Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Bone function and bone-derived...
Extraskeletal functions of bone...
Conclusion and Perspective
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):776-787. doi:10.7150/ijbs.27063 This issue Cite
Review
Bone function, dysfunction and its role in diseases including critical illness
Nan Su1, Jing Yang1, Yangli Xie1, Xiaolan Du1, Hangang Chen1, Hong Zhou2, Lin Chen1 ![]()
1. Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China.
2. Bone Research Program, ANZAC Research Institute, The University of Sydney, Hospital Road, Sydney, NSW 2139, Australia.
Received 2018-5-4; Accepted 2019-1-4; Published 2019-1-29
Abstract

The skeleton is one of the largest organs in the human body. In addition to its conventional functions such as support, movement and protection, the skeleton also contributes to whole body homeostasis and maintenance of multiple important non-bone organs/systems (extraskeletal functions). Both conventional and extraskeletal functions of the skeleton are defined as bone function. Bone-derived factors (BDFs) are key players regulating bone function. In some pathophysiological situations, including diseases affecting bone and/or other organs/systems, the disorders of bone itself and the subsequently impaired functions of extraskeletal organs/systems caused by abnormal bone (impaired extraskeletal functions of bone) are defined as bone dysfunction. In critical illness, which is a health status characterized by the dysfunction or severe damage of one or multiple important organs or systems, the skeleton shows rapid bone loss resulting from bone hyper-resorption and impaired osteoblast function. In addition, the dysfunctions of the skeleton itself are also closely related to the severity and prognosis of critical illness. Therefore, we propose that there is bone dysfunction in critical illness. Some methods to inhibit osteoclast activity or promote osteoblast function by the treatment of bisphosphonates or PTH1-34 benefit the outcome of critical illness, which indicates that enhancing bone function may be a potential novel strategy to improve prognosis of diseases including critical illness.
Introduction
The skeleton constitutes up to approximately 15% of total body weight and is among the largest organs/systems in the human body [1]. Adult bone structure mainly includes cortical bone, cancellous bone (trabecular bone) and bone marrow cavity. Bone consists of three compartments: bone cells, extracellular organic matrix including collagen fibers and amorphous matrix, and extracellular minerals [2, 3]. There are three major types of bone cells in bone tissue: osteoblasts, osteoclasts, and osteocytes. Osteoblasts are the major cells responsible for bone formation, whereas osteoclasts mainly resorb bone. Bone homeostasis is maintained by the coupling between bone-forming and bone-resorbing (bone turnover). Osteocytes, which make up the majority of bone cells in the adult [4], are terminally differentiated osteolineage cells. Osteocytes are now recognized as important cells playing essential roles in skeletal development and especially homeostasis, including bone modeling and remodeling that was previously thought to be largely controlled by the osteoblasts and osteoclasts [5].
Bone function and bone-derived factors
The conventional function of the skeleton is as a static structural organ supporting body movement, protecting the internal organs, and as a reservoir of minerals [2].
The skeleton is an important organ for the support of the body and for the attachment of muscles and tendons, as well as body movement. The skeleton protects the organs of the cranial and thoracic cavities from injuries, and it houses and protects the bone marrow within its cavities. Additionally, the skeleton has an important role as a reservoir of minerals such as calcium and phosphate, which can be released when demands are increased in the body, and in maintaining serum homeostasis [2]. These conventional functions of skeleton depend on the homeostasis of bone itself.
Recently, increasing studies have revealed that the skeleton contributes to whole body homeostasis and the maintenance of multiple important organs/systems such as hematopoiesis, immune activity, energy metabolism, and brain function. These functions affecting non-bone tissues are called extraskeletal functions [6, 7].
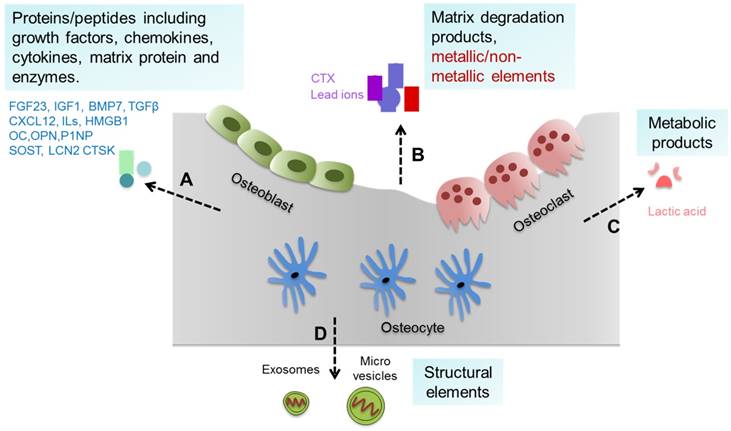
Bone tissue and cells can generate a variety of substances such as proteins/peptides including growth factors, chemokines, cytokines etc., as well as matrix degradation products, metallic/non-metallic elements, metabolic products, and extracellular vesicles (such as exosomes and micro vesicles) [8, 9], which can be collectively defined as bone-derived factors (BDFs) (Figure 1). In addition to their roles in regulating bone modeling and remodeling, these BDFs are the major factors mediating the extraskeletal functions of bone [1, 7, 8].
Bone-derived factors (BDFs) from bone. Bone tissue and cells can generate a variety of substances. (A) Proteins/peptides including growth factors, chemokines, cytokines, matrix protein and enzymes etc. such as FGF23, CXCL12, ILs, OC and others. (B) Matrix degradation products and metallic/non-metallic elements released during bone resorption such as CTX and lead ions. (C) Metabolic products of bone cells such as lactic acid. (D) Structural elements secreted by bone cells such as exosomes and micro vesicles.
Extraskeletal functions of bone mediated by BDFs
The effect of bone on the hematopoietic and immune systems
Hosted by bone tissue, hematopoietic stem cells (HSCs) in bone marrow are able to differentiate into all hematopoietic lineages. The functional communication among different bone cell types and hematopoiesis in the bone marrow has been extensively studied in recent decades. Mesenchymal stem cells, osteoblasts, and other cells in the bone marrow form a special local environment termed a “niche,” which not only nests the HSCs but also helps to maintain the stemness of the resident HSCs [10-12] (Figure 2A).
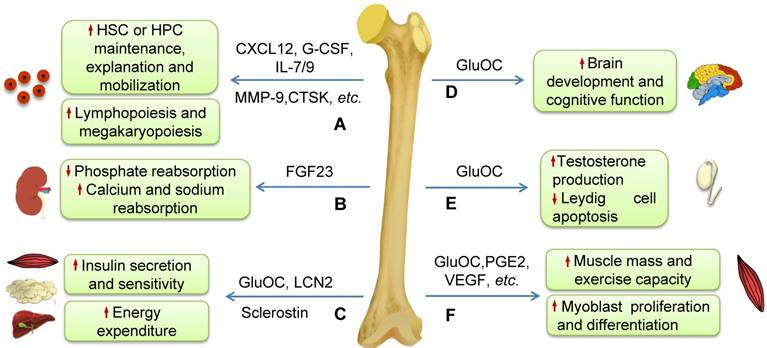
Schematic diagram illustrating the effects of some molecules generated by bone on systemic homeostasis. (A) Some molecules secreted by osteoblasts, osteocytes and osteoclasts such as CXCL12, CTSK, G-CSF, MMP-9, IL-7 and IL-9 play very important roles in regulating HSCs maintenance, expansion or mobilization, and thus influence the hematopoietic and immune systems such as lymphopoiesis and megakaryopoiesis. (B) FGF23 inhibits phosphate reabsorption and promotes calcium and sodium reabsorption in the kidney. (C) GluOC and LCN2 induce insulin production and improve insulin sensitivity and then regulate energy expenditure. Sclerostin secreted by osteocytes regulates insulin sensitivity and metabolism of adipocytes. (D) GluOC promotes brain development and cognitive function. (E) GluOC can regulate male fertility by promoting testosterone production and inhibiting the apoptosis of Leydig cells in testes. (F) Bone regulates muscle mass and exercise capacity through secretion of GluOC, PGE2, VEGF, and others.
Osteoblasts are indispensable for the quiescent status and self-renewal of HSCs. The absence of osteoblasts leads to decreased self-renewal of HSCs in the bone marrow [13]. Activation of parathyroid hormone (PTH) signaling in osteoblasts results in increased numbers of both osteoblasts and HSCs [10]. Spindle-shaped N-cadherin positive (N-cadherin+) osteoblasts (known to enrich osteoprogenitors) are mainly located on the surface of cancellous/trabecular bone and are directly attached to long-term HSCs (LT-HSCs) and maintain HSC quiescence [14, 15]. In addition to directly attaching to HSCs, some molecules secreted from osteoblasts can support HSCs. For example, Wnt11, one of the osteoblast derived Wnt ligands, activates non-canonical Wnt signaling mediated by Fmi and Fz8 in HSCs and maintains the quiescent status of HSCs in the bone marrow niche [16]. Thrombopoietin and angiopoietin-1 secreted by osteoblasts can also regulate HSC quiescence [17, 18]. In addition, osteoblasts play an important role in lymphopoiesis, myelopoiesis and megakaryopoiesis. C-X-C motif chemokine ligand 12 (CXCL12) is a chemokine that regulates HSC maintenance and retention [19, 20] and is required for lymphopoiesis and myelopoiesis [21, 22]. The deletion of CXCL12 from osteoblasts led to depletion of certain early lymphoid progenitors but not HSCs, suggesting that osteoblast CXCL12 mainly promotes the maintenance of early lymphoid progenitors [19]. The deletion of interleukin 7 (IL-7) in osteoblasts led to a lymphopenic phenotype by decreasing common lymphoid progenitor number but not HSCs, indicating the important effect of osteoblast IL-7 on lymphopoiesis [23]. Furthermore, m-TORC1 signaling inhibits megakaryopoiesis through decreasing the osteoblast derived IL-9 levels [24].
Osteoclasts are another important cells regulating hematopoiesis in bone. Engraftment of HSCs to bone marrow depends on local level of calcium ion elevated during bone resorption [25]. Cathepsin K, the major bone-resorbing proteinase secreted by osteoclasts, cleaves CXCL12 and stem cell factor (SCF) in the stem cell niche, which is associated with promoted mobilization of hematopoietic progenitor cells (HPCs) into the systemic circulation [11]. Matrix metalloproteinase 9 (MMP-9) secreted by osteoclasts induces the release of SCF from bone marrow stromal cells to promote hematopoietic reconstitution, suggesting an important role of osteoclasts in HSC niche maintenance and mobilization [26]. Administration of receptor activator of nuclear factor-κ B ligand (RANKL) increases concomitant mobilization of HSCs to the systemic circulation by promoting bone resorption [11], while inhibition of osteoclast function by bisphosphonates treatment (clinical drugs used to treat osteoporosis) reduced HSC numbers in mice [27]. The absence of osteoclast activity results in a defective HSC niche associated with an increased proportion of mesenchymal progenitors but reduced osteoblastic differentiation, leading to impaired HSC homing to the bone marrow [28]. It is suggested that bone marrow B lymphopoiesis is also regulated by osteoclast activity. Inhibiting osteoclast activity by zoledronic acid injections caused a decrease in the number of B-cells in the bone marrow in mice, resulting from decreased expression of CXCL12 and IL-7 by stromal cells, associated with reduced osteoblastic engagement [29].
Several studies have revealed that osteocytes also regulate the endosteal microenvironment and hematopoiesis. The ablation of osteocytes led to severe lymphopenia due to the lack of lymphoid-supporting stroma in both the bone marrow for B-cell precursors and the thymus for T-cell precursors, and a marked loss of white adipose tissues [30]. Osteocytes produce granulocyte-colony stimulating factor (G-CSF), an important factor promoting myeloid expansion in the bone marrow. The deletion of Gs alpha specifically in osteocytes resulted in enhanced G-CSF production [31].
The effect of bone on mineral metabolism
In the last 10 years, bone has been recognized as an important endocrine organ regulating multiple metabolic processes. Mineral metabolism is very important for homeostasis. In addition to storing minerals, the skeleton also regulates systemic mineral metabolism through an endocrine manner (Figure 2B).
Fibroblast growth factor 23 (FGF23), a member of the FGF family, is a bone-derived hormone secreted by osteoblasts and osteocytes. Mutations in FGF23 prevent its cleavage, resulting in autosomal-dominant hypophosphatemic rickets (ADHR), which is characterized by low serum phosphorus concentrations, rickets, osteomalacia, etc. [32-35]. The overproduction of FGF23 by tumors [36] and osteogenic cells in fibrous dysplastic lesions [37] is responsible for the hypophosphatemia in tumor-induced osteomalacia (TIO) and fibrous dysplasia. These studies help to define the critical role of FGF23 in regulating phosphate metabolism.
FGF23 inhibits phosphate reabsorption by inhibiting expression of the type IIa sodium-phosphate co-transporter (NaPi-2a) in the proximal tubules of the kidney through binding to a FGFR1-α-Klotho co-receptor complex [38-40], leading to phosphate loss. FGF23 also regulates calcium and sodium reabsorption of the kidney [41, 42]. FGF23 promotes renal calcium reabsorption through the transient receptor potential vanilloid-5 (TRPV5) channel in the distal renal tubules [41], and FGF23 promotes sodium reabsorption through the sodium-chloride co-transporter (NCC) in the renal distal renal tubules and leads to volume expansion and hypertension [42].
In addition, FGF23 systematically regulates serum calcium and phosphate by regulating levels of PTH and the active form of vitamin D (1,25-dihydroxyvitamin D3, 1,25(OH)2D3). PTH promotes calcium release from bone by stimulating bone resorption and calcium absorption in the kidney and intestinal calcium absorption via an increase in the 1,25(OH)2D3 level. PTH also inhibits phosphate reabsorption in the kidney [43]. 1,25(OH)2D3 promotes calcium absorption in the small intestine and phosphate reabsorption by suppressing the PTH level in the kidney [44].
FGF23 inhibits PTH synthesis and secretion in the parathyroid in a Klotho-dependent or independent manner [45, 46] and suppresses the synthesis of 1,25(OH)2D3 by inhibiting 25-hydroxyvitamin D-1a-hydroxylase (1a-OHase) and increasing 25-hydroxyvitamin D-24-hydroxylase (24-OHase) in the kidney [47]. These studies demonstrate that FGF23 regulates mineral metabolism locally in the kidney and systematically by regulating PTH and 1,25(OH)2D3.
The effect of bone cells on glucose and energy metabolism
As an endocrine organ, bone also regulates glucose and energy metabolism through different pathways (Figure 2C).
Osteocalcin (OC) is another osteoblast-secreted multifunctional hormone and is embedded in the bone matrix during bone formation [5]. There are two forms of OC: uncarboxylated OC (γ-carboxylated OC, GlaOC) and undercarboxylated OC (GluOC). During bone resorption, the acidic environment in the bone resorption pits generated by osteoclasts promotes the decarboxylation of GlaOC embedded in bone matrix to GluOC, which has a lower affinity for hydroxyapatite and therefore is more easily released from bone tissue into the circulation [48, 49]. GluOC can promote β-cell proliferation, stimulate insulin synthesis and secretion, and increase the insulin sensitivity of adipose tissue, muscle, and liver, leading to increased energy expenditure [50].
Changing osteoblast or osteoclast activity alters glucose metabolism by affecting GluOC levels. A clinical study shows that patients treated with preotact (PTH1-84), a bone formation-promoting drug for osteoporosis by increasing osteoblastic activity, resulted in an increased serum GluOC level and decreased blood glucose level [51]. Animal experiments show similar results. Conditional deletion of the insulin receptor (IR) in osteoblasts led to impaired osteoblast differentiation and decreased OC production, and increased expression of osteoprotegerin (OPG) in IR-deficient osteoblasts hampers osteoclast differentiation and bone resorption, resulting in a reduced serum GluOC level. The decreased serum GluOC level leads to impaired glucose tolerance, insulin secretion and sensitivity [48, 52]. It is well known that long-term glucocorticoid treatment induces insulin resistance and glucose intolerance. Interestingly, it is reported that these glucocorticoid side effects are partially mediated through osteoblasts. As a glucocorticoid target gene, the total serum OC levels can be suppressed within 3 days after glucocorticoid treatments [53, 54]. The deletion of glucocorticoid signaling in osteoblasts attenuated the glucocorticoid suppressed OC synthesis and prevented the development of insulin resistance and glucose intolerance [53]. The deletion of OPG in mice led to an increased osteoclast number and an improved serum GluOC level, resulting in higher glucose tolerance compare to wild-type animals, suggesting that osteoclasts control glucose metabolism through the regulation of OC decarboxylation.
In addition to OC, recent studies also implicate an OC-independent influence of bone on energy metabolism. For example, ablation of osteoblasts in adult mice or conditional deletion of β-catenin or Lrp5 (a coreceptor of Wnt-β-catenin signaling) in osteoblasts/osteocytes leads to systemic metabolic alterations, which could not be fully reversed by OC treatment or explained by increased or unchanged serum OC level [55-57]. These results suggest that there are some other unknown factors or mechanisms involved in mediating energy metabolism by bone.
Lipocalin 2 (LCN2), a newly identified hormone secreted by osteoblasts, also regulates energy metabolism (Figure 2C). LCN2 maintains glucose homeostasis by inducing insulin secretion and improving glucose tolerance and insulin sensitivity, as well as inhibiting food intake [58]. Sclerostin is a secreted glycoprotein specifically generated by osteocytes to inhibit Wnt signaling [59] and plays an endocrine function to influence body composition by regulating adipocyte metabolism [60, 61] (Figure 2C). The deletion of SOST in mice or sclerostin-neutralizing antibody treatment led to reduced mass of white adipose tissue and corresponding enhancements in insulin sensitivity and fatty acid [60], indicating a positive role of sclerostin on anabolic metabolism in adipocytes. However, another study showed the opposite results. Enhanced sclerostin levels in mice with deficiency of Gsα in mature osteoblasts and/or osteocytes also lead to the loss of white adipose tissue [61]. These inconsistent results might be due to the different mouse models.
A recently study shows that the deletion of von Hippel-Lindau (Vhl), a hypoxia signaling pathway component, in osteolineage cells led to hypoglycemia and increased glucose tolerance in mice, which is caused by increased glucose uptake and glycolysis of osteoblasts, but not caused by OC because the serum OC level was decreased in these mice [62]. This result suggests that osteoblasts regulate whole-body glucose homeostasis by regulating their own cellular glucose metabolism.
Other extraskeletal functions of bone
Embryonic brain development and adult brain function are regulated by bone via GluOC [63] (Figure 2D). Mouse maternal GluOC can cross the placenta and blood-brain barrier to directly promote brain development and the acquisition of cognitive function in the fetus [64]. In adult mice, GluOC can also pass through the blood-brain barrier to promote brain-derived neurotrophic factor expression and affect cognitive function of the brain. OC absence in mice resulted in deficiency of spatial learning and memory and a worsened anxiety-like behavior. Delivering GluOC can correct age-related cognitive decline and decrease anxiety-like behavior and functions [63,64].
In addition, GluOC can regulate male fertility by promoting testosterone production and inhibit the apoptosis of Leydig cells in testes [65] (Figure 2E).
Bone also has important effects on muscle. GluOC increases IL-6 production of muscle, which enhances glucose and fatty acid uptake of myofibers, and then promotes adaptation to exercise [66]. In addition, a comparatively high concentration of GluOC administered exogenously increased muscle mass in aged mice [67]. Moreover, in vitro studies have shown that bone marrow mesenchymal stromal cells stimulate myoblast proliferation through the paracrine release vascular endothelial growth factor (VEGF) [68], and osteocytes can support myogenesis and muscle function by secreted prostaglandin E2 (PGE2) [69]. These results indicate the positive influences of bone on muscle (Figure 2F).
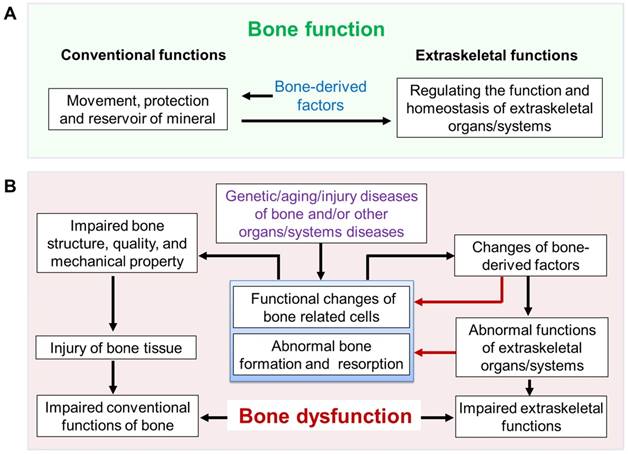
Based on these data described above, in addition to the classic functions of movement and protection, the skeleton has extraskeletal functions that play important roles in the maintenance of vital organs/systems and the general health (Figure 2). We, therefore, define these classic and extraskeletal functions of skeleton as BONE FUNCTION (Figure 3A). The impaired bone function may lead to bone dysfunction.
Bone function and hypothesis of bone dysfunction. (A) Bone function includes conventional functions (such as movement, protection and reserve of minerals) and extraskeletal functions (regulation of the function and homeostasis of extraskeletal organs/systems such as hematopoietic and immune systems, mineral and metabolism, etc.). (B) Hypothesis of bone dysfunction. In some pathophysiological situations, including genetic/aging/injury diseases of bone and/or other organs/systems diseases, the disorders of bone itself and the subsequently impaired function of extraskeletal organs/systems caused by abnormal bone (impaired extraskeletal function of bone) are defined as BONE DYSFUNCTION.
Bone dysfunction
The body is composed of multiple organs/systems and exists and functions as a whole system. There is elegant coordination and crosstalk among different organs/systems, which makes our bodies functional in physiological situations and might also accelerate the dysfunction or failure of multiple organs/systems during severe diseases. For example, chronic renal failure can lead to heart failure.
As for the skeleton, in some pathophysiological situations including diseases affecting bone tissue (such as genetic bone diseases or osteoporosis) and/or other organs/systems (such as systemic inflammation, chronic kidney disease (CKD), diabetes, etc.), the impaired functions of bone cells might lead to abnormal bone formation or resorption, and then result in bone disorders characterized by impaired bone microstructure and decreased bone strength, which increases bone fragility and fracture. Furthermore, the abnormalities of the skeleton might also injure the homeostasis of other organs/systems by changing the production and metabolism of BDFs, which might subsequently lead to dysregulated or impaired function of extraskeletal organs/systems (Figure 3B).
For example, osteoporosis is the most common bone disease in humans characterized by bone loss, microarchitectural deterioration, and compromised bone strength [70,71]. Osteoporosis leads to increasing bone fragility and propensity for fracture, particularly in postmenopausal women [70-72]. Recent studies also show that there is close relationship between osteoporosis (Or low bone mass) and other system diseases or mortality. Osteoporosis is associated with an increased risk of the incidence of Alzheimer's disease dementia [73]. Low bone mass is also associated with increased all-cause mortality of the elderly, and is also an independent risk factor for mortalities of stroke, chronic lung and cardiovascular diseases [74-77]. Osteoporosis treatment can reduce mortality in elderly and frailer individuals with osteoporosis who are at high risk of fracture [78].
Osteopetrosis is a kind of bone genetic diseases sharing the hallmark of a generalized increase in bone mass [79]. Many patients with osteopetrosis have more brittle bones and increased incidence of anemia and recurrent infections [79]. Activating mutation of β-catenin in osteoblasts led to osteosclerosis and the development of acute myeloid leukemia (AML) in mice, and thirty-eight percent of patients with myelodysplastic syndrome (MDS)/AML showed increased β-catenin signaling in osteoblasts [80].
Therefore, as mentioned above, we define these disorders of bone itself, and the impaired extraskeletal functions of bone as BONE DYSFUNCTION (Figure 3B).
Bone dysfunction in critical illness
Critical illness describes a health status with dysfunction or severe damages of one or multiple important organs/systems and includes conditions such as sepsis, shock, acute respiratory distress syndrome (ARDS), acute renal failure, heart failure, disorders of the immune and hematopoietic systems, endocrine and metabolic disturbances, and electrolyte and acid-base imbalance [81-84], etc.
However, as one of the largest organs with an emerging important role in regulating multiple organs/systems, changes of the skeleton and its functional role during critical illness remain poorly understood. We previously proposed a relationship between bone and critical illness and the potential important role of bone dysfunction in critical illness [85], which has been supported by increasing evidences.
Changes of bone in critical illness
The most significant pathological changes in bone during critical illness are accelerated bone loss and increased risk of fracture, especially in patients with osteoporosis [86-88] and especially following a prolonged stay in an intensive care unit (ICU) [89, 90]. These changes in bone are ultimately due to enhanced bone resorption and impaired bone formation.
Bone resorption activity is significantly increased in critically ill patients. C-telopeptide of type I collagen (CTX) and N-telopeptide of type I collagen (NTX) are degradation products of type I collagen during bone resorption. Serum levels of CTX or urine NTX are recommended as biochemical markers of bone resorption and have been used in clinical diagnosis [9]. Patients with critical illness had obvious enhanced bone resorption activity with elevated serum CTX and urine NTX levels [90-94].
In addition, bone formation is also profoundly affected by critical illness. N-terminal propeptide of type I procollagen (P1NP) is cleaved from procollagen molecules in the extracellular matrix before the assembly of type I collagen molecules into fibers [9]. P1NP, OC and serum bone alkaline phosphatase (bALP) secreted by mature osteoblasts are used as biochemical markers to assess osteoblast function and bone formation [9]. Patients with critical illness had a significant increase in P1NP and a decrease in OC levels, which suggests an increase in the activity of immature osteoblasts, with low activity of mature osteoblasts [95]. We also found that serum bALP levels were lower in infected newborns than that in the non-infected group [96]. These changes in osteoblasts might be a possible cause for bone loss and demineralization of bone in critically ill patients [88]. Our animal study also showed decreased bone mass and mRNA expression of OC in mice with systemic inflammation and sepsis caused by lipopolysaccharide (LPS) treatment [97]. Other animal experiments showed that the increased apoptosis or acute deficiency of osteoblasts might be another reason causing impaired of bone formation [23, 98].
In addition to the enhanced bone resorption and impaired bone formation, some molecules secreted from bone cells are also influenced by critical illness. For example, LPS stimulates osteoblasts to generate IL-1, IL-6, and tumor necrosis factor α (TNF-α), which are important cytokines regulating inflammation. Furthermore, researchers also found that pro-inflammatory factors promoted osteocyte secretion of FGF23 [99], and plasma FGF23 levels were elevated in critically ill patients [100].
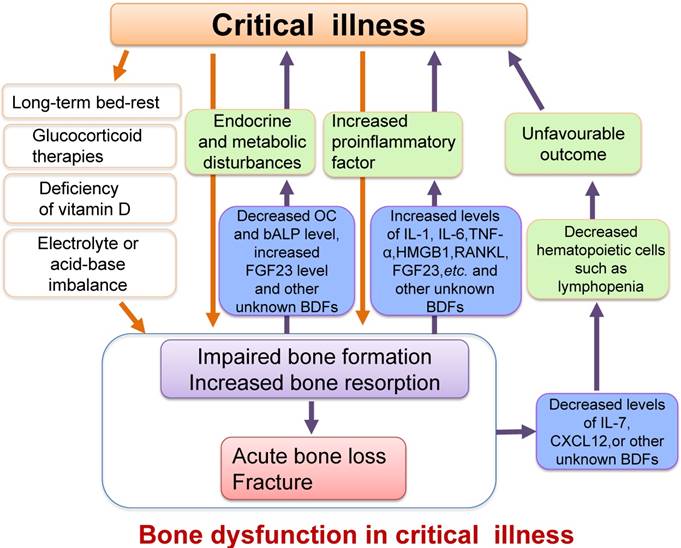
The causes of these changes in bone have not been fully elucidated but are intimately connected with extremely stressful situations such as vitamin D deficiency, glucocorticoid therapies [94, 101], long-term bed-rest, inflammatory environments, and disorders of metabolism such as glucose and hormones in critical illness [88, 94, 102, 103] (Figure 4).
Schematic diagram illustrating the relationship between critical illness and bone dysfunction. Many pathological conditions such as inflammation, endocrine and metabolic disturbances, electrolyte and acid-base imbalance or vitamin D deficiency, as well as other causes such as long-term bed-rest or glucocorticoid therapies in critical illness could result in impaired osteoblast function and increased osteoclast activity, ultimately leading to acute bone loss and even bone fracture. Furthermore, the impaired osteoblast function and enhanced bone resorption could also cause increased secretion of proinflammatory factors, which might exacerbate the systemic inflammatory response. The changes of some BDFs secreted by osteoblasts such as the decreased serum levels of OC, IL-7 and CXCL12 or increased FGF23 level in critical illness may also worsen metabolic or hematopoietic disorders. We define these damages or changes in the structure and functions of bone as bone dysfunction in critical illness.
The effects of changed bone on progress and prognosis of critical illness
The important role of bone in the maintenance of vital organs and systems described above strongly suggests that bone might be a critical player in the initiation, progress and prognosis of critical illness.
Patients in the ICU who died showed a greater degree of bone loss [104]. Low osteoblast activity (characterized by decreased bALP serum levels) was inversely correlated with the scores for neonatal acute physiology-perinatal extension II (SNAPPE-II, an index assessing neonatal illness severity in the neonatal ICU population) of infected neonates [96]. Higher serum FGF23 levels are also independently associated with greater mortality in critically ill patients with or without CKD [100, 105]. Animal experiments showed that osteoblast ablation in sepsis led to a high mortality rate in mice [23]. These studies provide important clinical and animal experimental evidences supporting the causal links between disorders of bone and the severity and outcome of critical illness.
The mechanisms underlying the important role of bone in the pathogenesis of critical illness have not been fully clarified to date. Changes of some BDFs might participate in the pathogenesis of critical illness. Decreased serum levels of OC might contribute to abnormalities of glucose metabolism, such as hyperglycemia in critical illness. Elevated serum level of FGF23 might contribute to hypophosphatemia during sepsis [99]. Meanwhile, the increased FGF23 level might be related to the impaired defense and inflammatory status of critically ill patients as FGF23 has been found to impair neutrophil activation and stimulate hepatic production of inflammatory cytokines [106, 107]. Furthermore, pro-inflammatory factors secreted by bone cells such as high mobility group box 1 protein (HMGB1), RANKL, TNF-α, and IL-1/6 might also aggravate the systemic inflammatory response in critical illness [108-110]. Osteoblast-derived IL-7 regulates early lymphopoiesis, and decreased IL-7 expression in osteoblasts resulted in lymphopenia in sepsis mice [23]. However, the detailed roles and underlying mechanisms of bone in critical illness need further studies.
Briefly, the above mentioned data suggest that damage or changes in bone function are highly related to the severity and prognosis of critical illness. Therefore, we hypothesize that there is bone dysfunction in critical illness, which is likely to be involved in the pathogenesis of critical illness and that improvement of bone function might benefit the critical illness prognosis (Figure 4).
Improvement of bone function may benefit the prognosis of critical illness
Enhancing osteoblast function is a potential mean to improve the prognosis of patients with critical illness. Pharmacological activation of osteoblasts using teriparatide (PTH1-34), a drug for osteoporosis that promotes bone formation, improves bone mass and sepsis-induced lymphopenia, and increases the survival rate of mice by increasing IL-7 generation [23]. We found that deletion of fibroblast growth factor receptor 1 (FGFR1) in mature osteoblasts leads to increased osteoblast activity and enhances the mobilization of endothelial progenitor cells (EPCs) into the peripheral blood by promoting CXCL12 secretion from osteoblasts, resulting in increased survival rate of mice with sepsis [111]. The inhibition of bone resorption might be another way to regulate critical illness prognosis. A recent study shows that preadmission treatment with bisphosphonates is associated with improved survival among critically ill patients, possibly by inhibiting bone resorption and increasing bone mass [104].
These studies suggest that improvement of bone mass or bone function is a potential novel strategy to improve the prognosis of critically ill patients, although the mechanisms and detailed approaches need to be further studied.
Conclusion and Perspective
As the one of the largest organs, the skeleton shows diverse important functions (bone function). Abnormalities of the skeleton will not only affect bone itself but will also profoundly influence other organs/systems. Dysfunction of bone might significantly affect the maintenance of our health. Improvement of bone function might benefit the function of vital organs and systems.
At present, the major clinical and experimental parameters used to evaluate bone status include imaging (X-ray, CT, MRI), ultrasound, mechanical properties, histology of bone and bone metabolic markers, which mainly focus on bone structure and bone turnover (bone forming and resorbing function). Few physiological parameters of bone such as intramedullary pressure, capillary permeability oxygen and water content, blood flow, PH value, etc. are evaluated in research and especially in clinics, and the effects of these physiological parameters on bone function are not clear. In addition, BDFs, the major mediators regulating bone function, are not well evaluated. Although some BDFs are known, more specific BDFs need to be further discovered using advanced techniques such as genomics, proteomics and metabonomics. Furthermore, more animal and clinical studies also should be conducted to reveal the production and metabolism of BDFs in disease states and to clarify the roles and underlying mechanisms of individual BDF in specific acute or chronic diseases.
To date, the parameters used to evaluate bone status during critical illness are mainly focused on bone mass and metabolic markers. The changes in some known and unknown BDFs, physiological parameters mentioned above, and the function of bone-related cells should be further explored during critical illness.
Previous studies indicate that bone dysfunction is closely related to the prognosis of critical illness, but more evidence is needed to further examine this relationship mechanistically. Many questions remain to be answered. For example, preadmission treatment with bisphosphonates is associated with improved survival of critically ill patients; however, it is unclear if this association is related to the blunted bone resorption due to the action of bisphosphonate itself, or to the increased bone mass, or to the molecular and cellular changes caused by bisphosphonates in other cell types. This mechanism needs to be clarified. It also remains unknown if a significantly increased serum CTX is a surrogate for the outcome of critical illness. In addition, the mechanism by which FGF23 affects the mortality of critically ill patients is not known.
Although only a few studies have shown that improvement of bone function might benefit the outcome of critical illness, considering the availability and efficacy of commonly used anti-osteoporosis drugs in the clinic, modulating bone structure and function might provide a novel strategy to improve the outcome of critical illness. In addition to the current drugs, further approaches with the potential to improve bone function, such as novel anti-resorptive drugs and rehabilitation measures (such as mechanical loading) should be studied for their potential beneficial effects on diseases including critical illness.
Abbreviations
ADHR: autosomal-dominant hypophosphatemic rickets; AML: acute myeloid leukemia; ARDS: acute respiratory distress syndrome; BDFs: bone-derived factors; bALP: bone alkaline phosphatase; CKD: chronic kidney disease; CXCL12: C-X-C motif chemokine ligand 12; CTX: C-telopeptide of type I collagen; EPCs: endothelial progenitor cells; FGF23: fibroblast growth factor 23; FGFR1: fibroblast growth factor receptor 1; G-CSF: granulocyte-colony stimulating factor; GlaOC: γ-carboxylated osteocalcin; GluOC: undercarboxylated osteocalcin; HMGB1: high mobility group box 1 protein; HPCs: hematopoietic progenitor cells; HSCs: haematopoietic stem cells; ICU: intensive care unit; IL: interleukin; IR: insulin receptor; LCN2: lipocalin 2; LPS: lipopolysaccharide; LT-HSCs: long-term HSCs; MMP-9: matrix metalloproteinase 9; MDS: myelodysplastic syndrome; NaPi-2a: type IIa sodium-phosphate co-transporter; NCC: sodium-chloride co-transporter; NTX: N-telopeptide of type 1 collagen; OC: osteocalcin; OPG: osteoprotegerin; P1NP: N-terminal propeptide of type I procollagen; PGE2: prostaglandin E2; PTH: parathyroid hormone; RANKL: receptor activator for nuclear factor-κ b ligand; SCF: stem cell factor; SNAPPE-II: neonatal acute physiology-perinatal extension II; TIO: tumor-induced osteomalacia; TNF-α: tumor necrosis factor α; TRPV5: transient receptor potential vanilloid-5; Vhl: von Hippel-Lindau; VEGF: vascular endothelial growth factor.
Acknowledgements
We thank Meng Xu for modifying the figures.
Consent for publication
All authors read and approved the final manuscript.
Funding
National Natural Science Foundation of China (No.81830075, 81870621), Independent Research Project of the State Key Laboratory of Trauma, Burns and Combined Injury, Third Military Medical University (No. SKLZZ(III)201601), Innovative Research Team in University (IRT1216).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizokami A, Kawakubo-Yasukochi T, Hirata M. Osteocalcin and its endocrine functions. Biochem Pharmacol. 2017;132:1-8
2. An YH, Martin KL. Handbook of Histology Methods for Bone and Cartilage. Totowa, USA: Humana Press. 2003:33-72
3. Boskey AL, Posner AS. Bone structure, composition, and mineralization. The Orthopedic clinics of North America. 1984;15:597-612
4. Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone. 2015;75:144-50
5. Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature. 2012;481:314-20
6. Asada N, Sato M, Katayama Y. Communication of bone cells with hematopoiesis, immunity and energy metabolism. BoneKEy reports. 2015;4:748
7. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone research. 2018;6:16
8. Xie Y, Chen Y, Zhang L, Ge W, Tang P. The roles of bone-derived exosomes and exosomal microRNAs in regulating bone remodelling. Journal of cellular and molecular medicine. 2017;21:1033-41
9. Vasikaran S, Eastell R, Bruyere O, Foldes AJ, Garnero P, Griesmacher A. et al. Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: a need for international reference standards. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2011;22:391-420
10. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841-6
11. Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y. et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nature medicine. 2006;12:657-64
12. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327-34
13. Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood. 2015;125:2678-88
14. Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D. et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457:97-101
15. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836-41
16. Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C. et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150:351-65
17. Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K. et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149-61
18. Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y. et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell stem cell. 2007;1:685-97
19. Tzeng YS, Li H, Kang YL, Chen WC, Cheng WC, Lai DM. Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood. 2011;117:429-39
20. Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L. et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nature immunology. 2002;3:687-94
21. Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. The Journal of experimental medicine. 2008;205:777-83
22. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y. et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635-8
23. Terashima A, Okamoto K, Nakashima T, Akira S, Ikuta K, Takayanagi H. Sepsis-Induced Osteoblast Ablation Causes Immunodeficiency. Immunity. 2016;44:1434-43
24. Xiao M, Wang Y, Tao C, Wang Z, Yang J, Chen Z. et al. Osteoblasts support megakaryopoiesis through production of interleukin-9. Blood. 2017;129:3196-209
25. Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC. et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599-603
26. Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR. et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625-37
27. Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood. 2011;117:1540-9
28. Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. The Journal of experimental medicine. 2012;209:537-49
29. Mansour A, Anginot A, Mancini SJ, Schiff C, Carle GF, Wakkach A. et al. Osteoclast activity modulates B-cell development in the bone marrow. Cell research. 2011;21:1102-15
30. Sato M, Asada N, Kawano Y, Wakahashi K, Minagawa K, Kawano H. et al. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab. 2013;18:749-58
31. Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu X. et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood. 2013;121:930-9
32. Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674-81
33. Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature genetics. 2000;26:345-8
34. Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K. et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179-82
35. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079-86
36. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S. et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500-5
37. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE. et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. The Journal of clinical investigation. 2003;112:683-92
38. Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Current opinion in nephrology and hypertension. 2006;15:437-41
39. Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95:5372-7
40. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T. et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409-14
41. Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R. et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. The EMBO journal. 2014;33:229-46
42. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B. et al. FGF23 regulates renal sodium handling and blood pressure. EMBO molecular medicine. 2014;6:744-59
43. Takashi Y, Fukumoto S. FGF23 beyond Phosphotropic Hormone. Trends in endocrinology and metabolism: TEM. 2018;29:755-67
44. Jacquillet G, Unwin RJ. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflugers Archiv: European journal of physiology. 2018
45. Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R. et al. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS genetics. 2013;9:e1003975
46. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M. et al. The parathyroid is a target organ for FGF23 in rats. The Journal of clinical investigation. 2007;117:4003-8
47. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y. et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429-35
48. Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A. et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296-308
49. Poser JW, Price PA. A method for decarboxylation of gamma-carboxyglutamic acid in proteins. Properties of the decarboxylated gamma-carboxyglutamic acid protein from calf bone. The Journal of biological chemistry. 1979;254:431-6
50. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C. et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456-69
51. D'Amelio P, Sassi F, Buondonno I, Spertino E, Tamone C, Piano S. et al. Effect of intermittent PTH treatment on plasma glucose in osteoporosis: A randomized trial. Bone. 2015;76:177-84
52. Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309-19
53. Brennan-Speranza TC, Henneicke H, Gasparini SJ, Blankenstein KI, Heinevetter U, Cogger VC. et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. The Journal of clinical investigation. 2012;122:4172-89
54. O'Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA. et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835-41
55. Frey JL, Li Z, Ellis JM, Zhang Q, Farber CR, Aja S. et al. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Molecular and cellular biology. 2015;35:1979-91
56. Yao Q, Yu C, Zhang X, Zhang K, Guo J, Song L. Wnt/beta-catenin signaling in osteoblasts regulates global energy metabolism. Bone. 2017;97:175-83
57. Yoshikawa Y, Kode A, Xu L, Mosialou I, Silva BC, Ferron M. et al. Genetic evidence points to an osteocalcin-independent influence of osteoblasts on energy metabolism. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2011;26:2012-25
58. Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature. 2017;543:385-90
59. Weivoda MM, Oursler MJ. Developments in sclerostin biology: regulation of gene expression, mechanisms of action, and physiological functions. Current osteoporosis reports. 2014;12:107-14
60. Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE. et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E11238-E47
61. Fulzele K, Lai F, Dedic C, Saini V, Uda Y, Shi C. et al. Osteocyte-Secreted Wnt Signaling Inhibitor Sclerostin Contributes to Beige Adipogenesis in Peripheral Fat Depots. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2017;32:373-84
62. Dirckx N, Tower RJ, Mercken EM, Vangoitsenhoven R, Moreau-Triby C, Breugelmans T. et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. The Journal of clinical investigation. 2018;128:1087-105
63. Obri A, Khrimian L, Karsenty G, Oury F. Osteocalcin in the brain: from embryonic development to age-related decline in cognition. Nature reviews Endocrinology. 2018;14:174-82
64. Oury F, Khrimian L, Denny CA, Gardin A, Chamouni A, Goeden N. et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell. 2013;155:228-41
65. Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE. et al. Endocrine regulation of male fertility by the skeleton. Cell. 2011;144:796-809
66. Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galan-Diez M. et al. Osteocalcin Signaling in Myofibers Is Necessary and Sufficient for Optimum Adaptation to Exercise. Cell metabolism. 2016;23:1078-92
67. Mera P, Laue K, Wei J, Berger JM, Karsenty G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Molecular metabolism. 2016;5:1042-7
68. Sassoli C, Pini A, Chellini F, Mazzanti B, Nistri S, Nosi D. et al. Bone marrow mesenchymal stromal cells stimulate skeletal myoblast proliferation through the paracrine release of VEGF. PloS one. 2012;7:e37512
69. Mo C, Romero-Suarez S, Bonewald L, Johnson M, Brotto M. Prostaglandin E2: from clinical applications to its potential role in bone- muscle crosstalk and myogenic differentiation. Recent patents on biotechnology. 2012;6:223-9
70. Siris ES, Miller PD, Barrett-Connor E, Faulkner KG, Wehren LE, Abbott TA. et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment. Jama. 2001;286:2815-22
71. Kanis JA, McCloskey EV, Johansson H, Cooper C, Rizzoli R, Reginster JY. et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2013;24:23-57
72. Chang B, Quan Q, Li Y, Qiu H, Peng J, Gu Y. Treatment of Osteoporosis, with a Focus on 2 Monoclonal Antibodies. Medical science monitor: international medical journal of experimental and clinical research. 2018;24:8758-66
73. Zhou R, Zhou H, Rui L, Xu J. Bone loss and osteoporosis are associated with conversion from mild cognitive impairment to Alzheimer's disease. Current Alzheimer research. 2014;11:706-13
74. Campos-Obando N, Castano-Betancourt MC, Oei L, Franco OH, Stricker BH, Brusselle GG. et al. Bone mineral density and chronic lung disease mortality: the rotterdam study. The Journal of clinical endocrinology and metabolism. 2014;99:1834-42
75. Johansson H, Oden A, Kanis J, McCloskey E, Lorentzon M, Ljunggren O. et al. Low bone mineral density is associated with increased mortality in elderly men: MrOS Sweden. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2011;22:1411-8
76. Nordstrom A, Eriksson M, Stegmayr B, Gustafson Y, Nordstrom P. Low bone mineral density is an independent risk factor for stroke and death. Cerebrovascular diseases. 2010;29:130-6
77. Qu X, Huang X, Jin F, Wang H, Hao Y, Tang T. et al. Bone mineral density and all-cause, cardiovascular and stroke mortality: a meta-analysis of prospective cohort studies. International journal of cardiology. 2013;166:385-93
78. Bolland MJ, Grey AB, Gamble GD, Reid IR. Effect of osteoporosis treatment on mortality: a meta-analysis. The Journal of clinical endocrinology and metabolism. 2010;95:1174-81
79. Palagano E, Menale C, Sobacchi C, Villa A. Genetics of Osteopetrosis. Current osteoporosis reports. 2018;16:13-25
80. Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N. et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506:240-4
81. Weiss AJ, Zaidi M, Mechanick JI. Pharmacological and Biological Therapies for Metabolic Bone Disease in Critical Illness: An Integrative Physiology Approach. Current Drug Therapy. 2010;5:48-57
82. Pfortmueller CA, Meisel C, Fux M, Schefold JC. Assessment of immune organ dysfunction in critical illness: utility of innate immune response markers. Intensive care medicine experimental. 2017;5:49
83. Russell JA. Management of sepsis. The New England journal of medicine. 2006;355:1699-713
84. Langouche L, Mesotten D, Vanhorebeek I. Endocrine and metabolic disturbances in critical illness: relation to mechanisms of organ dysfunction and adverse outcome. Verhandelingen - Koninklijke Academie voor Geneeskunde van Belgie. 2010;72:149-63
85. Zhang Y, Du X, Chen L. Is there a relationship between bone and critical illness? Medical Hypotheses. 2011;76:615-7
86. Owen HC, Vanhees I, Gunst J, Van Cromphaut S, Van den Berghe G. Critical illness-induced bone loss is related to deficient autophagy and histone hypomethylation. Intensive care medicine experimental. 2015;3:52
87. Owen HC, Vanhees I, Solie L, Roberts SJ, Wauters A, Luyten FP. et al. Critical illness-related bone loss is associated with osteoclastic and angiogenic abnormalities. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27:1541-52
88. Griffith DM, Walsh TS. Bone loss during critical illness: a skeleton in the closet for the intensive care unit survivor? Critical care medicine. 2011;39:1554-6
89. Orford NR, Bailey M, Bellomo R, Pasco JA, Cattigan C, Elderkin T. et al. The association of time and medications with changes in bone mineral density in the 2 years after critical illness. Critical care. 2017;21:69
90. Orford NR, Lane SE, Bailey M, Pasco JA, Cattigan C, Elderkin T. et al. Changes in Bone Mineral Density in the Year after Critical Illness. American journal of respiratory and critical care medicine. 2016;193:736-44
91. Nierman DM, Mechanick JI. Bone hyperresorption is prevalent in chronically critically ill patients. Chest. 1998;114:1122-8
92. Nierman DM, Mechanick JI. Biochemical response to treatment of bone hyperresorption in chronically critically ill patients. Chest. 2000;118:761-6
93. Gavala A, Makris K, Korompeli A, Myrianthefs P. Evaluation of Bone Metabolism in Critically Ill Patients Using CTx and PINP. BioMed research international. 2016;2016:1951707
94. Van den Berghe G, Van Roosbroeck D, Vanhove P, Wouters PJ, De Pourcq L, Bouillon R. Bone turnover in prolonged critical illness: effect of vitamin D. The Journal of clinical endocrinology and metabolism. 2003;88:4623-32
95. Orford N, Cattigan C, Brennan SL, Kotowicz M, Pasco J, Cooper DJ. The association between critical illness and changes in bone turnover in adults: a systematic review. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2014;25:2335-46
96. Zhang Y, Xue C, Zhu T, Du X, Su N, Qi H. et al. Serum bone alkaline phosphatase in assessing illness severity of infected neonates in the neonatal intensive care unit. International journal of biological sciences. 2012;8:30-8
97. Yang J, Su N, Du X, Chen L. Gene expression patterns in bone following lipopolysaccharide stimulation. Cellular & molecular biology letters. 2014;19:611-22
98. Guo C, Yang RJ, Jang K, Zhou XL, Liu YZ. Protective Effects of Pretreatment with Quercetin Against Lipopolysaccharide-Induced Apoptosis and the Inhibition of Osteoblast Differentiation via the MAPK and Wnt/beta-Catenin Pathways in MC3T3-E1 Cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;43:1
99. Ito N, Wijenayaka AR, Prideaux M, Kogawa M, Ormsby RT, Evdokiou A. et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Molecular and cellular endocrinology. 2015;399:208-18
100. Leaf DE, Siew ED, Eisenga MF, Singh K, Mc Causland FR, Srivastava A. et al. Fibroblast Growth Factor 23 Associates with Death in Critically Ill Patients. Clinical journal of the American Society of Nephrology: CJASN. 2018;13:531-41
101. Atalan HK, Gucyetmez B. Serum Vitamin D Level at ICU Admission and Mortality. Turkish journal of anaesthesiology and reanimation. 2017;45:193-6
102. Van den Berghe G, Wouters P, Weekers F, Mohan S, Baxter RC, Veldhuis JD. et al. Reactivation of pituitary hormone release and metabolic improvement by infusion of growth hormone-releasing peptide and thyrotropin-releasing hormone in patients with protracted critical illness. The Journal of clinical endocrinology and metabolism. 1999;84:1311-23
103. Via MA, Potenza MV, Hollander J, Liu X, Peng Y, Li J. et al. Intravenous ibandronate acutely reduces bone hyperresorption in chronic critical illness. Journal of intensive care medicine. 2012;27:312-8
104. Lee P, Ng C, Slattery A, Nair P, Eisman JA, Center JR. Preadmission Bisphosphonate and Mortality in Critically Ill Patients. The Journal of clinical endocrinology and metabolism. 2016;101:1945-53
105. Xue C, Yang B, Zhou C, Dai B, Liu Y, Mao Z. et al. Fibroblast Growth Factor 23 Predicts All-Cause Mortality in a Dose-Response Fashion in Pre-Dialysis Patients with Chronic Kidney Disease. American journal of nephrology. 2017;45:149-59
106. Rossaint J, Oehmichen J, Van Aken H, Reuter S, Pavenstadt HJ, Meersch M. et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. The Journal of clinical investigation. 2016;126:962-74
107. Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S. et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney international. 2016;90:985-96
108. Yang J, Shah R, Robling AG, Templeton E, Yang H, Tracey KJ. et al. HMGB1 is a bone-active cytokine. Journal of cellular physiology. 2008;214:730-9
109. Yu K, Ma Y, Li X, Wu X, Liu W, Li X. et al. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to up-regulate RANKL gene expression in MLO-Y4 cells. Cell biology international. 2017;41:84-92
110. Nagao M, Tanabe N, Manaka S, Naito M, Sekino J, Takayama T. et al. LIPUS suppressed LPS-induced IL-1alpha through the inhibition of NF-kappaB nuclear translocation via AT1-PLCbeta pathway in MC3T3-E1 cells. Journal of cellular physiology. 2017;232:3337-46
111. Zhang Y, Su N, Luo F, Wen X, Tang Y, Yang J. et al. Deletion of Fgfr1 in osteoblasts enhances mobilization of EPCs into peripheral blood in a mouse endotoxemia model. International journal of biological sciences. 2014;10:1064-71
Author contact
![]() Corresponding author: Lin Chen, MD, PhD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991. Fax: 86-23-68702991. E-mail: linchen70com
Corresponding author: Lin Chen, MD, PhD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991. Fax: 86-23-68702991. E-mail: linchen70com