is also necessary to store the post-transcriptional modifications of stable mRNAs, synthesize protein, and regulate post-translational modifications [57]. As for mitochondrial alterations, oocyte mitochondria are redistributed before progressing to MII, where altered mitochondrial redistribution is closely related to lower potentials in oocyte development [63,69-71]. In different mammalian species, the developmental competence of oocytes has been shown to be acquired progressively during oocyte growth [72].
3.3 Follicular atresia
Follicular atresia denotes the breakdown of ovarian follicles and occurs throughout the lifetime of women [73]. While a number of follicles do eventually develop into corpus luteum, the large majority (99%) undergo follicular atresia [74]. Follicular atresia is a hormonally regulated apoptotic process [75] that depends predominantly on the apoptosis of GCs. As such, GCs apoptosis is now widely considered an underlying mechanism of follicular atresia. Follicular atresia is also strictly inhibited by follicle-stimulating hormone (FSH), which is beneficial for the development of follicles. Once the follicle has developed, it secretes estrogen, which in high levels decreases the secretions of FSH [76]. Therefore, the hormonal regulation of follicular development and atresia is a threshold-depending mechanism. Follicular atresia has also been associated with several ligand-receptor systems involved in cell death [77,78], including TNF-α, Fas ligand, TNF-related apoptosis-inducing ligand (TRAIL), APO-3 ligand, PFG-5 ligand, and receptors [74].
It is now widely recognized that follicular atresia is not a redundant process and is in fact absolutely necessary for women to maintain a healthy reproductive system. According to recent studies, the inability to regulate GC apoptosis and follicular atresia has been linked to the development of hormone turbulence and chemo-resistant cancers[79]. A recent study demonstrated that the accumulation of autophagosomes induced apoptotic cell death of GCs through the decreased expression of Bcl-2 and the subsequent activation of caspases. These findings demonstrate that autophagy mainly occurs in GCs, suggesting that it leads to follicular atresia [79]. In brief, emerging evidence is highlighting the close relationship between autophagy, follicular growth, and atresia.
4. Autophagy in ovarian follicular development and atresia
4.1 The regulation of autophagy in follicular development and atresia
As the functional unit of ovaries, follicles support the development and maturation of oocytes. In mammalian follicles, oocytes are derived from primordial germ cells. Subsequently, through the maturation of primary follicles into mature follicles, mature oocytes undergo ovulation. However, it is widely recognized that follicular atresia occurs in about 99% of primordial follicles directly after birth in mammals by a mechanism which has yet to be elucidated. The drying of primordial follicle “pools” results in the senescence of ovaries, while atresia takes places in the primordial follicles to sustain the development of preponderant follicles and thus of the maturation of oocytes. As such, understanding the mechanisms of follicular atresia directly after birth will be beneficial to promote reproductive quality in women.
Autophagy and its related genes and proteins play a critical and significant role in follicular development. The downregulation of autophagy in GCs in the presence of gonadotropins has been previously demonstrated in vivo [80]. The expression of ATG7, a key autophagy-related gene involved in the formation of autophagosomes, is present at a constant level during all oocytogenesis stages. Mice with Atg7 gene knockdown typically produce small litters and become sterile over time [81]. The ovaries of Atg7-deficient mice always contain fewer germ cells and primordial follicles. Among these, many follicles are structurally altered or non-functional, which suggests that ATG7 may play a critical role in germ cell survival. In another study, the expression of LC3 was investigated and showed its importance in follicular development and atresia in a rat model. This study found that although LC3 was present in primary GCs and pre-antral follicles, it was absent in primordial follicle cells and oocytes [79]. Treatment with oxidized low-density lipoprotein (OxLDL) was found to induce the accumulation of autophagosomes, leading to autophagy of GCs in human ovaries, which provides a possible explanation for the fertility issues observed in obese women, who typically produce higher levels of oxidized low-density lipoprotein. When exposed to OxLDL in humans, GCs induce autophagy, resulting in cell death, that is the mechanism in follicular atresia [82]. All these findings suggest that follicular development is significantly altered by autophagy in follicles and systemic metabolic conditions with profound consequences on reproductive outcomes.
On the other hand, autophagy plays a key role in follicular atresia. According to a number of studies [83,84], a large proportion of follicular atresia via oocyte autophagy occurs during puberty. To investigate the reason for the occurrence of follicular atresia after birth [83], electron microscopy was used to show the abundant presence of lysosomes and autophagosomes in the cellular matrix of oocytes in spiny mouse. Another comprehensive study confirmed that autophagy and not apoptosis lead to the loss of germ cells after birth [82]. However, Escobar observed a number of different programmed cell death pathways in oocytes when investigating the mechanisms within follicular atresia and the death of oocytes in adolescent rats [85,86]. These findings suggested that autophagy led to programmed cell death, in which the regulation of follicular atresia induced by oocytes may vary in different species.
Meantime, GCs are closely associated with follicular atresia. Accordingly, although GC apoptosis is involved in the process of follicular atresia [87,88], numbers of studies have showed the participation of autophagy in GCs during follicular atresia. For example, under oxidative stress, melatonin mediated the survival of GCs through repressing FOXO1 during follicular atresia [89]. In another research, autophagy was induced by FSH, which is critical in maintaining follicular atresia and GC proliferation [90]. Moreover, the activation of mTOR mediated by AKT suppresses GC autophagy during follicular atresia [91]. These findings come to a conclusion that autophagy is specifically crucial in GCs during follicular atresia, which may play a significant role in the regulation of GC death during follicular atresia (Figure 5).
The regulation of autophagy in follicular development and atresia. The development of mammalian follicles involves several steps, from the primordial germ cells (PGC) to the oocytes. Autophagy performs a multifunctional role throughout this process, in which several autophagy-associated proteins or pathways participate. The induction of autophagy in granulosa cells leads to follicular atresia. Similarly, the balance between oocyte and follicular atresia may be maintained by autophagy. Consequently, autophagy influences folliculogenesis during the entire development process of follicular development and atresia.
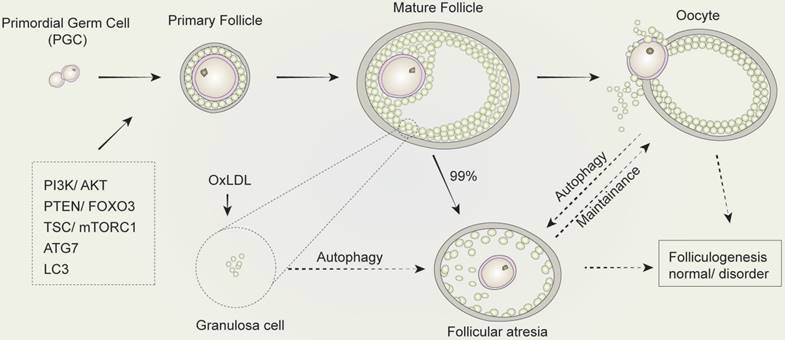
4.2 Factors inducing follicular atresia mediated by autophagy
Numerous factors are believed to link autophagy and follicular atresia. We will first discuss the signaling factors involved. Voltage-dependent anion-selective channel protein (VDAC2) is believed to regulate autophagy in the ovaries [92]. Moreover, VDAC2 may enhance the interaction between Beclin-1 and Bcl-2-like 1 (BCL2L1) to inhibit the involvement of autophagy in regulating the development of follicles. The accurate regulation of follicular atresia through VDAC2 also assists the development of preponderant follicles into ova, which is critical for oogenesis and for improving the reproductive ability of women. In the research conducted by Lee et al [93,94], autophagy was found to be dynamic during oocyte maturation of pigs in vitro. Notably, rapamycin was found to increase the expression of Beclin-1 and LC3-II, which promoted the maturation of oocytes by inducing autophagy. In addition, autophagy sustained the survival of apoptotic unfertilized ova in sea urchins mediated by mitogen-activated protein kinase 1/3 (MAPK 1/3) [95]. In the research conducted by Song et al [96], autophagy was found to up-regulate the transfection level of the apoptosis-related factor B cell lymphoma-extra large (Bcl-xL) through the induction of rapamycin in immature pig oocytes; this increased the formation and survival rate of blastocysts, hence promoting the maturation of the nucleus and cytoplasm of oocytes in vitro.
Furthermore, accumulating evidence is elucidating the role of LC3 in follicular atresia. LC3 expression is barely detectable in ovarian oocytes, whereas a large amount of LC3 immunoreactivity is present in GCs throughout all the stages of follicular development [80]. Furthermore, the induction of autophagy in GCs has been found to be closely related to the onset of apoptosis in a model of follicular development where pregnant mare serum gonadotropin (PMSG) were primed in rat ovaries [97]. Moreover, co-localization of active caspase 3 and LC3 was not detected in primordial, primary, or pre-antral follicles due to the inactivation of caspase 3 at these stages. On the contrary, cleaved caspase 3 was detected in the cells of antral follicles, but not in cells expressing inactive LC3, demonstrating the strong immunoreactivity of LC3II. Moreover, because the formation of autophagosomes is suppressed by FSH treatment, the autophagy observed in GCs seems to be dependent on gonadotropin levels [80]. Recent studies have demonstrated the critical role of FSH in protecting GCs from oxidative injury, which triggers GC death during follicular atresia [98]. Specifically, the protection of FSH-mediated GCs against oxidative damage is induced by the suppression of autophagic cell death through the PI3K-AKT-mTOR pathway, which suggests that FSH promotes follicular atresia by influencing autophagy in GCs [90].
Both genetic factors and environmental risk factors contribute to the occurrence of reproductive system diseases [99-101]. Genetic mutations can induce hereditary diseases of the genitals, where some are promoted and accelerated by environmental risk factors. For example, nutrient depletion can lead to autophagy in the ovaries of Drosophila, consequently reducing the production of ovum [102]. In addition, egg chamber degeneration took place in the matrix follicles of autophagy-deficient Drosophila, indicating that autophagy takes part in the regulation of follicular atresia [103]. A large amount of follicular atresia, as well as the generation of immature oocytes and programmed cell death, could be induced by certain environmental toxics. Melamine mediates the death of oocytes by promoting the expression of ATG14 and LC3, which results in the abnormal distribution of mitochondria and autophagy, respectively [104]. A previous study found a reduced number of offspring in melamine-treated pregnant rats, which resulted in the abnormal growth of embryos and fetuses, demonstrating the toxic effect of melamine [105]. Similarly, in humans, toxins such as deoxynivalenol and HT-2, acquired through the diet, could hinder the maturity of oocytes by affecting cytoskeleton dynamics, apoptosis and autophagy, oxidative stress, and epigenetic modification in cells [105,106]. On the other hand, by reducing toxicity in oocytes, resveratrol promoted the quality of oocytes by up-regulating the ATP content and DNA copies of mitochondria, lowering the activation of mTOR pathway, and subsequently inducing autophagy.
Altogether, these results suggest that autophagy plays an important role in the development of follicles from atresia to ovulation during follicular development in mammalian ovaries. Therefore, the development of follicles is partly dependent on autophagy in oocytes mediated by autophagy-related signaling molecules [103-107]. As a consequence, preponderant oocytes develop into ova. Because environmental risk factors can influence apoptosis and autophagy, they can accelerate the death of oocytes [108,109].
4.3 The relationship between autophagy and apoptosis in follicular atresia
According to the morphological criteria, both apoptosis and autophagy are programmed cell death processes. Karyopyknosis and DNA fragmentation are the main morphological changes that cells undergo during apoptosis which do not entail any alterations in the ultrastructure of organelles [110,111]. However, after the initiation of autophagy and the generation of phagophores, autophagosomes accumulate in the cellular matrix and combine with lysosomes before progressing to degradation [112,113]. Based on recent studies [114,115], despite their evident differences with programmed cell death, several interactive similarities exist between autophagy and apoptosis. Both processes are activated by multiple stress stimulations and are regulated by multiple shared molecules. The accumulation of autophagosomes is likely to be a critical procedure in the inhibition of programmed death of apoptotic cells, as mentioned above [79,86]. GCs are indispensable for the structure of ovarian follicles, which constitute membrane granulosa, therefore additional analyses of the molecular events during the autophagy of these cells will be necessary to understand how the cell death of GCs is maintained during follicular atresia.
Although follicular atresia is induced by autophagy in the ovaries directly after birth, apoptosis contributes to follicular atresia in the days that follow. According to one study, the use of hexadecadrol before labor causes injuries to the follicles in the spiny mouse, inducing autophagy or a combined effect of autophagy and apoptosis to enhance follicular atresia [83]. On the other hand, under conditions of nutrient or growth factor shortages, protective autophagy might be activated by oocytes in the fetus to sustain the survival of the oocytes, where anti-apoptosis factors Bcl-2, Bcl-X, and MCL-1 (induced myeloid leukemia cell differentiation protein) inhibit the expression of pro-apoptosis protein Bcl-2-like protein 4 (BAX) [86]. In addition, most oocytes mediate follicular atresia through the combined effect of apoptosis and autophagy in the ovaries of immature rats [86]. Moreover, both heterophagy and autophagy regulate follicular atresia in the ovaries of freshwater fish, eliminating metamorphic oocytes and dead follicles efficiently [84]. In conclusion, autophagy and apoptosis appear to co-exist during follicular development and atresia in oocytes and GCs in different situations. In the case of a shortage of growth factors, autophagy is induced through a signaling cascade in order to sustain the survival of cells. However, under conditions of extreme stress, excessive autophagy may induce cell death through a combined effect of autophagy and apoptosis.
4.4 Ovarian diseases mediated by the autophagy of follicles
The signaling pathways of the ovaries can be negatively affected by the environment, inducing follicular atresia and resulting in the advanced consumption of follicle “pools.” The most harm is often done to the primordial follicles, the consequences of which are only discovered many years later due to the absence of detection methods. Infertility diseases such as premature ovarian failure (POF) are generally diagnosed late. Similarly, smoking can lead to follicular atresia in ovaries and decreases the reserve of follicle “pools,” resulting in premature menopause [116]. In a study published by Gannon [117], exposure to tobacco smoke was found to induce oxidative and endoplasmic reticulum (ER) stress, leading to the loss of follicles. Several pathways are involved in this stress reaction, one of which unfolds proteins in response to ER stress induced by oxidative stress to protecting against injuries [118,119]. In another pathway, ER stress activates C/EBP homologous protein (CHOP) and c-Jun N-terminal kinases (JNKs), both of which are believed to inhibit the anti-apoptotic effect of Bcl-2 [120]. Bcl-2 and Beclin-1 are believed to inhibit the generation of the membrane in autophagosomes.
On the other hand [117], exposure to tobacco smoke in mice was found to reduce superoxide dismutase 2 (SOD 2) and B-cell lymphoma 2 (Bcl-2), whereas Beclin-1 and LC3 were up-regulated in the GCs, resulting in autophagy mediated by the dysfunction of mitochondria, leading to cell death. By inducing autophagy in the GCs and by consuming the follicular reserve of the ovaries, tobacco smoke caused alterations in circulating hormone levels and a reduction in the number of primordial follicles in mice. Autophagy and Beclin-1 may be crucial for the generation of certain hormones [121], including progesterone and estrogen, which are involved in primordial germ cell growth and follicle maturation. Therefore, autophagy of follicle cells is an adaptive response to external stress factors. Moreover, non-physical stimulus such as chemotherapy, which is the leading cause for reproductive system diseases, causes cell death induced by autophagy. Thus, in GCs and oocytes, research on the regulation of autophagy in follicular development and atresia could provide a great contribution to the treatment of ovarian diseases.
4. Perspective
In this review, we discussed the regulation, function, and relationship of autophagy in ovarian follicular development and atresia. Autophagy plays an important role in follicular development and atresia. The ovaries are at their most dynamic before puberty, when 99% of the follicles are degraded through a process known as follicular atresia. Inhibiting follicular atresia through the regulation of autophagy could be carried out to promote the reproductive capacity of women. The in vitro culture of human and animal gametes using the current understanding of autophagy in follicular development highlighted in this review could prove to be very beneficial. Autophagy take places at all stages of follicular development, however, the mechanisms through which this occurs are variable. Thus, the differences between autophagy in oocytes and GCs at different stages, or in different species, is in urgent need of study in order to improve our understanding of its regulation and to perform the accurate modulation of follicular atresia for the relevant diseases. Autophagy in pre-antral follicles mainly occurs in oocytes, whereas autophagy in antral follicles mainly takes place in the GCs of mammals and is intimately related to cell apoptosis. As a result, autophagy directly induces different outcomes at every stage of follicular development, whether follicular atresia or ovulation. However, there is currently a lack of research concerning the role of autophagy in the development of the ovaries and many regulatory mechanisms remain to be elucidated. At present, several factors, including the PI3K/PKA/mTOR pathways, calcium signal transduction, mitochondria, and the alteration of cytoskeletons, are all believed to participate in the regulation of autophagy and as a result in the process of follicular development. Consequently, the study of the relationship between autophagy, follicular development, and follicular atresia will not only contribute to our general understanding of the mechanism of the follicular development, but also lay the foundations of premature ovarian failure and ovarian cancer, which could in turn provide theoretical evidence for infertility and promising relevant therapeutic methods for reproductive issues in women.
Abbreviations
CMA: chaperone-mediated autophagy; EGA: embryo genome activation; FSH: follicle-stimulating hormone; GCs: granulosa cells; GSH: glutathione; MII: metaphase II; PGCs: primordial germ cells; PMSG: pregnant mare serum gonadotropin; POF: premature ovarian failure.
Acknowledgements
This work was financially supported by the Trans-gene Key Project of China (2018ZX08009-27B), the National Natural Science Foundation (31501924), the Hubei Technical Innovation Project (2017ABA134), the Hubei Natural Science Foundation (2018CFA014), the Innovation Team of the Hubei Agricultural Science and Technology Innovation Center (2016-620-000-001-022), and the Hubei Academy of Agricultural Sciences Youth Science Fund (2018NKYJJ09).
Author contributions
J.Z. and S.M. planned and wrote the manuscript. X.P. helped with the planning and writing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fortune JE. The early stages of follicular development: activation of primordial follicles and growth of preantral follicles. Anim Reprod Sci. 2003;78:135-163
2. Skinner MK. Regulation of primordial follicle assembly and development. Hum Reprod Update. 2005;11:461-471
3. Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740-743
4. Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: xenophagy. Methods. 2015;75:120-127
5. Kobayashi S. Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy. Biol Pharm Bull. 2015;38:1098-1103
6. Nakashima A, Aokin A, Kusabiraki T. et al. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J Obstet Gynaecol Res. 2017;43:633-643
7. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-741
8. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107-132
9. Cleau D, Jobard JM, Alves T. et al. Cholestatic hepatitis induced by the amoxicillin-clavulanic acid combination. A case and review of the literature. Gastroenterol Clin Biol. 1990;14:1007-1009
10. Thumm M, Egner R, Koch B. et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275-280
11. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169-174
12. Fleming A, Noda T, Yoshimori T. et al. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9-17
13. Ravikumar B, Sarkar S, Davies JE. et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383-1435
14. Hara T, Takamura A, Kishi C. et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008
15. Itakura E, Kishi C, Inoue K. et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360-5372
16. Russell RC, Tian Y, Yuan H. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741-750
17. Kang R, Zeh HJ, Lotze MT. et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571-580 doi:10.1038/cdd.2010.191
18. Di Bartolomeo S, Corazzari M, Nazio F. et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155-168
19. Hanada T, Noda NN, Satomi Y. et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298-37302
20. Fujita N, Hayashi-Nishino M, Fukumoto H. et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2004;19:4651-4659
21. Kabeya Y, Mizushima N, Yamamoto A. et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805-2812
22. Park S, Choi SG, Yoo SM. et al. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy. 2014;10:1906-1920
23. Satoo K, Noda NN, Kumeta H. et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28:1341-1350
24. Furuta N, Fujita N, Noda T. et al. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21:1001-1010
25. Yang Z, Huang J, Geng J. et al. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094-5104
26. Kroemer G, Galluzzi L, Vandenabeele P. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3-11
27. Huber TB, Edelstein CL, Hartleben B. et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009-1031
28. Cuervo AM, Bergamini E, Brunk UT. et al. Autophagy and aging: the importance of maintaining "clean" cells. Autophagy. 2005;1:131-140
29. Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26:16-23
30. Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925-935
31. Sancak Y, Bar-Peled L, Zoncu R. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290-303
32. Kim J, Kundu M, Viollet B. et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132-141
33. Egan DF, Shackelford DB, Mihaylova MM. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456-461
34. Avruch J, Khokhlatchev A, Kyriakis JM. et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127-155
35. Derijard B, Hibi M, Wu IH. et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025-1037
36. Tracy K, Dibling BC, Spike BT. et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229-6242
37. Bellot G, Garcia-Medina R, Gounon P. et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570-2581
38. Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175-33182
39. Zalckvar E, Berissi H, Eisenstein M. et al. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5:720-722
40. Hansen M, Chandra A, Mitic LL. et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24
41. Van Meter M, Simon M, Tombline G. et al. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016;16:2641-2650
42. Vlahopoulos S, Zoumpourlis VC. JNK: a key modulator of intracellular signaling. Biochemistry (Mosc). 2004;69:844-854
43. Wang MC, Bohmann D, Jasper H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell. 2003;5:811-816
44. Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115-125
45. Tasdemir E, Maiuri MC, Galluzzi L. et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676-687
46. Adams DJ, Beveridge DJ, van der Weyden L. et al. HADHB, HuR, and CP1 bind to the distal 3'-untranslated region of human renin mRNA and differentially modulate renin expression. J Biol Chem. 2003;278:44894-44903
47. Lee IH, Cao L, Mostoslavsky R. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374-3379
48. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253-295
49. Minois N. Molecular basis of the 'anti-aging' effect of spermidine and other natural polyamines - a mini-review. Gerontology. 2014;60:319-326
50. Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217-224
51. Lapierre LR, Kumsta C, Sandri M. et al. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy. 2015;11:867-880
52. Matsuda-Minehata F, Inoue N, Goto Y. et al. The regulation of ovarian granulosa cell death by pro- and anti-apoptotic molecules. J Reprod Dev. 2006;52:695-705
53. McNatty KP, Fidler AE, Juengel JL. et al. Growth and paracrine factors regulating follicular formation and cellular function. Mol Cell Endocrinol. 2000;163:11-20
54. McLaughlin EA, McIver SC. Awakening the oocyte: controlling primordial follicle development. Reproduction. 2009;137:1-1
55. Picton HM. Activation of follicle development: the primordial follicle. Theriogenology. 2001;55:1193-1210
56. Young JM, McNeilly AS. Theca: the forgotten cell of the ovarian follicle. Reproduction. 2010;140:489-504
57. Picton H, Briggs D, Gosden R. The molecular basis of oocyte growth and development. Mol Cell Endocrinol. 1998;145:27-37
58. Sutton ML, Gilchrist RB, Thompson JG. Effects of in-vivo and in-vitro environments on the metabolism of the cumulus-oocyte complex and its influence on oocyte developmental capacity. Hum Reprod Update. 2003;9:35-48
59. Griffin J, Emery BR, Huang I. et al. Comparative analysis of follicle morphology and oocyte diameter in four mammalian species (mouse, hamster, pig, and human). J Exp Clin Assist Reprod. 2006;3:2
60. Sturmey RG, Reis A, Leese HJ. et al. Role of fatty acids in energy provision during oocyte maturation and early embryo development. Reprod Domest Anim. 2009;44(Suppl 3):50-58
61. Fair T, Hulshof SC, Hyttel P. et al. Oocyte ultrastructure in bovine primordial to early tertiary follicles. Anat Embryol (Berl). 1997;195:327-336
62. Jansen RP, de Boer K. The bottleneck: mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol Cell Endocrinol. 1998;145:81-88
63. Dumollard R, Duchen M, Sardet C. Calcium signals and mitochondria at fertilisation. Semin Cell Dev Biol. 2006;17:314-323
64. Binelli M, Murphy BD. Coordinated regulation of follicle development by germ and somatic cells. Reprod Fertil Dev. 2010;22:1-12
65. Eppig JJ. Coordination of nuclear and cytoplasmic oocyte maturation in eutherian mammals. Reprod Fertil Dev. 1996;8:485-489
66. Tripathi A, Kumar KV, Chaube SK. Meiotic cell cycle arrest in mammalian oocytes. J Cell Physiol. 2010;223:592-600
67. Ajduk A, Malagocki A, Maleszewski M. Cytoplasmic maturation of mammalian oocytes: development of a mechanism responsible for sperm-induced Ca2+ oscillations. Reprod Biol. 2008;8:3-22
68. Ferreira EM, Vireque AA, Adona PR. et al. Cytoplasmic maturation of bovine oocytes: structural and biochemical modifications and acquisition of developmental competence. Theriogenology. 2009;71:836-848
69. Dumollard R, Carroll J, Duchen MR. et al. Mitochondrial function and redox state in mammalian embryos. Semin Cell Dev Biol. 2009;20:346-353
70. Dumollard R, Duchen M, Carroll J. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol. 2007;77:21-49
71. Van Blerkom J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion. 2011;11:797-813
72. Trounson A, Anderiesz C, Jones G. Maturation of human oocytes in vitro and their developmental competence. Reproduction. 2001;121:51-75
73. Rolaki A, Drakakis P, Millingos S. et al. Novel trends in follicular development, atresia and corpus luteum regression: a role for apoptosis. Reprod Biomed Online. 2005;11:93-103
74. Manabe N, Matsuda-Minehata F, Goto Y. et al. Role of cell death ligand and receptor system on regulation of follicular atresia in pig ovaries. Reprod Domest Anim. 2008;43(Suppl 2):268-272
75. Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annu Rev Physiol. 1997;59:349-363
76. Manabe N. et al. Regulation mechanism of selective atresia in porcine follicles: regulation of granulosa cell apoptosis during atresia. J Reprod Dev. 2004;50:493-514
77. Jerrell JM. Behavior and symptom identification scale 32: sensitivity to change over time. J Behav Health Serv Res. 2005;32:341-346
78. Manabe N, Goto Y, Matsuda-Minehata F. et al. cFLIP regulates death receptor-mediated apoptosis in an ovarian granulosa cell line by inhibiting procaspase-8 cleavage. J Reprod Dev. 2008;54:314-320
79. Choi J, Jo M, Lee E, Choi D. Induction of apoptotic cell death via accumulation of autophagosomes in rat granulosa cells. Fertil Steril. 2011;95:1482-1486
80. Choi JY, Jo MW, Lee EY. et al. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil Steril. 2010;93:2532-2537
81. Song ZH, Yu HY, Wang P. et al. Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death Dis. 2015;6:e1589
82. Duerrschmidt N, Zabirnyk O, Nowicki M. et al. Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology. 2006;147:3851-3860
83. Hulas-Stasiak M, Gawron A. Follicular atresia in the prepubertal spiny mouse (Acomys cahirinus) ovary. Apoptosis. 2011;16:967-975
84. Santos HB, Thome RG, Arantes FP. et al. Ovarian follicular atresia is mediated by heterophagy, autophagy, and apoptosis in Prochilodus argenteus and Leporinus taeniatus (Teleostei: Characiformes). Theriogenology. 2008;70:1449-1460
85. Escobar ML, Echeverria OM, Garcia G. et al. Immunohistochemical and ultrastructural study of the lamellae of oocytes in atretic follicles in relation to different processes of cell death. Eur J Histochem. 2015;59:2535
86. Escobar ML, Echeverria OM, Ortiz R. et al. Combined apoptosis and autophagy, the process that eliminates the oocytes of atretic follicles in immature rats. Apoptosis. 2008;13:1253-1266
87. He Y, Deng H, Jiang Z. et al. Effects of melatonin on follicular atresia and granulosa cell apoptosis in the porcine. Mol Reprod Dev. 2016;83:692-700
88. Zhao F, Zhao W, Ren S. et al. Roles of SIRT1 in granulosa cell apoptosis during the process of follicular atresia in porcine ovary. Anim Reprod Sci. 2014;151:34-41
89. Shen M, Cao Y, Jiang Y. et al. Melatonin protects mouse granulosa cells against oxidative damage by inhibiting FOXO1-mediated autophagy: Implication of an antioxidation-independent mechanism. Redox Biol. 2018;18:138-157
90. Shen M, Jiang Y, Guan Z. et al. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy. 2017;13:1364-1385
91. Choi J, Jo M, Lee E. et al. AKT is involved in granulosa cell autophagy regulation via mTOR signaling during rat follicular development and atresia. Reproduction. 2013;147:73-80
92. Yuan J, Zhang Y, Sheng Y. et al. MYBL2 guides autophagy suppressor VDAC2 in the developing ovary to inhibit autophagy through a complex of VDAC2-BECN1-BCL2L1 in mammals. Autophagy. 2015;11:1081-1098
93. Lee J, Park JI, Yun JI. et al. Rapamycin treatment during in vitro maturation of oocytes improves embryonic development after parthenogenesis and somatic cell nuclear transfer in pigs. J Vet Sci. 2015;16:373-380
94. Lee S, Hiradate Y, Hoshino Y. et al. Quantitative analysis in LC3-II protein in vitro maturation of porcine oocyte. Zygote. 2014;22:404-410
95. Houel-Renault L, Philippe L, Piquemal M. et al. Autophagy is used as a survival program in unfertilized sea urchin eggs that are destined to die by apoptosis after inactivation of MAPK1/3 (ERK2/1). Autophagy. 2013;9:1527-1539
96. Song BS, Kim JS, Kim YH. et al. Induction of autophagy during in vitro maturation improves the nuclear and cytoplasmic maturation of porcine oocytes. Reprod Fertil Dev. 2014;26:974-981
97. Pua HH, Dzhagalov I, Chuck M. et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25-31
98. Shen M, Jiang Y, Guan Z. et al. FSH protects mouse granulosa cells from oxidative damage by repressing mitophagy. Sci Rep. 2016;6:38090
99. Huo X, Chen D, He Y. et al. Bisphenol-A and Female Infertility: A Possible Role of Gene-Environment Interactions. Int J Environ Res Public Health. 2015;12:11101-11116
100. Mahalingaiah S, Winter MR, Aschengrau A. Association of prenatal and early life exposure to tetrachloroethylene (PCE) with polycystic ovary syndrome and other reproductive disorders in the cape cod health study: A retrospective cohort study. Reprod Toxicol. 2016;65:87-94
101. Ziv-Gal A, Flaws JA. Evidence for bisphenol A-induced female infertility: a review (2007-2016). Fertil Steril. 2016;106:827-856
102. Hou YC, Chittaranjan S, Barbosa SG. et al. Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008;182:1127-1139
103. Barth JM, Hafen E, Kohler K. The lack of autophagy triggers precocious activation of Notch signaling during Drosophila oogenesis. BMC Dev Biol. 2012;12:35
104. Duan X, Dai XX, Wang T. et al. Melamine negatively affects oocyte architecture, oocyte development and fertility in mice. Hum Reprod. 2015;30:1643-1652
105. Kim SH, Lee IC, Lim JH. et al. Effects of melamine on pregnant dams and embryo-fetal development in rats. J Appl Toxicol. 2011;31:506-514
106. Han J, Wang QC, Zhu CC. et al. Deoxynivalenol exposure induces autophagy/apoptosis and epigenetic modification changes during porcine oocyte maturation. Toxicol Appl Pharmacol. 2016;300:70-76
107. Zhu CC, Zhang Y, Duan X. et al. Toxic effects of HT-2 toxin on mouse oocytes and its possible mechanisms. Arch Toxicol. 2016;90:1495-1505
108. Makker A, Goel MM, Mahdi AA. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: an update. J Mol Endocrinol. 2014;53:R103-118
109. Omari S, Waters M, Naranian T. et al. Mcl-1 is a key regulator of the ovarian reserve. Cell Death Dis. 2015;6:e1755
110. Samplaski MK, Dimitromanolakis A, Lo KC. et al. The relationship between sperm viability and DNA fragmentation rates. Reprod Biol Endocrinol. 2015;13:42
111. Abdelhalim MA, Jarrar BM. Gold nanoparticles induced cloudy swelling to hydropic degeneration, cytoplasmic hyaline vacuolation, polymorphism, binucleation, karyopyknosis, karyolysis, karyorrhexis and necrosis in the liver. Lipids Health Dis. 2011:10 166
112. Bauvy C, Gane P, Arico S. et al. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res. 2001;268:139-149
113. Longo L, Platini F, Scardino A. et al. Autophagy inhibition enhances anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol Cancer Ther. 2008;7:2476-2485
114. Livesey KM, Kang R, Vernon P. et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72:1996-2005
115. Marino G, Niso-Santano M, Baehrecke EH. et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81-94
116. Klinger FG, Rossi V, De Felici M. Multifaceted programmed cell death in the mammalian fetal ovary. Int J Dev Biol. 2015;59:51-54
117. Gannon AM, Stampfli MR, Foster WG. Cigarette smoke exposure leads to follicle loss via an alternative ovarian cell death pathway in a mouse model. Toxicol Sci. 2012;125:274-284
118. Brookheart RT, Michel CI, Listenberger LL. et al. The non-coding RNA gadd7 is a regulator of lipid-induced oxidative and endoplasmic reticulum stress. J Biol Chem. 2009;284:7446-7454
119. Sorensen S, Ranheim T, Bakken KS. et al. Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J Biol Chem. 2006;281:468-476
120. Szegezdi E, Macdonald DC, Ni Chonghaile T. et al. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941-953
121. Gawriluk TR, Ko C, Hong X. et al. Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc Natl Acad Sci USA. 2014;111:E4194-4203
Author contact
![]() Corresponding author: Shuqi Mei, Hubei Key Lab for Animal Embryo Engineering and Molecular Breeding, Wuhan 430064, China. E-mail: msqpapercom
Corresponding author: Shuqi Mei, Hubei Key Lab for Animal Embryo Engineering and Molecular Breeding, Wuhan 430064, China. E-mail: msqpapercom