Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
P53 related lncRNAs that act as...
P53 related lncRNAs that act as...
Conclusion and future perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(6):1287-1298. doi:10.7150/ijbs.33218 This issue Cite
Review
Emerging Roles of p53 Related lncRNAs in Cancer Progression: A Systematic Review
Tian Lin1,2*, Ping-Fu Hou1,2,3*, Sen Meng1,2*, Fang Chen1,2, Tao Jiang1,3, Min-Le Li1,2, Mei-Lin Shi4, Jin-Jin Liu1,2, Jun-Nian Zheng1,2,3 ![]() , Jin Bai1,2
, Jin Bai1,2 ![]()
1. Cancer Institute, Xuzhou Medical University, Xuzhou 221002, Jiangsu Province, China;
2. Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Xuzhou Medical University, Xuzhou 221002, Jiangsu Province, China;
3. Center of Clinical Oncology, Affiliated Hospital of Xuzhou Medical University, Xuzhou 221002, Jiangsu Province, China;
4. School of Medical Imaging, Xuzhou Medical University, Xuzhou, Jiangsu, China.
* These authors contributed equally to this work.
Received 2019-1-16; Accepted 2019-3-12; Published 2019-5-12
Abstract

p53 is the major mediator of the tumor suppressor response. It participates in apoptosis and senescence and can respond to DNA damage. As a crucial sequence-specific transcription factor, p53 regulates the expression of many genes, such as small noncoding RNAs (ncRNAs), microRNAs, and long ncRNAs (lncRNAs). Given the emergence of novel and high-throughput sequencing technologies, many lncRNAs have been discovered. LncRNAs may function as vital gene regulators in a variety of biological processes through extensive mechanisms. Recently, lncRNAs have been demonstrated to be associated with the p53 regulatory pathway. In this review, we discuss the current and fast growing knowledge about the influence of lncRNAs to the p53 signaling pathway, the different mechanisms by which they affect gene expression in cancer. Our findings show that p53-associated lncRNAs may be used as biomarkers for cancer diagnosis or targets for disease therapy.
Keywords: lncRNA, p53, cancer, therapy
Introduction
The development of RNA-Seq and microarray technology has revealed the marvelous complexity of the human transcriptome[1, 2]. The proportion of genome is less than 2% of the human genome, of which nearly 75% is energetically transcribed into noncoding RNAs [1]. Among these RNAs are lncRNAs [3]. They are longer than 200 nucleotides (nt), mostly have no protein-coding potential, and are transcribed by RNA polymerase II in a similar manner to mRNAs [4, 5]. Compared with known microRNAs (miRNAs) and Piwi-interacting RNAs, which regulate gene expression by base pairing with their targets. Some of the definite mechanisms of how lncRNAs regulate gene expression are still unknown, but some we have known [6-8]. Although the function of 15,941 lncRNAs (ENCODE v24), which are transcribed from the human genome, is unclear, an increasing number of evidence imply that many lncRNAs are continually dysregulated in numerous cancers [9] and can regulate vital cellular processes [10, 11], including cellular proliferation, differentiation, development, and genomic stability [12-19].
The function of lncRNAs mainly includes four archetypes of molecular mechanisms. First, the expression of lncRNAs can serve as markers of gene regulation in space, time, developmental stage, and expression [20]. Second, lncRNAs act as decoys and can bind and titrate transcription factors and other protein targets without acting as a “molecular sink” for RNA binding proteins (RBP) or as a sponge for miRNAs [21, 22]. Third, lncRNAs can serve as guides for recruiting ribonucleoprotein complex to target genes and regulate distant gene expression in trans [23] or neighboring gene expression in cis [24-26]. Fourth, as scaffolds, lncRNAs can bind unique effector molecules through diverse domains and aggregate proteins, which may have transcriptional activating or repressive effects to form ribonucleoprotein complexes and regulate the expression of target genes in time and space [27, 28]. Also, some additional mechanisms may need to be further studied.
An increasing number of evidence shows that the expression of lncRNAs, which promoter regions were bound by p53, can drive p53-dependent reporter activity. Such activity requires the p53-binding motif to actively influence the p53 pathway, suggesting that lncRNAs are key functional components of the p53-dependent transcriptional pathway [4]. The master regulatory transcription factor p53 is the important DNA damage response (DDR) component and is mutated in more than 50% of human cancers. It is an excellent tumor suppressor. In response to stress, including replicative stress, oxidative stress, hypoxia, DNA damage, nutrient deprivation, and telomere shortening [29, 30], p53 directly activates the transcription of many protein-coding genes. These genes can control numerous cellular processes, including the induction of cell cycle arrest, apoptosis, autophagy, and senescence and inhibition of invasion, migration, stemness, and metabolic reprogramming, which all together result in tumor suppression [29, 31]. As an important component of DDR, p53 activates a gene expression program to induce cell cycle arrest, leading to DNA repair or eliminating damaged cells via induction of apoptosis [32, 33]. Some specific lncRNAs can function as master gene regulators in the p53 signaling pathway or serve as p53 transcriptional targets. In this review, we provide an update on the role of lncRNAs on p53 regulation (induced or activated) in cancers, focusing on the mechanism of mutual regulation of p53 and lncRNAs.
P53 related lncRNAs that act as oncogenes
Oncogenetic lncRNAs that regulate p53 through MDM2
PVT1
The production of the MYC protein in 8q24-amplified cancer cells is positively correlated with PVT1 expression, and the PVT1 amplification number is co-enhanced in over 98% of MYC-copy- increase cancers [34]. PVT1 is a predictive indicator for colorectal cancer (CRC) and can inhibit CRC cell apoptotic capacity. Furthermore, PVT1 also promotes the growth and stem cell-like dimension of malignant hepatoma cells by promoting the expression of NOP2 nucleolar protein [35, 36]. PVT1 recruits EZH2 to the large tumor suppressor kinase 2 (LATS2) promoter and restricts LATS2 transcription. The novel formulation of LATS2 increases apoptosis and suppresses LAD cell proliferation via the MDM2-p53 pathway [37]. A previous study indicated that MDM2 could be bound to LATS2, which could suppress its E3 ligase movement and activate p53 [38]. The study showed that the expression of MDM2 was low. When LATS2 expression was high, the p53 expression was increased in non-small cell lung cancer (NSCLC) cells, corresponding to the intervention of si-PVT1 in NSCLC cells. PVT1 can partake in the MDM2-p53 pathway, which enhances p53 stimulation by inhibiting MDM2-driven p53 regression epigenetically [37]. Taken together, PVT1 might serve as new target for cancer diagnosis and therapy. The PVT1/p53/MDM2 signaling pathway was illustrated in figure 1.
LncRNAs regulate p53 signaling pathway through MDM2. LncRNA-PRA promotes the combination of HSP90 and p53 and therefore the interaction of the p53-MDM2 is decreased. Thus, LncRNA-PRA can competitively suppress the ubiquitination of p53 to enhance the p53 protein levels. ANRIL represses the expression of INK4 family members and impair the p53 signaling pathway through influence the p14ARF-MDM2-p53 axis. MEG3 may facilitate p53 through suppressing the expression level of MDM2 or increasing the protein stability of p53 by RNA-protein interaction. LOC572558 may increase the p53 signaling pathway through decreasing phosphorylation of MDM2 and enhancing the phosphorylation of p53.
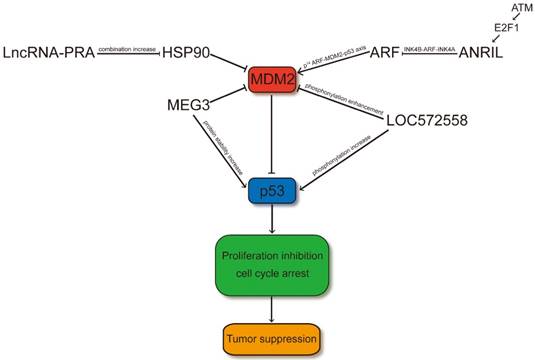
ANRIL
ANRIL, also designated as CDKN2B-AS1, is a long noncoding RNA that is involved in the repression of the INK4B-ARF-INK4A locus [39]. The ANRIL gene may share a bidirectional promoter because the 300 bp upstream of p14ARF gene's transcription start site (TSS) was occupied with the 5′end of the first exon of the ANRIL gene. P14ARF is a positive p53 regulator that inhibits MDM2-p53 interaction. The INK4b-ARF-INK4a locus is protected against DNA methylation in a poised conformation through binding the CpG island's CCCTC-binding factor, which overlaps with the ANRIL-p14ARF promoters. Emerging evidence has shown that ANRIL can bind directly to the INK4b transcript and recruit polycomb repressor complex (PRC) 1 and 2, thereby negatively regulating the transcription of the entire INK4B-ARF-INK4A locus (including p15, p14ARF, and p16 genes) at the late stage of DNA damage response; such regulation allows the cell to return to normal upon completion of DNA repair and simultaneously modulates the p53 and pRb pathways [40]. The downregulation of ANRIL is associated with decreased proliferation [41]. Therefore, in other cancer types, the overexpression of this lncRNA may play a procarcinogenic role [42-44]. High ANRIL expression can be detected in the aggressive TNC subtype in the IBC series [40]. Moreover, the single nucleotide polymorphisms in ANRIL has been shown to be associated with diverse types of cancer and some degenerative diseases (such as type-2 diabetes and atherosclerosis). ANRIL overexpression induces CDKN2A/CDKNB/ARF under-expression in prostate cancer despite having a strong positive link between ANRIL and CDKN2A/CDKNB/ARF [45]. As p14ARF binds to MDM2 and facilitates its degradation, the expression of p53 is upregulated. A previous study showed that the E2F1 transcription factor may cause the overexpression of ANRIL after DNA damage depending on ATM and that INK4B- ARF-INK4A was restrained by ANRIL overexpression at the late stage of DDR, forming a negative feedback loop to the DDR [46].
The primary pathways that can regulate p53 contain the p14ARF-MDM2-p53 axis [47]. The excellent tumor-suppressive INK4-ARF locus can encode p14ARF, which is a protein that can communicate with MDM2 and control its influence on p53, resulting in apoptosis or cell cycle arrest [48]. In rs2151280 TT patients, p14ARF can induce cell cycle arrest or apoptosis, which is dependent on p53 through inhibition of ARF-BP1. ARF-BP1 directly binds and ubiquitinates p53, thereby affecting the p53 cell cycle regulatory pathway in the nucleus. In melphalan therapy of patients carrying the rs2151280 TT genotype, the INK4-ARF locus may be reduced because of the overexpression of ANRIL, which may affect cell proliferation and apoptosis through diverse and unknown pathways. In ESCC, gastric cancer, and NSCLC [43, 49, 50], increasing evidence shows the strong prognostic association between ANRIL upregulation and poor patient survival. Therefore, the original capacity of p53 protein may be influenced through the ANRIL-regulated p14ARF-MDM2-p53 cascade (Figure 1). ANRIL probably plays a role in MM and clinical outcomes of melphalan-based treatment [51]. These data suggest that the expression of ANRIL should be considered to evaluate anti- tumoral drugs, in particular the BET bromodomain inhibitors.
Oncogenetic lncRNAs that regulate p53 stability
PURPL
PURPL is an intergenic lncRNA that can suppress basal p53 levels and is itself under p53 transcriptional control. Through RNA sequencing (RNA-seq), PURPL was found to be upregulated after DNA damage induced by doxorubicin (DOXO) for 16 h at a final concentration of 300 nM in a p53-dependent manner in HCT116, RKO, and SW48 cells. The mRNA and protein expression levels of several p53 target genes, including CDKN1A, MDM2, and TP53INP1, were upregulated due to the LincRNA-PURPL knockdown. In addition, basal PURPL loss resulted in the overrepresentation of p53 signaling and substantial increase in p53 transcriptional activity. PURPL-KO cells not only enhanced the basal p53 expression levels but also resulted in substantial growth defects in vitro. PURPL loss impaired tumor growth in vivo. Such loss also led to growth defects, hypersensitivity to DNA damage, and knockdown of p53. A remarkably decreased cell death was observed after p53 knockdown and DOXO treatment. PURPL loss partially restored the cell proliferation defect, indicating that the elevated basal p53 levels resulting from the loss play a role in growth defects and hypersensitivity to DNA damage. In DLD1- expressing mutant p53, the expression of PURPL was not upregulated after DNA damage, and p53RE mutation did not influence PURPL expression. However, unlike DLD1, when p53RE in PURPL was mutated in RKO and SW48, the basal and DOXO-induced PURPL expression levels were reduced. PURPL loss also upregulated the basal p53 levels in RKO and SW48, but not in DLD1.
However, PURPL does not result in a considerable difference in p53 mRNA expression levels in the presence or absence of DOXO, indicating posttranscriptional regulation. PURPL knockdown resulted in remarkable upregulation of p53-regulated p21 and TP53I3 mRNAs. RNA pull-down using biotinylated PURPL identified its interaction with MYBBP1A that occurred mainly in the nucleoplasm [52-54]. Moreover, HuR-RIP assays from UV-crosslinked HCT116 cells suggested that HuR directly binds to the ARE at the 3′ end of PURPL RNA [55]. RNA pull-downs using purified recombinant glutathione S-transferase-HuR confirmed that MYBBP1A is associated with PURPL RNA by directly binding to the adaptor protein (HuR). Silencing MYBBP1A expression resulted in decreased levels of basal p53 proteins and p21 mRNAs and increased proliferation in DOXO-treated cells. Therefore, an autoregulatory feedback loop seems to be present between p53 and PURPL (Figure 2). In general, p53 activation can transcriptionally upregulate PURPL expression under basal conditions and after DNA damage; moreover, increased PURPL expression downregulates p53 protein levels through a novel negative feedback loop [56]. Therefore, PURPL may be toxic to cells when p53 is induced as unwanted for maintaining cellular homeostasis. Also, identifying and deter- mining the role of other PURPL targets in mediating the effects of this lncRNA will be necessary to fully understand how PURPL controls basal p53 levels and in determining its role in tumor initiation and progression.
LncRNAs influence p53 stability and modification. DNA damage induced p53 activation could transcriptionally regulate PURPL expression and PURPL could repress the protein level of p53 through inhibiting the p53-MYBBP1A interaction, and therefore formed a novel negative feedback loop. DINO induced by p53 in turn enhanced the stability of p53 through RNA-protein interaction to enlarge DNA damage response. MALAT1 could reduce the acetylation of p53 medicated by SIRT1 through binding with DBC1 to inhibit the interaction between DBC1 and SIRT1.
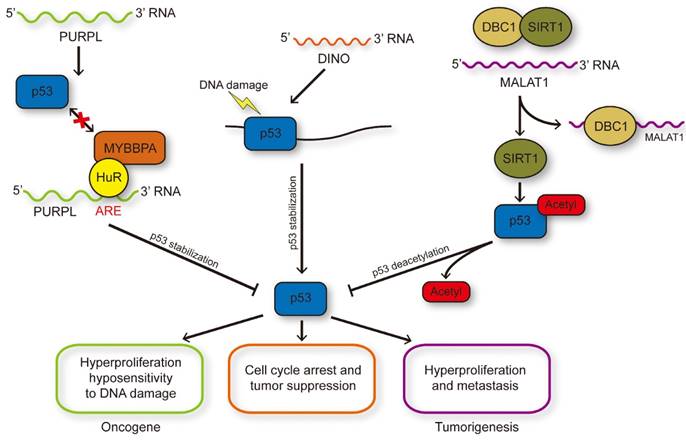
Oncogenetic lncRNAs that regulate posttranslational modification of p53
MALAT1
MALAT1, also known as nuclear-enriched abundant transcript 2 or alpha, is a broadly expressed lncRNA with a length of ~8000 nt. MALAT1 was initially identified from early-stage NSCLC cells, indicating its potential as a prognostic sign for NSCLC metastasis [57]. After this discovery, many studies revealed that MALAT1 is a nuclear lncRNA that is highly regulated in various types of cancers, including endometrial cancer, breast cancer, and cervical cancer [58-60]. The overexpression of MALAT1 is related to hyperproliferation and metastasis in lung cancer [61]. The ectopic expression of MALAT1 promotes cell proliferation and enhances invasion and migration in nude mice. A recent study confirmed that MALAT1 downregulation by infolding an antisense oligonucleotide targeting it represses cancer metastasis through regulation of a group of metastasis-associated genes [62]. Recent study shows that MALAT1 regulates alternative RNA splicing of endogenous target genes through coordinating SR protein's phosphorylation levels [63], or interacting with numerous splicing factors such as serine/ arginine-rich splicing factor 1 (SRSF1) and SRSF3 [15, 64]. Moreover, in HeLa cells, the activities of cell cycle genes are associated with MALAT1, which can communicate with unmethylated PC2 protein- mediated SUMOylation of E2F1 [65]. In human diploid fibroblasts, MALAT1 regulates cell cycle succession, and without it, the G1 phase cannot proceed [14].
MALAT1 has also been implicated in regulating the activity of E2F1 transcription factor by modulating PC2. However, whether a reduction in E2F1 activity or activation of p53 can elicit cell cycle arrest remains unknown [65]. MALAT1 regulated PC2 to possess the activity of E2F1 transcription factor. Nevertheless, whether cell cycle arrest can be caused by the decrease in E2F1 activity or activation of p53 still remains to be elucidated. Time course experiments revealed that the double-stranded DNA damage response may result in the decrease of p53 upon MALAT1 depletion [14].
The high-throughput quantitative proteomics analysis, RIP, electrophoretic mobility shift assay, and surface plasmon resonance revealed the interaction of MALAT1 with DBC1, which can regulate multiple biological processes, including DNA damage response, apoptosis, and tumorigenesis [66]. DBC1 can bind numerous epigenetic modifiers containing SIRT1, HDAC3, and methyltransferase SUV39H1 to modulate epigenetics [67, 68]. The reciprocity between DBC1's LZ domain and SIRT1's catalytic domain restrains the deacetylation of p53, which is the substrate of SIRT1. The 6918-8441 nt fragment of MALAT1 can bind the aa120-280 region of DBC1 involving the LZ domain and prevent the combination of SIRT1 and DBC1. Compared with control cells, MALAT1 overexpression can enhance the deacetylation activity of SIRT1 and downregulate the p53 acetylation level. This lncRNA suppressed the transcription levels of p53 target genes (p21, Bax, Puma, Stat3, Cyclin D, and Cyclin E). In summary, MALAT1 reduced p53 acetylation by interacting with DBC1 [69]. The checkpoint responses to DNA damage and activated anti-oncogenes of p53 may be enhanced by the acetylation of p53, improving its firmness and binding ability with low-affinity promoters [70]. The transcription level of MALAT1 could be inhibited by p53, which can directly bind to the two p53 binding motifs on the promoter region of MALAT1 in erythroid myeloid lymphoid cells. In general, p53 activation can decrease MALAT1 expression. The increased MALAT1 expression can suppress p53 through an inhibitory feedback loop, thus inhibiting cell proliferation and inducing cell apoptosis (Figure 2).
Oncogenetic lncRNAs that Regulated by p53
GUARDIN
Array profiling revealed that lncRNA GUARDIN (RP3-510D11.2-1) is responsive to p53. This finding was validated by qPCR in TP53-null H1299 human lung adenocarcinoma cells. Furthermore, GUARDIN expression is increased due to the activation of DNA damage response with DOXO or overexpression of the oncogenic H-RASV12 mutant in malignant cells and in untransformed adult foreskin fibroblasts [71]. In HCT116 cells expressing wild-type p53, GUARDIN downregulation induced decreased cell viability and caspase3 activation. The GUARDIN gene contains a p53-binding region in the promoter site, and its induction during the DDR is p53-dependent. The location of GUARDIN between the genes encoding miR-34a and hexose-6-phosphate dehydrogenase/glucose-1-dehydrogenase, which is part of the FRA1A fragile site, indicates its susceptibility to repression by genomic instability. Knockdown-induced apoptosis and senescence are commonly related with DDR activation and are typical manifestations of p53 responses to severe DNA damage [72]. Nevertheless, the influence of p53 downregulation on cell survival was not associated with GUARDIN inhibition even if it decreased the expression of GUARDIN. The pro-survival mechanism that was controlled through the p53 pathway was inhibited due to GUARDIN suppression. However, the integrated anti-survival signaling was not influenced. Meanwhile, p53 downregulation restrained anti-survival signaling [73]. Automatically regulated feedback loops between p53 and GUARDIN were found in a previous study. Furthermore, p53 regulates the expression of GUARDIN so as to influencing the function of GUARDIN, nevertheless, GUARDIN in turn keeps cells away from the cytotoxic impression of p53. By RNA pull-down using biotin-labeled GUARDIN followed by mass spectrometry, Wang Lai Hu et al., 2017 identified that BRCA1 and BARD1 are involved in homologous recombination (HR) associated with GUARDIN. GUARDIN also promotes the interaction between BRCA1 and BARD1 by operating as an RNA scaffold. GUARDIN knockdown upregulates the expression of TRF2, which along with TRF1 is directly responsible for double-stranded DNA binding [74]. Such binding was mediated through relieving miR-23a sequestration by GUARDIN. Similar to other lncRNAs, GUARDIN functions as an intrinsic molecular sponge for miRNAs [75]. Wang Lai Hu et al., 2017 also demonstrated that GUARDIN inhibition results in cell cycle arrest and apoptosis by downregulating TRF2 expression and initiating decreased activation of HR and NHEJ by BRCA1 [76]. Inhibiting HR and NHEJ can prevent chromosome end-to-end fusions to sustain genome stability. In summary, GUARDIN is a p53-regulated lncRNA and is essential for sustaining genome stability (Figure 3). Also, a practical implication of GUARDIN is the potential application in cancer.
LncRNAs regulated by p53. GUARDIN maintains genomic integrity and prevents chromosome end-to-end fusion under steady-state conditions and after exposure to exogenous genotoxic stress through maintaining the expression of TRF2 by sequestering microRNA-23a. GUARDIN facilitates the heterodimerization of BRCA1 and BARD1 through serving as an RNA scaffold to sustain the stability of BRCA1. In senescent cells, SAFA-PRC2-EZH2 and SAFA-BMI1-PRC1 complexes are disrupted and the expression of PANDA and pro-senescence genes are increased. PANDA can now interact with NF-YA and block proliferation by secluding NF-YA that is required for survival of senescent cells away from occupying E2F/NF-YA target gene promoters.
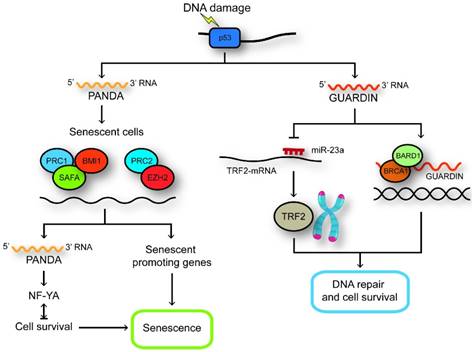
NEAT1
To search for new target genes of the p53 tumor suppressor pathway, the authors utilized p53 ChIP-seq and RNA-seq in wild-type mouse embryonic fibroblasts (MEFs) treated with doxorubicin, and identified the long noncoding RNA (lncRNA) NEAT1 [77]. NEAT1 is a fundamental constituent of paraspeckle nuclear bodies, which is usually induced by mouse and human p53 in different cell types and NEAT1 is a direct transcriptional target of p53. Recent studies on NEAT1 in cancer have demonstrated that NEAT1 can promote the tumorigenesis and the high level of NEAT1 is in positive correlation with poor prognosis in diverse types of cancer [78-80]. Also, NEAT1 can increase cell proliferation and migration of human cancer cell lines and act as an oncogene in mice subjected to the DMBA-TPA skin carcinogenesis protocol[81]. In NEAT1 KO mice, the formation of paraspeckle were blocked and induced the preneoplastic cells sensitive to double-strand break (DSB) and cell death and impaired skin tumorigenesis. Mechanistically, NEAT1 may activate the ATR-CHK1 signaling in response to replication stress through mediating paraspeckle assembly. Interesting, there is an auto-regulatory negative feedback loop that weaken oncogene- dependent activation of p53 between p53 and NEAT1. P53 accelerates the expression of NEAT1 to promote paraspeckle formation and NEAT1 paraspeckle nevertheless inhibited p53 activity [81].
However, some evidences confirmed that NEAT1 might also function as tumor suppressor [82, 83]. NEAT1 is essential to restrain transformation in response to oncogenic signals and cancer initiation on the basis of the function of NEAT1 in differentiation. NEAT1 deficiency enhances both transformation in oncogene-expressing fibroblasts and pancreatic cancer initiation and facilitates the advance of pancreatic intraepithelial neoplasias (PanINs) and cystic lesions reminiscent of intraductal papillary mucinous neoplasms (IPMNs) in KrasG12D- expressing mice[82]. The loss of NEAT1 in oncogene- expressing fibroblasts may promote neoplasia and result in global changes in gene expression. Additionally, low expression of NEAT1 may result in poor prognosis in diverse cancers types[83].
NEAT1 targeting aggravates the vulnerability of human cancer cells to genotoxic chemotherapeutics, including PARP inhibitors through enhancing the sensitivity of cancer cells to respond to S-phase checkpoint activation. Moreover, NEAT1 targeting can also induce synthetic lethal with nongenotoxic reactivation of p53 due to the feedback loop with p53. These data further illustrate that the sensitivity of an extensive series of human cancers cells to chemotherapy or tumors expressing WT or mutant p53 to p53 reactivation therapy may be enhanced by NEAT1 targeting [82].
P53 related lncRNAs that act as tumor suppressors
lncRNAs that regulate p53 through MDM2 and act as tumor suppressors
LOC572558
LOC572558 was identified via microarray of four pairs of human bladder cancer and matched normal bladder tissues in a previous study. In this study, the authors observed that the expression levels of LOC572558 were markedly linked with p53, bladder cancer, cell cycle, propanoate metabolism pathway, and gene expression in the bladder cancer group compared with the normal tissue group, indicating that LOC572558 might participate in the carcinogenesis and progression of bladder cancer [84].
The LOC572558 gene is located in chr9q13 and contains 2,652 nucleotides. Further researches implied that LOC572558 may function as a tumor negative regulator to modulate the p53 signaling and cell cycle pathway in bladder cancer (Figure 1). Furthermore, the lncRNA may serve as a therapeutic target in bladder tumor. Yiping Zhu et al., 2017 perform a phosphoprotein antibody array to recognize the proteins from bladder carcinoma cells after LOC572558 overexpression. They discovered that some proteins for cell proliferation, migration, invasion, and apoptosis were discrepantly phosphorylated after LOC572558 overexpression. Western blot analysis confirmed that the upregulation of LOC572558 caused the decreased phosphorylation of Akt and MDM2 and enhanced phosphorylation of p53. Yiping Zhu et al., 2017 confirmed that LOC572558 upregulation can inhibit proliferation due to the increase of p53 signaling pathway in bladder cancer [85]. However, the specific mechanisms of how LOC572558 regulates p53 phosphorylation are still unknown.
LncRNA-PRA
LncRNA-PRA is a lncRNA located at chromosome 17p13.1. Genomic alterations of lncRNA-PRA have been found to be associated with the decreased survival rate of HCC patients. Through cytogenetic studies and array-based profiling, researchers have observed genomic variations of cancer cell samples. Recurrent SCNVs have been recognized through targeted exome capture and may be related with cancer [86]. LncRNA-PRA was distinguished by published (GSE38323) and lncRNA expression microarray data (GSE27462) in a previous study[87]. To eliminate germline CNVs, the authors of the study measured the periodicity of SCNVs (Somatic copy number variations) in hepatocellular carcinoma by subtracting SCNVs from paired non-tumor liver tissues. In this study, lncRNA-PRAL was transcribed with a poly A+ tail and localized to the cytoplasmic and nuclear regions by RNA fluorescence in situ hybridization. The overexpression of lncRNA-PRA in HCCLM3 and SMMC-7721 cells can promote cell apoptosis and inhibit cell proliferation. However, in p53-deficient (Hep3B) or p53-mutant (Huh7) HCC cells, lncRNA-PRA was under-expressed. LncRNA- PRAL may directly bind to HSP90 in vitro, and the HSP90-p53 interaction increased in SMMC-7721 cells, thereby overexpressing lncRNA-PRAL [88]. Furthermore, the ectopic expression of lncRNA-PRAL in SMMC-7721 cells substantially increased the protein levels of p53 but did not affect the RNA levels. Moreover, it stimulated the upregulation of apoptosis related to p53 target genes (BAX, PUMA, NOXA, and FAS). The mechanism of p53 regulation by lncRNA- PRAL is unclear. However, subsequent findings showed that the combination of HSP90 and p53 was promoted by the three stem-loop motifs of lncRNA- PRAL at the 5′ end. The p53-MDM2 interaction is decreased with increased HSP90-p53 interaction in lncRNA-PRAL-overexpressed SMMC-7721 cells. These results revealed that lncRNA-PRAL can considerably suppress the MDM2-dependent ubiquitination of p53. However, the specific mechanism needs further investigations.
LncRNAs that regulate p53 stability and act as tumor suppressors
MEG3
The segmental DLK1-MEG3 locus is located at human chromosome 14q32 and at mouse chromosome 12 [89]. This locus was thought to be a tumor inhibitor [90]. MEG3 can be expressed aberrantly in diverse cancer types, including bladder tumor, glioma, and hepatocellular carcinoma [91-93]. The tumor grade of meningioma is associated with the expression level of MEG3 [94]. In NSCLC cells, the MEG3 levels are high. MEG3 regulates cell proliferation and apoptosis in a p53-dependent manner [95]. Furthermore, it can function in a p53-dependent and independent manner. The overexpression of MEG3 RNA in MCF7 cells can transcriptionally downregulate the mRNA levels of MDM2 without p53. Accordingly, the mRNA levels of p53 are stable, but its protein levels are increased, suggesting that the stability of p53 may be modulated through indirect transcriptional management of MDM2 by MEG3 (Figure 1). Chromatin immunoprecipitation (ChIP) assay demonstrated that MEG3 RNA can bind to the promoter region of its target gene (p21) to accumulate p53 [96]. MEG3 overexpression can motivate the expression of the GDF15-p53 target by enhancing the p53 binding to the p53 response element in its promoter region. MEG3 is not correlated with the p21 level in HCT116 cells.
As shown in Figure 1, the MDM2 level can be downregulated by MEG3 overexpression, and this may be one of the mechanisms whereby MEG3 can influence p53. However, the specific mechanism underlying the modulation of p53 by MEG3 is unclear. Further studies are needed to investigate whether MEG3 directly interacts with MDM2 and p53 and regulate their expression or indirectly modulate p53 via MDM2 [97]. These data suggested that MEG3 non-coding RNA may function as a tumor suppressor, whose action is mediated by both p53-dependent and p53-independent pathways.
DINO
The DINO lncRNA was identified in a study where the authors discovered a DNA-damage- induced transcription unit upstream of CDKN1A in screening for transcribed regions in human cell cycle promoters by ChIP [98]. DINO is a 951-base RNA transcribed divergently from CDKN1A by RACE and is situated adjacent to a p53-binding site. In human fibroblasts, the induction of DINO was abrogated due to the downregulation of p53 expression. Only TP53-null H1299 human lung adenocarcinoma cells together with wild-type p53 can induce DINO; the p53 mutant obtained from cancer-prone Li-Fraumeni syndrome did not exert any effects. In the eight inspected categories, DINO may be an underlying conserved transcriptional responding to DNA damage [55]. DINO participates in regulating cell cycle arrest in response to DNA damage. Human osteosarcoma U2OS cells with DINO knockdown can continue dividing by a greater degree than DINO-perfect cells in response to DNA damage. Moreover, genes that are influenced by DINO depletion, such as emblematic p53-responsive genes CDKN1A, DDB2, and GADD45A, may fail to respond to DNA damage. Using the assay of transposase accessible chromatin by sequencing, a previous study revealed that DINO-depleted human fibroblasts decreased the chromatin approachability in isogenous p53 binding sites [99]. The authors showed that DINO depletion abrogated the binding capacity of p53 with its isogenous target genes, including CDKN1A and attached genes, by using chromatin immunoprecipitation (ChIP) and quantitative PCR (qPCR). DINO can better interact with recombinant purified p53 than PRC2. A discrete DINO domain can be combined with the p53 C terminus that also participates in massive posttranslational revisions. Chromatin isolation by RNA purification revealed that the p53 target genes, namely, CDKN1A, GADD45A, and DDB2, were integrated with DINO at the p53 binding elements. Cycloheximide chase assay showed that without DINO binding to p53, the p53 protein stability and p21 protein levels induced by DNA damage were decreased even with the preservation of p53 phosphorylation at Ser9. Furthermore, DINO overexpression can stimulate DNA damage signaling and stabilize p53. A set of p53 target genes, such as RRM2, DDB2, and GADD45A, were induced by DINO through RRM2, DDB2, and GADD45A [100].
A previous study found that p53 protein expression levels can regulate the organism's reaction to DNA damage and lethal irradiation[101]. The authors surmised that DINO depletion in mice may be prone to tumors such as Trp53+/- and epi-allelic p53 hypomorphs [101]. They found that the response of mice and cells with Dino depletion are phenotypically analogous to that of Trp53+/- mice following serious DNA damage. In MEFs, Dino influences DNA damage-derivable genes independently of p21. Furthermore, Dino can directly interact with p53 and affect its stability in MEFs similar to human cells. The MEFs with Dino promoter depletion weaken the DNA damage induced by cell cycle arrest, and the cells may continue to proliferate without replicative senescence compared with DINO-proficient cells.
In summary, as shown in Figure 2, DINO can interact with p53 protein and functionally regulate the p53 signaling response to DNA damage, analogous to the maternally imprinted lncRNA MEG3, which can upregulate the activity of p53.
LncRNAs that Regulated by p53 and act as tumor suppressors
PANDA
PANDA (p21 associated ncRNA DNA damage activated) is a non-spliced 1.5-kb ncRNA and located approximately 5 kb upstream of the CDKN1A TSS and evolutionarily conserved. PANDA was identified from 216 transcribed regions encoding putative lncRNAs that recognized from an ultrahigh-density array involving the promoters of 56 cell cycle genes in 108 samples representing diverse perturbations. Also, the expression of these lncRNAs is associated with specific oncogenic stimuli, stem cell differentiation or DNA damage [98]. The p53 binding site was located at the upstream of the CDKN1A TSS that was demonstrated by CHIP analysis [102]. Knockdowning p53 can inhibit the induction of PANDA after DNA damage; however, the down-regulating CDKN1A has no influence on PANDA expression. Human primary breast tumors with inactivating mutation in TP53 show lower expression of PANDA expression [103]. Moreover, reintroducing wild-type p53 into p53-null H1299 lung carcinoma cells can restore DNA damage-inducible expression of PANDA. Knocking down PANDA can significantly induce numerous genes involved in apoptosis including APAF1, BIK, FAS and LRDD. PANDA interacts with the nuclear transcription factor NF-YA and consequently blocks binding of NF-YA to the chromatin to restrict NF-YA activation of apoptotic gene expression program [98].
Cellular senescence can limit the proliferation of pre-cancerous cells through a stable cell cycle arrest. PANDA may be identified as a 'locking device' that keeps cells in proliferative state. PANDA could interact with scaffold-attachment-factor A (SAFA) to recruit polycomb repressive complex (PRC) proteins BMI1-PRC1 and EZH2- PRC2 to repress the senescence-promoting genes in proliferating cells [104]. In senescent cells, SAFA-PANDA-PRC interactions are disrupted and PANDA sequesters the transcription factor NF-YA from occupying E2F/ NF-YA target gene promoters to inhibit the expression of proliferation-promoting genes regulated by E2F [104]. Moreover, the expression of PANDA in HCCs is remarkable low and consistently the expression levels of SAFA, BMI1 and EZH2 that are repressors of PANDA were increasing. Knocking down PANDA in U2OS cells could repress the protein level of p53 through reducing the stability of p53 protein without influencing the expression of p53 mRNA [105]. These results showed that PANDA might influence the p53 pathway to contribute to cancer development (Figure 3).
Conclusion and future perspective
In normal proliferating cells, the protein expression levels of p53, which serves as a master gene regulator, are low. Such levels are poorly regulated because the dysregulation of p53 may be harmful to the cells. The key role of p53, which is mutated or inactivated by multifarious mechanisms, including overexpression of MDM2 in more than 50% of all human cancers, is ascribed to its effect in maintaining cell homeostasis by activating genes that protect the genomic stability and survival of healthy cells and exhibit remarkable tumor suppressor activity. Many studies demonstrated the increasing importance and complexity of the p53 pathway. Recent studies focused on some new components and layers of regulation. For example, lncRNAs are referred to various biological progresses and often serve as effector molecules to coordinate transcription regulation containing p53 activity by interacting with epigenetic machinery and joining the p53 pathway [77, 106, 107]. On the one hand, lncRNAs are involved in the sophisticated regulatory system concerning the maintenance of p53 stability. On the other hand, the expression levels of lncRNAs are regulated by p53, and these lncRNAs can act as p53 effectors regulating cell cycle arrest, DNA damage repair, chromosomal stability, and apoptosis. The expression levels of lncRNAs in various cancers are low even after induction because of DNA damage and are usually dysregulated. However, for the substantial quantity of lncRNAs, we still want to increase our understanding of lncRNAs, which may function as crucial regulators of the p53 pathway, in order to expand the repertoire of p53 targets. LncRNAs can seclude transcription factors by serving as “molecular decoys” or “sponges” [108, 109] or by regulating specific gene expression in cis [110] or trans [111] by constituting ribonucleoprotein complexes.
In this review, we highlight only the role of some lncRNAs that are related to the mechanisms of the p53 pathway and its regulation to sustain p53 at a certain level or lncRNAs that serve as p53 effectors. The effect of lncRNAs on the regulation of p53 has been described in this review. Figures 1 , 2 and 3 present the summary of such effect. We have tried to categorize lncRNAs according to their individual regulatory mechanisms with p53. However, most transcripts adapt to numerous classifications, suggesting that lncRNAs may constitute crucial regulatory networks interacting with p53 to mediate its roles in driving many important cancer phenotypes. A clear distinction between oncogenic and tumor suppressor lncRNAs or p53-regulators and effectors cannot be made (Table 1). Given the importance and complexity of the network linking lncRNAs, p53, and tumor suppressor response, lncRNAs may constitute considerable regulatory networks to coordinate multiple aspects of cancer progression.
LncRNAs that regulating p53 or regulated by p53 in cancer progression
| lncRNA | Mechanistic theme(s) | Tumor suppressor /oncogene | disease type | References |
|---|---|---|---|---|
| PVT1 | Induced by p53,disturbs MDM2-driven p53 degradation | oncogene | CRC, hepatoma | 34-37 |
| ANRIL | may improve the protein levels of p53 through influence the p14ARF-MDM2-p53 cascade | oncogene | ESCC, gastric cancer and NSCLC | 39-51 |
| PURPL | Induced by p53,reduces the stability of p53 through inhibiting the p53-MYBBP1A interaction | oncogene | Colorectal cancer | 52-56 |
| MALAT1 | inhibited by p53,weaken the acetylation of p53 by interacting with DBC1 | oncogene | NSCLC, breast cancer, cervical cancer | 57-70 |
| GUARDIN | p53 regulates the expression of it,protects cells from the cytotoxic effect of p53 | oncogene | lung adenoidarcinoma, colorectal cancer | 71-76 |
| NEAT1 | regulated through p53, forms an autoregulatory negative feedback loop with p53 | oncogene/tumor suppressor | osteosarcoma, breast cancer, lung cancer, colorectal cancer | 78-82 |
| LOC572558 | might enhance the phosphorylation of MDM2 ,Akt,and p53 | tumor suppressor | bladder cancer | 84-85 |
| lncRNA-PRA | may increase the protein levels of p53 depending on MDM2 | tumor suppressor | HCC | 86-88 |
| MEG3 | may strengthen the protein stability of p53 by down-regulate the mRNA level of MDM2 | tumor suppressor | bladder tumor, glioma, and hepatocellular carcinoma | 89-97 |
| DINO | induced by p53,stabilizes p53 through RNA-protein interaction | tumor suppressor | lung adenocarcinoma | 98-101 |
| PANDA | regulated by p53, influence the protein level of p53 without mRNA level | tumor suppressor | osteosarcoma, lung carcinoma | 98-105 |
Therefore, an in-depth research on the regulatory mechanism of specific p53-regulated lncRNAs that may serve as p53-regulators and effectors might be essential for future clinical applications of lncRNAs. Many lncRNAs are exceptionally expressed in cancers. Thus, these lncRNAs may function as cancer biomarkers or potential therapeutic targets [112]. We can enhance our understanding of the localization, structure, and function of lncRNAs in cancer by detecting novel lncRNAs using new tools, which may help elucidate the role of lncRNAs in cancer. Furthermore, organismal models will be particularly important for clarifying the emerging physiological roles of lncRNAs in some central factors that are crucial for the connection between cancer cells and stromal cells, which support tumor growth and development.
Acknowledgements
This study was funded by grants from the National Natural Science Foundation of China (No. 81874183, 81602533, 81672845 and 81872304), the Education Department of Jiangsu Province (No. 15KJA320006), the Jiangsu Provincial Medical Youth Talent Project (QNRC2016776) and the Project of Invigorating Health Care through Science, Technology and Education from Jiangsu.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A. et al. Landscape of transcription in human cells. Nature. 2012;489:101-8
2. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915-27
3. Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799-816
4. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223-7
5. Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240-51
6. Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773-84
7. Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637-43
8. Whitehead J, Pandey GK, Kanduri C. Regulation of the mammalian epigenome by long noncoding RNAs. Biochim Biophys Acta. 2009;1790:936-47
9. Sanchez Y, Huarte M. Long non-coding RNAs: challenges for diagnosis and therapies. Nucleic Acid Ther. 2013;23:15-20
10. Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199-208
11. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26-46
12. Lee S, Kopp F, Chang TC, Sataluri A, Chen B, Sivakumar S. et al. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell. 2016;164:69-80
13. Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435-9
14. Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X. et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:e1003368
15. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT. et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925-38
16. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7-21
17. Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C. et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973
18. Arun G, Diermeier S, Akerman M, Chang KC, Wilkinson JE, Hearn S. et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016;30:34-51
19. Redis RS, Vela LE, Lu W, Ferreira de Oliveira J, Ivan C, Rodriguez-Aguayo C. et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol Cell. 2016;61:640
20. Tian D, Sun S, Lee JT. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell. 2010;143:390-403
21. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667-78
22. Postepska-Igielska A, Giwojna A, Gasri-Plotnitsky L, Schmitt N, Dold A, Ginsberg D. et al. LncRNA Khps1 Regulates Expression of the Proto-oncogene SPHK1 via Triplex-Mediated Changes in Chromatin Structure. Mol Cell. 2015;60:626-36
23. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59
24. Wang X, Arai S, Song X, Reichart D, Du K, Pascual G. et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126-30
25. Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K. et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell. 2014;54:777-90
26. Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z. et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513-31
27. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667-72
28. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F. et al. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science. 2010;329:689-93
29. Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935
30. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275-83
31. Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22:97-106
32. Majidinia M, Yousefi B. DNA damage response regulation by microRNAs as a therapeutic target in cancer. DNA Repair (Amst). 2016;47:1-11
33. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594-604
34. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H. et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82-6
35. Takahashi Y, Sawada G, Kurashige J, Uchi R, Matsumura T, Ueo H. et al. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br J Cancer. 2014;110:164-71
36. Wang F, Yuan JH, Wang SB, Yang F, Yuan SX, Ye C. et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology. 2014;60:1278-90
37. Wan L, Sun M, Liu GJ, Wei CC, Zhang EB, Kong R. et al. Long Noncoding RNA PVT1 Promotes Non-Small Cell Lung Cancer Cell Proliferation through Epigenetically Regulating LATS2 Expression. Mol Cancer Ther. 2016;15:1082-94
38. Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687-700
39. Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP. et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202-6
40. Meseure D, Vacher S, Alsibai KD, Nicolas A, Chemlali W, Caly M. et al. Expression of ANRIL-Polycomb Complexes-CDKN2A/B/ARF Genes in Breast Tumors: Identification of a Two-Gene (EZH2/CBX7) Signature with Independent Prognostic Value. Mol Cancer Res. 2016;14:623-33
41. Chen D, Zhang Z, Mao C, Zhou Y, Yu L, Yin Y. et al. ANRIL inhibits p15(INK4b) through the TGFbeta1 signaling pathway in human esophageal squamous cell carcinoma. Cell Immunol. 2014;289:91-6
42. Hua L, Wang CY, Yao KH, Chen JT, Zhang JJ, Ma WL. High expression of long non-coding RNA ANRIL is associated with poor prognosis in hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:3076-82
43. Lin L, Gu ZT, Chen WH, Cao KJ. Increased expression of the long non-coding RNA ANRIL promotes lung cancer cell metastasis and correlates with poor prognosis. Diagn Pathol. 2015;10:14
44. Qiu JJ, Lin YY, Ding JX, Feng WW, Jin HY, Hua KQ. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int J Oncol. 2015;46:2497-505
45. Rodriguez C, Borgel J, Court F, Cathala G, Forne T, Piette J. CTCF is a DNA methylation-sensitive positive regulator of the INK/ARF locus. Biochem Biophys Res Commun. 2010;392:129-34
46. Wan G, Mathur R, Hu X, Liu Y, Zhang X, Peng G. et al. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25:1086-95
47. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011;50:5566-82
48. Sherr CJ. Ink4-Arf locus in cancer and aging. Wiley Interdiscip Rev Dev Biol. 2012;1:731-41
49. Zhang EB, Kong R, Yin DD, You LH, Sun M, Han L. et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget. 2014;5:2276-92
50. Nie FQ, Sun M, Yang JS, Xie M, Xu TP, Xia R. et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14:268-77
51. Poi MJ, Li J, Sborov DW, VanGundy Z, Cho YK, Lamprecht M. et al. Polymorphism in ANRIL is associated with relapse in patients with multiple myeloma after autologous stem cell transplant. Mol Carcinog. 2017;56:1722-32
52. Kumazawa T, Nishimura K, Katagiri N, Hashimoto S, Hayashi Y, Kimura K. Gradual reduction in rRNA transcription triggers p53 acetylation and apoptosis via MYBBP1A. Sci Rep. 2015;5:10854
53. Kuroda T, Murayama A, Katagiri N, Ohta YM, Fujita E, Masumoto H. et al. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J. 2011;30:1054-66
54. Ono W, Hayashi Y, Yokoyama W, Kuroda T, Kishimoto H, Ito I. et al. The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J Biol Chem. 2014;289:4928-40
55. Schmitt AM, Garcia JT, Hung T, Flynn RA, Shen Y, Qu K. et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet. 2016;48:1370-6
56. Li XL, Subramanian M, Jones MF, Chaudhary R, Singh DK, Zong X. et al. Long Noncoding RNA PURPL Suppresses Basal p53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep. 2017;20:2408-23
57. Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM. et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031-41
58. Yamada K, Kano J, Tsunoda H, Yoshikawa H, Okubo C, Ishiyama T. et al. Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci. 2006;97:106-12
59. Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ. et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163
60. Guo F, Li Y, Liu Y, Wang J, Li Y, Li G. Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim Biophys Sin (Shanghai). 2010;42:224-9
61. Gutschner T, Hammerle M, Diederichs S. MALAT1 - a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl). 2013;91:791-801
62. Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G. et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180-9
63. Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z. et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082-93
64. Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185-93
65. Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD. et al. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773-88
66. Chini EN, Chini CC, Nin V, Escande C. Deleted in breast cancer-1 (DBC-1) in the interface between metabolism, aging and cancer. Biosci Rep. 2013:33
67. Chini CC, Escande C, Nin V, Chini EN. HDAC3 is negatively regulated by the nuclear protein DBC1. J Biol Chem. 2010;285:40830-7
68. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583-6
69. Chen R, Liu Y, Zhuang H, Yang B, Hei K, Xiao M. et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017;45:9947-59
70. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612-26
71. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638-42
72. Wynford-Thomas D. p53: guardian of cellular senescence. J Pathol. 1996;180:118-21
73. Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321-5
74. Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer. 2007;7:152
75. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-52
76. Hu WL, Jin L, Xu A, Wang YF, Thorne RF, Zhang XD. et al. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat Cell Biol. 2018;20:492-502
77. Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA. et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016-31
78. Zhen L, Yun-Hui L, Hong-Yu D, Jun M, Yi-Long Y. Long noncoding RNA NEAT1 promotes glioma pathogenesis by regulating miR-449b-5p/c-Met axis. Tumour Biol. 2016;37:673-83
79. Guo S, Chen W, Luo Y, Ren F, Zhong T, Rong M. et al. Clinical implication of long non-coding RNA NEAT1 expression in hepatocellular carcinoma patients. Int J Clin Exp Pathol. 2015;8:5395-402
80. Pan LJ, Zhong TF, Tang RX, Li P, Dang YW, Huang SN. et al. Upregulation and clinicopathological significance of long non-coding NEAT1 RNA in NSCLC tissues. Asian Pac J Cancer Prev. 2015;16:2851-5
81. Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P. et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. 2016;22:861-8
82. Mello SS, Sinow C, Raj N, Mazur PK, Bieging-Rolett K, Broz DK. et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017;31:1095-108
83. Idogawa M, Ohashi T, Sasaki Y, Nakase H, Tokino T. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int J Cancer. 2017;140:2785-91
84. Zhu YP, Bian XJ, Ye DW, Yao XD, Zhang SL, Dai B. et al. Long noncoding RNA expression signatures of bladder cancer revealed by microarray. Oncol Lett. 2014;7:1197-202
85. Zhu Y, Dai B, Zhang H, Shi G, Shen Y, Ye D. Long non-coding RNA LOC572558 inhibits bladder cancer cell proliferation and tumor growth by regulating the AKT-MDM2-p53 signaling axis. Cancer Lett. 2016;380:369-74
86. Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T. et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58:706-17
87. Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX. et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679-89
88. Zhou CC, Yang F, Yuan SX, Ma JZ, Liu F, Yuan JH. et al. Systemic genome screening identifies the outcome associated focal loss of long noncoding RNA PRAL in hepatocellular carcinoma. Hepatology. 2016;63:850-63
89. Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, Aisaka K. et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000;5:211-20
90. Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, Johnson SR. et al. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab. 2003;88:5119-26
91. Ying L, Huang Y, Chen H, Wang Y, Xia L, Chen Y. et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst. 2013;9:407-11
92. Wang P, Ren Z, Sun P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem. 2012;113:1868-74
93. Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A. et al. Loss of imprinting and allelic switching at the DLK1-MEG3 locus in human hepatocellular carcinoma. PLoS One. 2012;7:e49462
94. Zhang X, Gejman R, Mahta A, Zhong Y, Rice KA, Zhou Y. et al. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res. 2010;70:2350-8
95. Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu WQ. et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461
96. Sun L, Li Y, Yang B. Downregulated long non-coding RNA MEG3 in breast cancer regulates proliferation, migration and invasion by depending on p53's transcriptional activity. Biochem Biophys Res Commun. 2016;478:323-9
97. Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R. et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731-42
98. Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD. et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621-9
99. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213-8
100. Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13-20
101. Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ, Cordon-Cardo C. et al. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet. 2003;33:396-400
102. Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T. et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207-19
103. Geisler S, Lonning PE, Aas T, Johnsen H, Fluge O, Haugen DF. et al. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001;61:2505-12
104. Puvvula PK, Desetty RD, Pineau P, Marchio A, Moon A, Dejean A. et al. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat Commun. 2014;5:5323
105. Kotake Y, Kitagawa K, Ohhata T, Sakai S, Uchida C, Niida H. et al. Long Non-coding RNA, PANDA, Contributes to the Stabilization of p53 Tumor Suppressor Protein. Anticancer Res. 2016;36:1605-11
106. Akdemir KC, Jain AK, Allton K, Aronow B, Xu X, Cooney AJ. et al. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014;42:205-23
107. Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, Rinn JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015;43:4447-62
108. Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT. Jpx RNA activates Xist by evicting CTCF. Cell. 2013;153:1537-51
109. Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L. et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69-80
110. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904-14
111. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y. et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120-4
112. Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108:2419-25
Author contact
![]() Corresponding authors: Jin Bai, Cancer Institute, Xuzhou Medical University, 84 West Huaihai Road, Xuzhou, 221002, Jiangsu Province, China. E-mail: bjedu.cn or Jun-Nian Zheng, Cancer Institute, Xuzhou Medical University, 84 West Huaihai Road, Xuzhou, 221002, Jiangsu Province, China. E-mail: jnzhengedu.cn.
Corresponding authors: Jin Bai, Cancer Institute, Xuzhou Medical University, 84 West Huaihai Road, Xuzhou, 221002, Jiangsu Province, China. E-mail: bjedu.cn or Jun-Nian Zheng, Cancer Institute, Xuzhou Medical University, 84 West Huaihai Road, Xuzhou, 221002, Jiangsu Province, China. E-mail: jnzhengedu.cn.