Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1276-1286. doi:10.7150/ijbs.32332 This issue Cite
Research Paper
A CRISPR Interference of CBP and p300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro
Jianfa Li1*, ChenChen Huang1,2*, Tiefu Xiong1, Changshui Zhuang1, Chengle Zhuang1, Yawen Li1,2, Jing Ye1 ![]() , Yaoting Gui1
, Yaoting Gui1 ![]()
1. Guangdong and Shenzhen Key Laboratory of Male Reproductive Medicine and Genetics, Institute of Urology, Peking University Shenzhen Hospital, Shenzhen-Peking University-the Hong Kong University of Science and Technology Medical Center, Shenzhen 518000, People's Republic of China
2. Anhui Medical University, Hefei 230000, Anhui Province, People's Republic of China
*These authors have contributed equally to this work.
Received 2018-12-16; Accepted 2019-3-13; Published 2019-5-11
Abstract

The transcriptional coactivator CREB-binding protein (CBP) and p300 are adenoviral E1A-binding proteins involved in various cellular processes, including embryonic development, homeostasis, cell differentiation and transcription activation. Previous study suggested that synthetic lethality between CBP and p300 inhibition in lung and hematopoietic cancers. However, the underlying mechanism of CBP and p300 paralog in bladder cancer remains unknown. In this study, we discovered that combined CBP and p300 inhibition impaired cell proliferation and induced apoptosis of bladder cancer cells and normal bladder urothelial cell via decreasing c-Myc expression. Then, we employed the dCas9-KRAB system, hTERT promoter and hUPII promoter to construct an CRISPR interference system which could specifically repress CBP and p300 expression and cause lethality in bladder cancer cells in vitro. The CRISPR interference system we constructed could specifically inhibit the progression of bladder cancer, providing a novel strategy to fight against bladder cancer.
Keywords: bladder cancer, CBP, p300, synthetic lethality, CRISPR interference system
Introduction
Bladder cancer is the most common malignant neoplasm in the urinary system [1]. Although the great progress has been made in the clinical treatments, the prognosis of bladder cancer patients has not significantly improved [2, 3]. Therefore, novel treatments and strategies to fight against bladder cancer is urgently needed.
Previous studies have identified an ocean of mutated genes in the progression of bladder cancer. The most frequently mutated genes encode chromatin modifiers in bladder cancer, and a large portion of chromatin modifiers genes combined with their functional or/and structural paralogs, such as BRM/SMARCA2, PBRM1/BAF180 and CBP/p300 [4, 5]. The chromatin remodeling protein CREB-binding protein (CREBBP/CBP) and its paralog p300/EP300 are two highly conserved nuclear proteins originally identified for their interaction with the transcription factor CREB and with adenovirus E1a protein, respectively [6-8]. They serve as key transcriptional co-activators to mediate interaction between important transcription factors and the transcriptional machinery, playing an irreplaceable role in gene transcription [9]. A series of signal transduction pathways are associated with activity of CBP and p300, including p53, c-MYC, NF-kB and E2Fs [10-14]. Therefore, both CBP and p300 have been implicated in a multitude of cellular processes, including homeostasis, embryogenesis, cell growth and tumorigenesis [9, 15]. Recent genome-wide sequencing research indicate that somatic mutations of CBP and p300 are prevalent in some cancers, including lung cancer, leukemia, prostate cancer and bladder cancer. In prostate cancer, CBP and p300 obviously have oncogenic effects. Using the small-molecular inhibitor targeting the bromodomain of CBP/P300 can significantly inhibit the growth of prostate cancer cells [16]. In addition, suppression of p300 in CBP-deficient cancer cells results in the death of lung and hematopoietic cancer cells through decreasing c-Myc expression [17].
Based on this paralog targeting strategy, a novel cancer therapy called synthetic lethality has strung up. Synthetic lethality is a kind of genetic interaction that simultaneous mutations in two or more genes cause an obviously decrease of cell viability or cell death, whereas mutation or deletion in a single gene does not cause cell death [18, 19]. Based on this therapy theory, Synthetic lethality is widely used to fight against cancer precisely. However, synthetic lethality anticancer therapy is not fit for all the paralog addition. For instance, combined CBP and p300 inhibition not only kill cancer cells but also hurt the normal cells, which may greatly affect the health of cancer patients [17]. KRAS and EGFR can control the growth of normal cells, and all mutations in KRAS and EGFR genes are regarded as oncogenic behavior. Simultaneous suppression of KRAS and EGFR can cause lethality of cancer cells, but the great damage of normal cells is inevitable [20]. Therefore, synthetic lethality therapy is mainly applied in cells with one paralog deficient. It means that synthetic lethality therapy is only suitable for specific cancer cells.
Recently, the CRISPR-Cas9 system is a functional RNA-guided platform for acting as a programmable modulator of gene expression that works without modulating DNA sequence [21]. Nuclease Cas9 protein can be precisely targeted to genomic DNA since it was combined with a single guide RNA (sgRNA) [22]. On the other hand, mutations occurred in the catalytic domain of Cas9 protein produced a nuclease-deficient Cas9 (dCas9) which can fuse to transcriptional activators or repressors to activate or suppress gene expression [23]. When the four concatenated mSin3 interaction domains (SID4X) or KRAB was fused to the carboxyl terminus of dCas9 protein, the dCas9-SID4X or dCas9-KRAB fusion proteins can lead to strong suppression of genes in mammalian cells [24, 25].
In this study, we firstly detected the role of CBP and p300 in bladder cancer progression and found that combined inhibition of CBP and p300 can cause cell proliferation suppression and apoptosis induction in bladder cancer cells and SV-HUC1 cell (normal bladder urothelial cell) via reducing c-Myc expression. To solve this fetal limitation, we used the CRISPR-dCas9 system, the cancer-specific promoter of human telomerase reverse transcriptase (hTERT) gene and the bladder-specific promoter of human uroplakin II (hUPII) gene to specifically distinguish bladder cancer cells. Decreased CBP and p300 expression were observed in the presence of CRISPR interference system in bladder cancer cells, whereas solely p300 expression was inhibited in SV-40 transformed human uroepithelial (SV-HUC1) cell. In short, the CRISPR interference system can selectively inhibit the progression of bladder cancer in vitro.
Material and methods
Cell culture
All the cell lines included in this study, 5637, T24 and SV-HUC1 were purchased from the American Type Culture Collection (ATCC) and cultured according to the vendor's instructions. Cell lines were grown in RPMI-1640 or DMEM medium supplemented with 10% fetal bovine serum (FBS) and cultured at 37C in a humidified atmosphere containing 95% air and 5% CO2. Exponentially growing cells were used to perform all in vitro studies.
siRNA transfection
Specific siRNAs targeting on CBP or p300 were provided by Genepharma (Suzhou, China). The sequence of CBP siRNA (si-CBP) was CGGCACAGCCTCTCAGTCA-3. The sequence of p300 siRNA (si-p300) was GCACGAACTAGGAAAGAAA [26]. The negative control siRNA (si-NC) was also synthesized by (Suzhou, China). 50%-70% confluence of 5637 or T24 cells were seeded in 6-well or 12-well plates before siRNAs transfection, then transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the protocol. The transfected cells were collected to perform further experiments at least 48 hours (h) after transfection.
RNA extraction and RT-qPCR
Total RNAs from cultured cells and transfected cells using Trizol regent (Invitrogen, Carlsbad, CA, USA) according to the protocol. cDNA was synthesized from 1 μg of total RNA by using a cDNA synthesis kit (TOYOBO, Osaka, Japan). qRT-PCR assay was performed with a standard SYBR Green PCR kit (TOYOBO, Osaka, Japan) in triplicate by using the ABI7300 system (Applied Biosystems, Foster City, CA, USA). GAPDH was chose as the endogenous control and the relative level of mRNA was analyzed by using 2-∆Ct method. The gene-specific primers were shown as follows: CBP forward: 5'-GTCCAGTTGCCACAAGCAC-3'; CBP reverse: 5'- CATTCGGGAAGGAGAAATGG-3'; p300 forward: 5'-GCCAAGTACTTCAGCTACCCAGT-3'; p300 reverse: 5'- GGCATCAGTGCCTGTCGTAG-3' [27].
Construction of stable transfected cell lines
A lentiviral vector encoding dcas9-KRAB was provided by SyngenTech (Beijing, China). Then the auxiliary plasmid liposomes (psPAX.2 and Pmd2.G) and lentivirus vector were transfected into 293T cells to produce lentivirus vectors by using Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA). After 48 and 72h transfection, the supernatant was collected and centrifuged to discard cell debris. The centrifuged supernatant was filtered by using a 0.45 µm polyvinylidene difluoride filter. 5637, T24 and SV-HUC1 cells were infected with viral suspension mixed with polybrene (Solarbio, Perking, China). After 48h infection, puromycin was added to screen the positive stably transduced cell lines.
Plasmid construction and transfection
The sequence of pLV-CMV-NLS-dcas9-NLS- KRAB-T2A-neo was inserted into pHS-ACR-LW388 vector (Addgene, Cambridge, MA, USA) at the restriction sites of CPPT/PacI. To design the original sgRNAs targeting on CBP or p300, we used the online design tool “CRISPR-ERA” (http://CRISPR-ERA. stanford.edu). The cDNA sequence for each sgRNA was inserted into pLVU6/purp vector. Then CBP sgRNA driven by hTERT promoter and p300 sgRNA driven by hUPII promoter was inserted into pHS-AVC vector digested with HH/HDV ribozyme. At last, the vector containing sgRNA was transfected into the stably transduced cell lines after they reached 70-80% confluency through using Lipofectamine 3000 according to manufacturer's protocols.
Cell proliferation assay
Cell proliferation was determined by using CCK-8 (Cell Counting Kit-8) assay and EDU assay kit according to the manufacture's protocol. For CCK-8 assay, the transfected cells were harvested after 48h transfection and then seeded in a 96-well plate for a day. At 0, 24, 48 and 72h after cell attachment, 10 µl of CCK-8 kit was added to each well. The OD values were measured by using a microplate reader (Bio-Rad, Hercules, CA, USA) at a wavelength of 450 nm after 1h cultivation. The EDU assay was performed by utilizing an EdU assay kit (Ribobio, Guangzhou, China) according to the manufacture's protocol. The transfected cells were collected after 48h transfection and then seeded in a 12-well plate for a day. Cells with fluorescence were observed under a microscope (Olympus, Tokyo, Japan). Cell proliferation rate was determined by the rate of EDU positive cells to DAPI-stained cells.
Cell apoptosis assay
At 48 h post-transfection, the caspase-3 ELISA assay and flow cytometry assay were used to detect cell apoptosis. Briefly, the transfected cells were executed using the caspase-3 ELISA kit according the instructions. OD values was measured by a microplate reader (Bio-Rad, Hercules, CA, USA). Moreover, we also performed the flow cytometry assay to examine the rate of apoptotic cells. After 48 h transfection, cells were trypsinized and washed in PBS, and then suspended in binding buffer. Afterwards, the cells were incubated with Annexin V-FITC (AV) and Propidium Iodide (PI) in the darkness for 15 minutes. The rate of apoptotic cells was observed by using a flow cytometer (EPICS, XL-4, Beckman, CA, USA). All experiments were performed at least three times.
Western blotting analysis
The transfected cells were harvested, centrifuged and washed in PBS, and then lysed using RIPA buffer (Beyotime, Perking, China) supplemented with protease inhibitor. The protein concentration was determined by using a pierce BCA protein assay kit (Thermo, USA). Equal amounts of protein extracts were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes (Millipore, USA). The membranes were blocked in 5% dry milk and incubated with primary antibodies against CBP or p300 overnight at 4°C. Then, the samples were washed and incubated with secondary antibodies in room temperature for 1 h. Finally, densitometry was quantified by using supersignal chemiluminescence reagents. All experiments were performed at least three times.
Statistical analysis
Data analysis was conducted using SPSS 20.0 (SPSS, Chicago, USA). Group differences in qRT-PCR and cell apoptosis assay were analyzed using independent samples t-test. Total differences between experimental group and negative control group in the CCK-8 assay were analyzed by using two-factors repeated measurement analysis of variance. In addition, the differences between experimental group and negative control group at each time point were analyzed by using independent samples t-test. Data based on three repeats were shown as mean±standard deviation (SD). Differences with p < 0.05 were considered statistically significant.
Results
CBP and p300 were overexpressed in 5637 and T24 cells compared to that in SV-HUC1 cell
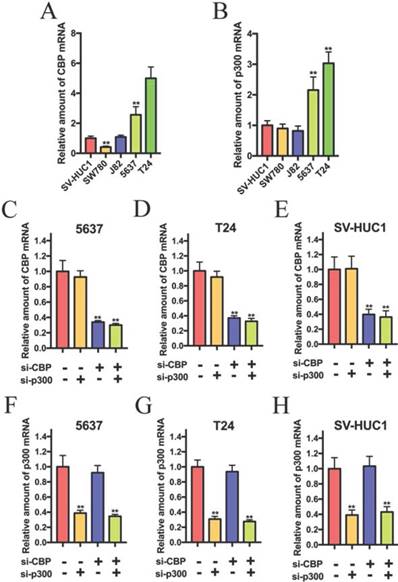
To investigate the role of CBP and p300 in the progression of bladder cancer, we detected the expression of CBP and p300 in bladder cancer cells. As shown in Fig.1A and B, CBP and p300 were up-regulated in 5637 and T24 cells compared to that in SV-HUC1 (normal bladder urothelial cell). 5637 and T24 cells were chosen for further experiments. Based on the previous study, we suspected that CBP combined with p300 to promote the progression of bladder cancer and investigated whether combined suppression of CBP and p300 could affect biological activity of bladder cancer cells. The siRNAs targeting on the coding region of CBP and p300 were used to suppress CBP and p300 expression. As shown in Fig. 1C, D and E, the relative expression of CBP in 5637, T24 and SV-HUC1 cells were significantly down-regulated by combined transfection of si-CBP and si-p300 and combined transfection of si-CBP and si-NC. In addition, co-transfection of si-NC and si-p300 and co-transfection of si-CBP and si-p300 obviously impaired the relative expression of p300 in 5637 (Fig. 1F), T24 (Fig. 1G) and SV-HUC1 cells (Fig. 1H). These results suggested that the siRNAs targeting CBP and p300 could be used in further experiments.
The relative expression of CBP and p300 mRNA in bladder cancer cell lines and SV-40 transformed human uroepithelial (SV-HUC1) cell. (A and B) CBP and p300 mRNA is overexpressed in 5637 and T24 cells compared to that in SV-HUC1 cell. (C-E) 5637, T24 and SV-HUC1 cells were transfected with CBP siRNA or non-sense control siRNA. (F-H) Relative mRNA expression levels of p300 in 5637, T24 and SV-HUC1 cells after transfection with p300 siRNA.
Combined inhibition of CBP and p300 results in decreased proliferation and increased apoptosis in bladder cancer cells and SV-HUC1
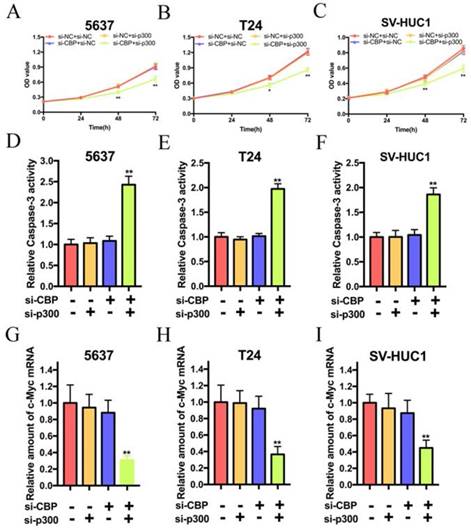
As shown in Fig. 2A, B and C, simultaneous suppression of CBP and p300 attenuated the proliferation of 5637, T24 and SV-HUC1 cells, whereas sole targeting either CBP or p300 had no effects on the proliferation of these cells. In consideration of the effects of CBP and p300 on the proliferation of these cells, we suspected that CBP and p300 can inhibit the apoptosis of these cell. As shown in Fig. 2D, E and F, the relative activity of caspase-3 in 5637, T24 and SV-HUC1 cells treated with si-CBP and si-p300 were significantly increased. However, either suppression of CBP or p300 could not change the activity of caspase-3 in these cells. Hence, simultaneous targeting of CBP and p300 triggers cell apoptosis of bladder cancer cells and SV-HUC1. By the way, we suspected that knockdown of CBP and p300 caused cell inhibition via decreasing c-Myc expression. As expected, synthetic lethality in loss of CBP and p300 expression resulted in c-Myc suppression in 5637, T24 and SV-HUC1 cells (Fig. 2G, H and I). These data indicated that lethality in bladder cancer and SV-HUC1 cells due to depletion of c-Myc was caused by simultaneous targeting of CBP and p300 with siRNA.
Combination of CBP siRNA and p300 siRNA impaired cell proliferation and induced apoptosis of bladder cancer cells and SV-HUC1 cell in vitro. (A-C) Simultaneous suppression of CBP and p300 mRNA expression significantly inhibited cell viability in bladder cancer cells and SV-HUC1 cell, whereas CBP or p300 expression was impaired individually showed no significant effects on growth inhibition. (D-F) Combined CBP and p300 inhibition promoted apoptosis of bladder cancer cells and SV-HUC1 cell, whereas either CBP deletion or p300 deletion exerted no significant effects on cell apoptosis. (G-H) Synthetic lethal combinations via targeting CBP and p300 via decreasing c-Myc mRNA expression in bladder cancer cells and SV-HUC1 cell.
Design and construction of element to specifically kill Bladder cancer cells
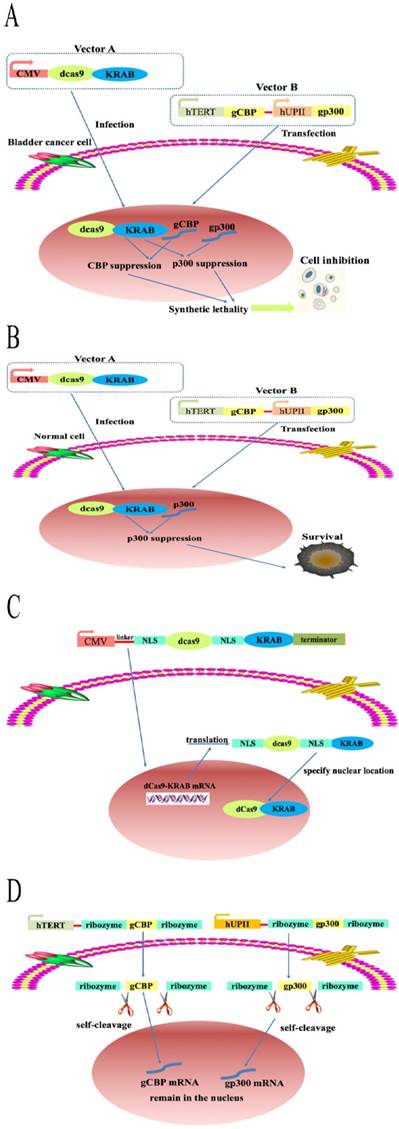
As expected, simultaneous inhibition of both CBP and p300 not only resulted in a lethal phenotype in bladder cancer cells but also in normal bladder cell, which greatly block the application of this paralog in fighting against bladder cancer. Moreover, knockdown of CBP and P300 may hurt a large variety of human cells. To solve this huge limitation, we constructed an artificial device based on dCas9-KRAB system by combining the hUPII and hTERT promoter. As shown in Fig. 3A, fusion of dCas9 protein to the Krüppel-associated box repressor (KRAB) protein driven by strong cytomegalovirus (CMV promoter) was encoded in vector A and produced in all cells. Besides, the mature dCas9 protein could be transported in to nucleus when the nuclear localization sequence (NLS) was inserted into both ends of the sequence of the dCas9 protein. The construction of vector B is based on the principle of logic AND gate and contains hUP II and hTERT promoter. As shown in Fig. 3B, the sgRNA targeting CBP (gCBP) was driven by hTERT promoter while the sgRNA targeting p300 (gp300) was controlled by hUPII promoter. The artificial device specifically activated the sgRNA of CBP and p300 gene in 5637 and T24 cells, whereas only sgRNA of p300 gene in SV-HUC1 cell (Fig, 3A and B). Moreover, dCas9 proteins were translated and modified in the cytoplasm while NLS sequence placed at both ends of dCas9 sequence (Fig. 3C). The expression of sgRNAs regulated by hUPII promoter or hTERT promoter is surrounded by two ribozymes located in both ends of sgRNAs. The primary transcript was self‐catalyzed cleavage to accurately produce the specific sgRNA (Fig. 3D). The hUPII and hTERT promoter are only simultaneously activated in bladder cancer cells. Hence, gCBP and gp300 are matured and expressed in the nucleus of bladder cancer cells, which suppress the transcription of CBP and p300 mRNA at the same time. To find optimal sgRNAs for repression of CBP and p300 by dCas9-KRAB, we designed a panel of three sgRNAs targeting CBP and p300, respectively (Supplementary Table 1 and Supplementary Fig. 1A, B).
A schematic of the mechanism of the CRISPR interference system. The combination of dCas9-KRAB system, hTERT and hUPII promoter to construct a CRISPR interference system that could selectively control the expression of the downstream output sgRNAs in bladder cancer cells. (A) CBP sgRNA and p300 sgRNA were matured and remained in the nucleus, then combined with dCas9-KRAB protein to suppress CBP and P300 transcription. (B) Only p300 sgRNA were matured and remained in the nucleus, then combined with dCas9-KRAB protein to inhibit the transcription of p300. (C and D) The mechanism of CRISPR-dCas9-KRAB system.
Specific inhibition gene expression of bladder cancer cells
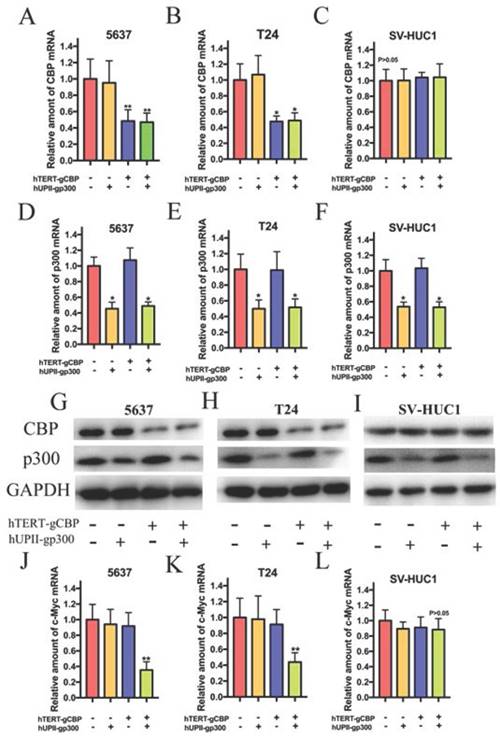
To verify the effects of the devices on gene expression in bladder cancer cells and SV-HUC1, we transfected the dCas9-KRAB plasmid into the cells and harvested the stably transduced cell lines. Total RNA and protein were extracted after 48 h transfection of vector B into the stably transduced cell lines. Since gCBP was regulated by hTERT promoter and gp300 was driven by hUPII promoter, we suspected that gCBP expression inhibition was observed in bladder cancer cells but not in SV-HUC1. qRT-PCR assay demonstrated that the CRISPR interference system attenuated the mRNA expression of CBP in 5637 and T24 cells (Fig. 4A and B). However, the hTERT- gCBP-hUPII-gNC group or hTERT-gNC- hUPII-gp300 group could only impair the mRNA expression of CBP in these cells. As we expected, the CRISPR interference system could not repress the mRNA expression of CBP for the low activity of hTERT promoter in SV-HUC1 (Fig. 4C). In addition, the CRISPR interference system could suppress p300 mRNA levels in 5637, T24 and SV-HUC1 cells (Fig. 4D, E and F). Further western blotting assay demonstrated that the CRISPR interference system could repress the protein levels of CBP and p300 in 5637 and T24 cells (Fig. 4G and H). Nevertheless, the CRISPR interference system could only impair the protein level of p300 in SV-HUC1 cell, whereas it could not attenuate CBP protein (Fig. 4I). Besides, the CRISPR interference system selectively inhibited the mRNA expression of c-Myc in 5637 and T24 cells (Fig. 4J, 4K) but not in SV-HUC1 (Fig. 4L). Therefore, the CRISPR interference system can synchronously inhibit the mRNA and protein level of CBP and p300 in bladder cancer cells, but solely suppress the expression of p300 mRNA and protein in SV-HUC1 cell. By the way, we predicted that this CRISPR interference system could play an important role of specific recognition and targeted lethality in bladder cancer cells.
The CRISPR interference system selectively impaired CBP and p300 expression in 5637 and T24 cells. (A, B and C) The CRISPR interference system selectively attenuated the mRNA expression of CBP in bladder cancer cells, but it could not decrease CBP mRNA expression in SV-HUC1 cell. (D, E and F) The artificial device. The mRNA levels of p300 in 5637, T24 and SV-HUC1 cell transfected with the CRISPR interference system were measured by qRT-PCR. (G and H) Western blotting analysis showed changes in CBP and p300 in 5637 and T24 cells transfected with this system. (I) Only p300 protein expression in SV-HUC1 cell was modulated while this cell was transfected with this system. (J and K) The CRISPR interference system specially restrained the mRNA levels of c-Myc in 5637 and T24 cells. (L) This system could not suppress the mRNA level of c-Myc in SV-HUC1 cell.
Specific cell proliferation of bladder cancer cells
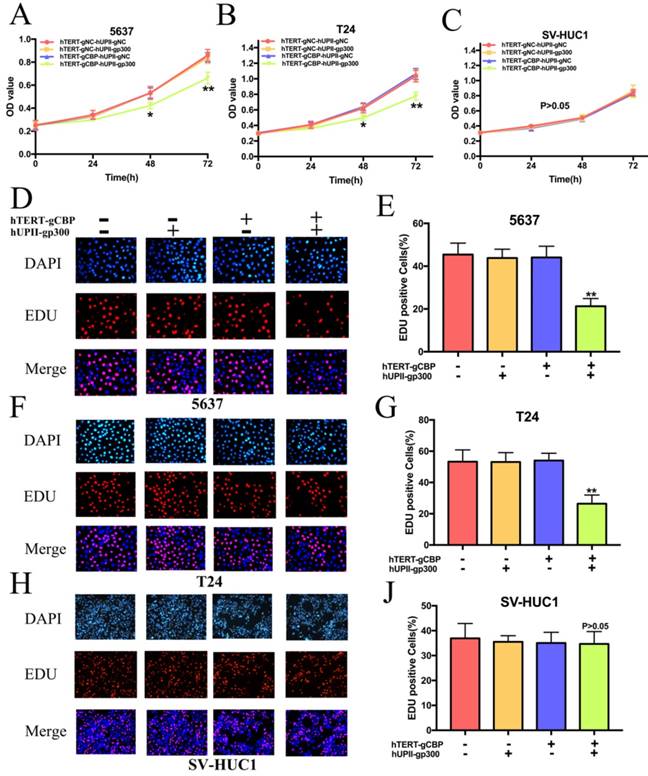
We next investigated the role of this system in the proliferation of bladder cancer cells. Cells overexpressing dcas9-KRAB were transfected with hTERT-gCBP-hUPII-gp300 vector or negative controls. As we guessed, the CRISPR interference system significantly slowed the growth rate of 5637 and T24 cells (Fig. 5A, 5B). However, it has no effects on the growth of SV-HUC1 (Fig. 5C). We further performed the EDU assay to detect the proliferation rate of cells. As shown in Fig. 5D and F, the percentage of EDU positive cells was significantly reduced in 5637 and T24 cells after transfection of this system. However, the percentage of EDU positive cells was not changed in SV-HUC1 cell (Fig. 5H). Further data analysis showed that the rate of EDU positive cells in the hTERT-gCBP-hUPII-gp300 vector group was reduced by 53.23% in 5637 cell (Fig. 5E) and 50.5% in T24 cell (Fig. 5G). There is no significant difference between hTERT-gCBP-hUPII-gp300 vector group and negative control groups in the EDU staining SV-HC1 cells (Fig. 5I). These data suggested that the CRISPR interference system could discriminate the bladder cancer cells and normal bladder cell and uniquely impair the proliferation of bladder cancer cells.
The CRISPR interference system specifically inhibited the proliferation of bladder cancer cells and it had no effects on the proliferation of SV-HUC1 cell. (A and B) CCK8 assay demonstrated that growth curves of 5637 and T24 cell lines transfected with the CRISPR interference system was slower that transfected with the negative control groups. (C) The CRISPR interference system could not affect the growth cure of SV-HUC1 cell. (D and E) EDU assay for 5637 cell proliferation in different treatment groups. This system leads to a significant decrease in 5637 cell proliferation. (F and G) EDU assays of T24 cell transfected with the CRISPR interference system and the other control groups. (H and I) The proliferation rate of SV-HU1 cell remained unchanged while this cell was transfected with this system.
Unique induced apoptosis of bladder cancer cells
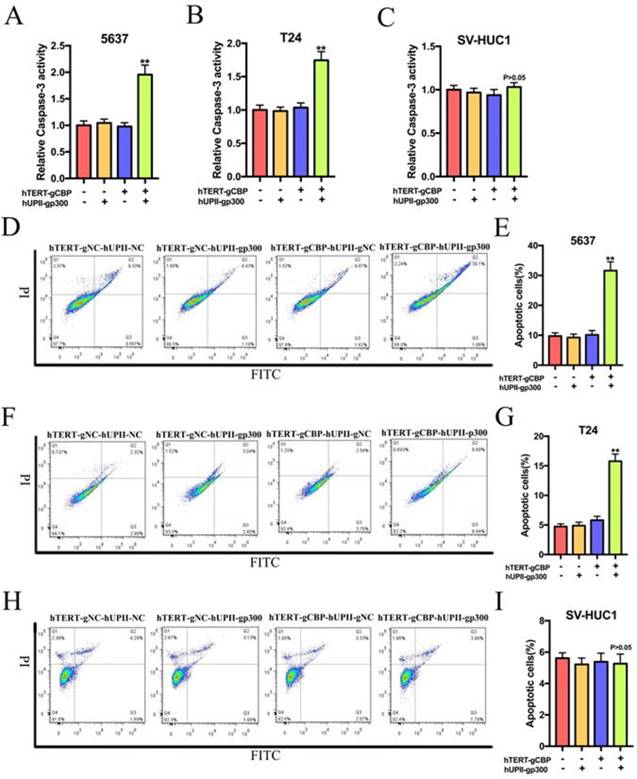
Based on the synthetic lethality role of CBP and p300 in bladder cancer cells, we speculated that this CRISPR interference system could uniquely induce bladder cancer cell apoptosis but not in SV-HUC1 cell. The cell-killing ability of this CRISPR interference system was depended on the ELISA assay and flow cytometry assay relative to the negative control group. As shown in Fig. 6A, B and C, the CRISPR interference system selectively increased the activity of caspase-3 in bladder cancer cells without significantly affecting the SV-HUC1 cells. Further flow cytometry assay demonstrated that the proportion of apoptotic cells in 5637 and T24 cells transfected with this device was obviously augmented compared to that in 5637 and T24 cells transfected with negative control groups (Fig. 6D and F). Nevertheless, no change was happened in the proportion of apoptotic cells in SV-HUC1 cell transfected with this system (Fig. 6H). Data based on three repeats demonstrated that the constructed CRISPR interference system could selectively promote the apoptosis of bladder cancer cells (Fig. 6E, G and I).
The CRISPR interference system selectively triggered the apoptosis of bladder cancer cells, whereas it had no effects on the apoptosis of SV-HUC1 cell. (A and B) The CRISPR interference system could increase the relative expression of caspase-3 of 5637 and T24 cells compared to that in the negative control groups. (C) This system could not change the caspase-3 level in SV-HUC1 cell. (D and E) Flow cytometry assay showed significant increases in the proportion of apoptotic cells in 5637 cell transfected with this system. (F and G) Changes in the cell apoptosis after the CRISPR interference system treatment in T24 cell measured by flow cytometry assay. (H and I) The CRISPR interference system could not affect the proportion of apoptotic cells in SV-HUC1 cell.
Discussion
Synthetic lethality therapy is a promising treatment for cancer as it provides a platform to selectively destroy cancer cells without hurting the normal cells. Besides, this precise method may be required lower dose of anti-cancer drug or inhibitors. Most of all, it will cause fewer side effects in the cancer patients, as the normal cells are isolated. Among so many synthetic lethality paralogs, CBP and p300 are identified as a promising paralog for targeting therapy in cancer treatment. CBP and p300 are homologous transcriptional coactivator which possesses histone acetyltransferase activity. Though this paralog is extensively investigated in various kinds of cancers, the role of CBP and p300 in cancer is not thoroughly and may depend on the physiologic background of cancer. In normal cells, CBP and p300 exert their functions by interacting with tumor suppressor p53, p21 and bax [28, 29]. In prostate cancer, CBP and p300 expression are associated the expression of androgen receptor (AR). Deletion of p300 resulted in a dramatically decreased cell proliferation and increased apoptosis via suppressing the nuclear factor kappa B (NF-kB) subunit p65 [30]. In lung adenocarcinoma, CBP promotes cell proliferation, migration and restrain apoptosis by activating the MAPK and inhibiting cytochrome C/caspase- dependent signaling pathways [31]. Moreover, Ogiwara et al. [17] identified the synthetic lethality role of CBP and p300 in lung cancer cells and lymphoma cells. p300 suppression in CBP-deficient cancer cells down-regulated c-Myc expression. Further experiments demonstrated that CBP and p300 were involved in the positive transcriptional regulation of the c-Myc gene through binding to the transcription start site of the c-Myc gene and acetylating histones H3K18 and H3K27, finally accelerating transcription initiation. Nevertheless, the role of CBP and p300 in the bladder cancer progression is still mysterious.
Our findings indicated that combined suppression of CBP and p300 expression inhibited cell growth and led to lethality in bladder cancer cells and SV-HUC1 through impairing c-Myc expression, whereas either decreased CBP or p300 expression has no effects on these cells. These findings are reasonable based on previous studies showing that CBP and p300 exhibit highly similar function as 63% homology and 72% similarity at the amino-acid level. Besides, the region of CBP gene location is highly homologous to p300 gene location [32, 33]. CBP protein can rescue some essential functions of p300 protein while p300 suppression. Similarity, p300 protein also can replace part of vital functions of CBP protein for CBP suppression. Nevertheless, synthetic lethal combinations via targeting CBP and p300 also impaired the growth of urothelial cells indicating that this synthetic lethal therapy was restricted in vivo assay and clinical applications. Then, we make use of the dcas9-KRAB system, hUPII and hTERT promoter to construct a CRISPR interference system which can efficiently settle this restriction.
However, one visible shortage is existed in this system. hUPII and hTERT promoters are defined as Pol II promoter, while U3 and U6 promoters (Pol III promoter) are usually used to control the transcription of sgRNA genes, which remain sgRNA in the nucleus. Unlike Pol III promoters, the Pol II promoters have critical defects, including a lack of cell specificity and tissue specificity. However, products of sgRNA genes will be actively transported from the nucleus into the cytoplasm after transcription while sgRNA genes are transcribed by Pol III promoters and the dCas9-gRNA only capture the genomic DNA inside the nucleus. One strategy to solve this limitation is to make use of the nuclease activity of ribozymes to produce the RNA molecule. Ribozyme sequences were inserted into both ends of the sgRNA. The primary transcripts of ribozyme-sgRNA- ribozyme suffer self-catalyzed cleavage to precisely release the specific sgRNA into nucleus [34].
The hTERT promoter is identified as a cancer-specific promoter for different kinds of cancer tissues and cell lines. The hUPII promoter, a bladder-specific promoter, is only expressed in urothelium cells. By combining dCas9-KRAB system and the two promoters, we supposed that the CRISPR interference system was able to selectively distinguish bladder cancer cells and simultaneously inhibit CBP and p300 expression.
To verify our hypothesis, we transfected the CRISPR interference system into the bladder cancer cells and SV-HUC1 and detected the expression of CBP and p300 in these cells. As expected, this system successfully inhibited CBP and p300 expression in bladder cancer cells, whereas only p300 repression was observed in SV-HUC1. Further experiments demonstrated that this system strongly and selectively impaired bladder cancer cell growth and induced apoptosis by regulating CBP and p300 at the same time. In addition, this system had no effects of the proliferation and apoptosis of SV-HUC1 for failure in synthetic lethal combinations between CBP and p300. The CRISPR interference system may be applied in the diagnosis and treatment in bladder cancer for efficiency and specificity. Intravesical injection therapy may be a suitable method for CRISPR interfere system delivery to bladder cancer tissues in vivo. Immediate postoperative intravesical instillation therapy is able to kill the tumor cells which were disseminated during surgery and remained in surgical wound, contributing to decrease the recurrence rate of non-muscle invasive bladder cancer. Therefore, intravesical our CRISPR interfere system may be used to combat bladder cancer cells in vivo.
However, one drawback should be addressed before the artificial device can be successfully applied in the bladder cancer treatment. First of all, the artificial device is consisted of two elements, dCas9-KRAB system and hTERT-gCBP-hUPII-gp300. In theory, the transfection efficiency of two elements mediately affect the anticancer effects. Therefore, integrating two elements into a vector can obviously enhance the efficiency of this device. Despite all this, the CRISPR interfere system may be widely used in the basic and clinical research, finally in cancer treatment in the future.
In general, we report for the first time that synthetic lethality via targeting CBP and p300 in bladder cancer cells and normal bladder urothelial cell. Moreover, we constructed a CRISPR interference system which can specifically inhibit CBP and p300 expression in bladder cancer cells, finally attenuating cell proliferation and promoting apoptosis in vitro. Hence, the CRISPR interference system can selectively kill bladder cancer cells without hurting normal bladder cell. This system provides a novel strategy to fight against bladder cancer and may be applied in anticancer drug in the future.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank all of the donors whose names were not included in the author list, but who participated in this program. This work was supported by grants from Guangdong Key Laboratories (2017B030314074) and the Shenzhen Project of Science and Technology (JCYJ20170413100245260).
Ethics approval and consent to participate
The study was approved by the Institutional Research Ethics Committee of Peking University Shenzhen Hospital.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, Oliver T. et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. 2005;23:4602-8
3. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
4. Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E. et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111:3128-33
5. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539-42
6. Arany Z, Sellers WR, Livingston DM, Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77:799-800
7. Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81-4
8. Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB. et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8:869-84
9. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553-77
10. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. 2013;70:3989-4008
11. Kim WJ, Rivera MN, Coffman EJ, Haber DA. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol Cell. 2012;45:587-97
12. Sankar N, Baluchamy S, Kadeppagari RK, Singhal G, Weitzman S, Thimmapaya B. p300 provides a corepressor function by cooperating with YY1 and HDAC3 to repress c-Myc. Oncogene. 2008;27:5717-28
13. Cras A, Politis B, Balitrand N, Darsin-Bettinger D, Boelle PY, Cassinat B. et al. Bexarotene via CBP/p300 induces suppression of NF-kappaB-dependent cell growth and invasion in thyroid cancer. Clin Cancer Res. 2012;18:442-53
14. Morris L, Allen KE, La Thangue NB. Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat Cell Biol. 2000;2:232-9
15. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225-31
16. Jin L, Garcia J, Chan E, de la Cruz C, Segal E, Merchant M. et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017;77:5564-75
17. Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y. et al. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016;6:430-45
18. Kamb A. Mutation load, functional overlap, and synthetic lethality in the evolution and treatment of cancer. J Theor Biol. 2003;223:205-13
19. Kroll ES, Hyland KM, Hieter P, Li JJ. Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics. 1996;143:95-102
20. Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907
21. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262-78
22. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15:387-95
23. Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5-15
24. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442-51
25. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH. et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647-61
26. Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang Y. et al. 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett. 2017;400:183-93
27. Hashwah H, Schmid CA, Kasser S, Bertram K, Stelling A, Manz MG. et al. Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A. 2017;114:9701-6
28. Kasper LH, Thomas MC, Zambetti GP, Brindle PK. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle. 2011;10:212-21
29. Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823-7
30. Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT. et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644-55
31. Tang Z, Yu W, Zhang C, Zhao S, Yu Z, Xiao X. et al. CREB-binding protein regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathway. Mol Oncol. 2016;10:317-29
32. Dutto I, Scalera C, Prosperi E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell Mol Life Sci. 2018;75:1325-38
33. Giles RH, Peters DJ, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998;14:178-83
34. Gao Y, Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol. 2014;56:343-9
Author contact
![]() Corresponding authors: Jing Ye, email: ye2013jcom and Yaoting Gui, email: guiyaoting2007com
Corresponding authors: Jing Ye, email: ye2013jcom and Yaoting Gui, email: guiyaoting2007com