Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):838-846. doi:10.7150/ijbs.31371 This issue Cite
Research Paper
The LIM-Only Protein FHL2 is involved in Autophagy to Regulate the Development of Skeletal Muscle Cell
Zihao Liu1*, Shunshun Han1*, Yan Wang1*, Can Cui1*, Qing Zhu1, Xiaosong Jiang2, Chaowu Yang2, Huarui Du2, Chunlin Yu2, Qingyun Li2, Haorong He1, Xiaoxu Shen1, Yuqi Chen1, Yao Zhang1, Lin Ye1, Zhichao Zhang1, Diyan Li1, Xiaoling Zhao1, Huadong Yin1 ![]()
1. Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu, Sichuan 611130, PR China
2. Animal Breeding and Genetics key Laboratory of Sichuan Province, Sichuan Animal Science Academy, Chengdu, Sichuan, 610066, PR China
* These authors contributed equally to this work.
Received 2018-11-9; Accepted 2019-1-21; Published 2019-3-1
Abstract

Scope: Four and a half LIM domain protein 2 (FHL2) is a LIM domain protein expressed in muscle tissue whose deletion is causative of myopathies. Although FHL2 has a confirmed important role in muscle development, its autophagy-related function in muscle differentiation has not been fully determined.
Methods: C2C12 cells were treated with FHL2-konwdown or FHL2-overexpression. The morphology of C2C12 cells was observed by transmission electron microscopy. The mRNA and protein abundances of muscle related genes and autophagy related genes were measured by RT-PCR and western blot. Immunofluorescence and co-immunoprecipitation assay were used to verify the interaction between FHL2 and LC3 protein.
Results: FHL2 silencing reduced LC3-Ⅱ protein expression and the amount of LC3 that co-immunoprecipitated with FHL2, indicating that FHL2 interacts with LC3-Ⅱ in the formation of autophagosomes. Moreover, the expression of muscle development marker genes such as MyoD1 and MyoG was lower in FHL2-silenced C2C12 cells but not in FHL2-overexpressing C2C12 cells. Electron microscopy analysis revealed large empty autophagosomes in FHL2-silenced myoblasts, while flow cytometry suggested that FHL2 silencing made cells more vulnerable to staurosporine-induced cell death.
Conclusion: These results suggest that FHL2 interacts with LC3-Ⅱ in autophagosome formation to regulate the development of muscle cells.
Keywords: FHL2, LIM-domain, autophagy, cell development, C2C12 cells
Introduction
Four and a half Lim domain protein 2 (FHL2) belongs to the FHL protein family which contains five members: FHL1, FHL2, FHL3, FHL4, and ACT [1]. Despite their high level of conservation among species, their expression levels differ from each other between tissues. FHL1, FHL2, and FHL3 are mainly expressed in skeletal and heart muscle [2], whereas FHL4 and ACT are highly expressed in the testis [3]. FHL2 has been shown to have a dual function in interacting with the cytoplasmic domain of several integrin chains [4], and also as a transcriptional coactivator of the androgen receptor [5]. Although FHL2 plays an important role in muscle development and its deletion was reported to lead to the development of myopathies [2, 6-10], the details of its function in skeletal muscle development are unclear.
Autophagy is the major intracellular degradation system by which cytoplasmic materials are delivered to and degraded in the lysosome, which also serves as a dynamic recycling system that produces new building blocks and energy for cellular renovation and homeostasis [11, 12]. Recently, the relationship between autophagy and muscle cell development has been shown to play an important role in muscle mass maintenance and integrity [13, 14]. As the main proteolytic system that controls protein degradation in skeletal muscle cells, the autophagy lysosome is activated in a number of catabolic disease states that lead to muscle loss and weakness [15, 16]. Excessive activation of autophagy aggravates muscle wasting by removing some cytoplasm, proteins, and organelles; conversely, the inhibition or alteration of autophagy can contribute to myofiber degeneration and weakness in muscle disorders [17]. Additionally, autophagy protects against apoptosis during myoblast differentiation [18].
Recently, a relationship between FHL2 and autophagy was identified, with involvement of the FHL2-activated nuclear factor-κB pathway reported in particulate matter 2.5-induced autophagy in mouse aortic endothelial cells [19]. Muscle Lim protein (MLP)/CSRP3 was reported to interact with microtubule-associated protein 1 light chain 3 (LC3) to regulate the differentiation of myoblasts and facilitate autophagy [20]. Moreover, Sabatelli et al found that the aggresome-autophagy pathway was involved in the pathophysiological mechanism underlying the muscle pathology of the C150R mutation in the second LIM domain of FHL1 [21]. Because FHL2 contains the LIM domain, similar to FHL1 and MLP/ CSRP3, it is reasonable to speculate that FHL2 is involved in autophagy to regulate the development of skeletal muscle. The present study examined this hypothesis.
Materials and Methods
Cell cultures
The mouse C2C12 myoblast cell line (Fuheng Cell Center, Shanghai, China) was maintained in growth medium composed of Dulbecco's Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS), and 1% Antibiotic-Antimycotic (ABAM) at 37 ℃ under 5% CO2. Differentiation into myotubes was activated by replacing the growth medium with differentiation medium composed of DMEM, 2% horse serum, and 1% ABAM.
FHL2 silencing and overexpression
C2C12 cells were cultivated in 6-well plates and transfected with siRNAs (sense: 5'-GCAAGGACUUGUCCUACAATT-3', antisense: 5'-UUGUAGGACAAGUCCUUGCTT-3'; Sangon Biotech, Shanghai, China) when grown to a density of approximate 70% in plates. In contrast, control cells were transfected with negative siRNA with same other condition. The transfection reagent was Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The knockdown efficiency was assessed by quantitative RT-PCR of FHL2 mRNA and western blot assay of FHL2 protein.
C2C12 cells were transfected with a plasmid pcDNA3.1 which was produced by cloning FHL2 cDNA into the pcDNA3.1 expression vector (Sangon Biotech, Shanghai, China). The transfection reagent Lipofectamine 3000 (Invitrogen) was mixed with optim-mem. Plasmid with optim-mem was mixed with Lip3000. Next, they were mixed all together at room temperature for 10 min. Finally, the mixture was added into C2C12 6-well plates at 37 ℃ under 5% CO2 for 48h.
RNA extraction, cDNA synthesis and RT-PCR
Total RNA was isolated using TRIzol (TAKARA, Dalian, China) reagent according to the manufacturers' instruction. RNA was reverse transcribed by TAKARA PrimeScriptTM RT reagent kit (TAKARA) according to the manufacturers' instruction. Quantitative RT-PCR assay was performed essentially as previously described [22].Primer are used in Table 1.
Primers used for quantitative real-time PCR
| Genes | Forward primer (5'-3') | Reverse primer (5'-3') |
|---|---|---|
| FHL2 | TGCGTGCAGTGCAAAAAG | TGTGCACACAAAGCATTCCT |
| MyoD1 | AGCACTACAGTGGCGACTCA | GGCCGCTGTAATCCATCA |
| MyoG | TACAGCGACCAACAGTACGC | TCTGCATTGTTTCCATCCTG |
| MYH3 | CGGCTGCCTAAAGTGGAGAT | AGGCCTGTAGGCGCTCAA |
| ATG5 | AGCAGCTCTGGATGGGACTGC | GCCGCTCCGTCGTGGTCTGA |
| ATG7 | GCTCCTCATCACTTTTTGCCAACA | GGAGCCACCACATCATTGC |
| β-actin | CGTGAAAAGATGACCCAGATCA | CACAGCCTGGATGGCTACGT |
Western blot assay
The cells were collected from the cultures, placed in the RIPA lysis buffer on ice (BestBio, Shanghai, China). The whole proteins were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride membranes (PVDF; Millipore Corporation, Billerica, MA, USA). The PVDF membrane was incubated with 5% defatted milk powder at room temperature for 1 h, then incubation with the following specific primary antibodies at 4℃ overnight: anti-FHL2 (Abcam, Cambridge, MA, USA), anti- MYOD1 (Abcam), anti-MyoG (Abcam), anti-MYH3 (Abcam), anti-ATG5 (Cell Signaling, Danvers, MA, USA), anti-ATG7 (Cell Signaling), and anti-β-Actin (Abcam). The secondary antibodies HRP-labeled mouse and rabbit IgG (Cell Signaling) were added at room temperature for 1h. Following each step, the membranes were washed five times with PBS-T for 3 min. The proteins were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, USA) with a Kodak imager (Eastman Kodak, Rochester, NY, USA). Quantification of protein blots was performed using the Quantity One 1-D software (version 4.4.0) (Bio-Rad, Hercules, CA, USA) on images acquired from an EU-88 image scanner (GE Healthcare, King of Prussia, PA, USA).
Microscopy
Cellular morphology was evaluated in proliferating myoblasts and differentiated myotubes by phase-contrast microscopy without preliminary fixation. Pictures were produced using the Olympus IX73 inverted microscope (OLYMPUS, Tokyo, Japan) and the Hamamatsu C11440 digital camera (HAMAMATSU, Shizuoka, Japan).
Transmission electron microscopy
C2C12 myoblasts or myotubes were detached from the plates using a manual scraper, washed with PBS for a while. Then the cells were suspended and fixed overnight at 4 °C in 2% glutaraldehyde with 1% tannic acid in 0.1M sodium cacodylate, pH 7.3. The cells were rinsed three times in the sodium cacodylate buffer and incubated in 2% osmium tetroxide in the same buffer for 2 h at room temperature. Afterwards, the cells were rinsed three times in distilled water and exposed to 1% uranyl acetate in water for 15 min at room temperature and twice in distilled water. Next, the cells were spun down into 3% agarose at 45 °C and cooled to form blocks. The agarose blocks were dehydrated in graded steps of acetone and embedded in Spurr's low-viscosity media. Following polymerization overnight at 65 °C, 80-nm sections were cut on a Reichert-Jung Ultracut E ultramicrotome and picked up on copper grids. The grids were post-stained in uranyl acetate and bismuth subnitrate. The sections were observed on a Philips CM-10 TEM (HT7700) and micrographs were recorded on a Kodak 4489 sheet film (Eastman Kodak).
Immunofluorescence and confocal microscopy
Cells were plated on glass cover slides in complete medium and incubated overnight at 37℃ and 5% CO2. Then the cells were washed with PBS and fixed in 4% paraformaldehyde for 30 min. After washing twice, the cells were permeabilized with 0.5% Triton X-100 for 6 min (Sigma). Then the cells were washed by PBS again and incubated with primary antibody diluted in PBS-1% sheep serum at 4℃. Next, the cells were washed three times for 5min each with PBS. After that, cells were incubated in the dark in fluorescent secondarty antibody for 90 min at room temperature and washed thrice with PBS. Subsequently, Coverslips were used to mount the antifade mountant. Finally, Fluorescence intensity was visualized with microscope Olympus FluoView FV1000 Confocal Microscope (Olympus, Melville, NY, USA).
Co-immunoprecipitation assay
Protein concentration was determined using the BCA Protein Quantitation Kit (BestBio). 1mg of lysate was mixed with lysis buffer including phosphatase inhibitor to a volume of 1ml. Then Lysates were precleared with 5 μg of appropriate control IgG (Santa Cruz Biotechnology) and 20 μl of protein A/G plus-agarose (Santa Cruz Biotechnology) for 1 h rotation at 4 °C. Lysates were centrifuged (500 × g for 5 min at 4 °C) and 5 μg of FHL2 (Abcam) or LC3-II antibody (Abcam) or corresponding IgG was added to the precleared lysates and kept on ice for ~ 4 h. After incubation, 30 μl of protein A/G plus-agarose was added to each tube and kept on a rotator overnight at 4 °C. Lysates were then centrifuged (500 × g for 5 min at 4 °C). The pellet fractions were washed four times with PBS-PI and then resuspended in 20 μl of loading buffer. Samples were electrophoresed on a 12% SDS-PAGE gel and immunoblotted with the appropriate antibody.
Flow cytometry assay
Apoptosis was induced by treating cells with 2uM Staurosporine (STS) (Selleckchem, Houston, TX, USA) in 12-well plates. Then cells were collected and digested by pancreatic enzyme to be cell suspension. Then the cells were washed twice with cold PBS and then resuspended cells in 1×binding buffer (BD Pharmingen, Santiago, CA, USA) at a concentration of 1×106 cells/mL. One hundred microliters of the solution were transferred to a 5-mL culture tube, and then 5 μL of Annexin V-FITC (BD Pharmingen) and 5 μL of PI (BD Pharmingen) were added. The cells were gently vortexed and incubated for 15 min at room temperature (25 ℃) in the dark. Four hundred microliters of 1×binding buffer was added to each tube and analyzed by flow cytometry (BD FACSCalibur, BD Pharmingen) within 1 h.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA). Data are presented as least squares means ± standard error of the mean (SEM), and values were considered statistically different at P<0.05.
Results
The role of FHL2 in skeletal muscle differentiation
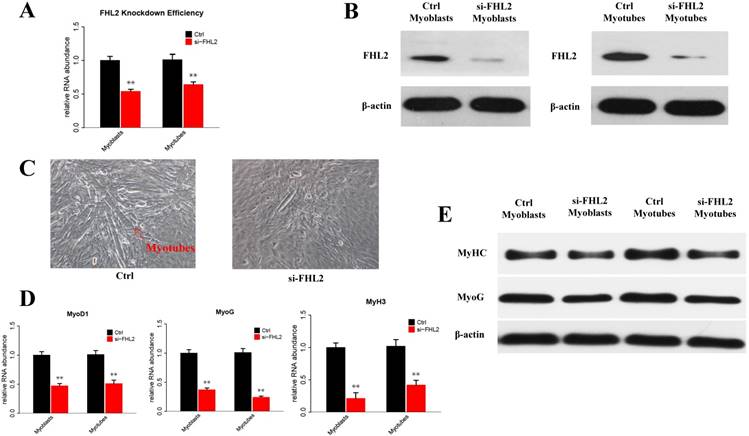
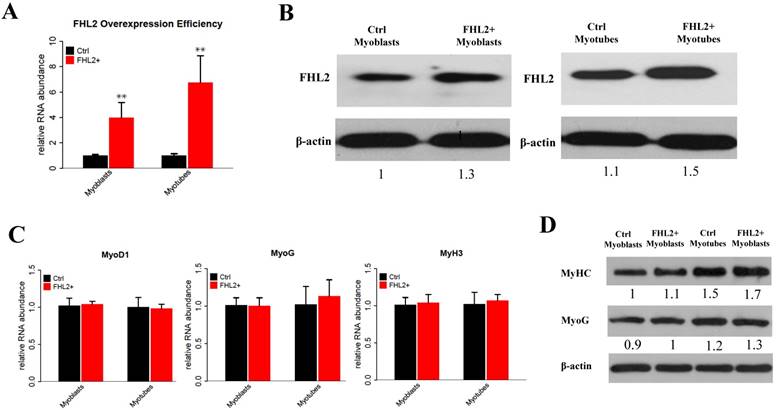
To explore the potential role of FHL2 in skeletal muscle differentiation, we performed a knockdown assay in C2C12 myoblasts derived from mouse satellite cells. C2C12 cells transfected with FHL2 siRNA or scrambled siRNA were induced to differentiate, and FHL2 mRNA expression was shown to be reduced significantly after knockdown in both myoblasts and myotubes compared with controls (Fig. 1A). Western blot analysis revealed a decrease in FHL2 protein in FHL2-silenced cells compared with control cells (Fig. 1B). Next, morphological differences between negative control and FHL2 siRNA- transfected groups were compared during C2C12 differentiation into myotubes. The FHL2-silenced group showed reduced myotube formation (Fig. 1C), and the expression of myogenic marker genes MyoD1, MYH3, and MyoG was significantly reduced in FHL2-silenced cells compared with controls (Fig. 1D). Moreover, western blotting revealed that MYHC and MyoG protein levels were reduced after FHL2 silencing (Fig. 1E). These results suggest that FHL2 siRNA was effective and that FHL2 plays an important role in muscle differentiation by regulating myogenesis-related genes. The overexpression of FHL2 in C2C12 myoblasts and myotubes significantly increased the FHL2 mRNA (Fig. 2A) and protein abundance (Fig. 2B), but which had no significant effect on MyoG, MyoD1, or MYH3 mRNA expression (Fig. 2C), and the protein levels of MyoG and MyHC (Fig. 2D).
The efficiency of FHL2 knockdown and its influence on muscle development-related genes. (A) FHL2 mRNA expression in C2C12 cells after knockdown by siRNA. (B) FHL2 protein expression after knockdown by siRNA in myoblasts and myotubes. (C) Cellular morphology of myotubes in control and si-FHL2 groups. Pictures of cells were taken at ×40 with digital camera. (D) MyoD1, MyoG, and MyH3 mRNA expression after FHL2 knockdown. (E) MyHC and MyoG protein expression after FHL2 knockdown. * P <0.05, ** P <0.01 compared with controls.
The efficiency of FHL2 overexpression and its influence on muscle development-related genes. (A) FHL2 mRNA expression after vector transfection into C2C12 cells. (B) FHL2 protein expression after vector transfection into myoblasts and myotubes. (C) MyoD1, MyoG, and MyH3 expression after FHL2 overexpression. (D) MyHC and MyoG protein expression after FHL2 overexpression. * P <0.05, ** P <0.01 compared with controls.
FHL2 regulated autophagy in skeletal muscle cells
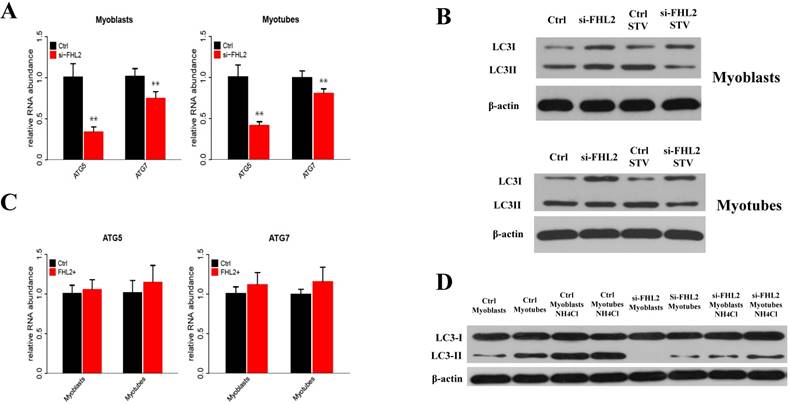
To determine whether FHL2 silencing in skeletal muscle influenced the induction of autophagy, the expression of autophagy genes ATG5 and ATG7 was measured and shown to be significantly reduced in myoblasts and myotubes from FHL2-silenced cells compared with control cells (Fig. 3A). Next, LC3 protein level changes were examined to monitor autophagy induction, and the ratio of LC3-Ⅱ to LC3-I protein was found to be reduced in FHL2-silenced myoblasts and myotubes compared with controls (Fig. 3B). Interestingly, the starvation of FHL2- silenced myoblasts and myotubes, which should have been able to activate autophagy, did not induce the accumulation of LC3-Ⅱ. Moreover, FHL2 overexpression did not significantly influence the expression of ATG5 or ATG7 (Fig. 3C). Furthermore, to determine whether the decrease of LC3 protein is due to low autophagy induction or high autophagic flux, the cells were treated with NH4CL for 17h. As a result, the LC3-Ⅱ level increased significantly in cells without FHL2-silencing, whereas the protein level remained unchanged in si-FHL2 cells (Fig. 3D).
FHL2 affects autophagy-related genes. (A) ATG5 and ATG7 expression in FHL2-silenced myoblasts and myotubes. (B) The LC3-II to LC3-I ratio in FHL2-silenced or starved myoblasts and myotubes. (C) ATG5 and ATG7 mRNA expression after FHL2 overexpression. (D) LC-I and LC-II protein expression in the cells were treated with NH4CL. * P <0.05, ** P <0.01 compared with controls.
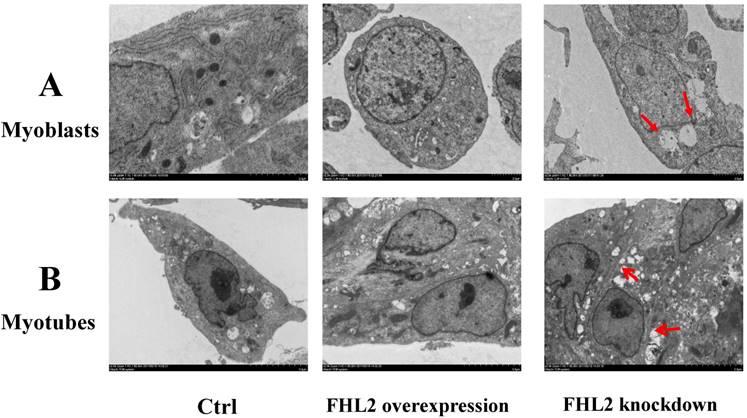
To further confirm our findings, we used TEM to observe the ultrastructure of myoblasts (Fig. 4A) and myotubes (Fig. 4B). The negative control, and FHL2-overexpressing myoblasts and myotubes were observed to contain normal autophagosomes whereas large empty autophagosomes were present in FHL2-silenced myoblasts and myotubes, which were indicative of impaired autophagy in myoblasts and myotubes, respectively.
Morphology of C2C12 cells. (A) Myoblasts and (B) myotubes from control, FHL2-overexpressing and siFHL2 cells were processed for transmission electron microscopy. Normal myoblasts with normal autophagosome-like vacuoles and mitochondria. FHL2-overexpressing myoblasts with autophagosome vacuoles and mitochondria resembling those of control cells. FHL2-silenced myoblasts show large empty autophagosomes.
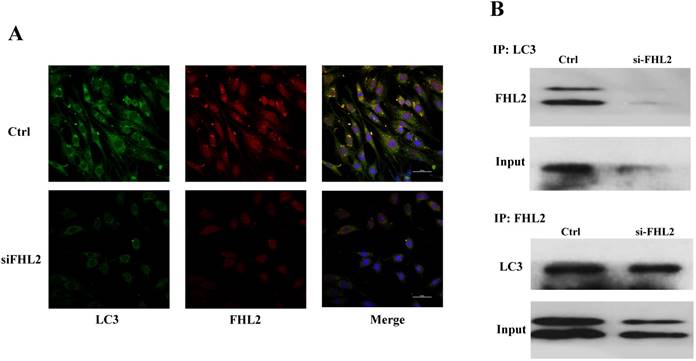
FHL2-LC3-Ⅱ co-localization
Immunofluorescence and co-immunoprecipitation assays were next performed to determine whether FHL2 co-localized with the autophagosomal marker LC3 during myoblast differentiation. Immunofluorescence analysis showed similar FHL2 and LC3 protein distribution in myoblasts, while FHL2 silencing decreased both FHL2 and LC3Ⅱ expression which was indicative of their co-expression (Fig. 5A). The co-immunoprecipitation assay revealed co-localization of FHL2 and LC3 proteins in C2C12 cells during myoblast differentiation, and reduced co-immunoprecipitation in FHL2-silenced myoblasts compared with control cells (Fig. 5B). These results support a role for FHL2 in the formation of autophagosomes by binding LC3 in an intracellular complex.
Colocalization of FHL2 and LC3 protein in C2C12 cells. (A) Co-localization of LC3 (green fluorescence) and FHL2 (red fluorescence) in basal and FHL2 knockdown conditions by immunofluorescence. (B) WT control and si-FHL2 cells were processed to obtain whole cell extracts. Upper panel: Cellular extracts were immunoprecipitated with an anti-LC3-II antibody and immunoblotted with an anti-FHL2 antibody. Lower panel: Cellular extracts were immunoprecipitated with an anti-FHL2 antibody and immunoblotted with an anti-LC3-II antibody.
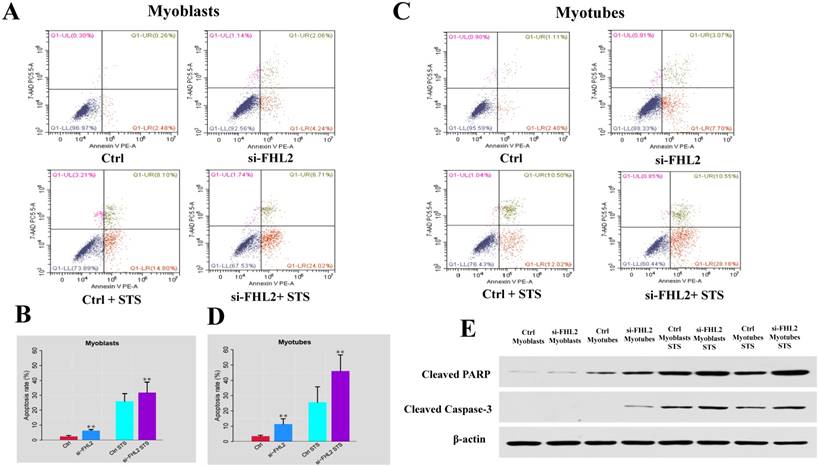
FHL2 silencing promoted apoptosis in both myoblasts and myotubes
Flow cytometry was next used to measure cell death after treatment of C2C12 cells with 2 μM STS for 12 h. Cell death was observed in 2.74% and 3.51% of control myoblasts and myotubes, respectively, compared with 6.30% and 10.77% in si-FHL2 groups, moreover, a decreased resistance to STS was detected in FHL2-silenced myoblasts and myotubes (Fig. 6 A-D). This indicated that FHL2 silencing enhanced apoptosis in both myoblasts and myotubes. Western blotting of PARP and caspase-3 proteins under different conditions revealed the increased accumulation of PARP in myoblasts following FHL2 silencing (Fig. 6E). Caspase-3 protein expression was not detected in WT cells, but was observed in si-FHL2 myotubes, suggesting an important role for FHL2 in protecting myotubes from apoptosis. We also found that myotubes were more resistant to STS-induced apoptosis than myoblasts.
siFHL2 myoblasts and myotubes were treated with STS. (A, B) Cell death proportions after FHL2 downregulation in untreated or STS-treated myoblasts. (C, D) Cell death proportions after FHL2 downregulation in untreated or STS-treated myotubes. (E) Control and si-FHL2 myoblasts and myotubes were left untreated (NT) or treated with 2 μM STS for 24 h. Then the protein expression of RARP, Caspase-3, β-actin were measured by western blot.
Discussion
Although previous studies suggested that FHL2 is involved with autophagy and may be associated with LC3 protein [19, 23], its autophagy-related role in muscle differentiation had not been fully determined. In this study, we explored the mechanism of FHL2 in muscle differentiation through autophagy induction.
We initially confirmed that FHL2 played a role in muscle development by measuring the gene and protein expression of muscle-related MyoD1, MyH3, and MyoG after FHL2 silencing and overexpression. FHL2 expression was significantly lower after siRNA transfection, indicating the efficiency of siRNA. Expression of the muscle-related genes was decreased in FHL2-silenced myoblasts or myotubes, and their protein expression was slightly reduced. However, no increase in their expression was detected in FHL2-overexpressing cells. In cell nucleus, FHL2 regulates muscle development through interacting with β-catenin and thus promoting the myogenic differentiation [24]. Therefore, one possible reason for the unchanged expression and protein level of muscle-related genes is that the overexpression of FHL2 doesn't affect the activity of β-catenin thus fail to upregulate the expression of MyoG in the downstream. Those results confirm that FHL2 play a part in muscle development.
Previous studies detected LC3 protein accumulation during muscle cell development by western blotting [25, 26]. We investigated the relationship between FHL2 and autophagy using two methods: first, measuring the expression of genes essential for autophagy such as ATG5 and ATG7 [27] following FHL2 knockdown, and second, measuring the correlation between FHL2 and LC3-Ⅱ. Knockdown, but not the overexpression, of FHL2 significantly changed the expression of ATG5 and ATG7 in both myoblasts and myotubes. As the autophagy is a complex process regulated by many factors, simply increasing one of the factors may not be able to promote the entire system. It may account for the unchanged expression of autophagy-related genes after FHL2 overexpression. However, the reduced autophagy level after FHL2 silencing may indicate that FHL2 participates the autophagy induction with an indispensable role in the system. Additionally, the autophagy level as indicated by the ratio of LC3-Ⅱ to LC3-I protein [28] was reduced in FHL2-silenced myoblasts and myotubes. These results indicate a role for FHL2 in autophagy induction.
It is reasonable to assume that the function of FHL2 in muscle development is associated with autophagy because similar effects on muscle-related genes and autophagy markers were detected following FHL2 silencing or overexpression. Moreover, the starvation that activated autophagy in control cells did not induce LC3-Ⅱ accumulation in either FHL2-silenced myoblasts or myotubes, indicative of a function for FHL2 in autophagy induction. We next determine autophagic flux by adding NH4Cl, which is able to inhibit the lysosome acidification and thus result in the accumulation of LC3-Ⅱ, into the cells for 17 h to clarify if LC3 decrease after FHL2 silencing is due to low autophagy induction or high autophagic flux. Our results suggest that FHL2 silencing accounts for the decrease of LC3-Ⅱ level after FHL2 silencing. Further TEM analysis showed that FHL2 knockdown caused organelle abnormalities and impaired autophagy in C2C12 cells, but not in control or FHL2- overexpressing C2C12 cells. The persistent activation of catabolic pathways in muscle causes atrophy and weakness, and autophagy is required to maintain muscle mass [17, 29]. Therefore, our results suggest that FHL2 has a role in myotube formation by activating cell autophagy to maintain cellular homeostasis.
MLP/CSRP3 containing a LIM domain has been reported to participate in muscle differentiation and autophagosome formation by interacting with LC3-Ⅱ [20]. FHL2 also contains four and a half LIM domains that act as a protein-protein binding interface involved in muscle differentiation [30]. Considering the potential of FHL2 to bind LC3-Ⅱ, its correlation with LC3-Ⅱ at the protein level in C2C12 cells, and its likely role in the assembly of extracellular membranes [31], we hypothesized that FHL2 participates in autophagosome formation by interacting with LC3-Ⅱ. We therefore next explored their localization in C2C12 cells. The protein distribution of FHL2 in myoblasts was shown to resemble that of LC3-Ⅱ, and immunoprecipitation revealed the co-localization of LC3-Ⅱ and FHL2. More importantly, FHL2 silencing simultaneously reduced both FHL2 and LC3 protein expression. Previous study reaveals that FHL2 regulates muscle development by interacting with β-catenin and activating the transcription of MyoG in cell nucleus [24]. The results of co-localization and co-immunoprecipitation in this study suggest that FHL2 interacts with LC3 in cytoplasm thus regulates the autophagy thus FHL2 may have different roles in nucleus and cytoplasm.
Insufficient autophagy contributes to the accumulation of waste material inside the cell and the induction of apoptosis, however, autophagy can also protect cells from apoptosis [32, 33]. Study has revealed autophagy protects myoblasts against apoptosis [34]. In a previous study, the specific autophagy inhibitor 3MA [35] was used to treat C2C12 cells, resulting in increased CASP3 activity which is a marker of increased apoptosis [18]. If FHL2 functions in the induction of autophagy, FHL2-silenced C2C12 cells would be expected to show more apoptosis. Indeed, we observed increased STS-induced death of both FHL2-silenced myoblasts and myotubes compared with controls. These results suggested that FHL2 knockdown reduces autophagy, indicating that it regulates muscle development by controlling autophagy and thereby FHL2 may regulate muscle development through autophagy against apoptosis. However, exactly how autophagy prevents apoptosis requires additional study. Furthermore, to validate the results of flow cytometry, the protein levels of cleaved caspase-3 and PARP, markers of apoptosis [36, 37], were measured in WT and STS-treated groups by western blotting. Increased cleavage of PARP and caspase-3 was observed in FHL2-silenced cells relative to controls, and increased PARP cleavage was also detected during the differentiation of FHL2-silenced cells. These results reflect increased apoptosis in the absence of FHL2, likely caused by repressed autophagy.
In conclusion, our findings suggest that FHL2 interacts with LC3-Ⅱ protein to regulate muscle development through autophagy. As a result, the deletion of FHL2 inhibited muscle development and damaged autophagosomes. However, further studies of mechanism of autophagy in muscle cells are needed to understand in detail how FHL2 regulates autophagy and its contribution to muscle development or myopathies.
Acknowledgements
This work was financially supported by the China Agriculture Research System (CARS-40), and the Thirteenth Five Year Plan for Breeding Program in Sichuan (2016NYZ0050). We thank Sarah Williams, PhD, from Liwen Bianji, Edanz Group China (www. liwenbianji.cn), for editing the English text of a draft of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fimia GM, Cesare DD, Sassonecorsi P. A Family of LIM-Only Transcriptional Coactivators: Tissue-Specific Expression and Selective Activation of CREB and CREM. Molecular & Cellular Biology. 2000;20:8613-8622
2. Chu PH, Ruizlozano P, Zhou Q, Cai C. et al. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mechanisms of Development. 2000;95:259-265
3. Fimia GM, De CD, Sassone-Corsi P. CBP-independent activation of CREM and CREB by the LIM-only protein ACT. Nature. 1999;398:165-169
4. Wixler V, Geerts D, Laplantine E, Westhoff D. et al. The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several alpha and beta integrin chains and is recruited to adhesion complexes. Journal of Biological Chemistry. 2000;275:33669-33678
5. Müller JM, Isele U, Metzger E, Rempel A. et al. FHL2, a novel tissue-specific coactivator of the androgen receptor. Embo Journal. 2000;19:359-369
6. Martin B, Schneider R, Janetzky S, Waibler Z. et al. The LIM-only protein FHL2 interacts with β-catenin and promotes differentiation of mouse myoblasts. Journal of Cell Biology. 2002;159:113
7. Liang Y, Bradford WH, Zhang J, Sheikh F. Four and a half LIM domain protein signaling and cardiomyopathy. Biophysical Reviews. 2018:1-13
8. Genini M, Schwalbe P, Scholl FA, Remppis A. et al. Subtractive cloning and characterization of DRAL, a novel LIM-domain protein down-regulated in rhabdomyosarcoma. Dna & Cell Biology. 1997;16:433-442
9. Kong Y, Shelton JM, Rothermel B, Li X. et al. Cardiac-Specific LIM Protein FHL2 Modifies the Hypertrophic Response to Beta-Adrenergic Stimulation. Circulation. 2001;103:2731-2738
10. Lange S, Auerbach D, McLoughlin P, Perriard E. et al. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. Journal of Cell Science. 2002;115:4925-4936
11. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728-741
12. Wang Y, Cai S, Yin L, Shi K. et al. Tomato HsfA1a plays a critical role in plant drought tolerance by activating ATG genes and inducing autophagy. Autophagy. 2015;11:2033-2047
13. Sandri M. Autophagy in skeletal muscle. Febs Letters. 2010;584:1411-1416
14. Neel BA, Lin Y, Pessin JE. Skeletal muscle autophagy: a new metabolic regulator. Trends in Endocrinology & Metabolism. 2013;24:635-643
15. Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nature Reviews Molecular Cell Biology. 2005;6:439-448
16. Wolfe RR. The underappreciated role of muscle in health and disease. American Journal of Clinical Nutrition. 2006;84:475-482
17. Masiero E, Agatea L, Mammucari C, Blaauw B. et al. Autophagy Is Required to Maintain Muscle Mass. Cell Metabolism. 2009;10:507-515
18. Mcmillan EM, Quadrilatero J. Autophagy is required and protects against apoptosis during myoblast differentiation. Biochemical Journal. 2014;462:267-277
19. Xia WR, Fu W, Wang Q, Zhu X. et al. Autophagy Induced FHL2 Upregulation Promotes IL-6 Production by Activating the NF-kB Pathway in Mouse Aortic Endothelial Cells after Exposure to PM2.5. International Journal of Molecular Sciences. 2017;18:1484
20. Rashid MM, Runci A, Polletta L, Carnevale I. et al. Muscle LIM protein/CSRP3: a mechanosensor with a role in autophagy. Cell Death Discovery. 2015;1:15014
21. Sabatelli P, Castagnaro S, Tagliavini F, Chrisam M. et al. Autophagy Involvement in a Sarcopenic Patient with Rigid Spine Syndrome and a p.C150R Mutation in FHL1 Gene. Frontiers in Aging Neuroscience. 2014;6:215
22. Han S, Wang Y, Liu L, Li D. et al. Influence of three lighting regimes during ten weeks growth phase on laying performance, plasma levels- and tissue specific gene expression- of reproductive hormones in Pengxian yellow pullets. Plos One. 2017;12:e0177358
23. Tran MK, Kurakula K, S.Koenis D, Vries CJMd. Protein-protein interactions of the LIM-only protein FHL2 and functional implication of the interactions relevant in cardiovascular disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863:219-228
24. Bernd M, Richard S, Stefanie J, Zoe W. et al. The LIM-only protein FHL2 interacts with beta-catenin and promotes differentiation of mouse myoblasts. Journal of Cell Biology. 2002;159:113-122
25. Fortini P, Ferretti C, Iorio E, Cagnin M. et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell death & disease. 2016;7:e2168
26. Pizon Vr, Rybina S, Gerbal F, Delort F. et al. MURF2B, a novel LC3-binding protein, participates with MURF2A in the switch between autophagy and ubiquitin proteasome system during differentiation of C2C12 muscle cells. PLoS One. 2013;8:e76140
27. Cao QH, Liu F, Yang ZL, Fu XH. et al. Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. American Journal of Translational Research. 2016;8:3831-3847
28. Wu J, Dang Y, Su W, Liu C. et al. Molecular cloning and characterization of rat LC3A and LC3B-two novel markers of autophagosome. Biochemical & Biophysical Research Communications. 2016;339:437-442
29. Gordon BS, Kelleher AR, Kimball SR. Regulation of muscle protein synthesis and the effects of catabolic states. International Journal of Biochemistry & Cell Biology. 2013;45:2147-2157
30. Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nature Reviews Molecular Cell Biology. 2004;5:920-931
31. Park J, Will C, Martin B, Gullotti L. et al. Deficiency in the LIM-only protein FHL2 impairs assembly of extracellular matrix proteins. Faseb Journal. 2008;22:2508-2520
32. Ravikumar B, Berger Z, Vacher C, O'kane CJ. et al. Rapamycin pre-treatment protects against apoptosis. Human molecular genetics. 2006;15:1209-1216
33. Wang Y, Zhou J, Jingquan YU. The critical role of autophagy in plant responses to abiotic stresses. Frontiers of Agricultural Science & Engineering. 2017;4:28-36
34. Boya P, Gonzálezpolo RA, Casares N, Perfettini JL. et al. Inhibition of Macroautophagy Triggers Apoptosis. Molecular & Cellular Biology. 2005;25:1025-1040
35. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445-544
36. Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Communication and Signaling. 2010;8:31
37. Choudhary GS, Al-harbi S, Almasan A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods in Molecular Biology. 2015;1219:1-9
Author contact
![]() Corresponding author: Huadong Yin, Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu, Sichuan 611130, PR China. E-mail: yinhuadongedu.cn
Corresponding author: Huadong Yin, Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu, Sichuan 611130, PR China. E-mail: yinhuadongedu.cn