Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Epidemiology
Histological Challenges for...
Genetic Evidence for Heredity
Autoimmune Association
Autoimmune & Genetic Targets...
Conclusion and Future Directions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(7):1429-1439. doi:10.7150/ijbs.34613 This issue Cite
Review
Lichen Sclerosus: An autoimmunopathogenic and genomic enigma with emerging genetic and immune targets
Davis A. Tran1, Xiaohui Tan1, Charles J. Macri2, Andrew T. Goldstein2,3 ![]() , Sidney W. Fu1
, Sidney W. Fu1 ![]()
1. Department of Medicine (Division of Genomic Medicine), and Department of Microbiology, Immunology and Tropical Medicine, The George Washington University School of Medicine and Health Sciences, Washington, DC
2. Department of Obstetrics and Gynecology, The George Washington University School of Medicine and Health Sciences, Washington, DC
3. Center for Vulvovaginal Disorders, Washington, DC
Received 2019-3-4; Accepted 2019-4-10; Published 2019-6-2
Abstract

Lichen sclerosus (LS) is an inflammatory dermatosis with a predilection for anogenital skin. Developing lesions lead to vulvar pain and sexual dysfunction, with a significant loss of structural anatomical architecture, sclerosis, and increased risk of malignancy. Onset may occur at any age in both sexes, but typically affects more females than males, presenting in a bimodal fashion among pre-pubertal children and middle-aged adults. A definitive cure remains elusive as the exact pathogenesis of LS remains unknown. A general review of LS, histologic challenges, along with amounting support for LS as an autoimmune disease with preference for a Th1 immune response against a genetic background is summarized. In addition to the classically referenced ECM1 (extracellular matrix protein 1), a following discussion of other immune and genetic targets more recently implicated as causative or accelerant agents of disease, particularly miR-155, downstream targets of ECM1, galectin-7, p53, and epigenetic modifications to CDKN2A, are addressed from the viewpoint of their involvement in three different, but interconnected aspects of LS pathology. Collectively, these emerging targets serve not only as inherently potential therapeutic targets for treatment, but may also provide further insight into this debilitating and cryptic disease.
Introduction
Lichen sclerosus (LS) is a chronic relapsing inflammatory dermatosis of cryptic etiology that mainly affects pre-pubertal children and middle-aged adults. While LS lesions may develop over any cutaneous tissue at any age in both sexes, there is a predilection for anogenital skin in 85%-98% of cases, with a higher incidence of onset among pre-menarche and post-menopausal females [1, 2]. LS patients suffer from patchy lesions that coalesce into sclerotic plaques, culminating in urinary and sexual dysfunction with a 4% elevated risk of squamous carcinoma [3]. Although LS is seen as a rare condition [4, 5], LS is among one of the most common referrals for vulvar pruritus and is the most common form of structural change to the vulvar region [6-8]. Yet, to date, much of LS is uncertain, as discourse concerning its pathogenesis, histological diagnostic criteria, and treatment remains contested even after more than a century since its first clinical description presented in 1887 [9-12]. However, there are emerging molecular targets that may provide better insight into the progression of LS and the onset of morphological features that lead to the diseased state. Nevertheless, since the pathology of LS involves intimate anatomical areas, patients are less likely to seek treatment; thus, further compounding the dynamics of this underrecognized and undertreated dermatosis.
Epidemiology
LS exhibits a bimodal distribution in incidence, with the first peak occurring before puberty in pre-adolescent teens and the second peak arising in middle to late adulthood, specifically after menopause for women and between 30-50 years of age in men [11, 13, 14]. In the literature, however, there is a lack of large-scale epidemiological studies that consider both the sex and age of onset. Therefore, the true incidence of LS is unknown. The estimated prevalence is most likely underestimated, as studies are limited to the confines of case-control studies, which mainly focuses on one demographic (i.e. men, women, boys, or girls)—few studies include a sample of all four age and sex groups.
Nevertheless, the classically cited prevalence approximates 0.1%-0.3% in a general hospital setting [14, 15], and LS is therefore recognized as a rare condition by the Genetic and Rare Diseases Information Center (GARD) of the National Institutes of Health (NIH) and the National Organization for Rare Disorders (NORD) [4, 5]. Most recently, a report on the prevalence of LS in Botswana amassed to 0.2%, falling within this range [16]. In contrast, however, the European Dermatology Forum classified LS as a common disease [17] and the prevalence of LS was observed to be 1.67% in a general gynecology clinic in the United States [18]. Extrapolated data from the Oxford clinic approximates 150 to 200 women per million population seek medical treatment for LS each year [19] and a more recent report suggests the incidence of LS is rising, from 7.4 to 14.6 women per 100,000 between 1991 and 2011 in the Netherlands [20]. Taken together from the literature, LS is reported to be more prevalent in women (3%) [21] than men (>0.07%) [22], and commonly affects more adults (1.5%) than children (0.3%) [23, 24] (Table 1).
Prevalence of LS in different age and sex groups. LS presents in a bimodal fashion, typically before puberty and during the middle-ages of life, with a higher incidence in menopausal women overall (3%) and among pre-pubertal boys when compared to their female counterparts (0.5%).
| Child | Adult | |
|---|---|---|
| Female | 0.11% | 3% |
| Male | 0.5% | >0.07% |
The sex ratio among women and men vary widely in the literature, from 10:1 [14], 6:1 [25], to 3:1 [2]. In children, the sex ratio is reversed [26], as there is a prevalence of 0.5% in boys [13, 27] and 0.11% in girls [13, 28]. This reversal in childhood prevalence may be due to a detection bias, as boys are often admitted to a clinical setting for LS-associated phimosis, whereas girls may be asymptomatic in early LS and develop characteristic lesions later in adulthood [13]. Additionally, the bimodal prevalence observed in females may also be skewed. Given that changing levels of estrogen is associated with changes in skin hydration, collagen content, and glycosaminoglycan concentrations [7], estrogen deficiency may therefore directly weaken the structural integrity of vulvar skin. Such changes may lead to worsening symptoms, causing pre-pubertal girls and menopausal women to more likely seek treatment than otherwise well estrogenzied female age groups. As such, the incidence is an underestimate given various additional compounding factors, such as under-reporting due to physician under-recognition and misdiagnosis, patient trepidation in seeking treatment (as LS may be easily mistaken for sexual abuse in premenarchal girls, and therefore may heighten hesitancy), LS management under different specialties, and asymptomatic cases that go unnoticed [18].
Histological Challenges for Diagnosis
Typical LS lesions are characterized by a zone of lymphatic infiltrate observed underneath a condensed region of sclerosis in the upper dermal region (comprised of hyalinized collagen and homogenized collagen fibrils), with flattening of Rete's pegs and hyperkeratosis [8, 29, 30]. As with much of LS, there is a lack of unanimity regarding the reliability of histological biopsy, especially in the early stages of disease. In contrast to many studies, Regauer and Liegl [31] did not find the characteristic zone of dermal sclerosis as an essential, early diagnostic feature [8].
A review of the literature shows inconsistences with the description of this sclerotic zone, as some authors have used “hyalinized collagen,” “homogenous collagen,” and “sclerotic band” interchangeably [29, 31-33]; whereas others seemingly viewed these descriptions as distinct histopathological features [30, 34, 35]. The usage of “hyalinized collagen” may indeed signify a zone of thickened, homogenous collagen fibrils in the presence of amorphous eosinophilic material as defined by Saluja and Iyer [36]. However, phrasing in the first manner, in which “homogenous collagen” is used with the omission of “hyalinized collagen,” regrettably draws into question whether or not the amorphous eosinophilic material is present. Furthermore, diagnostic criteria failed to distinguish early LS from eczema [8] and due to overlapping features to other forms of sclerosis, early vulvar LS is often misdiagnosed as “non-specific vulvitis” [31]. Finally, the various subtypes [37, 38], additional hodgepodge of other dermal changes (e.g. edema, abnormal extracellular structures, basement membrane alterations), and morphological changes due to self-treatment medication, considerably widens the spectrum of disease and blurs the histological picture of LS [8].
In spite of such challenges in establishing a full diagnostic picture, some recommend a biopsy to confirm diagnosis as well as to exclude potential vulvar intraepithelial neoplasia or malignancy in suspicious lesions or areas [17, 39]. Multiple sections [31] with repeated biopsies in subsequent follow-ups, are encouraged, particularly in evolving lesions resistant to treatment [10]. Kato et al. [40], however, placed high reliability on histological diagnosis, as clinical suspicion of LS in males alone had a high rate of inaccuracy (59%). The difference in the cohort demographic studied with its focus on LS in males, in part, may account for such discrepancies compared to the majority of studies on LS in females. Taken together, such complexity reinforces the importance of making a diagnosis in alignment with a true, clinicopathologic correlation as opposed to any one clinical finding or isolated biopsy.
Genetic Evidence for Heredity
Familial Occurrence
Familial studies show a positive family history of LS [6, 41-43]. In particular, Sherman et al. [43] found 12% of female LS patients had a first-degree female relative also diagnosed with LS. Case reports of LS in monozygotic and dizygotic twins, siblings, and mothers-daughters further support a genetic component. However, the mode of inheritance has not yet been established [44-46].
Immunogenetics (HLA Class II Antigens)
LS genetic susceptibility is also strengthened by a significant positive association with genes regulating HLA class II antigens, which are critical regulators of humoral immunity. Much study has focused on the prevalence of HLA-DQ7 in LS patients, which appears in 50% of adult females, 45% adult males, and 66% prepubertal females, suggesting HLA-DQ7 may play a role in LS susceptibility [47-49]. In addition, HLA-DR12 and haplotype DRB1*12/DQB1*0301/04/09/010 are shown to appear more frequently in LS patients than controls [48, 50]. In contrast, DR17 has a negative association and therefore, may prove to confer protection against LS [50]. Interestingly, when considering autoimmune diseases with LS, it appears DRB1*13 is more prominent in those with LS alone than those with a comorbidity of both LS and autoimmune disease [50]. Such data suggests that while DRB1*13 may not be protective of LS, it may be protective against the comorbidity of LS and onset of autoimmune disease.
Autoimmune Association
Autoimmune Disease Comorbidity
Although not universally accepted [42], the association between LS and autoimmune disease has been well-established in women, but less so in men. A large, retrospective review of 532 LS patients comprised of both men and women by Kreuter et al. [2] revealed that while 82% of the total cohort had at least one type of autoimmune disease, more women significantly (p<0.0001) showed comorbidity for an autoimmune disease compared to men (18.9% vs. 5.1%). Such findings further confirmed prior case-control reports by Harrington and Dunsmore [51] and Cooper et al. [52], citing 34% and 28.4% of LS women having comorbidity with at least one other autoimmune disease compared to healthy controls, respectively. These figures are comparable to case series studies exclusive to women, citing 21.5%-25% of LS female patients with at least one other autoimmune disease [48, 53]. In stark contrast, only 3%-7% LS male patients presented comorbidity with an autoimmune disease, with figures comparable to the prevalence of autoimmune diseases in populations without LS [47, 54, 55]. This suggests there is a strong, positive association between LS and autoimmune disease in women and in contrast, a weak association in men.
Thyroid Disease
Various autoimmune diseases, both cutaneous (e.g. vitiligo, alopecia areata, localized scleroderma, systemic sclerosis, psoriasis) and interestingly, extracutaneous autoimmune diseases such as autoimmune thyroid diseases (e.g. Hashimoto thyroiditis, Graves' disease), inflammatory bowel disease (e.g. Crohn's disease, ulcerative colitis), rheumatoid arthritis, and pernicious anemia can be found positive in LS patients [2, 17]. Among these, thyroid disease appears to be the most common, comprising 12.2%-16.3% of the overall cohort studied as reported by Kreuter et al. [2] and Cooper et al. [52], respectively. Other cases of autoimmune disease amassed only to 3.3% [2]. Furthermore, not only is thyroid disease more common in patients with LS, it is also more prevalent among LS women. Kreuter et al. [2] reported 60/65 cases of autoimmune thyroid diseases were found in female LS patients compared to male LS patients (15.2% vs. 3.8%). A recent cross-sectional study by Kantere et al. [55] noted of 100 male LS patients studied, none reported thyroid disease and only 5 presented cases with mild abnormal thyroid function. Therefore, while comorbidity of LS with an autoimmune disease appears infrequent in males, females diagnosed with LS should be screened for other autoimmune diseases, particularly for thyroid disease.
Autoantibodies to Associated Autoimmune Diseases
In alignment with the clinical profile of autoimmune diseases, autoantibodies were found to be in more female LS patients than male. Goolamali et al. [56], credited as the first to suggest LS “may be related to or caused by autoimmune processes,” suggested 40% (n=25) female LS patients had autoantibodies against thyroid cytoplasm compared to 12% (n=443) of healthy female controls [57]. Recent reports by Kreuter et al. [2] confirmed this trend with more anti-thyroid antibodies (11.1% vs. 4.4%) and antinuclear antibodies (9.6% vs. 0.7%) found in LS females when compared to their male counterparts. Kantere et al. [55] further determined that LS males positive for antinuclear antibodies (6%) was lower than that of the general population, suggesting autoimmunity may not play a significant role in LS etiology in males as it does in females.
Interestingly, Goolamali et al. [56] noted the presence of these organ-specific autoantibodies did not correlate with either the severity or duration of disease, and therefore concluded these were not a consequence of LS. In fact, Cooper et al. [52] noted little difference in anti-thyroid autoantibodies between LS and controls in spite of a higher prevalence of thyroid disease in the LS group, citing treatment of hypothyroidism with thyroxine as a potential explanation for the unexpected decrease in anti-thyroid autoantibodies.
Autoimmune & Genetic Targets of Lichen Sclerosus
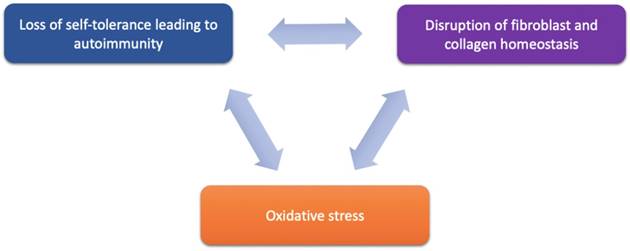
Despite the strong association with autoimmune disease, the etiology of LS remains a large mystery. However, potential therapeutic targets in recent years have been implicated in LS pathology and these emerging targets are discussed below and may be further categorized based on their functional roles in LS pathogenesis and progression via their involvement in (1) activating autoimmunogenic mechanisms, (2) inducing sclerotic tissue formation, or (3) triggering oxidative stress (Figure 1 and Table 2).
Proposed components of LS pathogenesis. Emerging immune and genetic targets implicated in mediation of LS can be divided based on their functional involvement into three proposed components of LS pathogenesis: (1) loss of self-tolerance to induce autoimmune mechanisms, (2) disruption of fibroblast and collagen homeostasis, and oxidative stress.
Expression levels of potential genetic/immune targets in LS. The overall genetic and molecular landscape of LS is complex. Expression profiles are largely immune related, with miR-155 implicated as an important upstream regulator of sclerotic tissue formation (via fibroblast proliferation and heightened collagen synthesis) and activation of a Th1 autoimmune response (by reduced Treg suppression activity). Downstream targets involved in sclerotic tissue formation include FOXO3, CDKN1B, collagen (type I, III, V), endothelial ECM1, galectin-7, and p53. Targets promoting autoimmunity include ECM1 autoantibodies, IL-10, TNF, and IL-6. Targets are listed in the order discussed.
| Genetic/Immune Target | Expression in LS | Effects and/or Significance in LS | References |
|---|---|---|---|
| ECM1 autoantibodies | Increased | • Disrupt scaffolding structure that holds extracellular matrix components together via promiscuous binding to multiple partners • Interfere with regulatory binding of ECM1 to MMP9 to suppress, otherwise, collagenase overactivity • Interfere with functional binding of ECM1 to perlecan and collagen IV | • Oyama et al. 2003 • Oyama et al. 2015 • Edmonds et al. 2011 • Fujimoto et al. 2006 • Sercu et al. 2006 |
| TNF-α | Increased | • Upstream mediator of Treg cell function | • Valencia et al. 2007 • Terlou et al. 2012 • Szabo et al. 2003 • Gambichler et al. 2013 • Farrell et al. 2006 |
| IL-6 | Increased | • Upstream mediator of Treg cell function | • Romero et al. 1992 • Gambichler et al. 2013 • Farrell et al. 2006 |
| TGF-β | Equal | • Originally seen as downstream indicator of Treg cell function | • Gambichler et al. 2013 • Fujimoto et al. 2006 |
| miR-155 | Increased | • Promote fibroblast proliferation • Reduce Treg cell suppressive function | • Terlou et al. 2012 • Gambichler et al. 2013 • Ren et al. 2018 |
| IL-10 | Decreased | • Downstream indicator of Treg cell function • Downregulated by overexpressed miR-155 | • Gambichler et al. 2013 • Corthay 2009 • Sakaguchi et al. 2008 |
| FOXO3 | Decreased | • Promote fibroblast proliferation and increased collagen synthesis • Downregulated by miR-155 | • Ren et al. 2018 |
| CDKN1B | Decreased | • Promote fibroblast proliferation and increased collagen synthesis • Downregulated by miR-155 | • Ren et al. 2018 |
| Galectin-7 | Increased | • Decrease fibroblast proliferation and increase collagen synthesis • Possible mediator of T cell response | • Zhao et al. 2018 • Luo et al. 2018 |
| Collagen I, III, V | Increased | • Promote deposition and development of sclerotic and hyalinized dermal tissue | • Godoy et al. 2015 • Ren et al. 2018 • Oikarinen et al. 1991 • Bharat et al. 2006 • Martin et al. 2012 |
| Elastic fibers | Decreased | • Triggers synthesis of collagen V and low endothelial ECM1 | • Godoy et al. 2015 |
| Endothelial ECM1 | Decreased | • Hyalinized dermal vessels | • Godoy et al. 2015 |
| P53 | Increased | • Response to ischemic stress from hyalinized dermal vessels • Somatic mutation as a later event involved in LS progression towards SCC | • Soufir et al. 2006 • Liegel et al. 2006 • Gambichler et al. 2011 • Vanin et al. 2002 • Sander et al. 2004 • Trietsch et al. 2014 • Tapp et al. 2007 • Soufir et al. 2007 • Marin et al. 2000 |
| CDKN2A | Hypermethylated | • Early event that may propagate LS progression towards SCC | • Sourir et al. 2006 • Gambrichler et al. 2013 • Lerma et al. 2002 |
Targets involved in an autoimmunogenic mechanism
ECM1 as an autoantigen for humoral autoimmunity
Given the familial occurrence of LS and the close, positive association between HLA class II antigens and autoimmune disease, many researchers speculated LS pathogenesis may progress along an immunogenetic route towards humoral autoimmunity as the development of autoantibodies to an unknown autoantigen may account for the histological changes seen as extensive extracellular remodeling. Dysfunction of extracellular matrix protein 1 (ECM1), found at the dermal-epidermis junction, has long been implicated in the pathogenesis of LS. Support comes from loss-of-function mutation studies in ECM1 gene in lipoid proteinosis (LiP), an autosomal recessive genodermatosis that features comparable clinical symptoms to that of LS and as such, implicates ECM1 as a strong putative autoantigen in LS autoimmunity [58].
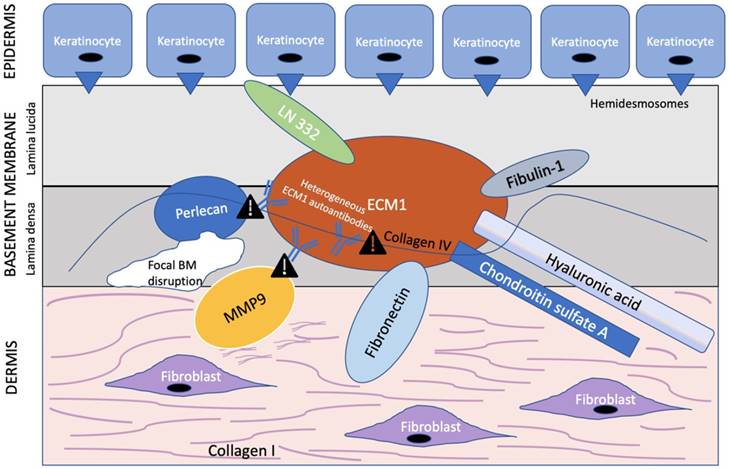
ECM1 is an 85 kDa soluble glycoprotein [59] with promiscuous extracellular binding targets that includes basement proteins (laminin 332, laminin 10, collage IV, fibulin polysaccharides (HA, heparin, CSA), proteoglycans such as perlecan; phospholipids (phospholipid scramblase 1), and proteolytic enzymes (MM9) [60, 61]. ECM1, therefore, appears to act as the “biological glue” that is responsible for the structural organization and integrity in human skin [32] (Figure 2). Thus, any disruption to this ECM1 scaffold would result in pathology.
ECM1 acts as a scaffold for multiple extracellular components. ECM1 serves as the ”biological glue” with multiple binding partners in the basement membrane at the dermal-epithelial junction. Heterogeneous ECM1 autoantibodies block the functional interaction of ECM1 to perlecan [61, 64], collagen IV [61], and MMP9 [60]. Such disruption in functional binding induces histological changes observed in LS. In particular, disruption of ECM1-MMP9 interaction leads to overactive collagenase activity of MMP9, resulting in focal basement membrane disruption. Exclamation point symbol indicates region of disrupted functional binding of extracellular component to ECM1. BM = basement membrane. Modified from Sercu et al. [61].
In spite of such supporting evidence for ECM1 as a potential autoantigen, Edmonds et al. [62] concluded ECM1 autoimmunity may only represent an epiphenomenon in response to the progression of disease as opposed to being a causative factor, citing the lack of predilection for genital skin in LiP when compared to LS. Further analysis showed ECM1 was significantly downregulated only in pediatric male LS lesions when compared to male adult and healthy controls, in spite of concordance of expression profiles between male age samples indicating pediatric LS and adult-onset LS consist of the same entity [63]. In addition, failure to establish complete LS mice models has hindered in vivo studies [64]. Thus, given such concerns, autoimmunity to ECM1 itself alone does not explain the full pathogenesis of LS.
Activation and maintenance of a Th1 response in LS progression
Gene expression profiles of LS have further suggested LS as an immunogenic disease, given increased expression in genes responsible for immune response [63, 65]. In particular, more pro-inflammatory cytokines were upregulated in LS (IL-1, IL-7, IL-15, IFN-γ, TNF-α) than anti-inflammatory cytokines (TNF-β), suggesting LS is specifically mediated by a T-helper type 1 (Th1) course of action [65, 66]. This observation further confirmed past findings of upregulated IFN-γ within the inflammatory infiltrate of LS, comprised largely of CD4+, CD8+, and FOXP3+ regulatory T (Treg) cells with notable expression of CXCR3 and CCR5, chemokine receptors known to characterize a Th1 response [65, 67]. While Th1 processes are known to direct immunity against intracellular pathogens, a Th1 response has also been associated with autoimmune disease [66].
miR-155 induces loss of immune tolerance as trigger for autoimmunity
MicroRNAs are small, endogenous RNA molecules that act as regulators of gene expression by binding to the 3'-untranslated region (3' UTR) of target mRNA transcript [68, 69]. An aberrant level of microRNA expression is therefore implicated in LS pathogenesis. miR-155 has been shown to be expressed in activated immune cells (i.e. macrophages, dendritic cells, B cells, and T cells) and plays a significant regulatory role in the production of cytokine, chemokine, and transcription factors towards promoting Th1 T-cell differentiation [65]. In the context of autoimmunity, overexpressed miR-155 levels are hypothesized to disrupt the suppressive function of T regulatory (Treg) cells, which are responsible for maintaining self-tolerance by downregulating the proliferation (via IL-10 and TGF-β cytokine production) [70] of autoreactive CD4+ effector T cells that would otherwise have triggered an immune response against self-antigens [65, 70, 71]. Reduction or inactivation of this critical Treg-cell-mediated suppression, either by downregulated Treg cells or altered functional phenotype, would therefore induce autoimmunity (Figure 3).
Pathological schema of LS with possible immune and genetic targets. Although the pathogenesis of LS is unknown, several mechanisms have been implicated involving immune and genetic targets. Each target can be viewed as promoting either fibroblast activity and/or mediation of disease through an autoimmunogenic route. Vascular changes, oxidative stress, inflammation, and loss of elastic fibers are downstream events that further complicate the etiology. miR-155 plays a dual role in inhibiting FOXO3 and CDKN1B to promote fibroblast growth and collagen synthesis as well as inhibiting the suppressive function of Treg T cells on CD4+ cells, triggering loss of self-tolerance and promotion of inflammation through a Th1 cytokine profile. There is also speculation regarding loss of Treg T cell suppression and onset of ECM1 autoantibodies. Inflammation releases reactive oxygen species (ROS) that, in turn, is predicted to allow autoantigen fragmentation to occur within the local region, thereby providing new autoepitopes for humoral autoimmunity. Galectin-7 expressed in the epidermis inhibits fibroblast growth, but, with the loss of elastic fibers, promotes collagen synthesis. In addition, galectin-7 may have an effect on promoting the ratio of Th1 cells as demonstrated in cardiac allografts. The cumulative effect of downregulated endothelial ECM1 and collagen deposition leads to the sclerotic scarring and constriction of dermal vessels with further oxidative stress. DNA damage ultimately triggers p53-induced apoptosis with the potential for carcinogenesis. A mechanism for advancement of LS lesions to carcinogenesis is presented at the epigenetic level. Normally, no p53 or CDKN2a mutations are found in normal LS. However, inactivation of these two tumor-suppressor genes downstream via p53 somatic mutation and CDKN2A hypermethylation eliminates normal growth limiting processes (indicated by gray box), which then leads to the promotion of cell proliferation and malignancy. * = LS to squamous cell carcinoma (SCC) progression, apparent in only 4% of all LS lesions.
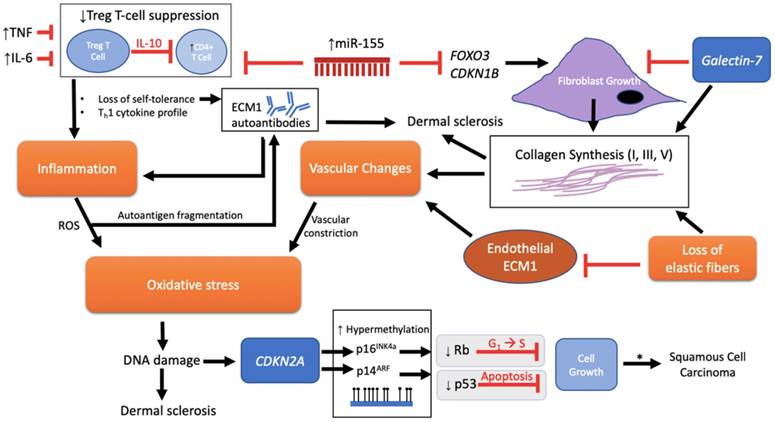
Specifically, miR-155 from LS lesions showed a 9.5-fold increase in expression compared to controls [65]. CD4+, CD8+, and FOXP3+ Treg cells were also present in significant counts, while IL-10 showed decreased expression with equal TGF-β levels compared to healthy controls [65, 72] (Table 2). Such data suggests high miR-155 expression is able to suppress Treg cell activity without reducing Treg cell numbers. Given that Treg cell exert their suppressive activity via IL-10 and TGF-β production [70], the low IL-10 found in LS may indicate reduced Treg cell activity in their suppressive role in protecting the body from autoimmunity. Insignificant differences in TGF-β, however, may suggest other inflammatory mediators are involved, such as TNF and IL-6, which are shown to be increased in LS [67, 72-74]. Taken together, Treg cell activity is mediated not by reduced cell counts in LS, but possibly by an altered Treg phenotype due to increased miR-155 expression as is the case in lupus erythematosus [65, 75]. Thus, miR-155 may serve as a critical regulator triggering the loss of normal Treg suppressive function and loss of CD4+ self-tolerance in this proposed autoimmunogenic mechanism for LS pathogenesis.
Targets affecting sclerotic tissue formation via fibroblast proliferation and collagen homeostasis
Overexpressed miR-155 promotes fibroblast proliferation via downregulation of FOXO3 and CDKN1B
In addition to its possible role for inducing autoimmunity, miR-155 is also associated with sclerotic tissue formation. Specifically, Ren et al. [76] found increased expression levels of miR-155 in LS tissues along with an associated decrease in FOXO3 and CDKN1B, two tumor suppressor genes involved in fibroblast cell proliferation and cell cycle suppression, respectively (Table 2). Reduction in FOXO3 and CDKN1B expression may promote fibroblast proliferation and persistence, thereby explaining the high level of collagen synthesis found in the zone of hyalinized and sclerotic dermal tissue in LS [76] (Figure 3).
Autoantibodies to ECM1, Interaction with MMP9, and Histological Changes
Of special interest to the histological changes in LS is the ECM1-mediated reduction in matrix metallopeptidase 9 (MMP9) proteolytic activity. Autoantibodies against ECM1 have been found in both LS females (74% vs. 7% controls) [30] and males [62]. Specifically, the majority of these autoantibodies found in LS females by Oyama et al. [30] targeted the c-terminal second-tandem repeat (exon 7) of recombinant ECM1, the same region to which MMP9 also binds as demonstrated by Fujimoto et al. [60]. Loss of this region, either by mutation to the ECM1 gene or autoantibodies to this particular ECM1 region, would in effect disrupt the usual regulatory binding of ECM1 to MMP9, resulting in overreactive MMP9 collagenase activity disrupting collagen homeostasis, which, in turn, may explain the focal basement membrane disruption observed in LS [60] (Figure 2). However, due to the existence of heterogeneous ECM1 antibodies targeting different ECM1 epitopes, Sercu et al. [61] and Oyama et al. [30] further speculated that anti-ECM1 antibodies in LS predominately affects the functional binding of ECM1 to collagen IV and perlecan more so than MMP9.
In other regions of the basement membrane, thickening may occur instead of degradation. Oikarinen et al. [77] demonstrated an increase in collagen synthesis by fibroblasts in LS lesions, albeit through a small sample size (n=4), when compared to non-affected samples and healthy controls. Such seemingly paradoxical phenomenon may also be due to MMP9's overactivity from its dysregulated binding to ECM1, as MMP9, in addition to its collagenase function, has also been shown to cleave latent transforming growth factor beta (TGF-β), a possible mechanism for TGF-β activation and therefore, enhances collagen synthesis and regeneration as seen in LiP [60]. High MMP activity, however, was linked with decreased dermal fibroblast activity and reduced collagen synthesis in photodamaged skin [78] and differences in lesional TGF-β levels were insignificant compared to healthy controls [72].
Galactin-7 inhibits fibroblast growth and increases collagen synthesis
The cellular activity of fibroblasts in LS was further investigated with the regulatory role of galectin-7, a keratinocyte protein under p53 regulation [79]. Zhao et al. [80] noted high galectin-7 expression levels in LS tissue that may exert a dual effect on dermal fibroblasts by (1) inhibiting growth and (2) promoting the transcription of type I and type III collagen, both primary collagen types seen to be overly expressed in LS (Figure 3). Even though galectin-7 expression was localized only to the epidermis of LS tissue, it is possible that galectin-7 may find its way via paracrine signaling from keratinized epithelial cells that originally synthesized galectin-7 in the epidermis, to a surface receptor of dermal fibroblast in the dermis in order to downregulate fibroblast growth and increase collagen I & III transcription [80]. In light of LS as a possible immunogenic disease, while prior reports of galectin-7 in cardiac allografts of mice suggested a promising role as a positive mediator of T cell response by increasing the ratio of Th1 cells and lowering IL-10, the role of galectin-7 in LS needs further investigation [81].
Increased collagen type V and decreased endothelial ECM1 implicated in sclerotic tissue formation
In addition to hyalinized and homogenized, sclerotic collagen fibrils, Godoy et al. [82] observed reduced elastic fibers in the upper dermis of LS lesions, which was found to be associated with aberrantly increased collagen V deposition (in addition to type I & III collagen) and low ECM1 blood vessel expression within the hyalinized LS zone (Table 2). Given that type V collagen is known to induce scleroderma in rabbits [83] and lung transplant rejection [84] it is possible that overexpression of collagen V may explain the observed collagen remodeling observed in LS [82].
Targets and markers involved in oxidative stress within LS lesions serve as a gateway for progression to squamous cell carcinoma
Overexpression of wild-type p53 as indicator of oxidative stress
In addition to its regulatory role on downstream tumor suppressors such as galectin-7, p53 has also been further implicated in LS biopathology; however, interestingly, as a triggered response to oxidative stress [85] (Figure 1 & 3). Reactive oxygen species (ROS) are suspected to interact with apoptotic macromolecules as a consequence of DNA damage, thereby creating new epitopes for autoimmunity activation, such as those found in ECM1 autoantibodies [86, 87]. The presence of marked inflammation may also further accelerate LS disease progression via production of more ROS [87]. Staining for oxidative DNA damage marker 8-hydroxy-2'-deoxyguanosine (8-OHG) was found in all skin layers, while protein oxidation staining showed significant activity in dermal sclerotic lesions, further supporting the role of ROS-induced DNA damage that trigger downstream dermal sclerosis [87]. Such findings collectively suggest an intricately complex and interwoven relationship between autoimmunity and oxidative stress in LS (Figure 3).
High p53 immunoreactivity in immunostaining, interpreted as p53 overexpression, was found in basal keratinocytes in LS [88, 89], LS' various subtypes (e.g. early and classical; pediatric and adult LS) [37] as well as in lesions of both sexes [37, 72, 88]. In addition to inflammation and its release of ROS, vascular changes due to dermal sclerosis and hyalinization lead to the development of sclerotic vessels, which restrict oxygen flow and induce ischemic stress [37, 87]. Such stress, in turn, is seen to enhance p53 protein stability and accumulation, possibly as a compensatory mechanism to counteract the damaging effects of oxidative stress (Figure 3). Further, the wild-type p53 protein is shown to be present in LS lesions as opposed to its mutant form [72, 88, 90], with only one study showing p53 mutations in as high as 70% of patients [85, 91]. Furthermore, Vanin et al. [89] demonstrated no association between loss of heterozygosity (LOH) and p53 mutations in the presence of high p53 expression in LS. In fact, an analysis of several studies pinpoints the mutation frequency of TP53 in LS to be 4% [85]. Given these collective findings, the accumulation and high expression of wild-type p53 in LS lesions strongly suggest the presence of oxidative stress in LS.
CDKN2A epigenetic modifications in LS progression towards squamous cell carcinoma (SCC)
Similar to p53, there were no mutations found in lesional CDKN2A, which normally codes for p16INK4a and p14ARF, two tumor suppressors that regulate the cell cycle [88]. In general, both protein products inhibit aberrant cell growth through different mechanisms: p16INK4a activates the Rb protein family to block G1 to S-phase progression, whereas p14ARF rescues p53 from MDM2 ubiquitin protein degradation [88, 92, 93] (Figure 3).
With the absence of p53 and CDKN2A mutations in LS, investigators considered the role of epigenetic modifications in LS, particularly for its potential to induce malignant transformation. Aberrant 5-hydroxymethylation and altered isocitrate dehydrogenase (IDH) enzymes [72] were associated with LS and up to 42.8% of lesions showed hypermethylation in p16INK4a gene promoters [88, 94], thus providing a basis for LS against an epigenetic background. More interestingly, while there were no p53 mutations found in LS alone, p53 mutations occurred much more frequently in LS associated with SCC. While much is unknown regarding the larger epigenetic changes in LS, a synergetic effect may occur between p16INK4a/p14ARF hypermethylation as an early event in tandem with p53 somatic mutations as a late event to promote cell immortality and tumor development in the malignant transformation of LS to SCC [88] (Figure 3).
Conclusion and Future Directions
Lichen Sclerosus (LS) is a relapsing inflammatory dermatosis with an unknown pathogenesis marked by intolerable pruritus, scarring, and drastic changes to genital anatomy in both sexes, often culminating in sexual and urinary dysfunction with an increased risk for SCC. Due to histological challenges, its diagnosis is often difficult. Patients are often plagued with reticence and embarrassment, and therefore, fail to seek treatment. The plague of detection biases, in addition to the lack of large-scale epidemiological studies, may render current rates of incidence as mere underestimates.
Although the etiology of LS remains elusive, there are several lines of evidence that suggest LS is an autoimmunogenic disease against a genetic background. LS is not only found more commonly in females than males, but there is also a strong positive association between LS and HLA class II antigens and comorbidity with autoimmune diseases, especially thyroid disease. The presence of autoantigens against ECM1, an essential scaffolding glycoprotein critical to dermal equilibrium, in both females and males, further provides a potential basis for autoimmunity, but remains insufficient in explaining the full pathology of LS alone. LS is also an inflammatory disorder, as genome-wide expression profiles showed differential increase in genes important to immune response, particularly towards Th1 differentiation. Therefore, the activation and maintenance of Th1 cytokine microenvironment plays a key role in disease progression.
In addition to ECM1, aberrant expression of various immune and genetic targets has been implicated in the pathogenesis of LS that are either involved in inducing an immunogenic mechanism and/or modulating fibroblast activity and collagen homeostasis (Table 2 and Figure 2). In particular, the upregulation of miR-155, TNF, IL-6, galectin-7, collagen (type I, III, V), p53, and ECM1 autoantibodies showed enhanced expression in LS lesions compared to controls, whereas FOXO3, CDKN1B, IL-10, and endothelial ECM1 were downregulated. Reduced Treg cell suppression activity is considered as a critical mechanism for loss of self-tolerance. Epigenetic modification in both promoters of CDKN2A and the role of oxidative stress from inflammation and vascular damage further add complexity to the already enigmatic pathogenesis of LS. Nevertheless, these markers serve as potential therapeutic targets that warrant further study and analysis in order to uncover the true onset and course of disease progression.
Finally, in an attempt to shed light on this disease described over a century ago, consensus must be reached on the histological diagnostic criteria for various LS subtypes and stages, with particular clarification on nomenclature and usage. Doing so will encourage more epidemiological studies to investigate aspects of LS across all ages and sex groups to better understand how the course of disease unfolds across different demographics. Such concerns, interestingly, are also listed among “top 10” priorities for future LS research as determined by a Priority Setting Partnership (from June 2017-July 2018) utilizing the James Alliance (JLA) methodology [11]. The top concern, above diagnostic criteria, was prevention and management options. With the long-term goal of finding a curative treatment, however, the numerous evidence gaps in the etiology of LS must first be filled through the lens of immunology and genetics in order to effectively solve this autoimmunopathogenic and genomic enigma.
Acknowledgements
This research was supported by the Gynecological Cancer Research Foundation (to SWF), and the Elaine H. Snyder Cancer Research Award (to SWF).
Competing Interests
Dr. Goldstein receives funding from Endoceutics, SST, Ipsen, Elen and Cellular Medicine Association. He also serves as a consultant for Ipsen, SST, Amag, Lupin, as well as the President of GCRF.
References
1. Sheinis M, Selk A. Development of the Adult Vulvar Lichen Sclerosus Severity Scale—A Delphi Consensus Exercise for Item Generation. J Low Genit Tract Dis. 2018;22:66-73
2. Kreuter A, Kryvosheyeva Y, Terras S, Moritz R, Mollenhoff K, Altmeyer P. et al. Association of Autoimmune Diseases with Lichen Sclerosus in 532 Male and Female Patients. Acta Dermato-Venereologica. 2013;93:238-41
3. Haefner HK, Aldrich NZ, Dalton VK, Gagné HM, Marcus SB, Patel DA. et al. The Impact of Vulvar Lichen Sclerosus on Sexual Dysfunction. J Womens Health (Larchmt). 2014;23:765-70
4. Lichen sclerosus. Genetic and Rare Diseases 2019 ed; 2019.
5. Lichen Sclerosus. NORD (National Organization for Rare Disorders); 2019.
6. Friedrich EG, MacLaren NK. Genetic aspects of vulvar lichen sclerosus. American Journal of Obstetrics & Gynecology. 1984;150:161-6
7. Lambert J. Pruritus in Female Patients. BioMed Research International. 2014
8. Niamh L, Naveen S, Hazel B. Diagnosis of Vulval Inflammatory Dermatoses: A Pathological Study With Clinical Correlation. International Journal of Gynecological Pathology. 2009;28:554-8
9. Lipscombe TK, Wayte J, Wojnarowska F, Marren P, Luzzi G. A study of clinical and aetiological factors and possible associations of lichen sclerosus in males. Australasian Journal of Dermatology. 1997;38:132-6
10. Murphy R. Lichen Sclerosus. Dermatologic Clinics. 2010;28:707-15
11. Simpson RC, Cooper SM, Kirtschig G, Larsen S, Lawton S, McPhee M. et al. Future research priorities for lichen sclerosus - results of a James Lind Alliance Priority Setting Partnership. British Journal of Dermatology. 2018
12. Smith YR, Haefner HK. Vulvar Lichen Sclerosus: Pathophysiology and Treatment. American Journal of Clinical Dermatology. 2004;5:105-25
13. Chi C-C, Kirtschig G, Baldo M, Brackenbury F, Lewis F, Wojnarowska F. Topical interventions for genital lichen sclerosus. Cochrane Database of Systematic Reviews. 2011
14. Wallace HJ. Lichen sclerosus et atrophicus. Transactions of the St John's Hospital Dermatological Society. 1971;57:9-30
15. Tasker GL, Wojnarowska F. Lichen sclerosus. Clinical and Experimental Dermatology. 2003;28:128-33
16. Madu PN, Williams VL, Noe MH, Omech BG, Kovarik CL, Wanat KA. Autoimmune skin disease among dermatology outpatients in Botswana: a retrospective review. International Journal of Dermatology. 2019;58:50-3
17. Kirtschig G, Becker K, Günthert A, Jasaitiene D, Cooper S, Chi CC. et al. Evidence-based (S3) Guideline on (anogenital) Lichen sclerosus. Journal of the European Academy of Dermatology and Venereology. 2015;29:e1-e43
18. Goldstein AT, Marinoff SC, Christopher K, Srodon M. Prevalence of vulvar lichen sclerosus in a general gynecology practice. J Reprod Med. 2005;50:477-80
19. Clayton R, Stewart E, Wonjnarowska F. Rising demand for the services of a dedicated dermatological vulval clinic without changes in disease profile. 2006.
20. Bleeker MC, Visser PJ, Overbeek LI, Beurden Mv, Berkhof J. Lichen Sclerosus: Incidence and Risk of Vulvar Squamous Cell Carcinoma. Cancer Epidemiol Biomarkers Prev. 2016;25:1224-30
21. Leibovitz A, Kaplun V, Saposhnicov N, Habot B. Vulvovaginal examinations in elderly nursing home women residents. Archives of Gerontology and Geriatrics. 2000;31:1-4
22. Kizer WS, Prarie T, Morey AF. Balanitis xerotica obliterans: epidemiologic distribution in an equal access health care system. South Med J. 2003;96:9-11
23. Fistarol SK, Itin PH. Diagnosis and Treatment of Lichen Sclerosus. American Journal of Clinical Dermatology. 2013;14:27-47
24. Yesudian PD, Sugunendran H, Bates CM, O'Mahony C. Lichen sclerosus. International Journal of STD & AIDS. 2005;16:465-73
25. Brownstein M. Lichen sclerosus et atrophicus [Comment]. 1973; 108: 433. Arch Dermatol. 1973
26. Kyriakis KP, Emmanuelides S, Terzoudi S, Palamaras I, Damoulaki E, Evangelou G. Gender and age prevalence distributions of morphea en plaque and anogenital lichen sclerosus. Journal of the European Academy of Dermatology and Venereology. 2007;21:825-6
27. KR S, AM R. The incidence of phimosis in boys. BJU International. 1999;84:101-2
28. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus: An increasingly common problem. Journal of the American Academy of Dermatology. 2001;44:803-6
29. McPherson T, Cooper S. Vulval lichen sclerosus and lichen planus. Dermatologic Therapy. 2010;23:523-32
30. Oyama N, Chan I, Neill SM, Hamada T, South AP, Wessagowit V. et al. Autoantibodies to extracellular matrix protein 1 in lichen sclerosus. The Lancet. 2003;362:118-23
31. Regauer S, Liegl B, Reich O. Early vulvar lichen sclerosus: a histopathological challenge. Histopathology. 2005;47:340-7
32. Chan I. Experimental dermatology • Review article The role of extracellular matrix protein 1 in human skin. Clinical & Experimental Dermatology. 2004;29:52-6
33. Hasegawa M, Ishikawa O, Asano Y, Sato S, Jinnin M, Takehara K. et al. Diagnostic criteria, severity classification and guidelines of lichen sclerosus et atrophicus. The Journal of Dermatology. 2018;45:891-7
34. Carlson BC, Hofer MD, Ballek N, Yang XJ, Meeks JJ, Gonzalez CM. Protein Markers of Malignant Potential in Penile and Vulvar Lichen Sclerosus. The Journal of Urology. 2013;190:399-406
35. PA N. Dermatosis associated with menopause. Journal Midlife Health. 2014;5:168-75
36. Saluja T, Iyer J. Unmasking the Grey Zone of Hyalinization with a Proposed Classification of Oral Hyalinizing Lesions. Journal of Interdisciplinary Histopathology. 2017:1
37. Liegl B, Regauer S. p53 immunostaining in lichen sclerosus is related to ischaemic stress and is not a marker of differentiated vulvar intraepithelial neoplasia (d-VIN). Histopathology. 2006;48:268-74
38. Nair PA. Vulvar Lichen Sclerosus et Atrophicus. J Midlife Health. 2017;8:55-62
39. Pérez-López FR, Vieira-Baptista P. Lichen sclerosus in women: a review. Climacteric. 2017;20:339-47
40. Kato T, Mizuno K, Nishio H, Moritoki Y, Nakane A, Kurokawa S. et al. Can lichen sclerosus be diagnosed by preputial appearance or symptoms? Res Rep Urol. 2018;10:89-94
41. Aslanian FMNP, Marques MTQ, Matos HJ, Pontes LFS, Porto LCS, Azevedo LMS. et al. HLA markers in familial Lichen Sclerosus. JDDG: Journal der Deutschen Dermatologischen Gesellschaft. 2006;4:842-7
42. Higgins CA, Cruickshank ME. A population-based case-control study of aetiological factors associated with vulval lichen sclerosus. Journal of Obstetrics and Gynaecology. 2012;32:271-5
43. Sherman V, McPherson T, Baldo M, Salim A, Gao XH, Wojnarowska F. The high rate of familial lichen sclerosus suggests a genetic contribution: an observational cohort study. Journal of the European Academy of Dermatology and Venereology. 2010;24:1031-4
44. Doulaveri G, Armira K, Kouris A, Karypidis D, Potouridou I. Genital Vulvar Lichen Sclerosus in Monozygotic Twin Women: A Case Report and Review of the Literature. Case Rep Dermatol. 2013;5:321-5
45. Lis-Święty A, Mierzwińska K, Wodok-Wieczorek K, Widuchowska M, Brzezińska-Wcisło L. Co-existence of lichen sclerosus and localized scleroderma in female monozygotic twins. J Pediatr Adolesc Gynecol. 2014;27:e133-6
46. Meyrick Thomas RH, Ridley CM, McGibbon DH, Black MM. Lichen sclerosus et atrophicus and autoimmunity in 250 women. British Journal of Dermatology. 1985;113:28-9
47. Azurdia R, Luzzi G, Byren I, Welsh K, Wojnarowska F, Marren P. et al. Lichen sclerosus in adult men: a study of HLA associations and susceptibility to autoimmune disease. British Journal of Dermatology. 1999;140:79-83
48. Marren P, Jell J, Charnock FM, Bunce M, Welsh K, Wojnarowska F. The association between lichen sclerosus and antigens of the HLA system. British Journal of Dermatology. 1995;132:197-203
49. Powell J, Wojnarowska F, Winsey S, Marren P, Welsh K. Lichen sclerosus premenarche: autoimmunity and immunogenetics. British Journal of Dermatology. 2000;142:481-4
50. Gao X-H, Barnardo MCMN, Winsey S, Ahmad T, Cook J, Agudelo JD. et al. The Association Between HLA DR, DQ Antigens, and Vulval Lichen Sclerosus in the UK: HLA DRB1*12 and its Associated DRB1*12/DQB1*0301/04/09/010 Haplotype Confers Susceptibility to Vulval Lichen Sclerosus, and HLA DRB1*0301/04 and its Associated DRB1*0301/04/DQB1*0201/02/03 Haplotype Protects from Vulval Lichen Sclerosus. Journal of Investigative Dermatology. 2005;125:895-9
51. Harrington CI, Dunsmore IR. An investigation into the incidence of auto-immune disorders in patients with lichen sclerosus and atrophicus. British Journal of Dermatology. 1981;104:563-6
52. Cooper SM, Ali I, Baldo M, Wojnarowska F. The Association of Lichen Sclerosus and Erosive Lichen Planus of the Vulva With Autoimmune Disease: A Case-Control Study. Arch Dermatol. 2008;144:1432-5
53. Meyrick Thomas RH, Ridley CM, McGibbon DH, Black MM. Lichen sclerosus et atrophicus and autoimmunity-a study of 350 women. British Journal of Dermatology. 1988;118:41-6
54. Edmonds EVJ, Hunt S, Hawkins D, Dinneen M, Francis N, Bunker CB. Clinical parameters in male genital lichen sclerosus: a case series of 329 patients. Journal of the European Academy of Dermatology and Venereology. 2012;26:730-7
55. Kantere D, Alvergren G, Gillstedt M, Pujol-Calderon F, Tunbäck P. Clinical Features, Complications and Autoimmunity in Male Lichen Sclerosus. Acta Dermato Venereologica. 2017;97:365-9
56. Goolamali SK, Barnes EW, Irvine WJ, Shuster S. Organ-specific Antibodies in Patients with Lichen Sclerosus. Br Med J. 1974;4:78-9
57. Guarneri F, Giuffrida R, Di Bari F, Cannavò SP, Benvenga S. Thyroid Autoimmunity and Lichen. Front Endocrinol (Lausanne). 2017:8
58. Hamada T. Lipoid proteinosis. Clinical and Experimental Dermatology. 2002;27:624-9
59. Smits P, Ni J, Feng P, Wauters J, Van Hul W, Boutaibi ME. et al. The Human Extracellular Matrix Gene 1 (ECM1): Genomic Structure, cDNA Cloning, Expression Pattern, and Chromosomal Localization. Genomics. 1997;45:487-95
60. Fujimoto N, Terlizzi J, Aho S, Brittingham R, Fertala A, Oyama N. et al. Extracellular matrix protein 1 inhibits the activity of matrix metalloproteinase 9 through high-affinity protein/protein interactions. Experimental Dermatology. 2006;15:300-7
61. Sercu S, Zhang M, Oyama N, Hansen U, Ghalbzouri AEL, Jun G. et al. Interaction of Extracellular Matrix Protein 1 with Extracellular Matrix Components: ECM1 Is a Basement Membrane Protein of the Skin. Journal of Investigative Dermatology. 2008;128:1397-408
62. Edmonds EVJ, Oyama N, Chan I, Francis N, McGrath JA, Bunker CB. Extracellular matrix protein 1 autoantibodies in male genital lichen sclerosus. British Journal of Dermatology. 2011;165:218-9
63. Edmonds E, Barton G, Buisson S, Francis N, Gotch F, Game L. et al. Gene expression profiling in male genital lichen sclerosus. International Journal of Experimental Pathology. 2011;92:320-5
64. Oyama N. Current Understanding of Molecular-based Immunopathology in Lichen Sclerosus. International Trends in Immunity. 2015:6
65. Terlou A, Santegoets LAM, van der Meijden WI, Heijmans-Antonissen C, Swagemakers SMA, van der Spek PJ. et al. An Autoimmune Phenotype in Vulvar Lichen Sclerosus and Lichen Planus: A Th1 Response and High Levels of MicroRNA-155. Journal of Investigative Dermatology. 2012;132:658-66
66. Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annual Review of Immunology. 2003;21:713-58
67. Farrell AM, Dean D, Millard PR, Charnock FM, Wojnarowska F. Cytokine alterations in lichen sclerosus: an immunohistochemical study. British Journal of Dermatology. 2006;155:931-40
68. Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nature reviews Genetics. 2016;17:719-32
69. Rieger JK, Klein K, Winter S, Zanger UM. Expression Variability of Absorption, Distribution, Metabolism, Excretion-Related MicroRNAs in Human Liver: Influence of Nongenetic Factors and Association with Gene Expression. Drug Metab Dispos. 2013;41:1752-62
70. Corthay A. How do Regulatory T Cells Work? Scand J Immunol. 2009;70:326-36
71. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell. 2008;133:775-87
72. Gambichler T, Belz D, Terras S, Kreuter A. Humoral and cell-mediated autoimmunity in lichen sclerosus. British Journal of Dermatology. 2013;169:183-4
73. Romero LI, Pincus SH. In situ Localization of lnterleukin-6 in Normal Skin and Atrophic Cutaneous Disease. IAA. 1992;99:44-9
74. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253-61
75. Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. Journal of Immunology. 2011;186:924-30
76. Ren L, Zhao Y, Huo X, Wu X. MiR-155-5p promotes fibroblast cell proliferation and inhibits FOXO signaling pathway in vulvar lichen sclerosis by targeting FOXO3 and CDKN1B. Gene. 2018;653:43-50
77. Oikarinen A, Sandberg M, Hurskainen T, Kinnunen T, Kallioinen M. Collagen biosynthesis in lichen sclerosus et atrophicus studied by biochemical and in situ hybridization techniques. Acta Derm Venereol Suppl (Stockh). 1991;162:3-12
78. Quan T, Little E, Quan H, Qin Z, Voorhees JJ, Fisher GJ. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: impact of altered extracellular matrix microenvironment on dermal fibroblast function. J Invest Dermatol. 2013;133:1362-6
79. Polyak K, Xia Y. A model for p53-induced apoptosis. Nature. 1997;389:300
80. Zhao Y, Zhao S, Li H, Qin X, Wu X. Expression of galectin-7 in vulvar lichen sclerosus and its effect on dermal fibroblasts. Oncol Lett. 2018;16:2559-64
81. Luo Z, Ji Y, Tian D, Zhang Y, Chang S, Yang C. et al. Galectin-7 promotes proliferation and Th1/2 cells polarization toward Th1 in activated CD4+ T cells by inhibiting The TGFβ/Smad3 pathway. Molecular Immunology. 2018;101:80-5
82. Godoy CAP, Teodoro WR, Velosa APP, Garippo AL, Eher EM, Parra ER. et al. Unusual remodeling of the hyalinization band in vulval lichen sclerosus by type V collagen and ECM 1 protein. Clinics. 2015;70:356-62
83. Martin P, Teodoro WR, Velosa APP, de Morais J, Carrasco S, Christmann RB. et al. Abnormal collagen V deposition in dermis correlates with skin thickening and disease activity in systemic sclerosis. Autoimmunity Reviews. 2012;11:827-35
84. Bharat A, Fields R, Steward N, Trulock E, Patterson G, Mohanakumar T. CD4+25+ Regulatory T Cells Limit Th1-Autoimmunity by Inducing IL-10 Producing T Cells Following Human Lung Transplantation. American Journal of Transplantation. 2006;6:1799-808
85. Trietsch MD, Nooij LS, Gaarenstroom KN, van Poelgeest MIE. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: A review of the current literature. Gynecologic Oncology. 2015;136:143-57
86. Rosenthal AN, Ryan A, Hopster D, Surentheran T, Jacobs IJ. High frequency of loss of heterozygosity in vulval intraepithelial neoplasia (VIN) is associated with invasive vulval squamous cell carcinoma (VSCC). International Journal of Cancer. 2001;94:896-900
87. Sander CS, Ali I, Dean D, Thiele JJ, Wojnarowska F. Oxidative stress is implicated in the pathogenesis of lichen sclerosus. British Journal of Dermatology. 2004;151:627-35
88. Soufir N, Queille S, Liboutet M, Thibaudeau O, Bachelier F, Delestaing G. et al. Inactivation of the CDKN2A and the p53 tumour suppressor genes in external genital carcinomas and their precursors. British Journal of Dermatology. 2007;156:448-53
89. Vanin K, Scurry J, Thorne H, Yuen K, Ramsay RG. Overexpression of Wild-type p53 in Lichen Sclerosus adjacent to Human Papillomavirus-negative Vulvar Cancer. Journal of Investigative Dermatology. 2002;119:1027-33
90. Tapp RA, Feng J, Wesley Jones J, Andrew Carlson J, Wilson VL. Single Base Instability Is Promoted in Vulvar Lichen Sclerosus. Journal of Investigative Dermatology. 2007;127:2563-76
91. Marin MC, Jost CA, Brooks LA, Irwin MS, O'Nions J, Tidy JA. et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nature Genetics. 2000;25:47-54
92. Inoue K, Fry E. Aberrant expression of p16INK4a in human cancers - a new biomarker?. Cancer Reports and Reviews. 2018:2
93. Larsen CJ. Contribution of the dual coding capacity of the p16INK4a/MTS1/CDKN2 locus to human malignancies. Prog Cell Cycle Res. 1997;3:109-24
94. Lerma E, Esteller M, Herman JG, Prat J. Alterations of the p16INK4a/Rb/cyclin-D1 pathway in vulvar carcinoma, vulvar intraepithelial neoplasia, and lichen sclerosus. Human Pathology. 2002;33:1120-5
Author contact
![]() Corresponding authors: Andrew T Goldstein, M.D., Department of Obstetrics and Gynecology, The George Washington University School of Medicine and Health Sciences, Washington, DC 20037. Email: drg.cvvdcom. Sidney W. Fu, M.D., Ph.D., Department of Medicine (Division of Genomic Medicine), The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C, Washington, DC 20037. Email: sfuedu; Tel: 202-994-4767.
Corresponding authors: Andrew T Goldstein, M.D., Department of Obstetrics and Gynecology, The George Washington University School of Medicine and Health Sciences, Washington, DC 20037. Email: drg.cvvdcom. Sidney W. Fu, M.D., Ph.D., Department of Medicine (Division of Genomic Medicine), The George Washington University School of Medicine and Health Sciences, 2300 Eye Street, N.W. Ross Hall 402C, Washington, DC 20037. Email: sfuedu; Tel: 202-994-4767.