Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):788-799. doi:10.7150/ijbs.30677 This issue Cite
Research Paper
Farrerol Ameliorates APAP-induced Hepatotoxicity via Activation of Nrf2 and Autophagy
Lidong Wang, Wei Wei, Qingfei Xiao, Huahong Yang, Xinxin Ci ![]()
Institute of Translational Medicine, The First Hospital of Jilin University, Changchun 130001, China
Received 2018-10-14; Accepted 2018-12-22; Published 2019-1-29
Abstract

Farrerol has been shown to have antioxidative potential via Nrf2 activation, which in turn is involved in the prevention of hepatotoxicity. The purpose of the current study was to explore the protective effect of farrerol against acetaminophen-induced hepatotoxicity and its underlying mechanisms. Mice were used to evaluate the hepatoprotective effect of farrerol on liver injury induced by acetaminophen in vivo. HepG2 cells were utilized to further determine the functional role and mechanisms by which Nrf2 and autophagy are involved in the hepatoprotective effect of farrerol in vitro. We found that treatment with farrerol leads to a significant reduction in acetaminophen-induced hepatotoxicity by decreasing mortality, histopathological liver changes, and ALT and AST levels. Furthermore, farrerol effectively suppressed mitochondrial dysfunction by reducing JNK phosphorylation, Bax mitochondrial translocation, AIF and cytochrome c release. Further investigations revealed that the activation of Nrf2 and the induction of autophagy via the AMPK/AKT pathway by farrerol contributed to its hepatoprotective activity in vitro. In addition, farrerol inhibited acetaminophen-induced the mortality and histopathological changes in WT mice were evidently alleviated but not abrogated in Nrf2 -/- mice, which attributed to the induction of autophagy. Together, farrerol has protective potential against acetaminophen-induced hepatotoxicity which may be associated with activation of Nrf2 and autophagy.
Keywords: Farrerol, APAP, Hepatotoxicity, Nrf2, Autophagy
1. Introduction
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug that can be easily obtained over-the-counter. Acute liver injury induced by APAP overdose is the leading cause of drug-induced acute liver failure in many developed countries 1. Mitochondrial oxidative stress and mitochondrial dysfunction are considered to be the predominant cellular processes that occur as a result of APAP hepatotoxicity 2. Accordingly, the inhibition of oxidative stress and mitochondrial dysfunction may play an essential role in attenuating APAP-induced acute liver injury. N-acetyl cysteine, which is a known antioxidant, is also known to be an effective treatment for APAP-induced acute liver injury when it is given at an early stage. However, the narrow therapeutic window and some side-effects limit its use 3. Hence, novel therapeutic approaches that can protect against APAP-induced acute liver injury are clearly needed.
Natural products, including herbal medicines, have contributed significantly to drug discovery, as they have many advantages over conventional chemical compound-based medications, such as fewer side effects, lower rates of toxicity with prolonged use, variable bioavailability, and biological activities 4. In recent years, there have been intensive studies demonstrating the protective effects of natural products against APAP-induced hepatotoxicity due to their multiple actions in inflammation, oxidant/antioxidant balance and damage response 5. Key mechanisms of APAP-induced liver injury, including APAP metabolism, oxidative stress, endoplasmic reticulum stress, autophagy, microcirculatory dysfunction and sterile inflammation have been shown to be regulated by natural products 6. Therefore, we propose that natural products can prevent APAP-induced hepatotoxicity by targeting multiple signaling pathways.
In a widely understood mechanism, APAP-induced hepatotoxicity is associated with overproduction of the reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI), which contributes to the depletion of glutathione (GSH) and the formation of ROS. This triggers oxidative stress and results in mitochondrial dysfunction, hepatocyte necrosis, and liver injury 7. To counteract the environmental stress caused by ROS, cells have developed dynamic responses that serve to maintain cellular redox homeostasis and reduce oxidative damage. This is accomplished through a series of antioxidant molecules and detoxifying enzymes. The Nrf2/ARE pathway is the major nuclear transcription factor that responds to reactive species and redox potentials by activating phase II detoxification enzymes 8. In the last few decades, several studies have demonstrated the benefits of using natural products to counteract oxidative stress via the Nrf2/ARE pathway 9. Farrerol, a new type of 2,3-dihydro-flavonoid, has been isolated from rhododendron. Our previous study has showed that farrerol reduced oxidative stress by activating Nrf2 and thereby inducing HO-1 expression 10. Given the importance of oxidative stress in APAP-induced hepatotoxicity, we speculate that farrerol, as an Nrf2 activator, might protect against this toxicity.
Although the detrimental mechanisms induced by APAP have been well studied, little is known about the cellular adaptive mechanisms that may attenuate APAP-induced liver injury. Cells may protect themselves by removing damaged mitochondria using a mechanism called autophagy. There is accumulating evidence indicating that pharmacological activation of autophagy protects against APAP- induced liver injury 11. This study aims to investigate the protective effects of farrerol on APAP-induced liver injury and to determine whether this is accomplished via the regulation of Nrf2 and the autophagy signal pathway.
2. Materials and methods
2.1 Reagents and chemical
Farrerol, ((S)-2,3-dihydro-5,7-dihydroxy-2-(4-hydroxyphenyl)-6,8-dimethyl-4-benzopyrone, analytical grade, purity P 98%) was obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). 3-(4,5-Dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT), LY294002 (Akt inhibitor) and DCFH-DA were purchased from the Sigma Chemical Co. (St. Louis, MO, USA). Antibodies directed against Nrf2, HO-1, NQO1, GCLC, and KEAP1 were purchased from Abcam (Cambridge, MA, USA). Anti-phospho-c-Jun NH2-terminal kinase (JNK) antibody and β-actin were obtained from SUNGENE BIOTECH (Tianjin, China). Antibodies against Cytochrome c, Bax, Bcl-2, LC3, Atg5, AMPK, phosphor-AMPK, AKT, phosphor-AKT, phospho-extracellular signal-regulated kinase (ERK), ERK, JNK, phospho-p38 as well as p38, were purchased from Cell Signaling (Boston, MA, USA). The horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgG were purchased from proteintech (Boston, MA, USA). ALT, AST, MDA, MPO, GSH, and SOD test kits were supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Additionally, Compound c (AMPK inhibitor) and chloroquine were obtained from MedChem Express (Monmouth, NJ, USA).
2.2 Animals
Female B6 mice, weighing approximately 18-20 g, were purchased from Liaoning Changsheng Technology Industrial, Co., Ltd. (Certificate SCXK2010-0001; Liaoning, China). Nrf2-deficient mice (Nrf2-/-) with a B6 background were provided by The Jackson Laboratory (Bar Harbor, ME, United States). All animals were raised under SPF-condition and received food and water ad libitum. All animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals, which was published by the U.S. National Institute of Health. This study was reviewed and approved by the Animal Welfare and Research Ethics Committee at Jilin University.
2.3 Experimental design and animal procedures
For detecting the preventive effect of farrerol against APAP-induced hepatotoxicity, mice were randomly divided into the following four groups (n=10): vehicle control group (0.5% DMSO), farrerol only group (40 mg/kg, dissolved in 0.5% DMSO), APAP only group (900 mg/kg or 400 mg/kg) and APAP (900 mg/kg or 400 mg/kg) + farrerol (40 mg/kg, dissolved in 0.5% DMSO) group. The mice were administered farrerol (40 mg/kg) i.p. injections two times, 12 h and 1 h before the injection of APAP (900 mg/kg). The survival rates of the animals were monitored every 8 h for 2 days. In addition, the mice were administered farrerol (40 mg/kg) i.p. injections two times, 12 h and 1 h before the injection of APAP (400 mg/kg). After treatment with APAP (400 mg/kg) for 6 h, the mice were euthanized, their livers were harvested and serum was collected.
For detecting the therapeutic effect of farrerol against APAP-induced hepatotoxicity, mice were randomly divided into the following four groups (n=10): vehicle control group (0.5% DMSO), farrerol (40 mg/kg, dissolved in 0.5% DMSO) only group, APAP only (900 mg/kg) group and APAP (900 mg/kg) + farrerol (40 mg/kg, dissolved in 0.5% DMSO) group. The mice were treated with APAP (900 mg/kg) for 1 h and subsequently treated with farrerol (40 mg/kg) to observe the mortality rate.
For detecting the critical role of Nrf2 involved in the preventive effect of farrerol against APAP-induced hepatotoxicity, wild-type or Nrf2 deficient mice were randomly divided into three groups, respectively (n=10): vehicle control group (0.5% DMSO), APAP only (900 mg/kg or 400 mg/kg) group and APAP (900 mg/kg or 400mg/kg) + farrerol (40 mg/kg, dissolved in 0.5% DMSO) group. The mice were administered farrerol (40 mg/kg) i.p. injections two times, 12h and 1h before the injection of APAP (900 mg/kg). Survival rates of animals were monitored every 8 h for 2 days. In addition, the mice were administered farrerol (40 mg/kg) i.p. injections two times, 12h and 1h before the injection of APAP (400 mg/kg). After treatment with APAP (400 mg/kg) for 6 h, mice were euthanized, harvested livers and collected serum.
2.4 Histopathology assessment
Histopathological evaluations were performed on mice. Fresh liver tissues were removed and fixed in Neutral-buffered formalin, embedded in paraffin and cut into 3 mm sections. The tissue sections were subsequently stained with hematoxylin and eosin (H & E) for pathological analysis.
2.5 Biochemical assay
Mice blood samples were kept at 4℃ for 24h, then collected after centrifugation at 1500g for 10min. The serum ALT and AST activities were measured using an assay kit according to the manufacturer's instructions. Mice liver tissues were homogenized and dissolved in extraction buffer to analyze the GSH, MPO, MDA and SOD levels using commercial kits. All results were normalized by the total protein concentration in each sample.
2.6 Cell Culture
The HepG2 cell line, obtained from the China Cell Line Bank (Beijing, China), were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS, 3 mM glutamine, 100 U/mL of penicillin, 100 U/mL of streptomycin and at 37 ℃ in a humidified atmosphere containing 5% CO2. In all experiments, cells were allowed to acclimate for 24 h before any treatments.
2.7 MTT assay for cell viability
Cell viability was measured by MTT assay in accordance with the manufacturer's instructions. HepG2 cells were seeded in 96-well plates at 1 × 104 cells/well and treated with farrerol (20μM) for 1 h, then exposed to H2O2 (300 μM) for 18 h. Subsequently, 20 μl of 5 mg/mL MTT was added to the cells and incubated for another 4 h. Cell-free supernatants were removed, and DMSO was added to resolve the formazans. The absorbance was measured at 570 nm using a microplate reader.
2.8 CRISPR/Cas9 knockout
HepG2 cells were grown in 24-well plates at 5 × 104 cells/well for 16 h, then transfected with a plasmid expressing Cas9 with Nrf2 sgRNA and a plasmid carrying a puromycin resistant gene using Viafect transfection reagent (Promega). After 36 h, 2 mg/ml puromycin was added to select cells. Two days later, live cells were seeded in 96-well plates at a density of 1 cell per well. Gene editing efficiency after clonal expansion was determined by immunoblotting. DNA sequencing was employed to verify that gene editing was successful.
2.9 Intracellular ROS Measurement
Cells (1 × 104 cells/well) were seeded into 96-well plates for 24 h and recovered in serum-free DMEM for another 6 h. Then, the cells were treated with different dosages of farrerol (5, 10 or 20 μM) for 18h, and H2O2 (300 μM) was added to each well for additional 5min. Next, the cells were incubated with 50 mM of DCFH-DA for 30 min and DCF fluorescence intensities were assessed by a multi-detection reader at excitation and emission wavelengths of 485 and 535 nm, respectively.
2.10 Immunoblot analyses
Liver tissues were collected 6h after APAP challenge, added to lysis buffer and homogenized. cells were plated into 6-well plates and pretreated with or without inhibitor for 18 h, followed by treatment with different concentrations of farrerol for the indicated durations. Total protein samples were prepared according to a standard protocol. protein concentrations were measured using a BCA protein assay kit (Beyotime, China). Equal amounts of proteins were separated by 10% SDS-poly-acrylamide gel, electrophoretically transferred to PVDF membranes and blocked with 5% BSA. Then, the membranes were washed and probed with corresponding primary antibodies and subsequently by secondary antibodies. Protein bands were visualized by ECL. The gray densities of protein bands were normalized by employed β-actin density as an internal control.
2.11 Statistical Analysis
Data were expressed as the means ± SEM and analyzed using SPSS19.0 (IBM). Comparisons between experimental groups were conducted using one-way ANOVA. Statistical significance was defined as p* < 0.05 or p** < 0.01.
3. Results
3.1 Farrerol alleviated acute liver failure (ALF) induced by APAP exposure in vivo
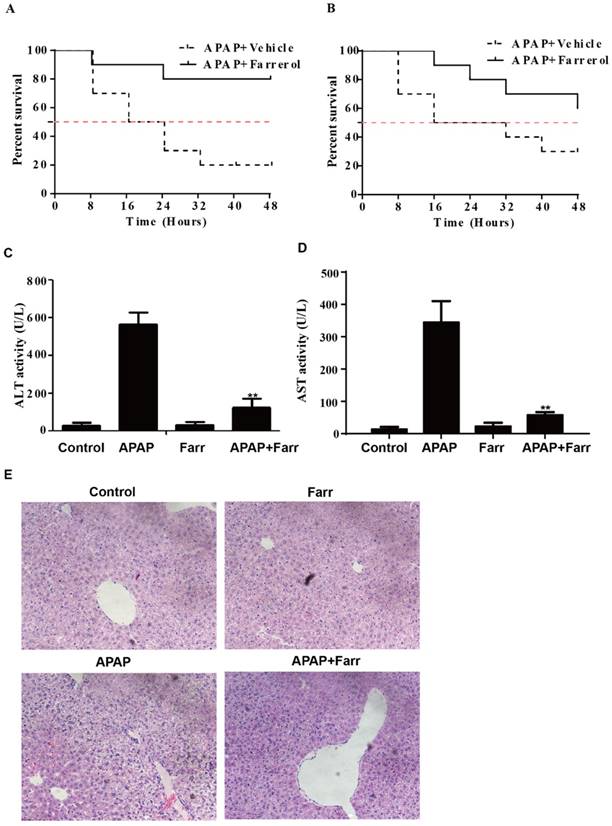
For detecting the preventive and therapeutic effect of farrerol against APAP-induced hepatotoxicity, we first assessed the effect of farrerol on survival rates in the setting of APAP-induced liver injury. As shown in Fig. 1A and 1B, the survival rate of the APAP (900 mg/kg) plus farrerol (40 mg/kg)-treated mice was much higher than that of the APAP (900 mg/kg)-treated mice. Then, we evaluated ALT and AST levels in the serum and found that APAP (400 mg/kg) dramatically increased the serum AST and ALT levels, whereas farrerol (40 mg/kg) reduced these (Fig. 1C and 1D). Furthermore, the results of the histological evaluation of the liver verify the protective effects of farrerol, showing that it effectively relieved APAP-induced liver damages, such as hepatocyte necrosis and hemorrhage (Fig. 1E).
Effects of farrerol on APAP-induced mortality and liver injury. (A) Mice were administered farrerol (40 mg/kg) i.p. two times at an interval of 12 h. The survival rate of mice who received the last dose of farrerol (40 mg/kg) at 1 h, followed by exposure to a lethal dose of APAP (900 mg/kg) is shown. The data are expressed as the percentage of surviving mice at each point in time, where n=10 in each group. (B) Mice were treated with APAP (900 mg/kg) for 1 h, followed by treatment with farrerol (40 mg/kg) to determine the mortality rate. The data are expressed as the percentage of surviving mice at each point in time point, n=10 in each group. (C, D) At 1 h after the last dose of farrerol, APAP (400 mg/kg) was administered for 3 h (n=10/group). Then, the serum levels of ALT and AST were measured, as shown. (E) Representative images of H & E staining of liver tissues. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the APAP group.
3.2 Farrerol ameliorated APAP-induced oxidative stress in vivo
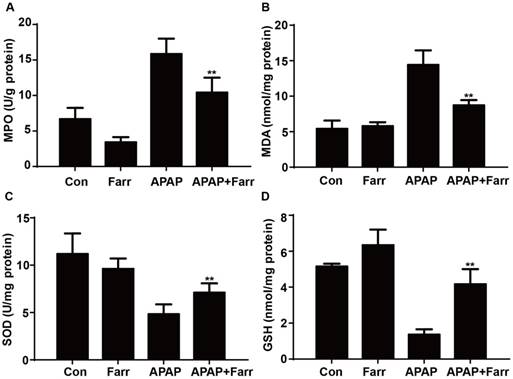
Next, we examined the effect of farrerol on APAP-induced oxidative stress. Our results showed that farrerol pretreatment significantly decreased MPO and MDA levels (Fig. 2A and B) and increased GSH and SOD levels (Fig. 2C and D) which play vital roles in protecting against APAP-induced oxidative stress.
Effect of farrerol treatment on APAP-mediated oxidative stress in mice. Mice were administered farrerol (40 mg/kg) i.p. two times. At 1 h after the last dose of farrerol, the mice were administered APAP (400 mg/kg) for 3 h. (A, B) The effects of farrerol on MPO and MDA activities in liver homogenates are shown. (C, D) The effects of farrerol on the SOD and GSH levels are shown. All of the data represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the APAP group.
3.3 Farrerol alleviated APAP-induced mitochondrial dysfunction and upregulated antioxidant signaling pathways in vivo
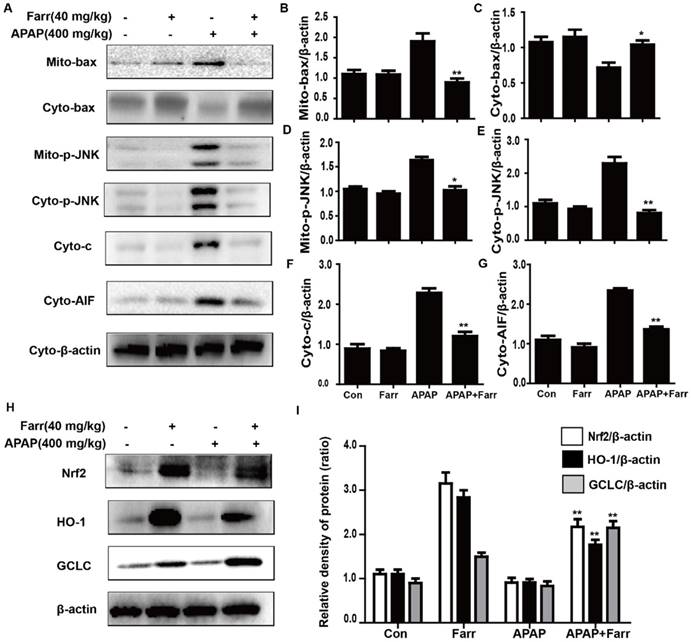
Mitochondrial dysfunction and cell apoptosis were regarded as two crucial factors in APAP-induced liver injury. As shown in Fig. 3A-G, APAP (400 mg/kg) significantly induced Bax mitochondrial translocation, the release of AIF and cytochrome c, and JNK activation; these effects were effectively inhibited by farrerol pretreatment (40 mg/kg). Furthermore, we evaluated the effects of farrerol on the activation of Nrf2. As shown in Fig. 3H-I, the APAP challenge mildly suppressed Nrf2, HO-1 and GCLC protein expression, whereas these changes were completely reversed by farrerol pretreatment.
Effect of farrerol treatment on APAP-induced mitochondrial dysfunction and antioxidant signaling pathways. (A-G) Immunoblot analysis was performed to determine the levels of Bax, AIF, cytochrome c and JNK in mitochondria isolated from liver tissues harvested at 3 h post-APAP injection. (H-I) Liver tissue lysates were examined for Nrf2, HO-1 and GCLC by western blot analysis. All of the data represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the APAP group.
3.4 Farrerol enhanced cell viability and alleviated oxidative stress in vitro
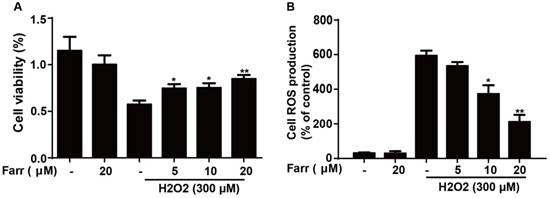
To explore the hepatoprotective potential of farrerol in vitro, we employed H2O2 to induce toxicity in HepG2 cells. The results indicated that farrerol pretreatment markedly suppressed cytotoxicity caused by H2O2 in a dose-dependent manner (Fig. 4A). Furthermore, farrerol pretreatment markedly decreased H2O2-induced ROS generation in a dose-dependent manner (Fig. 4B).
Effect of farrerol on H2O2-induced cell viability and oxidative stress. (A) HepG2 cells were treated with increasing concentrations of farrerol (5, 10 and 20 μM) for 1 h, followed by treatment with H2O2 (300 μM) for 24 h, and then cell viability was evaluated by MTT assay. (B) HepG2 cells were cultured with farrerol (5, 10 and 20 μM) for 18 h, followed by exposure to H2O2 (300 μM) for 5 min, and then the cells were harvested and were stained with 50 mM of DCFH-DA for 30 min to induce ROS generation. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the H2O2 group.
3.5 Farrerol activated Nrf2 and antioxidant enzyme expression in vitro
Farrerol significantly upregulated Nrf2 and antioxidant enzyme expression including HO-1, GCLC and GCLM in vitro in a dose-dependent manner (Fig. 5A) and in a time-dependent manner (Fig. 5B), respectively. Next, we examined whether the upregulation of HO-1, GCLC and GCLM expression was mediated by Nrf2 using HepG2 Nrf2-/- cells. As shown in Fig. 5C, Nrf2, HO-1, GCLC and GCLM protein expression induced by farrerol was markedly suppressed in Nrf2-/- cells. Furthermore, the protective effect of farrerol against H2O2-induced cell death in the HepG2 Nrf2-/- cells was lower than that in the HepG2 WT cells (Fig. 5D).
Effect of Farrerol on Nrf2 and antioxidant enzyme expression. (A-B) HepG2 cells were treated with farrerol for the indicated durations, and then the protein levels were examined by western blot analysis. (C) HepG2 WT and Nrf2-/- cells were cultured with or without farrerol (20 μM) for 18 h, and then immunoblotting was performed to detect Nrf2, HO-1, GCLC and GCLM expression. (D) HepG2 cells and Nrf2-/- cells were pretreated with farrerol for 1 h, followed by treatment with H2O2 (300 μM) for 24 h, and then cell viability was assessed by MTT assay. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the control or H2O2 group.
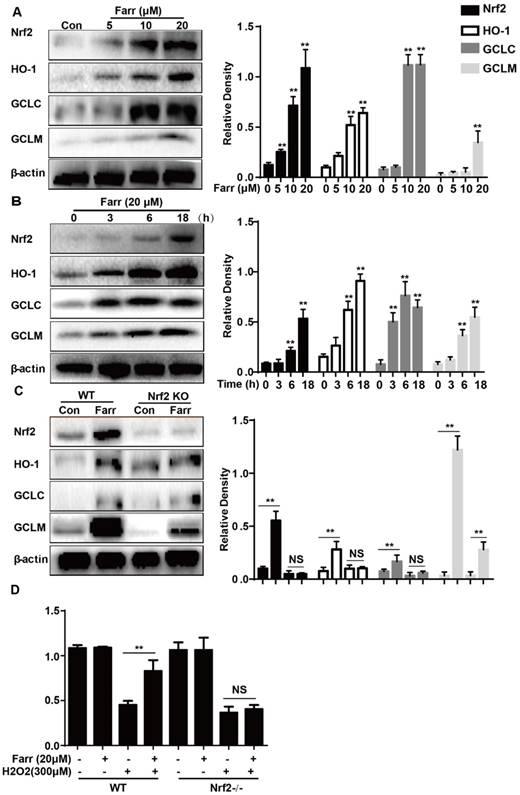
3.6 Farrerol enhanced cell viability through an AMPK/AKT/Nrf2-dependent mechanism
We further examined the effect of farrerol on PI3K and AKT phosphorylation and found that farrerol significantly increased AKT and PI3K phosphorylation in a dose-dependent manner (Fig 6A). AMPK acts upstream of AKT and can be activated by farrerol. In addition, preincubation of cells with LY294002 (an AKT inhibitor) or compound c (an AMPK inhibitor) prevented the farrerol-induced Nrf2/ HO-1 activation and cytoprotection (Fig 6C-6H).
The effect of farrerol on AMPK/AKT activation is necessary for Nrf2 activity and cytoprotection. (A-B) HepG2 cells were incubated with farrerol for 18 h, and then the expression of AMPK, AKT and PI3K were detected by immunoblotting using specific antibodies. (C-D) Cells were treated with 20 μM LY 294002 for 1 h, followed by treatment with farrerol for 18 h, and then cell lysates were immunoblotted to assess Akt, Nrf2 and HO-1 expression. (E-F) Cells were treated with 3 μM compound c for 1 h, followed by treatment with farrerol for 18 h, and then cell lysates were immunoblotted to assess AMPK, Akt, Nrf2 and HO-1 expression. (G-H) Cells were preincubated with LY 294002 (20μM) or compound c (3 μM) for 1 h, incubated with farrerol for another 1 h, and then incubated with H2O2 (300μM) for 24 h. Subsequently, cell viability was assessed by MTT assay. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the control or H2O2 group.
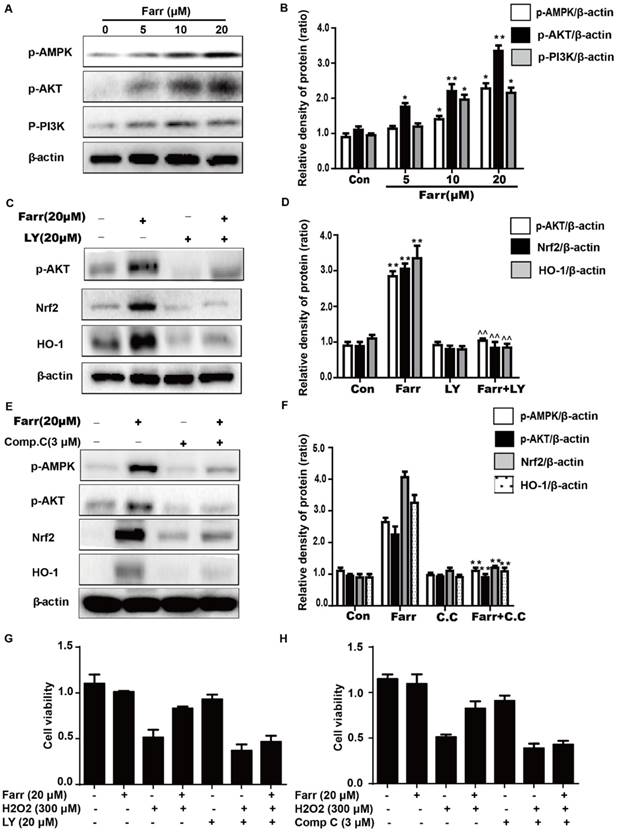
3.7 Farrerol exhibited hepatoprotective effects, which may be associated with autophagy activation in vitro
We next explored whether autophagy was involved in the hepatoprotective activity of farrerol. Autophagic markers, LC3, Atg5 and Atg7, were analyzed by western blot. As shown in Fig 7A, treatment with farrerol led to a significant increase in LC3Ⅱ, Atg5 and Atg7 protein expression in a dose-dependent manner in vitro. Moreover, preincubation of cells with chloroquine (CQ, an autophagy inhibitor) prevented the farrerol-induced LC3, Atg5 and Atg7 activation and cytoprotection (Fig 7C-7E).
The activation of autophagy by farrerol is necessary for cytoprotection. (A-B) HepG2 cells were incubated with farrerol for 18 h, and then the expression of Atg5, Atg7 and LC3 were detected by immunoblotting by using specific antibodies. (C-D) Cells were treated with 20 μM CQ for 1 h, followed by treatment with farrerol for 18 h, and then cell lysates were immunoblotted to assess Atg5, Atg7 and LC3 expression. (E) Cells were preincubated with CQ (20 μM) for 1 h, incubated with farrerol for another 1 h, and then incubated with H2O2 (300 μM) for 24 h. Subsequently, cell viability was assessed by MTT assay. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the control or H2O2 group.
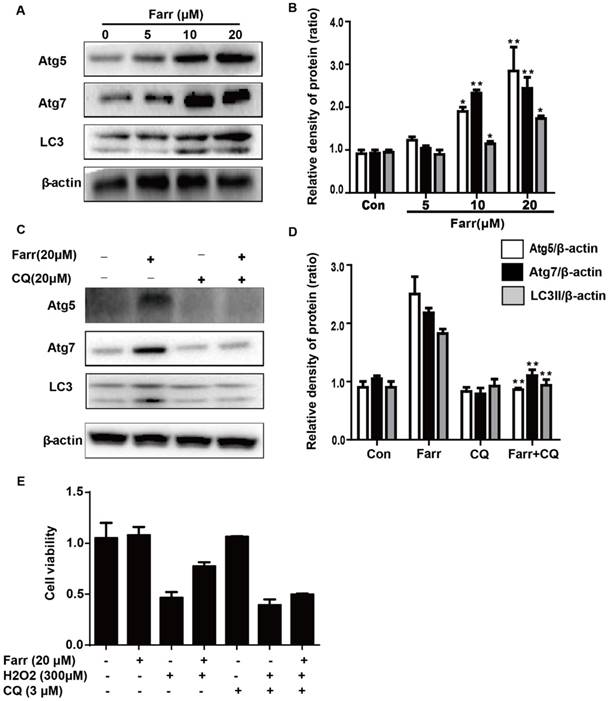
3.8 The suppressive effects of farrerol on APAP-induced liver injury were dependent on Nrf2 and autophagy
The dependency of the hepatoprotective role of farrerol on Nrf2 was further assessed in WT and Nrf2-/- mice. First, as shown in Fig 8A, Nrf2-/- mice appeared to be more vulnerable to APAP-induced lethality than WT mice, with survival rates being 0% and 20%, respectively. Second, for WT mice, the final survival rate was 20% (vehicle group) versus 80% (farrerol-treated group). Meanwhile, for Nrf2-/- mice, the final survival rate was 0 (vehicle group) versus 30% (farrerol-treated group). We further examined serum ALT and AST levels as well as histopathological changes in the liver. As shown in Fig 8B-8D, the inhibitory effects of farrerol were alleviated but not abolished in Nrf2-/- mice. Farrerol still downregulated JNK activation in Nrf2-/- mice, which may be attributed to activation of LC3, Atg5 and Atg7 (Fig 8E).
The suppressive effects of farrerol on APAP-induced liver injury depended on Nrf2 and autophagy. (A) The survival rates of the WT and Nrf2 -/- mice (n = 10/group) were observed within 48 h after APAP exposure. (B-C) Serum samples were collected from the mice after exposure to APAP (400 mg/kg) for 6 h in order to measure the ALT and AST levels. (D) Livers from each experimental group were processed for histological evaluation at 6 h after the APAP (400 mg/kg) challenge. (E) Livers from each experimental group were immunoblotted to assess Nrf2, JNK, Atg5, Atg7 and LC3 expression. All of the data shown represent the average from three independent experiments. *p < 0.05 and **p < 0.01 versus the APAP group.
4. Discussion
In clinical and animal studies, farrerol has been shown to have multiple biological activities, including antitussive, antibacterial, anti-inflammatory and anti-oxidant effects 10 12. Recent studies suggest that mitochondrial-targeted antioxidants can be viable therapeutic agents against hepatotoxicity induced by APAP overdose 3. Moreover, growing evidence reveals that Nrf2, a key coordinator of oxidative stress, plays an indispensable role in ameliorating acute liver injury via activating antioxidant enzymes to detoxify NAPQI 13. Farrerol has antioxidant potential via activation of Nrf2, but the hepatoprotective activity has not previously been reported. In the current study, we aimed to test the hepatoprotective effects of farrerol by using animal and cell models and to further investigate its underlying mechanisms.
APAP-induced acute liver failure was characterized by mortality, high ALT and AST in serum and pathological changes in the liver 14. In our current study, farrerol treatment significantly suppressed APAP-induced hepatic damage in mice. Massive evidence proved that oxidative stress contributes to histopathologic changes by inducing malondialdehyde (MDA) and myeloperoxidase formation (MPO) and decreasing hepatic levels of SOD and glutathione (GSH) 15. In the current study, farrerol remarkably reduced the formation of MDA and MPO and decreased the depletion of GSH and SOD, indicating that farrerol treatment dramatically alleviated APAP-induced oxidative injuries in mice. Oxidative stress-mediated JNK activation plays a central role in APAP-induced hepatic injury 16. Furthermore, pharmacological inhibition of JNK or silencing the expression of the JNK gene markedly attenuate hepatic injury after APAP overdose17. In this study, farrerol treatment effectively suppressed phosphorylation of JNK, which was triggered by APAP overdose. Mitochondrial proteins are the main targets of NAPQI during APAP-associated hepatotoxicity 18. Accumulating evidence has shown that APAP provokes mitochondrial dysfunction by enhancing release of cytochrome c and AIF from the mitochondria, leading to Bax translocation to mitochondria 19. As shown in Fig 3A-B, Farrerol inhibited Bax mitochondrial translocation, AIF and cytochrome c release. Together, these results indicate that the hepatoprotective effects of farrerol involve the inhibition of oxidative stress and attenuation of mitochondrial dysfunction.
Nrf2, being a regulator for multiple signaling pathways, improves oxidative stress-induced cell apoptosis and mitochondrial dysfunction 20 21 and exhibits hepatoprotective effects in APAP-induced hepatotoxicity. This is accomplished by having antioxidant enzymes detoxify NAPQI 13,22. Our data showed that farrerol induced activation of Nrf2 and antioxidant enzymes, including causing HO-1, GCLC and GCLM activation both in vivo and in vitro. Under conditions of Nrf2 deficiency, farrerol failed to activate antioxidant enzymes expression and exert the protective effect on H2O2-induced hepatotoxicity in HepG2 cells, clearly supporting the idea that there is involvement of an Nrf2 pathway in the hepatoprotective effect of farrerol. We further delineated the mechanisms of Nrf2 activation by farrerol in vitro. It is also noteworthy that AMPK, a sensor of the cellular energy status, regulates cell survival and death in the presence of oxidative stress 23. Our previous study has shown that natural products activate Nrf2 and protect mitochondria against oxidative stress through the AMPK/Akt/GSK3β pathway 24. In this study, farrerol treatment increased the phosphorylation of AMPK, AKT and PI3K, which might contribute to the activation of Nrf2. This hypothesis was verified by our result that Nrf2 activation and the hepatoprotective effect of farrerol were abolished by AKT or AMPK inhibition (Fig 6 C-H). Together, our results suggest that farrerol exerts antioxidant effects through Nrf2 activation via the AMPK/AKT pathway against H2O2-induced toxicity in vitro.
AMPK, as an evolutionarily conserved protein, plays an important role in cell survival and organism longevity through modulation of energy homeostasis and autophagy. Activation of AMPK prevents drug-induced mitochondrial and hepatocellular damage through regulation of mitochondrial fusion and autophagy, making it a potentially valuable approach to the treatment of drug-induced liver injury 25. Since farrerol activated AMPK in a dose-dependent manner, we further explored whether autophagy was also involved in the hepatoprotective activity of farrerol. Our results demonstrated that farrerol treatment increased LC3Ⅱ, Atg5 and Atg7 protein expression in a dose-dependent manner. Moreover, preincubation of cells with chloroquine (CQ, an autophagy inhibitor) prevented the farrerol-induced LC3, Atg5 and Atg7 activation and decreased the cytoprotective effect, which suggested that farrerol exerts hepatoprotective effects through the autophagy pathway.
Given all of these results, Nrf2-deficient mice were used to further elucidate whether farrerol decreased APAP-induced hepatotoxicity. For WT mice, farrerol prevented the mortality associated with the APAP challenge. This inhibitory effect of farrerol was clearly attenuated but not abolished in Nrf2-deficient mice. Moreover, ALT and AST levels in the serum and histopathological changes in the liver were alleviated but not abolished in Nrf2-/- mice by farrerol treatment. Farrerol still downregulated JNK activation in Nrf2-/- mice, which may be attributed to activation of LC3, Atg5 and Atg7. Taken together, our results support the idea that the hepatoprotective effect of farrerol in hepatotoxicity is, at least partially, based on the activation of both Nrf2-mediated antioxidative cascades and autophagy.
Acknowledgements
This work was in part supported by the National Natural Science Foundation of China (Grant No. 81603174) and the General Financial Grant from the China Postdoctoral Science Foundation (Grant No. 168847).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Budnitz DS, Lovegrove MC, Crosby AE. Emergency department visits for overdoses of acetaminophen-containing products. American journal of preventive medicine. 2011;40(6):585-592
2. Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug metabolism reviews. 2012;44(1):88-106
3. Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox biology. 2016;10:148-156
4. Kim BM. The Role of Saikosaponins in Therapeutic Strategies for Age-Related Diseases. Oxidative medicine and cellular longevity. 2018;2018:8275256
5. Lv H, Xiao Q, Zhou J. et al. Licochalcone A Upregulates Nrf2 Antioxidant Pathway and Thereby Alleviates Acetaminophen-Induced Hepatotoxicity. Front Pharmacol. 2018;9:147
6. Yan M, Huo Y, Yin S. et al. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox biology. 2018;17:274-283
7. McGill MR, Williams CD, Xie Y. et al. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicology and applied pharmacology. 2012;264(3):387-394
8. Ahmed SM, Luo L, Namani A. et al. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta. 2017;1863(2):585-597
9. Kumar H, Kim IS, More SV. et al. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Natural product reports. 2014;31(1):109-139
10. Ci X, Lv H, Wang L. et al. The antioxidative potential of farrerol occurs via the activation of Nrf2 mediated HO-1 signaling in RAW 264.7 cells. Chem Biol Interact. 2015;239:192-199
11. Ni HM, McGill MR, Chao X. et al. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol. 2016;65(2):354-362
12. Qiu J, Xiang H, Hu C. et al. Subinhibitory concentrations of farrerol reduce alpha-toxin expression in Staphylococcus aureus. FEMS Microbiol Lett. 2011;315(2):129-133
13. Goldring CE, Kitteringham NR, Elsby R. et al. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology. 2004;39(5):1267-1276
14. Brodsky M, Hirsh S, Albeck M. et al. Resolution of inflammation-related apoptotic processes by the synthetic tellurium compound, AS101 following liver injury. J Hepatol. 2009;51(3):491-503
15. Wu H, Zhang G, Huang L. et al. Hepatoprotective Effect of Polyphenol-Enriched Fraction from Folium Microcos on Oxidative Stress and Apoptosis in Acetaminophen-Induced Liver Injury in Mice. Oxidative medicine and cellular longevity. 2017;2017:3631565
16. Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicology and applied pharmacology. 2010;246(1-2):8-17
17. Henderson NC, Pollock KJ, Frew J. et al. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56(7):982-990
18. Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273(28):17940-17953
19. Bajt ML, Farhood A, Lemasters JJ. et al. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324(1):8-14
20. Lv H, Liu Q, Zhou J. et al. Daphnetin-mediated Nrf2 antioxidant signaling pathways ameliorate tert-butyl hydroperoxide (t-BHP)-induced mitochondrial dysfunction and cell death. Free Radic Biol Med. 2017;106:38-52
21. Zeng X, Li X, Xu C. et al. Schisandra sphenanthera extract (Wuzhi Tablet) protects against chronic-binge and acute alcohol-induced liver injury by regulating the NRF2-ARE pathway in mice. Acta Pharm Sin B. 2017;7(5):583-592
22. Ye D, Wang Y, Li H. et al. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1alpha-mediated antioxidant capacity in mice. Hepatology. 2014;60(3):977-989
23. Shirwany NA, Zou MH. AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed). 2014;19:447-474
24. Lv H, Liu Q, Wen Z. et al. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox biology. 2017;12:311-324
25. Kang SW, Haydar G, Taniane C. et al. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS One. 2016;11(10):e0165638
Author contact
![]() Corresponding author: Tel.: +86 431 88783044. E-mail address: cixinxinedu.cn (Xinxin Ci). Full postal address: Dongminzhu road 519, The First Hospital of Jilin University, Changchun, Jilin, 130001, China.
Corresponding author: Tel.: +86 431 88783044. E-mail address: cixinxinedu.cn (Xinxin Ci). Full postal address: Dongminzhu road 519, The First Hospital of Jilin University, Changchun, Jilin, 130001, China.