Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1215-1224. doi:10.7150/ijbs.29741 This issue Cite
Research Paper
The Nuclear Export and Ubiquitin-Proteasome-Dependent Degradation of PPARγ Induced By Angiotensin II
Li Sun1,2, Ka Bian3 ![]()
1. Tianjin Medical University General Hospital; Tianjin Neurological Institute; Key Laboratory of Post-trauma Neuro-repair and Regeneration in Central Nervous System, Ministry of Education and Tianjin City, Tianjin, 300052, PR China
2. Murad Research Institute for Modernized Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, PR China
3. Department of Biochemistry and Molecular Medicine, The George Washington University, Ross Hall 2300 Eye Street, NW, Washington, DC 20037, USA
Received 2018-9-6; Accepted 2019-3-12; Published 2019-5-11
Abstract

Evidence has documented local angiotensin II (Ang II) as a pro-oxidant and pro-inflammatory molecule contributes to progressive deterioration of organ function in diseases. Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated transcription factor, plays crucial roles in protection against oxidative stress and inflammation. Ang II stimulation decreases PPARγ protein in multiple types of cells, while the regulatory role of Ang II on PPARγ is not clear. Here we show that Ang II down-regulated PPARγ in ECV304 cells through 2 actions, inducing nuclear export and loss of protein. The nuclear export of PPARγ occurred transiently in the early phase, while the reduction in PPARγ protein happened in the later phase and was more persistent. Both alterations in PPARγ were accompanied by the decrease in PPARγ-DNA binding activity. Reduction of PPARγ protein levels was also coupled with the inhibition of PPARγ target genes. In addition, activation of PPARγ by its ligand troglitazone could completely counteract both 2 actions of Ang II on PPARγ. Further studies demonstrated that the decline of PPARγ protein was in association with ubiquitin-proteasome-dependent degradation, which was supported by the increase in polyubiquitin-PPARγ conjugates and the inhibitory effect of lactacystin, a specific proteasome inhibitor, on the loss of PPARγ. Taken together, this study uncovers a novel means by which Ang II down-regulates PPARγ. This down-regulation disrupts nuclear PPARγ function, which may lead to the loss of beneficial effects of PPARγ in response to Ang II stress.
Keywords: angiotensin II, peroxisome proliferator-activated receptor γ, nuclear export, proteasome, ubiquitin
Introduction
The renin-angiotensin system (RAS) is a hormonal cascade that functions in the homeostatic control of arterial pressure, tissue perfusion, and extracellular volume. The classical view of the RAS pathway begins with renin cleaving its substrate, angiotensinogen, to produce the inactive peptide, angiotensin I, which is then converted to angiotensin II (Ang II) by angiotensin-converting enzyme (ACE). ACE activation of Ang II occurs most extensively in the lung. However, it is now recognized that there are also several tissue (local) renin-angiotensin systems. In particular, Ang II generation at the tissue level by these local systems appears to have physiologic effects that are as important as circulating Ang II and, under some circumstances, more important than circulating Ang II [1]. The discovery of local and intracellular RAS highlights several prominent non-haemodynamic effects of Ang II including pro-inflammatory, pro-oxidative, proliferative and pro-fibrotic activities. Ang II promotes reactive oxygen species (ROS) production, cell apoptosis, extracellular matrix remodeling, mediates several key events of the inflammatory processes leading to tissue injury [2-5].
Peroxisome proliferators-activated receptor γ (PPARγ) belongs to ligand-activated nuclear receptors that act as transcription regulators of genes encoding proteins involved in glucose and lipid metabolism. Recent studies have shown that, in addition to its classical role, PPARγ exerts significant anti-inflammatory and anti-oxidative properties in multiple cells and tissues. For example, activation of PPARγ either by the ligand thiazolidinediones (TZDs) or by the overexpression of a constitutively active mutant of PPARγ in cultured endothelial cells (ECs) significantly reduced NF-κB activation and intercellular adhesion molecule-1 (ICAM-1) expression [6-7]. In PC12 neuronal cell, PPARγ loss of function increases susceptibility to H2O2- or β-amyloid (Aβ)-induced oxidative toxicity, whereas PPARγ overexpression can prevent H2O2- or Aβ-induced ROS production and cell insult [8]. More recently, down-regulation of PPARγ protein and activity by cigarette smoke promotes inflammatory pathways in pulmonary epithelial cells [9]. These data suggest that PPARγ is important in cellular defense against inflammatory and oxidative injuries. It has been reported that Ang II stimulation can decrease the expression of PPARγ in various cells, such as endothelial progenitor cells (EPCs) [10], vascular smooth muscle cells (VSMCs) [11], cardiac myofibroblasts [12], hepatocytes [13], and macrophages [14], while the regulatory role of Ang II on PPARγ is not yet clearly explored.
To address this issue, we investigated the effect of Ang II on PPARγ using ECV304 cell line. We provide evidence that Ang II regulates endogenous PPARγ through two actions: (1) inducing the early phase of PPARγ nuclear export into cytoplasm, and (2) causing the late phase of PPARγ protein degradation via ubiquitin-proteasome-dependent mechanism. These findings provide new clues in understanding the role of Ang II in PPARγ signaling regulation.
Materials and Methods
Materials
Angiotensin II, troglitazone, lactacystin, and trypan blue dye were purchased from Sigma-Aldrich (St. Louis, MO). Anti-PPARγ protein polyclonal antibody (used for immunoprecipitations) and monoclonal antibody (used for western immunoblots), anti-Histone H1 antibody were from Santa Cruz Biotechnology Inc (Santa Cruz, CA). α-Tubulin antibody was obtained from Sigma-Aldrich (St. Louis, MO). Ubiquitin antibody was purchased from Cayman Chemical (Ann Arbor, MI).
Cell culture
ECV304 cells were obtained from Shanghai Institute of Cell Biology in the Chinese Academy of Sciences. In our experiments, the cells were cultured at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (containing 10% fetal calf serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 2 mM glutamine, purchased from Invitrogen). Cells were switched to serum-free medium for 24 h when the confluence was about 95% and then received treatments in serum-free medium.
Nuclear and cytoplasmic extraction
After treatments, the cells were collected and washed twice with cold PBS. Nuclear and cytoplasmic proteins were separated with the Active Motif nuclear extract kit (Active Motif, Carlsbad, CA, USA) as we described [15].
Western blot analysis
Cells were washed in ice-cold PBS and then treated with a RIPA protein lysis buffer (Beyotime) to prepare total protein lysates. The protein content was determined by Lowry assay. After boiling in loading buffer, samples were analyzed by loading on a 10% SDS-PAGE, which was then transferred onto a nitrocellulose membrane. PPARγ was detected with its antibody (Santa Cruz, CA) at 4°C overnight. Antibody reactivity was visualized by chemiluminescence (ECL kit, Amersham).
PPARγ-DNA binding assay
To detect PPARγ nuclear activity in response to Ang II, PPARγ-DNA binding was monitored by using nuclear fraction from cells incubated 10 min or 4 h with Ang II (0.001-1 μM). Quantitative measurement of PPARγ binding to the specific PPRE was performed using an ELISA-based assay (TransAM PPARγ transcription factor assay kit, Active Motif, Carlsbad, CA, USA) as we described [15]. Nuclear proteins were isolated with the Active Motif nuclear extract kit (Carlsbad, CA, USA). Immunoblotting of α-tubulin was used to detect cytoplasmic contamination in nuclear extracts.
Cell viability assay
Cell viability was determined by Trypan blue dye exclusion assay. After treatment, cells were resuspended and a small aliquot of cell suspension (0.3 ml) was harvested from each culture, labeled with 0.4 % w/v trypan blue and then counted using a haemocytometer to determine the percentage of cells that have clear cytoplasm (viable cells) versus cells that have blue cytoplasm (nonviable cells). Cell survival was also measured by 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) assay. The experimental procedure using MTT solution was performed exactly as we described recently [16].
Total RNA isolation and RT‑PCR amplification
Cells were harvested 10 min or 4 h after Ang II exposure. RNA extraction and RT-PCR procedures were carried out as we reported [17]. In brief, total RNA was isolated from ECV304 cells with Tri reagent (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. Total RNA was quantified by the absorbance at OD260. cDNA was converted from 1.5 μg total RNA. The primers used for PCR amplification were as follows: PPARγ, 5'-TCTCTCCGTAATGGAAGACC-3' and 5'-GCATTATGAGACATCCCCAC-3'; PPARγ target gene fatty acid-binding protein 4 (FABP4), 5'-GCCAGGAATTTGACGAAG-3' and 5'-ATCCCACAGAATGTTGTAGAGT-3'; and PPARγ target gene cytochrome P450 family 1 subfamily A member 1 (CYP1A1), 5'-TCTTTCTCTTCCTGGCTTC-3' and 5'-CTGTCTCTTCCCTTCACTCT-3'. Housekeeping genes GAPDH or β-actin was used for normalizing gene expression.
Immunoprecipitation
Cells were washed twice in ice-cold phosphate- buffered saline (PBS), then lysed on ice with PBS containing 1% Triton X-100, 1 mM EDTA, 1 mM PMSF, 1 μM pepstatin, 10 μM leupeptin and freshly prepared 10 mM N-ethylmaleimide [18]. PPARγ was immunoprecipitated from whole cell lysates with polyclonal anti-PPARγ antibody followed by rotating for 3 h at 4 °C. Immunocomplex was captured by adding protein G agarose bead slurry (Santa Cruz, CA) and rocked overnight at 4°C. Then, agarose beads were collected by centrifugation and washed in PBS for three times. Immunocomplexes were dissociated from agarose beads by boiling with SDS-PAGE sample buffer for 5 min. PPARγ-ubiquitin conjugates were analyzed by immunoblotting after SDS-PAGE using monoclonal anti-PPARγ and polyclonal anti-ubiquitin antibodies. A HRP-conjugated VeriBlot for IP secondary antibody (Abcam, Cambridge, MA, USA) was used to exclude interference from the antibody heavy and light chains.
Statistical analysis
The experimental data are expressed as mean ± SEM and SPSS 11.0 software package was used for data processing. One-way ANOVA was used to compare the means of different groups. Comparisons between two groups were conducted by t-test. A P value less than 0.05 was considered as statistically significant.
Results
Ang II down-regulates PPARγ via both nuclear export and decrease in protein expression
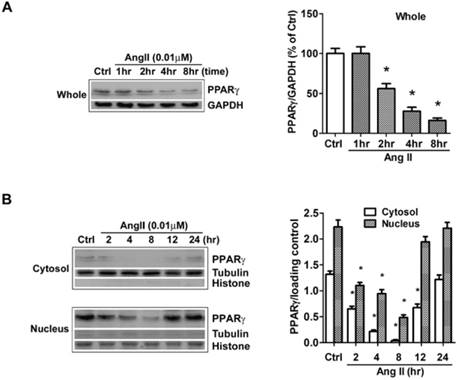
The effects of Ang II on PPARγ expression and subcellular distribution were examined by Western blot in our study. Clearly, the analysis of whole cell extracts revealed that PPARγ levels were stable for approximately 1 hour after cells were exposed to Ang II, then declined markedly thereafter (Fig. 1A). As PPARγ is a nuclear receptor, we then dissected the impact of Ang II on PPARγ in both cytoplasmic and nuclear fractions (Fig. 1B). The purity of cell fractionation was verified by the clear separation between the nuclear histone H1 and the cytosolic tubulin. Under the basal condition, PPARγ proteins were observed in both subcellular fractions with a much higher level in the nucleus. Upon the stimulation with Ang II, PPARγ proteins decreased in both cytosol and nuclear compartments at 2 h, reached the maximal level at about 4-8 h, and gradually returned to the basal level within 12-24 h.
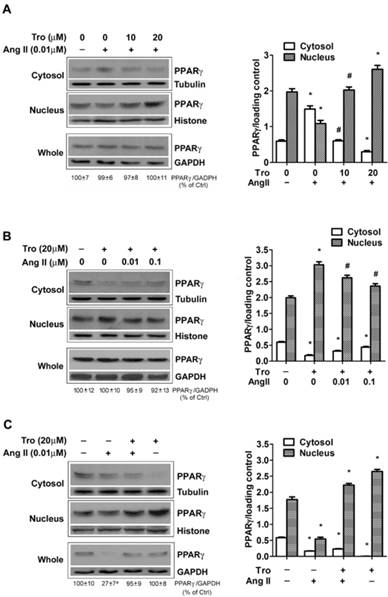
Ang II reduces the levels of PPARγ protein in both cytosol and nuclear fractions. The cells were switched to serum-free medium for 24 hours, and then treated with 0.01 μM Ang II for indicated time periods. PPARγ protein expression in (A) total cell lysates (GAPDH loading control), (B) nuclear (Histone H1 loading control) and cytoplasmic fractions (Tubulin loading control) were analyzed with anti-PPARγ antibody by Western blotting. The bar graph illustrates the densitometrical analysis of the related bands. Values were normalized to corresponding loading control and are mean ± SEM (n = 6). *P<0.05 versus corresponding control (Ctrl).
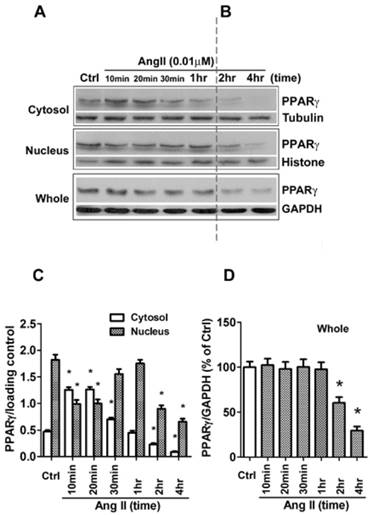
Besides the level of protein expression, the control of nuclear localization is also an important mechanism to regulate PPARγ activity [19]. Nucleo-cytoplasmic shuttling of PPARγ has been noticed in cells stimulated with mitogen tetradecanoyl phorbol acetate (TPA) [20]. To detect whether Ang II provokes shuttling of PPARγ from the nucleus to the cytosol, we analyzed the Ang II-driven dynamic of the subcellular distribution of PPARγ by further subdividing the exposure time of Ang II. As demonstrated in Figs. 2A and 2B (quantified in Figs. 2C and 2D), there are two time-phases of the change in intracellular PPARγ. The early phase (0-1 h) showed that the addition of Ang II resulted in a rapid increase in cytosolic PPARγ 10 and 20 min with a concomitant decrease in nuclear PPARγ (Fig. 2A), suggesting an induction of nuclear export of PPARγ. Then, the nuclear content of PPARγ started to recover at 30 min and reverted to basal levels within 60 min (1 h) (Fig. 2A). These changes indicated that nuclear export of PPARγ was a rapid and transient process under the stimulation of Ang II. No detectable alteration in whole PPARγ expression was observed in the early phase. In contrast, as shown in Fig. 2B, a time-dependent repression of PPARγ protein was observed in both nucleus and cytoplasm at later phase, which was consistent with the results in Fig. 1B. A similar decrease of whole PPARγ was found in this later phase. In addition, the effect of Ang II was dose-dependent in inducing both nuclear export (Fig. 3A) and reduction in PPARγ (Fig. 3B).
Ang II induces nuclear export of PPARγ. The cells were switched to serum-free medium for 24 hours, then treated with 0.01 μM Ang II for indicated times. (A, B) PPARγ protein expression in total cell lysates (GAPDH loading control), nuclear (Histone H1 loading control), and cytoplasmic fractions (Tubulin loading control) were determined by Western blot analysis. (C, D) Quantitative analysis of A and B. At the indicated time points, the average grey scale of each band representing PPARγ in cytosol and nucleus (C), or whole PPARγ (D) was determined, respectively (n = 6). *P<0.05 versus corresponding control (Ctrl).
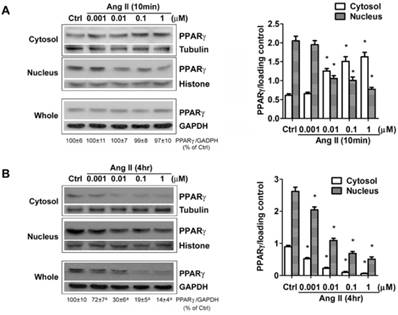
The down-regulation of PPARγ by Ang II is dose-dependent. Cells were serum-deprived for 24 hours and treated with Ang II (0.001, 0.01, 0.1, and 1μM) for 10 minutes (A) and 4 hours (B), respectively. PPARγ protein expression in total cell lysates (GAPDH loading control), nuclear (Histone H1 loading control) and cytoplasmic fractions (Tubulin loading control) were analyzed by anti-PPARγ antibody. Values were normalized to corresponding loading control and are mean ± SEM (n = 6). *P<0.05 versus corresponding control (Ctrl). aP<0.05 versus Ctrl.
The down-regulation of PPARγ by Ang II is associated with reduction in PPARγ nuclear activity
The net effect of Ang II on PPARγ was the reduction in PPARγ nuclear protein. As a nuclear receptor, the level of PPARγ in the nucleus is closely correlated with its transcriptional activity. To confirm whether exposure to Ang II inhibits PPARγ nuclear activity, an ELISA-based assay was carried out to examine PPARγ specific DNA binding activity. Cell cultures were incubated for 10 min or 4 h with increasing concentrations of Ang II. Nuclear proteins were extracted, then incubated in a 96-well plate immobilized synthetic oligonucleotides containing the specific PPRE sequences. As expected, PPARγ-DNA binding activity was inhibited in a concentration-dependent manner (Figs. 4A and 4B), which was in parallel with the alterations of nuclear PPARγ protein induced by 10 min- (Fig. 3A) and 4 h- (Fig. 3B) treatment of Ang II, respectively.
Ang II treatment suppresses nuclear activity of PPARγ. Cells were serum-deprived for 24 hours and treated with Ang II (0.001, 0.01, 0.1, and 1μM) for 10 minutes and 4 hours, respectively. (A, B) Nuclear proteins were extracted and nuclear activity of PPARγ was quantified by PPARγ-DNA binding activity utilizing the PPARγ transcription factor assay kit. Data are expressed as mean ± SEM (n = 6). *P<0.05 versus Ctrl (control). (C) Ang II decreases PPARγ regulated gene transcription. PPARγ target genes (CYP1A1 and FABP4) mRNA expression was evaluated by RT-PCR. Gene expression was normalized to GAPDH. Data are expressed as mean ± SEM (n = 6). *P<0.05 versus control (Ctrl). (D) Cells viability was measured by the trypan blue exclusion and MTT assay. The percentage of surviving cells compared with that of vehicle-treated control cells is shown from six independent experiments.
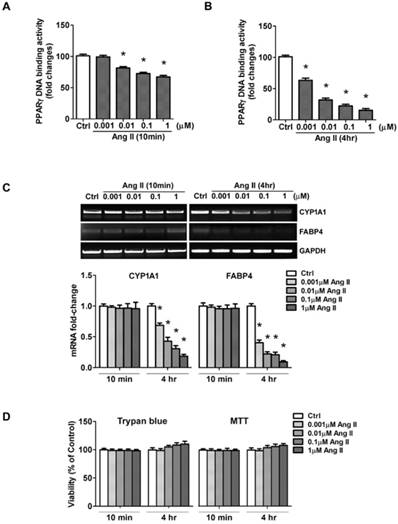
In addition, we performed the functional analysis of PPARγ in Ang II-treated cells by assessing transcription of the confirmed gene targets of PPARγ, CYP1A1 and FABP4. No significant alterations were detected 10 min after exposure to Ang II, whereas a dose-dependent inhibition in CYP1A1 and FABP4 mRNA was observed by the 4 hours exposure to Ang II (Fig. 4C). These data suggested Ang II-induced functional inactivation of PPARγ.
To rule out the possibility that the loss of PPARγ activity could be due to a loss of cell viability, we measured cell viability by trypan blue dye exclusion and MTT assay. As shown in Fig. 4D, Ang II slightly increased cell survival rate, but the difference was not statistical significant. These results suggested the loss of PPARγ activity mediated by acute exposure of cells to Ang II was not due to damage to cell viability.
PPARγ ligand reverses Ang II-induced down-regulation of PPARγ
We previously reported that PPARγ ligand activates PPARγ and induces PPARγ nuclear translocation [15, 21], we next examined whether exposing cells to PPARγ ligand could counteract the action of Ang II on PPARγ down-regulation. In the first set of test, to observe the effect of Tro on Ang II-induced PPARγ nuclear export, cells were treated with a synthetic ligand of PPARγ troglitazone (Tro) 3 h prior to the addition of Ang II for 10 min (Figs. 5A and 5B). Cultures treated with Ang II alone showed marked nucleo-cytoplasmic translocation (Fig. 5A). Significantly, this PPARγ nuclear export was prevented by pre-incubation with different concentrations of Tro (10 and 20 μM). Furthermore, the higher concentration of Tro (20 μM) not only completely blocked the effect of Ang II, but further promoted PPARγ nuclear translocation. This counteracting state between Ang II and PPARγ ligand was also evidenced by the result that Tro-induced PPARγ nuclear translocation could be partially inhibited by further increasing the concentration of Ang II (Fig. 5B). In the second set of test, we evaluated the effect of Tro on Ang II-induced reduction in PPARγ protein by extending Ang II exposure time to 4 hours. As illustrated in Fig. 5C, Tro pretreatment not only promoted PPARγ translocation into the nucleus but effectively maintained PPARγ protein in cells treated with the combination of Tro and Ang II. Overall, these findings suggested that PPARγ ligand could completely counteract both 2 actions of Ang II on PPARγ.
PPARγ ligand reverses Ang II-induced down-regulation of PPARγ. Cells were cultured in serum-free medium for 24 hours, and then challenged to Ang II and the PPARγ ligand troglitazone (Tro) treatment. PPARγ protein expression in total cell lysates (GAPDH loading control), nuclear (Histone H1 loading control), and cytoplasmic fractions (Tubulin loading control) were analyzed by anti-PPARγ antibody. (A) Cells were treated with Tro (0, 10, or 20 μM) for 3 hours, and then incubated with Ang II (0.01 μM) for 10 minutes. Data are expressed as mean ± SEM (n = 6). *P<0.05 versus all other groups. #P<0.05 versus all other groups except control (untreated). (B) Cell cultures were exposed to Tro (20 μM) 3 hours prior to the addition of Ang II (0, 0.01, 0.1 μM) for 10 minutes. *P<0.05 versus all other groups. #P<0.05 versus control (untreated) and Tro alone. (C) Cells were treated with Tro (20 μM) for 3 hours, and then incubated with Ang II (0.01 μM) for additional 4 hours. Data are expressed as mean ± SEM (n = 6). *P<0.05 versus all other groups. aP<0.05 versus control (Ctrl).
Ang II induces PPARγ degradation by increasing polyubiquitination of PPARγ
Ang II-induced rapid loss of PPARγ protein occurred within 2 hours and no alteration in PPARγ mRNA was observed by RT-PCR (Fig. 6A), both of which suggested a possible involvement of protein degradation. We presumed the Ang II-induced loss of PPARγ may due to the activity of ubiquitin- proteasome system. To verify this hypothesis, we first examined whether the decline of PPARγ by Ang II is related to proteasome by treating cells with a specific proteasome inhibitor, lactacystin [22]. As shown in Fig. 6B, pretreatment of cells with 10 μM of lactacystin did not alter the basal level of PPARγ, however, the proteasome inhibitor completely prevented Ang II-induced decline in PPARγ protein expression.
Ang II induces ubiquitin-proteasome-dependent degradation of PPARγ. (A) mRNA expression of PPARγ in Ang II-treated cells. Cells were treated with Ang II (0.01 μM) for 30 minutes, 1 hour, and 2 hours, respectively. Total RNA was isolated and PPARγ mRNA expression was assessed by RT-PCR. Housekeeping genes GAPDH and β-actin were used for normalizing gene expression. Data are expressed as mean ± SEM (n = 6). (B, C) Cell cultures were pretreated with 10 μM lactacystin for 90 minutes and then incubated in the presence or absence of 0.01 μM Ang II for 2 hours. (B) The proteasomal inhibitor lactacystin prevents Ang II-induced depletion of PPARγ. Whole cell lysates were processed for immunoblotting with anti-PPARγ antibody. The bar graph illustrates the densitometrical analysis of the related bands. Values were normalized to GAPDH and are mean ± SEM (n = 6). *P<0.05 versus all other groups. (C) Ang II treatment is associated with an increase in PPARγ-ubiquitin conjugates. Whole cell extracts were immunoprecipitated (IP) with Ab specific to PPARγ. The PPARγ immunoprecipitations were then immunoblotted (IB) with PPARγ Ab (left panel) or ubiquitin Ab (middle panel). For comparison, both PPARγ immunoprecipitations (middle panel) and 25 μg aliquots of whole cell lysates (right panel) were probed for ubiquitin. Molecular mass is indicated in kilodaltons on the left.
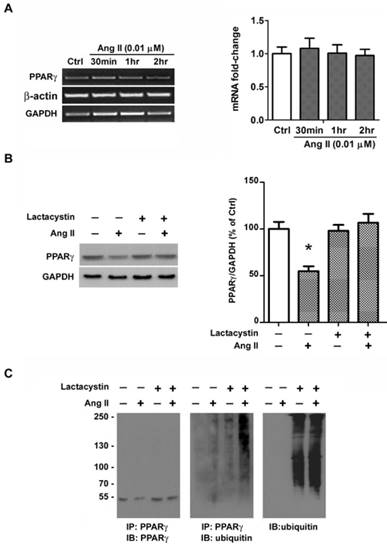
The recognition of protein substrates by proteasome requires covalent bond between polyubiquitin chain and the proteins. We next explored the effect of Ang II on PPARγ ubiquitylation with or without the pretreatment of proteasome inhibitor lactacystin to preserve short-lived ubiquitin (Ub) conjugates (Fig. 6C). PPARγ proteins were immunoprecipitated from whole cell extracts and the immunoprecipitations were then analyzed by immunoblotting using either an anti-PPARγ antibody (Fig. 6C, left panel) or an anti-ubiquitin antibody (Fig. 6C, middle panel). As shown in Fig. 6C, the loss of PPARγ was reversed by lactacystin pretreatment (Fig. 6C, left panel). Although treatment of cells with lactacystin alone had little effect on the level of ubiquitinated PPARγ, co-treatment with Ang II and lactacystin increased the amount of ubiquitination of PPARγ (Fig. 6C, middle panel). The Ub reactivity of the PPARγ immunoprecipitations showed a series of Ub Ab-reactive bands at higher molecular weight, indicating the polyubiquitinated PPARγ molecule (Fig. 6C, middle panel). We also examined the effect of lactacystin on the ubiquitination of proteins in whole cell lysates and found that lactacystin treatment, as expected, enhanced the accumulation of ubiquitinated proteins (Fig. 6C, right panel). These findings suggest that Ang II-stimulated loss of PPARγ protein may due to the ubiquitination of the protein and degradation by the proteasome pathway.
To rule out the possibility that PPARγ was degraded by lysosomal proteases, cells were treated with Ang II alone or with Ang II plus the lysosomal protease inhibitors NH4Cl [23]. NH4Cl (20 mM) pretreatment failed to show any significant effects on Ang II-induced PPARγ decline (data not shown), indicating the loss of PPARγ was not a result of the degradation by lysosomes.
Discussion
The data from our report revealed that Ang II regulated PPARγ through 2 modes of action, the early and rapid phase was characterized with exporting PPARγ from the nucleus to the cytoplasm while the later phase was composed with reduction in the protein levels of PPARγ. Concomitantly, the two actions of Ang II were accompanied by the suppression of PPARγ-DNA binding activity, and the decrease in PPARγ protein was also coupled with the inhibition of PPARγ target genes. Thus, the net effect of Ang II on PPARγ is the down-regulation of nuclear PPARγ protein and activity.
The regulation of transcription factor activity plays an important role in many biological processes. The expression levels regulation, post-translational modification, and protein stability affection are all key mechanisms known to influence transcription factor activity [24-25]. Nevertheless, the control of nuclear localization is another strategy used for a particular transcription factor to regulate its nuclear activity [19]. PPARγ was shown in our data and several other studies [26-27] to localize mainly in the nucleus of resting cells. However, the high cytosolic content of PPARγ can be achieved upon stimulations, such as the mitogen TPA [20]. This process probably plays an important regulatory role in terminating the nuclear activity of PPARγ [28]. Indeed, in our study, we found decreased PPARγ-DNA binding activity (Fig. 4A) concomitant with increased cytoplasmic PPARγ (Fig. 3A) in Ang II (10 min)-exposed cells. However, so far no nuclear export signals (NES) have been identified in PPARγ [29], and therefore, the mechanism that allows PPARγ cytoplasmic localization upon stimulation is not clear. Recently, Burgermeister et al. [20] observed that MEK (MAP kinase kinase) functioned as a shuttle protein for PPARγ and modulated redistribution of PPARγ from the nucleus to the cytoplasm. They found the mitogen TPA-induced export of PPARγ from the nucleus via PPARγ interaction with MEK. TPA-induced nuclear shuttling of MEK was demonstrated in their study to assist PPARγ export to the cytosol. Similar to TPA, Ang II is also a mitogenic stimulator [30], and can stimulate MEK activation in a variety of cells [31-34]. Among them, in pancreatic beta cells, there was a significant increase in phosphorylated MEK at 5 minutes after Ang II treatment [34], suggesting the rapid MEK activation by Ang II. Actually, we observed the similar activation of MEK 10 min after initiation of exposure to Ang II in ECV304 (data not shown). Moreover, in both Burgermeister's data [20] and our findings, PPARγ was observed to be exported rapidly from the nucleus toward the cytosol upon stimulation for 10-15 min and the relocalization was most pronounced within 60 min. Therefore, we speculate that in our study Ang II might, like TPA, evoke MEK- dependent redistribution of PPARγ at the early phase.
In our study, we also observed that PPARγ ligand Tro could completely inhibit the 2 actions of Ang II on PPARγ. MEK, the key enzyme in the MAP kinase cascade, phosphorylates and activates ERK1/2. It has been reported that PPARγ ligands inhibited ERK1/2 phosphorylation induced by Ang II or other stimulators [35-36], implying the possible inhibition of MEK under PPARγ ligands. Thus, in our study the repression of Ang II-induced PPARγ export by PPARγ ligand might proceed through inhibiting MEK and MEK-PPARγ interaction. Alternatively, the activation of PPARγ by ligands and thereby increase in PPARγ association with DNA may result in a reduction in the free PPARγ available for the interaction with MEK in the nucleus, thus inhibiting MEK-mediated PPARγ nuclear export.
The ubiquitin-proteasome system (UPS) catalyzes the breakdown of the majority of cellular proteins [37]]. In this system, ubiquitin (Ub), containing 76 amino acids, is conjugated to proteins destined for degradation [38]]. This process is repeated as multiple Ub molecules are added to form an Ub chain. Ub protein conjugates are recognized and rapidly degraded by a 26S proteasome complex in an ATP-dependent manner. Ang II interacts with the ubiquitin-proteasome system in multiple ways. Ang II directly stimulates protein degradation in myotube cultures through an increased expression and activity of key components of the ubiquitin-proteasome proteolytic pathway [39]. Also, Ang II activates the 26S proteasome in human umbilical vein endothelial cells via peroxinitrite-induced tyrosine nitration of PA700, a 26S proteasome regulatory subunit. The subsequent enhanced GTP cyclohydrolase I degradation results in nitric oxide signaling pathway dysfunction [40]. Very recently, it was reported that Ang II regulates Th1 T cell differentiation through the activation of AT1R/PKA signaling pathway and the consequent increases in the activities and expression of proteasome catalytic subunits [41]. In our study, we demonstrated for the first time that Ang II activates ubiquitination-proteasome system, which promotes degradation of PPARγ. Our finding showed that lactacystin could block the effect of Ang II on the decline of PPARγ protein. As lactacystin is a highly specific proteasome inhibitor, this data indicate that the loss of PPARγ following Ang II treatment was mediated by targeting of PPARγ to the proteasome. Our experiments with Ub Ab demonstrating the accumulation of high molecular weight polyubiquitinated species of PPARγ in Ang II-treated cells, which suggested Ang II enhancement of Ub conjugation to PPARγ, also supports PPARγ degradation by Ang II through ubiquitination -proteasome system.
To date, the ubiquitin/proteasome system- mediated degradation occurs primarily in the cytoplasm. However, evidence has reported this pathway- dependent degradation of certain proteins can also occur in the nucleus [42-44]. For example, the promyelocytic leukemia (PML)-nuclear bodies has been implicated in the nuclear degradation of transcription factor Nrf2. Nrf2 is recruited to PML-nuclear bodies where it undergoes polyubiquitylation and subsequent proteasomal degradation [44]. In our study, PPARγ was predominantly present in the nucleus under the basal condition. PPARγ degradation does not occur immediately following its translocation from nucleus to cytoplasm. Instead, the degradation was observed after the elimination of PPARγ nuclear export and the restoration to basal nuclear distribution of PPARγ, suggesting the possibility of nuclear degradation of PPARγ. PML-nuclear bodies are dynamic structures in the nucleus and assembled under conditions of oxidative stress [45], a recognized factor contributing to Ang II-induced injury [2-5]. Furthermore, Ang II has been documented to significantly trigger the assembly of PML-nuclear bodies in neonatal mouse cardiac fibroblasts [46]. Accordingly, we speculate that PPARγ may, as in the case of Nrf2, be degraded in PML-nuclear bodies in our study. Future works using nuclear fraction to explore PPARγ degradation and using live cell imaging to monitor the kinetics of PPARγ trafficking to PML-nuclear bodies are needed to provide direct evidence for nuclear degradation of PPARγ and the involvement of PML-nuclear bodies.
Regarding the relationship between the two actions of Ang II, we do not currently know whether Ang II-induced PPARγ nuclear export is a prerequisite for the ubiquitin-dependent degradation of PPARγ or whether the two events are independent. However, our results that PPARγ ligand Tro increased PPARγ nuclear translocation and reversed the loss of PPARγ induced by Ang II (Fig. 5C) suggested that both events may be relevant. It is possible that PPARγ nuclear export in response to Ang II might function as a signal for subsequent PPARγ degradation. The modulation of pathways involved in Ang II-induced PPARγ nuclear export would make it possible to clarify this point.
Our data demonstrated that both 2 actions of Ang II on PPARγ protein were accompanied by the decrease in PPARγ-DNA binding activity determined 10 min and 4 h after initiation of exposure to Ang II (Figs. 4A and 4B). The alteration in PPARγ activity suggests the inhibition of nuclear function of this transcription factor. Indeed, 4 h after Ang II exposure we observed reduced PPARγ target genes (Fig. 4C) concomitant with decreased PPARγ nuclear activity. However, we failed to detect any alterations in these target genes 10 min after Ang II stimulation (Fig. 4C). The reason for the discrepancy between PPARγ activity and function at this time point may be due to the fact that 10 min treatment is too brief for the observation of changes in the downstream effect of PPARγ. e.g. In Burgermeister et al's study showing TPA-induced export of PPARγ from the nucleus, the earliest reduction in PPARγ target genes were detected at about 1 h after TPA stimulation [20]. Alternatively, it is possible that PPARγ activity regulated by 10-min exposure of Ang II may not drop to the level sufficient to affect the downstream function. In line with this, our data showed that the decline of PPARγ activity 4 h after Ang II treatment was more pronounced than that of 10 min and was accompanied by decreased PPARγ-regulated genes expression. In addition, the complexity of PPARγ activity regulation, which is controlled by DNA binding activity and multiple nuclear receptor cofactors (co-repressors and co-activators) may also result in the mismatch between Ang II's effect on PPARγ-DNA binding activity and on target genes expression. However, this possibility can be ruled out by the parallel reduction in PPARγ activity and targeted gene expression observed 4 h after Ang II treatment.
Accumulating evidence has proven the anti-oxidant and anti-inflammatory effects of PPARγ in diverse cells and tissues, such as brain [7], lung [8], liver [47], and intestine [48]. Consistently, the anti-inflammatory effects of PPARγ through suppression of pro-inflammatory cytokines have also been identified in both vascular endothelial cells [49-50] and bladder epithelial cells [51], though whether ECV304 cells are endothelial cells or epidermal cells is still controversial. The down- regulation of PPARγ by Ang II disrupts PPARγ nuclear function, which may lead to the loss of beneficial effects of PPARγ on controlling oxidant- and inflammatory-related signaling. On the other hand, local Ang II as a pro-oxidant and pro- inflammatory molecule contributes to progressive deterioration of organ function in multiple diseases [2-5]. Moreover, Ang II stimulation reduces the expression of PPARγ in various cells, such as endothelial progenitor cells, vascular smooth muscle cells, cardiac myofibroblasts, hepatocytes, and macrophages [9-13]. These data suggested that inhibition of PPARγ pathway may be common for local Ang II signals in different cells. No matter whether ECV304 cells represent endothelial or epidermal cells, the regulatory mechanism of Ang II on PPARγ described in our study may provide new clues in understanding how Ang II negates PPARγ.
In summary, we demonstrated Ang II not only induced the redistribution of PPARγ from the nucleus to the cytosol but targeted this transcription factor to degradation by the ubiquitin-proteasome system. This presents a novel means of Ang II-induced down- regulation of PPARγ, and may shed some light on the mechanism underlying local Ang II signals.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81571201 and 81601039).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Miyazaki M, Takai S. Tissue angiotensin II generating system by angiotensin-converting enzyme and chymase. J Pharmacol Sci. 2006;100:391-7
2. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247-57
3. Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17:2985-91
4. Marchesi C, Paradis P, Schiffrin EL. Role of the rennin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367-74
5. de Cavanagh EM, Inserra F, Ferder M, Ferder L. From mitochondria to disease: role of the rennin-angiotensin system. Am J Nephrol. 2007;27:545-53
6. Liu Y, Zhu Y, Rannou F, Lee TS, Formentin K, Zeng L, Yuan X, Wang N, Chien S, Forman BM, Shyy JY. Laminar flow activates peroxisome proliferator-activated receptor-gamma in vascular endothelial cells. Circulation. 2004;110:1128-33
7. Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, Neves MF, Schiffrin EL. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation. 2002;105:2296-302
8. Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, Inestrosa NC, Bronfman M. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006-15
9. Lakshmi SP, Reddy AT, Zhang Y, Sciurba FC, Mallampalli RK, Duncan SR, Reddy RC. Down-regulated peroxisome proliferator-activated receptor γ (PPARγ) in lung epithelial cells promotes a PPARγ agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. 2014;289:6383-93
10. Han T, Liu M, Yang S. DJ-1 Alleviates Angiotensin II-Induced Endothelial Progenitor Cell Damage by Activating the PPARγ/HO-1 Pathway. J Cell Biochem. 2018;119:392-400
11. Li HY, Yang M, Li Z, Meng Z. Curcumin inhibits angiotensin II-induced inflammation and proliferation of rat vascular smooth muscle cells by elevating PPAR-γ activity and reducing oxidative stress. Int J Mol Med. 2017;39:1307-1316
12. Hao GH, Niu XL, Gao DF, Wei J, Wang NP. Agonists at PPAR-gamma suppress angiotensin II-induced production of plasminogen activator inhibitor-1 and extracellular matrix in rat cardiac fibroblasts. Br J Pharmacol. 2008;153:1409-19
13. Zhao J, Liu J, Pang X, Zhang X, Wang S, Wu D. Rosiglitazone attenuates angiotensin II-induced C-reactive protein expression in hepatocytes via inhibiting AT1/ROS/MAPK signal pathway. Int Immunopharmacol. 2016;31:178-85
14. Meng Y, Chen C, Tian C, Du J, Li HH. Angiotensin II-induced Egr-1 expression is suppressed by peroxisome proliferator-activated receptor-γ ligand 15d-PGJ₂ in macrophages. Cell Physiol Biochem. 2015;35:689-98
15. Sun L, Xu YW, Han J, Liang H, Wang N, Cheng Y. 12/15-Lipoxygenase metabolites of arachidonic acid activate PPARγ: a possible neuroprotective effect in ischemic brain. J Lipid Res. 2015;56:502-14
16. Sun L, Xu YW, Han J, Xiao C, Cao SS, Liang H, Cheng Y. Hydroxysafflor Yellow A Shows Protection against PPARγ Inactivation in Nitrosative Neurons. Oxid Med Cell Longev. 2018;2018:9101740
17. Han J, Sun L, Xu Y, Liang H, Cheng Y. Activation of PPARγ by 12/15-lipoxygenase during cerebral ischemia-reperfusion injury. Int J Mol Med. 2015;35:195-201
18. Floyd ZE, Wang ZQ, Kilroy G, Cefalu WT. Modulation of peroxisome proliferator-activated receptor gamma stability and transcriptional activity in adipocytes by resveratrol. Metabolism. 2008;57:S32-8
19. Xu L, Massagué J. Nucleocytoplasmic shuttling of signal transducers. Nat Rev Mol Cell Biol. 2004;5:209-19
20. Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol cell biol. 2007;27:803-17
21. Xu YW, Sun L, Liang H, Sun GM, Cheng Y. 12/15-Lipoxygenase inhibitor baicalein suppresses PPAR gamma expression and nuclear translocation induced by cerebral ischemia/reperfusion. Brain Res. 2010;1307:149-57
22. Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726-31
23. Ward WF, Moss AL. Effects of lysosomal inhibitors on 125I-insulin and 125I-asialofetuin degradation by the isolated, perfused rat liver and isolated rat hepatocytes. Diabetes. 1985;34:446-51
24. Whitmarsh AJ, Davis RJ. Regulation of transcription factor function by phosphorylation. Cell Mol Life Sci. 2000;57:1172-83
25. Gill G. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev. 2003;13:108-13
26. Berger J, Patel HV, Woods J, Hayes NS, Parent SA, Clemas J, Leibowitz MD, Elbrecht A, Rachubinski RA, Capone JP, Moller DE. A PPARgamma mutant serves as a dominant negative inhibitor of PPAR signaling and is localized in the nucleus. Mol Cell Endocrinol. 2000;162:57-67
27. Gurnell M, Wentworth JM, Agostini M, Adams M, Collingwood TN, Provenzano C, Browne PO, Rajanayagam O, Burris TP, Schwabe JW, Lazar MA, Chatterjee VK. A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J Biol Chem. 2000;275:5754-9
28. Burgermeister E, Tencer L, Liscovitch M. Peroxisome proliferator-activated receptor-gamma upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene. 2003;22:3888-900
29. Burgermeister E, Seger R. MAPK kinases as Nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle. 2007;6:1539-48
30. Su EJ, Lombardi DM, Wiener J, Daemen MJ, Reidy MA, Schwartz SM. Mitogenic effect of angiotensin II on rat carotid arteries and type II or III mesenteric microvessels but not type I mesenteric microvessels is mediated by endogenous basic fibroblast growth factor. Circ Res. 1998;82:321-7
31. Zhao Y, Liu J, Li L, Liu L, Wu L. Role of Ras/PKCzeta/MEK/ERK1/2 signaling pathway in angiotensin II-induced vascular smooth muscle cell proliferation. Regul Pept. 2005;128:43-50
32. Li X, Lerea KM, Li J, Olson SC. Src kinase mediates angiotensin II-dependent increase in pulmonary endothelial nitric oxide synthase. Am J Respir Cell Mol Biol. 2004;31:365-72
33. Aroor A, Zuberek M, Duta C, Meuth A, Sowers JR, Whaley-Connell A, Nistala R. Angiotensin II Stimulation of DPP4 Activity Regulates Megalin in the Proximal Tubules. Int J Mol Sci. 2016:17 (5) doi: 10.3390/ijms17050780
34. Lyu J, Imachi H, Fukunaga K, Sato S, Ibata T, Kobayashi T, Dong T, Yoshimoto T, Yonezaki K, Nagata H, Iwama H, Murao K. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J Lipid Res. 2018;59:1906-1915
35. Benkirane K, Amiri F, Diep QN, El Mabrouk M, Schiffrin EL. PPAR-gamma inhibits ANG II-induced cell growth via SHIP2 and 4E-BP1. Am J Physiol Heart Circ Physiol. 2006;290:H390-7
36. Kim J, Han DC, Kim JM, Lee SY, Kim SJ, Woo JR, Lee JW, Jung SK, Yoon KS, Cheon HG, Kim SS, Hong SH, Kwon BM. PPAR gamma partial agonist, KR-62776, inhibits adipocyte differentiation via activation of ERK. Cell Mol Life Sci. 2009;66:1766-81
37. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895-9
38. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425-79
39. Sanders PM, Russell ST, Tisdale MJ. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br J Cancer. 2005;93:425-34
40. Xu J, Wang S, Wu Y, Song P, Zou MH. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension. 2009;54:625-32
41. Qin XY, Zhang YL, Chi YF, Yan B, Zeng XJ, Li HH, Liu Y. Angiotensin II Regulates Th1 T Cell Differentiation Through Angiotensin II Type 1 Receptor-PKA-Mediated Activation of Proteasome. Cell Physiol Biochem. 2018;45:1366-1376
42. Geoffroy MC, Jaffray EG, Walker KJ, Hay RT. Arsenic-induced SUMO-dependent recruitment of RNF4 into PML nuclear bodies. Mol Biol Cell. 2010;21:4227-39
43. Sharma P, Murillas R, Zhang H, Kuehn MR. N4BP1 is a newly identified nucleolar protein that undergoes SUMO-regulated polyubiquitylation and proteasomal turnover at promyelocytic leukemia nuclear bodies. J Cell Sci. 2010;123:1227-34
44. Burroughs AF, Eluhu S, Whalen D, Goodwin JS, Sakwe AM, Arinze IJ. PML-Nuclear Bodies Regulate the Stability of the Fusion Protein Dendra2-Nrf2 in the Nucleus. Cell Physiol Biochem. 2018;47:800-816
45. Sahin U, Ferhi O, Jeanne M, Benhenda S, Berthier C, Jollivet F, Niwa-Kawakita M, Faklaris O, Setterblad N, de Thé H, Lallemand-Breitenbach V. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J Cell Biol. 2014;204:931-45
46. Liu Y, Zhao D, Qiu F, Zhang LL, Liu SK, Li YY, Liu MT, Wu D, Wang JX, Ding XQ, Liu YX, Dong C1, Shao XQ, Yang BF, Chu WF. Manipulating PML SUMOylation via Silencing UBC9 and RNF4 Regulates Cardiac Fibrosis. Mol Ther. 2017;25:666-678
47. Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Zingarelli B, Lentsch AB. Peroxisome proliferator-activated receptor-gamma protects against hepatic ischemia/reperfusion injury in mice. Hepatology. 2008;47:215-24
48. Nakajima A, Wada K, Miki H, Kubota N, Nakajima N, Terauchi Y, Ohnishi S, Saubermann LJ, Kadowaki T, Blumberg RS, Nagai R, Matsuhashi N. Endogenous PPAR gamma mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120:460-9
49. Hu C, Lu KT, Mukohda M, Davis DR, Faraci FM, Sigmund CD. Interference with PPARγ in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Genomics. 2016;48:124-34
50. De Silva TM, Hu C, Kinzenbaw DA, Modrick ML, Sigmund CD, Faraci FM. Genetic Interference With Endothelial PPAR-γ (Peroxisome Proliferator-Activated Receptor-γ) Augments Effects of Angiotensin II While Impairing Responses to Angiotensin 1-7. Hypertension. 2017;70:559-565
51. Lee J, Hong EM, Byun HW, Choi MH, Jang HJ, Eun CS, Kae SH, Choi HS. The effect of PPARalpha and PPARgamma ligands on inflammation and ABCA1 expression in cultured gallbladder epithelial cells. Dig Dis Sci. 2008;53:1707-15
Author contact
![]() Corresponding author: Dr. Ka Bian, Department of Biochemistry and Molecular Medicine, The George Washington University, Ross Hall 2300 Eye Street, NW, Washington, DC 20037, USA. Tel: USA-202-994-5824; E-mail address: bcmkxbedu
Corresponding author: Dr. Ka Bian, Department of Biochemistry and Molecular Medicine, The George Washington University, Ross Hall 2300 Eye Street, NW, Washington, DC 20037, USA. Tel: USA-202-994-5824; E-mail address: bcmkxbedu