Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(5):1080-1090. doi:10.7150/ijbs.31484 This issue Cite
Research Paper
ASB6 Promotes the Stemness Properties and Sustains Metastatic Potential of Oral Squamous Cell Carcinoma Cells by Attenuating ER Stress
Kai-Feng Hung1,2,4, Po-Chen Liao3, Chih-Kai Chen3, Yueh-Ting Chiu3, Dong-Hui Cheng4, Masaoki Kawasumi5, Shou-Yen Kao2,4 ![]() , Jeng-Fan Lo2,3,6,7,8
, Jeng-Fan Lo2,3,6,7,8 ![]()
1. Department of Medical Research, Division of Translational Research, Taipei Veterans General Hospital, Taipei, Taiwan
2. Department of Dentistry, School of Dentistry, National Yang-Ming University, Taipei, Taiwan
3. Institute of Oral Biology, National Yang-Ming University, Taipei, Taiwan
4. Department of Stomatology, Taipei Veterans General Hospital, Taipei, Taiwan
5. Division of Dermatology, Department of Medicine, University of Washington, Seattle, Washington 98109, USA.
6. Graduate Institute of Chinese Medical Science and Institute of Medical Science, China Medical University, Taichung, Taiwan
7. Genome Research Center, National Yang-Ming University, Taipei, Taiwan
8. Cancer Progression Center of Excellence, National Yang-Ming University, Taipei, Taiwan
Received 2018-11-13; Accepted 2019-2-9; Published 2019-4-22
Abstract

Up-regulation of ASB6 has been previously associated with late-stage and poor prognosis of oral squamous cell carcinoma (OSCC) patients. To explore the cellular and molecular basis of how ASB6 enhances the malignancy of OSCC, we employed the clonogenicity and migration assays, murine pulmonary metastasis model, Western blot, and immunofluorescence microscopy to characterize the phenotypes of OSCC cells with lentiviral-based stable overexpression or knockdown of ASB6. We found that ASB6 overexpression increases, whereas ASB6 knockdown decreases, the potential of tumor-sphere formation, colony formation, and expression of Oct-4 and Nanog. While knockdown of ASB6 decreases cell migration in vitro and lung metastasis in mice, the migratory potential was however not promoted by ASB6 overexpression. ASB6 knockdown down-regulates the level of vimentin, and the loss of filopodia formation became more prominent following CRISPR/Cas9-directed knockout of ASB6. Moreover, ASB6 was up-regulated when cells were grown in selective condition featured with a collateral effect of enhancing intracellular stress, and the level of endoplasmic reticulum (ER) stress was further increased by knockdown of ASB6. Thus, ASB6 may attenuate ER stress that would otherwise accumulate and subsequently impede the potential of cells to acquire or sustain the stemness properties and metastatic capacity, thereby enhancing the malignancy of OSCC by increasing the population of cancer stem or stem-like cells.
Keywords: ASB6, stemness, metastasis, endoplasmic reticulum (ER) stress, oral squamous cell carcinoma (OSCC)
Introduction
Oral squamous cell carcinoma (OSCC), like most solid cancers, is composed of a heterogeneous population of tumor cells, among which a portion of cancer cells, termed cancer stem cells (CSCs) or stem-like cells, are believed to be the main subpopulation responsible for sustained tumor growth [1, 2]. CSCs share the expression of stemness genes with adult or embryonic stem cells [2]. Not only are these cells extremely tumorigenic, CSCs also often harbor plasticity of differentiation as well as resistance to conventional chemotherapies and radiation [3]. CSCs have also been associated with epithelial-mesenchymal transition (EMT) [4], a developmental program hijacked by cancer cells to lose their epithelial polarity and concomitantly gain mesenchymal properties that reportedly promote metastasis, although the effect of stemness on EMT, as well as the contribution of EMT to metastasis, are still heavily debated [5, 6]. More recently, emerging evidence demonstrates epithelial-mesenchymal plasticity of cells during metastasis and unlinks the stemness and EMT, thus implying that tumorigenesis and metastasis can be separately regulated in CSCs [7]. Since relapse or recurrence following treatment is at least partly attributable to CSCs, investigation of the molecular mechanism underlying cancer cells to acquire or maintain stemness and metastatic potential is important.
ASB6 is a member of a protein family harboring two functional domains, the ankyrin repeat that mediates specific protein-protein interaction and serves as a platform for substrate recruitment, as well as the suppressor of cytokine signaling (SOCS) box motifs that in turn associates with cullin scaffold proteins (Cullin-2 or Cullin-5) and a RING finger protein (Roc1 or Rbx1) to form an ECS protein complex [8]. This ECS protein complex functions as an E3 ubiquitin ligase that, together with a ubiquitin- activating enzyme (E1) and ubiquitin-conjugating enzyme (E2), mediates the degradation of proteins that are recruited by ankyrin repeat [9]. ASB6 was found to be involved in signaling of insulin or insulin-like growth factor receptors and decrease the level of adaptor protein containing PH and SH2 domains (APS) in response to insulin stimulation [10]. Interestingly, we previously demonstrated that ASB6 is induced by areca nut extract, the major carcinogen in betel quid, in normal oral keratinocytes and OSCC cell lines, consistent with the clinicopathological analysis of OSCC patients showing that betel quid consumption positively correlates with increased ASB6 in tissue [11]. Importantly, ASB6 up-regulation is associated with reduced overall survival [11]. However, it is currently unknown whether, and if so, how ASB6 enhances the malignancy of OSCC and thus compromise the prognosis of cancer patients.
In cancers, aberrant activation of oncogenes often results in perturbation of protein homeostasis as well as dysregulation of DNA replication, causing so-called endoplasmic reticulum (ER) stress and replication stress [12, 13]. Nutrient deprivation and hypoxia that commonly occur during tumor expansion may also lead to accumulation of metabolic and oxidative stress [14, 15]. These cellular stresses likely function as a double-edged sword for CSCs, as the evidence suggesting promotive and inhibitory effects of cellular stress on CSCs are both provided [16-20]. Clearly, a further investigation of how CSCs fine-tune the stress response in order to enable the acquisition of stemness and alleviate stress-associated cytotoxicity may allow more effective targeting of this subpopulation.
In this study, we aim to determine the involvement of ASB6 in the malignancy of OSCC. We first determined whether ASB6 augments the stemness properties and is required to sustain the migratory and metastatic potential of OSCC cells. We also determined the circumstances in which ASB6 is preferentially induced, as well as how stem-like OSCC cells may benefit from ASB6 induction. As the clinical significance of ASB6 up-regulation has been reported [11], the current study revealing the role of ASB6 in modulation of malignant phenotype may possibly serve as a molecular basis for how up-regulation of ASB6 compromises the outcome of OSCC patients.
Results
ASB6 enhances the ability of OSCC cells to form single-cell colonies and tumor spheres
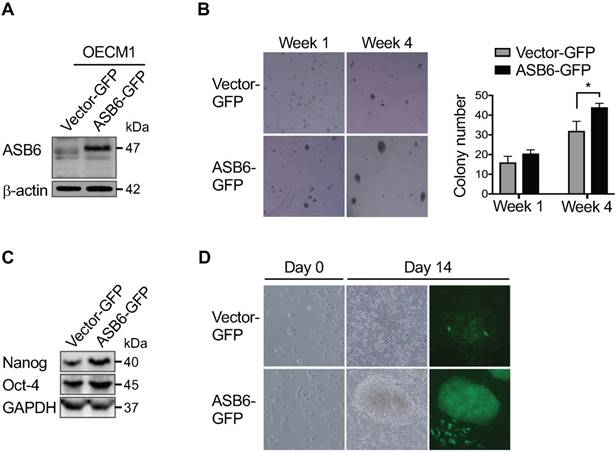
Our previous RNA-sequencing profiling of several distinct HN cancer cell lines identified ASB6 as a gene preferentially amplified as the stem-like subpopulation is enriched using serum-free EGF/ bFGF-supplemented selective media (Figure S1). To further determine the role of ASB6 in the promotion or maintenance of stemness properties, we established ASB6-overexpressing sublines of SAS and OECM1 cells that inherently express low levels of endogenous ASB6 (Figure S2). We found that cells with an overexpression of ASB6 formed significantly more soft agar colonies (Figure 1A and 1B), and the levels of pluripotency markers including Oct4 and Nanog were up-regulated compared to control cells (Figure 1C). In addition, we found that ASB6-overexpressing cells under defined culture condition formed sizable tumor spheres in 10-14 days, during which the comparable tumor-sphere formation by control cells remains rare (Figure 1D). Notably, the majority of tumor spheres were enriched with cells expressing GFP presumably with accumulated ASB6 protein, suggesting that ASB6 is involved in stem-like cell formation. Conversely, we employed lentiviral-based stable knockdown of ASB6 in SAS-M5, the cell line derived from pulmonary metastatic nodules of its tumorigenic parental SAS cells. We found that the depletion of ASB6 by either of the two different shRNA (shASB6#1 and #2) significantly decreased the in vitro soft agar colony-forming ability (Figure 2A & 2B), as well as the levels of Oct4, Nanog, and Bmi1 (Figure 2C). We also engineered CRISPR/Cas9-directed gene editing to knockout the ASB6 in SAS cells (Figure S3). Interestingly, we found that while the cell viability and proliferation are not affected by stable knockdown of ASB6 (Figure S4), the ASB6 knockout cells are neither steadily dividing nor viable particularly when plated at low density or grown in selective media. For this reason, we only obtained ASB6-knockout cell pools rather than single cell clones for subsequent experiments. Collectively, these results indeed support the notion that ASB6 is essential under certain circumstances and may play an intimate role in promotion or maintenance of clonogenic potential and tumorigenicity of OSCC cancer cells.
The effect of ASB6 overexpression on soft agar colony formation, stemness genes expression, and tumor sphere formation of OSCC cells. The OECM1 cells with stable overexpression of green fluorescent protein (vector GFP) or GFP-tagged ASB6 (ASB6-GFP) were validated by western blot for ASB6 (with as β-actin as the loading control) (A), and were subjected to anchorage-independent growth analysis by the soft agar assay (B), western blots analysis for Nanog and Oct-4 (with as the GAPDH as loading control) (C), and tumor sphere formation analysis (D).* P < 0.05.
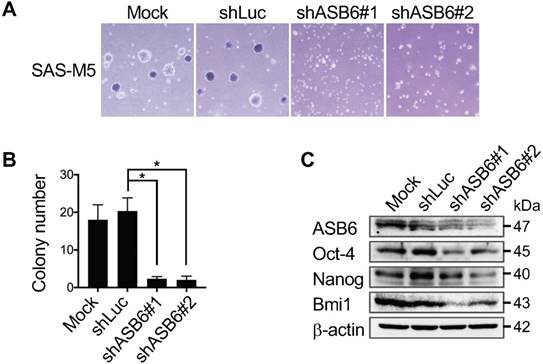
The effect of ASB6 knockdown on soft agar colony formation and stemness gene expression of OSCC cells. The mock-, control shRNA- (shLuc), or ASB6 shRNA- (shASB6#1 and shASB6#2) transduced SAS-M5 cells were subjected to anchorage-independent growth analysis by the soft agar assay (A), and western blots analysis for ASB6, Oct-4, Nanog, and Bmi1 (with as β-actin as the loading control) (B).* P < 0.05.
ASB6 sustains the migratory and metastatic potential of highly malignant OSCC cells
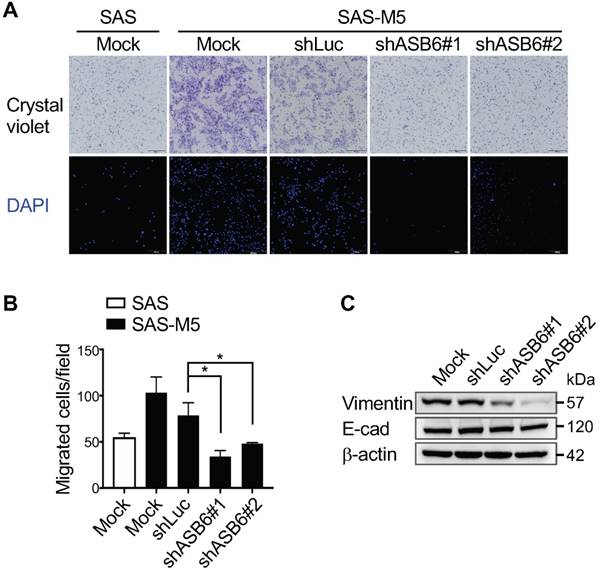
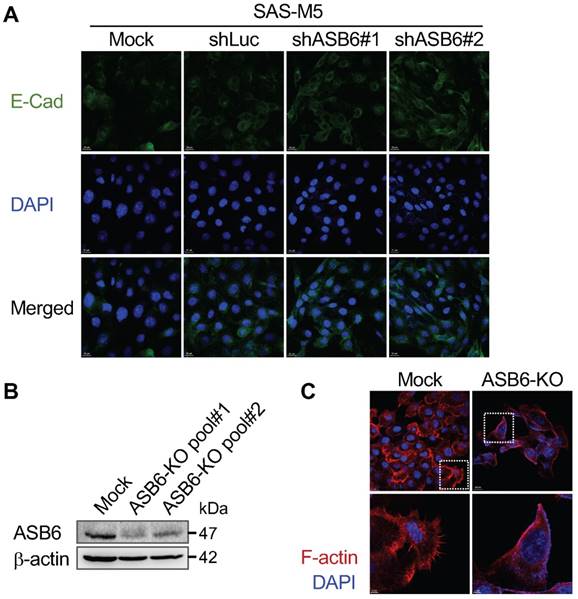
Given that the stemness properties of cancers are often characterized by both enhanced tumorigenic and metastatic potential, our finding that ASB6 promotes clonogenicity led us to examine its role in cell migration. We found that, compared to the parental or vector control (shLuc) cells, the migratory capacity of highly metastatic SAS-M5 cells assessed by the transwell assay is significantly suppressed following knockdown of ASB6 (shASB6#1 and #2) (Figure 3A and 3B). In addition, the level of vimentin that correlates with mesenchymal cell shape and motility was decreased in the ASB6 stable knockdown clones (Figure 3C). While a concomitant increase in cellular E-cadherin that more convincingly indicates the EMT was not demonstrated, the staining intensity of membranous E-cadherin in these cells appeared to be slightly higher than the parental or control cells (Figure 4A). Moreover, the loss of filopodia formation that has been implicated in reduced cell migration and tumor metastasis was noted following ASB6 knockdown and become more evident in the ASB6-knockout SAS cell pools (Figure 4B and 4C). Intriguingly, the overexpression of ASB6 in SAS was unable to augment in vitro migration of several cell lines examined (Figure 5A and 5B), and the expression levels of vimentin and E-cadherin were essentially unchanged (Figure 5C). Thus, the function of ASB6 is more likely to sustain than to establish cancer cell migratory capacity.
The effect of ASB6 knockdown on cell migration. The mock-transduced SAS cells, as well as the mock-, control shRNA- (shLuc), or ASB6 shRNA- (shASB6#1 and shASB6#2) transduced SAS-M5 cells were analyzed by transwell migration assay (A, B), and the transduced SAS-M5 cells were analyzed by western blots examining the levels of vimentin and E-cadherin (with as β-actin as the loading control) (C).
The effect of ASB6 knockdown or knockout on E-cadherin expression and filopodia formation. The mock-, control shRNA- (shLuc), or ASB6 shRNA- (shASB6#1 and shASB6#2) transduced SAS-M5 cells were analyzed by confocal microscopy immunofluorescence staining of E-cadherin (FITC, green) and nuclei (DAPI, blue) (A). The mock-transfected cells (Mock) and cell pools with different CRISPR/Cas9-directed knockout of ASB6 (ASB6-KO pool#1 and pool#2) were validated by western blots for ASB6 (B), and analyzed by confocal microscopy immunofluorescence staining of F-actin (Phalloidin, red) and nuclei (DAPI, blue). Enlarged images of the regions in the white boxes are shown in the lower panels (C).
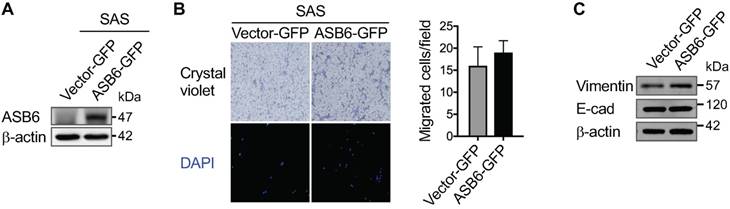
The effect of ASB6 overexpression on cell migration. The SAS cells with stable overexpression of GFP (vector GFP) or GFP-tagged ASB6 (ASB6-GFP) were validated by western blots examining the level of ASB6 (with as β-actin as the loading control) (A), and subjected to transwell migration assay analysis (B). * P < 0.05. The levels of vimentin and E-cadherin following ASB6 overexpression were examined by the Western blot analysis (C).
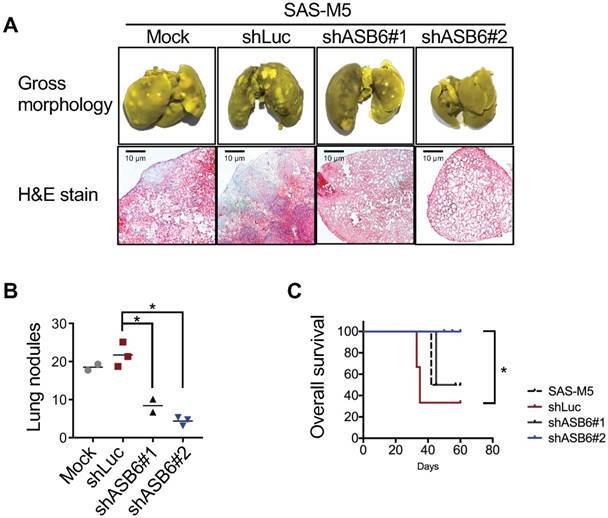
To further substantiate the role of ASB6 in metastatic colonization, four groups of nude mice were inoculated with parental SAS-M5 (Mock), vector control (shLuc), or ASB6-stable knockdown (shASB6#1 and #2) cells, respectively, via the lateral tail vein and were subsequently monitored for the incidences of lung metastasis. Lungs were harvested when mice develop the signs of severe dyspnea, pain or distress, weight loss, and/or inability to walk, and overt surface pulmonary nodules were counted and subjected to histological analysis. We showed that ASB6 down-regulation markedly reduced the formation of lung metastasis from these two stable knockdown clones (Figure 6A upper panel and 6B). Histologically, all lungs in mice inoculated with parental SAS-M5 or vector control cells were packed with tumor nests accompanied by inflammatory cellular infiltrates. In comparison, the pulmonary tumor masses and inflammatory infiltrates were decreased, and even more significantly, in mice inoculated with a particular ASB6 knockdown stable clone (shASB6#2) (Figure 6A, lower panel). Accordingly, the overall survival of these mice was significantly improved compared to the remaining groups (Figure 6C). Taken together, these results indeed suggest that ASB6 is essential for the distant colonization of at least the highly metastatic SAS-M5 OSCC cell line.
The effect of ASB6 knockdown on the in vivo metastasis. The mock-, control shRNA- (shLuc), or ASB6 shRNA- (shASB6#1 and shASB6#2) transduced SAS-M5 cells were injected into the tail vein of nude mice, and the lung of mice fixed with Bouin's solution were photographed and microscopic examined after H&E staining (magnification 50x) (A), and the pulmonary nodules were quantitated (B). The tumor-free survival of mice was examined by Kaplan-Meier survival analysis (C). * P < 0.05.
ASB6 is up-regulated as cellular stress is increased to attenuate ER stress
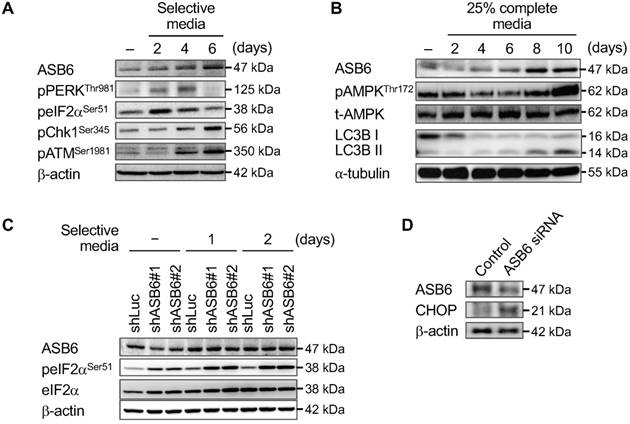
Since ASB6 is up-regulated as stem-like subpopulation is enriched by selective media, characterization of cellular response to the given media may potentially point to the foundation upon which ASB6 promotes stemness and sustains the invasive phenotype. We found that the phosphorylation of PERKThr981 and eIF2Ser51 that reflect the level of ER stress, as well as the phosphorylation of Chk1Ser345 and ATMSer1981 that reveal the level of replication stress are continuously increased in cells maintained in selective media, implying that up-regulation of ASB6 is possibly involved in certain cellular stress responses (Figure 7A). Interestingly, the ASB6 was also up-regulated following the induction of metabolic stress through nutrient deprivation as evidenced by both phosphorylation of AMP-activated protein kinase (AMPKThr172) and conversion of LC3 autophagy markers (Figure 7B). On the other hand, a moderate knockdown of ASB6 in cells grown in regular media sufficiently led to a marked increase in the levels of eIF2Ser51 phosphorylation and CHOP expression, and the phosphorylation of eIF2Ser51 was further augmented as these cells were transferred to selective media (Figure 7C). Moreover, we found that the ASB6-knockout SAS cell pools were rarely viable when cultured in selective media or subjected to nutrient deprivation (data not shown). Taken together, although whether and how ASB6 directly initiates cell migration and stemness acquisition has yet to be clearly demonstrated, our results indeed support the idea that ASB6 functions as an essential mediator to alleviate accumulation of cellular stress in the process whereby the stem-like and/or metastatic phenotype are promoted.
The effect of selective media, nutrient-deprivation media, and/or knockdown of ASB6 on the induction of ASB6 and cellular stresses. The SAS cells were grown in selective media (A) or nutrient-deprivation media containing 25% of ingredient of complete media (B) for 0 to 6 or 10 days, and the time-dependent induction of ASB6, ER stress, replication stress, metabolic stress, and autophagy were analyzed by western blots. The control shRNA- (shLuc), or ASB6 shRNA- (shASB6#1 and shASB6#2) transduced SAS-M5 cells were grown in regular or selective media for up to 2 days, and the induction of ASB6 and ER stress were analyzed by western blots. The β-actin and α-tubulin were used as the loading control.
Discussion
In this study, we found that ASB6 promotes and sustains the tumorigenicity and stemness properties when cells are cultured under the defined condition known to have a collateral effect of raising cellular stress. We also found that knockdown of ASB6 induces ER stress and decreases stemness, and the level of stress is further augmented as these ASB6-knockdown cells are cultured in selective media. These results imply that ASB6 provides endurance for cells to enhance their stemness in response to increased cellular stress. Corresponding to this notion are the findings that cancer progression to an advanced stage with an increase in highly malignant stem-like cells is often accompanied by the accumulation of cellular stress as a consequence of tumor expansion in the adverse environment [21, 22], and that ASB6 was found to be increased in the tissues of late-stage OSCC patients [11].
The ER stress has been closely associated with cancer stemness at different levels. Indeed, the induction of ER stress was demonstrated to force the differentiation of intestinal epithelial stem cells and colon cancer stem cells [19, 23]. On the other hand, the GRP78 that has been regarded as an ER stress mediator was found to be up-regulated in ovarian, oral, and breast cancer stem-like cells [24-26]. Notably, the increased expression of GRP78 can be, but not limited to, a consequence following ER stress elevation. As the ER stress response involves the activation of several pro-survival/apoptotic and phenotypic transitioning pathways, the effect of ER stress on cancer stemness is certainly determined by the balance between multiple signaling. Nonetheless, the ER stress has to be confined to the extent that cells are not overwhelmed by excess damage, thus providing opportunities for cancer cells to increase their stemness and enhance survival under stress.
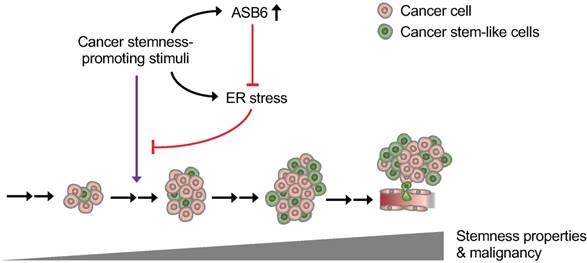
Graphical Abstract depicting the role of ASB6 in cancer progression. As cancer progression, the external stimuli (such as nutrient deprivation and hypoxia) that possibly exert a stemness-promotive effect on cancers may not only induce ASB6 but also cause ER stress. An increase in ASB6 subsequently attenuates the accumulation of ER stress, thus allowing cells to acquire stemness properties and sustain metastatic potential.
Since the cancer stemness is related to both the ASB6 and ER stress, whether and how ASB6 mediates the response to ER stress needs to be discussed. Thus far, the direct link between ASB6 and ER stress has yet well recognized; however, the studies on other members of this protein family may provide a hint of the potential involvement of ASB6 in ER stress. With this respect, the ASB11 was identified as an ER resident ubiquitin ligase [27], and the ASB8 was characterized as the ER metabolism-related gene [28]. Moreover, the ASB7 is recently identified as an ER stress responsive protein [29], and the involvement of several other ubiquitin E3 ligases in the regulation of CSCs are also reported [30]. As such, attenuation of ER stress may be the basis by which ASB6 promotes and sustains the cancer stemness.
While our results suggest a promotive role of ASB6 in tumorigenicity and stemness, it has to be noted that this effect becomes prominent only when cells are grown in the selective media. The growth of cells with or without an overexpression or knockdown of ASB6 is nearly indistinguishable if cells are cultured in regular media. These findings imply that ASB6 alone may be less efficient to initiate the acquisition of stemness properties. In fact, various conditions such as chemotherapy and radiation have been reported to induce stemness genes in cancers and facilitate tumor cells to transform into high-stemness cancer cells [31, 32], suggesting that the development of CSCs is indeed triggered by an interaction of multiple proteins rather than by a single gene. Notably, we found that knockout, but not knockdown, of ASB6 significantly compromises cell growth even under the normal condition. Because presumably the difference between knockout and knockdown of coding genes is that the latter may retain low levels of protein expression [33], this finding implies that a minimum level of ASB6 is required for cancer cells to sustain their growth. That the vulnerability of ASB6-knockout cells to any further insults also provides a plausible explanation as to why these cells are not viable when they are individually seeded, thus preventing us to isolate a clone derived from a single cell with knockout of ASB6.
Given that knockdown of ASB6 decreases cancer cell migration and metastasis, our finding that ASB6 overexpression, however, does not increase the migratory potential is intriguing. Metastatic progression requires a tumor cell to gain motility, invasiveness, and anoikis resistance [34]. Regardless of whether EMT is a prerequisite for metastasis, the dissolution of cadherin-mediated intercellular adhesion that allows separation from primary tumor remains the beginning and a crucial step of tumor spreading [35]. Since we found that neither an overexpression nor knockdown of ASB6 significantly affects the levels of E-cadherin and β-catenin, ASB6 is likely less involved in the regulation of cell-cell adhesion molecules. Subsequently, knockdown of ASB6 that reduces vimentin expression and filopodia formation certainly decreases the migratory capability [36, 37]. On the other hand, even if ASB6 overexpression may possibly increase the expression of vimentin filaments and the amount of filopodial outgrowth, the adhesion junctions that anchor cells between neighboring cells and extracellular matrix may still hamper individual cell migration. Therefore, our findings showing that cell migration is decreased by ASB6 knockdown but not increased by ASB6 overexpression seems to be reasonable and probable.
Although the presented results favor the notion that the promotive role of ASB6 in stemness properties is attributed to its function in the regulation of ER stress response, a limitation of this study is the lack of identification of its interacting proteins. In fact, our attempts to identify the substrates of this E3 ligase using co-immunoprecipitation and mass spectrometry were not successful, which was presumably due to the unavailability of ASB6 antibodies applicable for co-immunoprecipitation. We also found that further overexpressing ASB6 in cells, such as SAS-M5, that already have a relatively high ASB6 expression level is extremely difficult. Possibly, the level of ASB6 reveals the status of cellular stress, and an exogenous protein overexpression that may perturb normal ER function tends to be interrupted or suspended in these cells that already have high ER stress.
In conclusion, we demonstrated that ASB6 promotes the tumorigenicity and stemness properties of OSCC cells. Whereas the exogenous expression of ASB6 is unable to further increase the migratory capacity, ASB6 is indeed needed for cells to sustain the metastatic potential. We also showed that ASB6 attenuates ER stress that would otherwise accumulate and subsequently impede the enrichment of stem-like OSCC cells (Figure 7). As the clinical significance of ASB6 up-regulation has been reported, our study focusing on the molecular basis by which ASB6 enhances the malignancy of OSCC sheds light on the translational potential of targeting the ASB6 as an additional therapeutic strategy.
Materials and Methods
Cells and culture conditions
The SAS cancer cell line derived from squamous cell carcinoma of a tongue cancer patient (Japanese Collection of Research Bioresources Cell Bank, JCRB0260), as well as the OECM1 cancer cell line derived from squamous cell carcinoma of a gingival cancer patient [38] were obtained from Dr. Jeng-Fan Lo [39]. The SAS-VO3 cell line was established by serial xenotransplantation of SAS cells in nude mice [40]. The SAS-M5 cancer cell line established by repeatedly injecting SAS-VO3 cells into the tail vein of nude mice was a kind gift from Dr. Pei-Fen Su [37]. The SAS and its derived cell lines were grown in Dulbecco's Modified Eagle Medium (DMEM; #11-0550, Biological Industries) supplemented with 10% Fetal Bovine Serum (FBS; #10438-028, ThermoFisher Scientific) and 1% Penicillin-Streptomycin (#15140-122, ThermoFisher Scientific). The OECM1 cells were grown in Roswell Park Memorial Institute Medium (RPMI; #11-100, Biological Industries) supplemented with 10% FBS, 1% penicillin/ streptomycin, and 1% glutamine.
For tumor sphere formation, OSCC cell lines were cultured in tumor sphere media consisting of serum-free DMEM/F12 media (#12500-062, Gibco), N2 supplement (#17502048, Gibco), 20 ng/mL human recombinant basic fibroblast growth factor (bFGF, #100-18B, PeproTech) and 20 ng/mL Epidermal Growth Factor (EGF, #AF-100-15, PeproTech), termed the selective media. Cells were plated at a density of 7.5X104 cells/10-mm dishes, and the media was changed every other day until the formation of tumor sphere [39, 41].
DNA construction and lentiviral vector for stable lines
cDNA of human ASB6 (GenBank Accession No. NM_017873.3) was subcloned into pLenti-EF1α-GFP-2A-Puro mammalian expression (Abm, #LV067) using In-Fusion® HD Cloning Kit (Takaro Bio USA, Inc, #121416). Specifically, EcoRI site was used to insert ASB6 cDNA into the expression vector. For constructing a plasmid harboring sequencing that facilitates subcloning, cDNA of ASB6 was first amplified by PCR using the following primers to add the restriction sites: forward 5'- GCCTGCAGGTGAATTGCCACCATGCCGTTCCTGCACGGC-3'; reverse 5'-CGGTACTAGTGAATTTCAGATGTCATCTTCCACGGAGC-3'. The Lenti-GFP vector (Abm, #LV011-a) was used as a control vector. The ASB6 knockdown lentiviral pLKO.1 construct harboring human ASB6- specific shRNA sequences (5'-GCCTTAACAAGGTCCTTATAT-3' or 5'-GCAGATCCACAATACTGAGAA -3') or control shRNA (5'-CCGGACTTACGCTGAGTACTTCGAACTCG-3' or 5'-AGTTCGAAGTACTCAGCGTAAGTTTTTTG-3') were obtained from Academia Sinica (Taipei, Taiwan).
The transfection experiments for packaging the ASB6 expression or shRNA lentiviral vector were performed using PolyJet transfection reagent (#SL100688, SignaGen) according to the manufacturer's instructions. Specifically, HEK293 cells were transfected with a combination of pCMVΔR8.91 or pCDH (System Biosciences), pMD2.G (http://www.addgene.org/Didier_Trono), and ASB6 shRNA or expression vectors. The supernatant containing lentiviral vectors was collected 2 days after transfection. Stable knockdown or overexpression of ASB6 in OSCC cell lines was obtained by infection of cells with lentiviral supernatant. The transduced cells were then selected by puromycin (1 µg/mL) treatment for 5 days.
For generation of a plasmid for clustered, regularly interspaced, short palindromic repeat RNA-guided (CRISPR) targeting ASB6 exon1, two sets of primers for gRNA expression (5'-CACCGGGTACTCGAAGATGATCCTC-3' and 5'-AAACGAGGATCATCTTCGAGTACCC-3') were cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Plasmid #62988, Addgene). Meanwhile, the surrogate reporter plasmid was constructed by introducing the surrogate sequence (5'-GATCTCCTGCACGGCTTCCGGAGGATCATCTTCGAGTACCAGCCGGGTAC-3') into pEGFPC1. The co-transfection of CRISPR plasmids and surrogate reporter plasmid was conducted using PolyJet transfection reagent (#SL100688, SignaGen) according to the manufacturer's instructions. Following transfection, cells were subjected to flow cytometric sorting for selection of cells with knockout of ASB6 [42].
Western Blot
Cells were lysed in cell lysis buffer (#9803S, Cell signaling) containing PhosSTOP mini (#04906837001, Roche) and protease inhibitor cocktail (#04693124001, Roche). Protein concentration was measured using the protein BCA assay kit (#23227, ThermoFisher Scientific) and equal amount of protein were boiled in NuPAGE 4X LDS Sample buffer (#NP0007, ThermoFisher Scientific), separated by 15% SDS /PAGE, and transferred onto nitrocellulose membrane (#10484060, Bio-Rad). The membrane was incubated for 1 hour in blocking buffer (Tris-buffered saline with 0.1% Tween (TBS-T) and 5% non-fat dry milk) and then probed by overnight incubation at 4 °C with the following primary antibodies: Anti-ASB6 (GTX45549, GeneTex; SC-515649, Santa Cruz), anti-OCT3/4 (SC-9081, Santa Cruz), anti-Nanog (#AB9220, Millipore), anti-Vimentin (#550513, BD Pharmingen), anti-Fibronectin (#GTX112794, GeneTex), anti-E-cadherin (#610182, BD Pharmingen), anti-Phospho-ATM (#13050, Cell Signaling), anti-Chk1 (#2348, Cell Signaling), anti-phospho-PERKThr981 (#SC-32577, Santa Cruz), anti-phospho-eIF2α (#3597, Cell Signaling), anti- eIF2α (#9722, Cell Signaling), anti-phospho-AMPKThr172 (#9101L, Cell Signaling), anti-AMPK (#9158S, Cell Signaling), anti-LC3B (#2775, Cell Signaling), anti-α-tubulin (#SC-5286, Santa Cruz), anti-β-action (MA5-15739, ThermoFisher Scientific), and/or anti-GAPDH (#MA1-16757, ThermoFisher Scientific) antibodies. After washing in TBS-T, the blot was incubated with horseradish peroxidase-conjugated secondary antibodies, and detection was performed using the enhanced chemiluminescence system (#WBKLS0500, Millipore) as described by the manufacturer.
In vitro cell migration assay
For the Transwell migration assay, 5 x 104 cells in media without serum were plated in the top chamber of a Transwell (#3422, Corning). Media supplemented with normal serum (10% FBS) was placed in the lower chamber. The cells were incubated for 24 hours at 37°C and cotton swabs were used to remove cells that did not migrate through the pores. The cells on the lower surface of the membrane were stained with DAPI and fluorescent cells were identified at a magnification of 100X using a fluorescence microscope. The number of fluorescent cells in a total of five randomly selected fields was counted.
Anchorage-independent growth assay
A total of 1 x 104 cells was mixed with 2 mL of DMEM containing 0.35% agarose (#15510-027, ThermoFisher Scientific) and 0.5% FBS, and were then poured onto 60-mm plastic culture dishes pre-solidified with 2 mL DMEM containing 0.7% agarose and 15% FBS. The plates were incubated at 37°C for 7 to 10 days and then stained with 0.005% crystal violet (#HT901-8FOZ, Sigma-Aldrich). The number of colonies with a diameter ≥100 μm was scored. Triplicate samples were performed for each experiment.
Immunofluorescence staining
The cells seeded on 4-well chamber slides (#154941, LAB-TEK) at density of 3.5 x 104 cells per well were washed with 1x PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, and subsequently permeabilized using 0.05% Trion-X100 in PBS for 5 min. The fixed cells were then incubated in blocking solution (PBS supplemented with 10% FBS) for 1 h, followed by overnight incubation at 4ºC with anti-E-cadherin (#610182, BD Pharmingen), anti-F-action or TRITC-conjugated phalloidin (Sigma-Aldrich) antibodies. Cells were then washed and incubated with the FITC-conjugated secondary anti-mouse antibodies (#554001, BD Pharmingen) if necessary. After removal and proper washing of secondary antibody, the nuclei counterstained with 4',6-diamino-3-phenylidole, dihydrochloride (DAPI, #D1306, ThermoFisher Scientific) and examined using an Olympus FV1000 confocal microscope.
In vivo pulmonary metastasis assay
All animal procedures were approved by the Institutional Animal Care and Use Committee of the National Yang-Ming University (IACUC1060501; date of approval: May 5, 2017) and the Taipei Veterans General Hospital (IACUC2017-238; date of approval: January 16, 2018). BALB/c nude mice were purchased from National Laboratory Animal Center (Taipei, Taiwan) and were randomly grouped into sets of 2~3 mice each. A total amount of 5 x 105 cells was individually injected into mice via the tail vein. The mice were weighed every three days. The date of animal death or euthanization was recorded, and the lung tissue from these mice was harvested and fixed with Bouin's solution (#HT10132, Sigma-Aldrich). Peripheral and superficial lung nodules were counted under a dissecting microscope before paraffin embedded. Lung tissue histology of H&E-stained sections was assessed by a pathologist.
WST8 assay
A total of 7 x 103 cells (100 μL/well) was seeded in a 96-well plate and incubated at 37°C, 5% CO2 incubator for 18 hours. Cells were then added with 10 μL of the Cell Counting Kit-8 reagent (CCK-8, #CK04, Dojindo Molecular Technologies) to each well of the plate. After 3 hours of incubation, the absorbance at 450 nm was measured using a microplate reader.
Statistics
The t-test was used for statistical analysis. Overall survival was estimated using the Kaplan- Meier method. Comparisons were considered to be statistically significant difference when P <0.05.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study is supported by MOST-106-2314-B-075-077-MY3 and MOST-107-2314-B-075-023-MY3 from Ministry of Science and Technology, CI-107-8 from Yen Tjing Ling Medical Foundation, and V107C-042 from Taipei Veterans General Hospital, Taiwan.
Authors' contribution
Hung KF, Kao SY, and Lo JF participated in the design of the study and the sample preparation. Hung KF and Liao PC performed statistical analysis. Liao PC, Chen CK, Chiu YT performed the experiments. Hung KF, Cheng DH, Kawasumi M, and JF Lo drafted the manuscript. All authors read and approve this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973-8
2. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
3. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
4. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E. et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256-60
5. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
6. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
7. Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, Maynard C. et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016;14:2281-8
8. Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS. et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114-9
9. Kohroki J, Nishiyama T, Nakamura T, Masuho Y. ASB proteins interact with Cullin5 and Rbx2 to form E3 ubiquitin ligase complexes. FEBS Lett. 2005;579:6796-802
10. Wilcox A, Katsanakis KD, Bheda F, Pillay TS. Asb6, an adipocyte-specific ankyrin and SOCS box protein, interacts with APS to enable recruitment of elongins B and C to the insulin receptor signaling complex. J Biol Chem. 2004;279:38881-8
11. Hung KF, Lai KC, Liu TY, Liu CJ, Lee TC, Lo JF. Asb6 upregulation by Areca nut extracts is associated with betel quid-induced oral carcinogenesis. Oral Oncol. 2009;45:543-8
12. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25:563-73
13. Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276-89
14. Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323-32
15. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479-96
16. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell. 2017;168:692-706
17. Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B. et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572-6
18. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R. et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066-77
19. Wielenga MCB, Colak S, Heijmans J, van Lidth de Jeude JF, Rodermond HM, Paton JC. et al. ER-Stress-Induced Differentiation Sensitizes Colon Cancer Stem Cells to Chemotherapy. Cell Rep. 2015;13:489-94
20. Chang CW, Chen YS, Tsay YG, Han CL, Chen YJ, Yang CC. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018;9:194
21. Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012;198:281-93
22. Chang SJ, Wang TY, Tsai CY, Hu TF, Chang MD, Wang HW. Increased epithelial stem cell traits in advanced endometrial endometrioid carcinoma. BMC Genomics. 2009;10:613
23. Heijmans J, van Lidth de Jeude JF, Koo BK, Rosekrans SL, Wielenga MC, van de Wetering M. et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013;3:1128-39
24. Mo L, Bachelder RE, Kennedy M, Chen PH, Chi JT, Berchuck A. et al. Syngeneic Murine Ovarian Cancer Model Reveals That Ascites Enriches for Ovarian Cancer Stem-Like Cells Expressing Membrane GRP78. Mol Cancer Ther. 2015;14:747-56
25. Wu MJ, Jan CI, Tsay YG, Yu YH, Huang CY, Lin SC. et al. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol Cancer. 2010;9:283
26. Bartkowiak K, Effenberger KE, Harder S, Andreas A, Buck F, Peter-Katalinic J. et al. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J Proteome Res. 2010;9:3158-68
27. Andresen CA, Smedegaard S, Sylvestersen KB, Svensson C, Iglesias-Gato D, Cazzamali G. et al. Protein interaction screening for the ankyrin repeats and suppressor of cytokine signaling (SOCS) box (ASB) family identify Asb11 as a novel endoplasmic reticulum resident ubiquitin ligase. J Biol Chem. 2014;289:2043-54
28. Civelek M, Manduchi E, Riley RJ, Stoeckert CJ Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453-61
29. Anasa VV, Manickam M, Talwar P, Ravanan P. Identification of ASB7 as ER stress responsive gene through a genome wide in silico screening for genes with ERSE. PLoS One. 2018;13:e0194310
30. Kang B, Sun XH. Regulation of cancer stem cells by RING finger ubiquitin ligases. Stem Cell Investig. 2014;1:5
31. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;7:e43628
32. Hu X, Ghisolfi L, Keates AC, Zhang J, Xiang S, Lee DK. et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle. 2012;11:2691-8
33. Xu XM, Yoo MH, Carlson BA, Gladyshev VN, Hatfield DL. Simultaneous knockdown of the expression of two genes using multiple shRNAs and subsequent knock-in of their expression. Nat Protoc. 2009;4:1338-48
34. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679-95
35. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
36. Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838-51
37. Chen YS, Huang WL, Chang SH, Chang KW, Kao SY, Lo JF. et al. Enhanced filopodium formation and stem-like phenotypes in a novel metastatic head and neck cancer cell model. Oncol Rep. 2013;30:2829-37
38. Yang CY, Meng CL. Regulation of PG synthase by EGF and PDGF in human oral, breast, stomach, and fibrosarcoma cancer cell lines. J Dent Res. 1994;73:1407-15
39. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH. et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14:4085-95
40. Chen CY, Chiou SH, Huang CY, Jan CI, Lin SC, Tsai ML. et al. Distinct population of highly malignant cells in a head and neck squamous cell carcinoma cell line established by xenograft model. J Biomed Sci. 2009;16:100
41. Lo JF, Yu CC, Chiou SH, Huang CY, Jan CI, Lin SC. et al. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res. 2011;71:1912-23
42. Ramakrishna S, Cho SW, Kim S, Song M, Gopalappa R, Kim JS. et al. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat Commun. 2014;5:3378
Author contact
![]() Corresponding authors: Kai-Feng Hung, Department of Medical Research, Division of Translational Research, Taipei Veterans General Hospital, No. 201, Sec. 2, Shipai Rd., Beitou Dist., Taipei, 11217, Taiwan. Telephone: 886-2-28757434 #7382; E-mail: kfhunggov.tw and Jeng-Fan Lo, Institute of Oral Biology, National Yang-Ming University, No. 155, Sec. 2, Li-Nong St., Taipei, 11221, Taiwan. Telephone: 886-2-28267222; Fax: 886-2-28264053; E-mail: jfloedu.tw.
Corresponding authors: Kai-Feng Hung, Department of Medical Research, Division of Translational Research, Taipei Veterans General Hospital, No. 201, Sec. 2, Shipai Rd., Beitou Dist., Taipei, 11217, Taiwan. Telephone: 886-2-28757434 #7382; E-mail: kfhunggov.tw and Jeng-Fan Lo, Institute of Oral Biology, National Yang-Ming University, No. 155, Sec. 2, Li-Nong St., Taipei, 11221, Taiwan. Telephone: 886-2-28267222; Fax: 886-2-28264053; E-mail: jfloedu.tw.