Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
SEC23 Regulation of...
SEC23 Regulation of Embryo...
SEC23 Mutations in Human Diseases
SEC23 Regulation of Autophagy in...
SEC23 Alterations in Human...
Summary and Future Direction of...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(11):2419-2426. doi:10.7150/ijbs.37008 This issue Cite
Review
The Functional Role of SEC23 in Vesicle Transportation, Autophagy and Cancer
Jingchen Jing1,2, Bo Wang1,2, Peijun Liu1,2 ![]()
1. Center for Translational Medicine, The First Affiliated Hospital, Xi'an Jiaotong University
2. The Key Laboratory for Tumor Precision Medicine of Shaanxi Province, The First Affiliated Hospital, Xi'an Jiaotong University, Xi'an, Shaanxi 710061, China
Received 2019-5-26; Accepted 2019-8-1; Published 2019-9-7
Abstract

SEC23, the core component of the coat protein complex II (COPII), functions to transport newly synthesized proteins and lipids from the endoplasmic reticulum (ER) to the Golgi apparatus in cells for secretion. SEC23 protein has two isoforms (SEC23A and SEC23B) and their aberrant expression and mutations were reported to cause human diseases and oncogenesis, whereas SEC23A and SEC23B may have the opposite activity in human cancer, for a reason that remains unclear. This review summarizes recent research in SEC23, COPII-vesicle transportation, autophagy, and cancer.
Keywords: SEC23, COPII-vesicle transportation, autophagy, cancer
Introduction
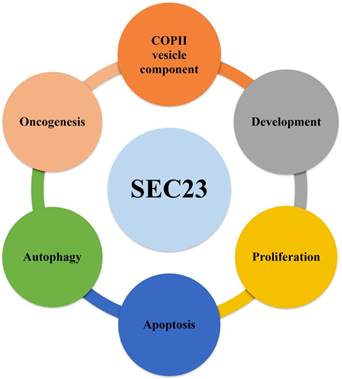
SEC23 is one of the components in the coat protein complex II (COPII) and regulates the transportation of proteins and lipids from the endoplasmic reticulum (ER) to the Golgi apparatus in cells. Approximately, one-third of all newly synthesized proteins will be transported from the ER to the Golgi through COPII-coated vesicles [1]. The SEC23 protein, as the core component of COPII, contains five distinct domains (i.e., the zinc finger, trunk domain, β-barrel, α-helix, and gelsolin domain) and has two isoforms (SEC23A and SEC23B). SEC23 is a GTPase-activating protein (GAP) that stimulates SAR1-GTP hydrolysis in order to facilitate vesicle transportation in vivo [2], which activity could be induced by SEC31 [3]. A previous study demonstrated that a SEC23B mutation causes congenital dyserythropoietic anemia type-II (CDAII) [4], while the SEC23A mutation causes cranio-lenticulo-sutural dysplasia (CLSD) due to abnormal endoplasmic-reticulum-to-Golgi trafficking [5]. Aberrant expression was reported to be associated with human cancer development, whereas SEC23A and SEC23B may have the opposite activity in human cancer, for a reason that remains unclear [1]. Thus, in this review, we summarized recent research involving SEC23, COPII-vesicle transportation, autophagy, and cancer development and progression (Fig. 1).
SEC23 function in cell proliferation, autophagy, and apoptosis. As a core component of COPII vesicles, SEC23 is not only involved in the protein transportation and secretion process in cells but also participates in autophagy and promotes the survival of cancer cells. Furthermore, SEC23B regulates development of the brain and pancreas in mice.
Structure of SEC23
There are two isoforms of SEC23, namely SEC23A and SEC23B, to interact with SEC24 [6] and SEC16 [7, 8]. Both hSEC23A and hSEC23B have a molecular mass of approximately 85 kDa (765 vs. 767 amino acids) with 85% homology [9], exceeding 95% similarities between mouse and human SEC23A and SEC23B. The functions of SEC23A and SEC23B in the COPII complex were interchangeable in vivo [1]. The human SEC23A gene is localized at chromosome 14q21.1 with 22 exons, while SEC23B is localized at chromosome 20p11.23 (https://www.genecards.org/). The SEC23 protein contains five distinct domains (i.e., the zinc finger, trunk domain, β-barrel, α-helix, and gelsolin-like domain; Fig. 2) [10, 11]. The zinc finger domain has 55 amino acids, containing a series of the zinc fingers that can interact with RNA and DNA, while the trunk domain consists of 250 amino acids and is able to interact with Sar1 and the β-barrel domain consists of 180 amino acids and is essential to form the inner coat. Moreover, the α-helical domain (with 105 amino acids) interacts with Sar1 [12] and the 105 amino acid gelsolin-like domain is able to bind to the C-terminus of SEC31, which shares the same triple-proline motifs with SEC16 [13], TANGO1, and cTAGE5 [14, 15]. However, after we searched and blasted the functional domains of SEC23A and SEC23B, we found that there are differences in the regions of their five functional domains, like the Trunk domain, which could lead to some different functions between these two proteins (Fig. 3). In contrast, these two proteins do have many identical regions and in some situations, they may compensate each other in cells. Thus, these two proteins are siblings, they do have differences in functions and expression in tissues; for example, the level of SEC23B protein in the pancreas was higher by approximately nine folds than SEC23A did, whereas SEC23A in the liver was much higher than SEC23B was. Thus, the differential expression of these two isoforms in a given organ or tissue could lead to their distinct phenotype or functions obviously.
Illustration of SEC23 protein functional domains. (Modified from Lee and Miller [10] and Yoshihisa et al. [11], using the tools from a previous study [85]). The SEC23 protein contains five functional domains, i.e., the zinc finger, trunk domain, β-barrel, α-helix, and gelsolin-like domain [10, 11].

Amino acid blast results of SEC23A and SEC23B. The five functional domains of SEC23B [41] are labeled by different colors, while SEC23A could not be labeled for its unknown domain location. Refer to the structure of SEC23 [12], the domain location of SEC23A may be similar to SEC23B.

SEC23 Regulation of COPII-vesicle Transportation
In normal eukaryotic cells, the ER functions to facilitate protein synthesis, folding, and dispatch in the rough ER or lipid synthesis in the smooth ER [16]. The newly translated proteins destined for secretion are transferred to the Golgi apparatus for post-translational modification and then either transferred to different cell compartments or secreted into the extracellular space [17], while lipids are also transported to other organelles or secreted [18]. However, before being transferred to the Golgi, they are selected and packaged into the COPII to make COPII-coated vesicles [19, 20], and the COPII physically deforms the ER membrane into vesicles and selects cargo molecules to transport them to the Golgi. To date, the COPII is shown to consist of five proteins (i.e., SAR1, SEC23, SEC24, SEC13, and SEC31). GTP-loaded SAR1, a small GTPase, will bind to the ER membrane and recruit the SEC23/SEC24 complex to form the inner coat. The SEC23/SEC24 complex will recruit the SEC13/SEC31 complex (the outer coat) for complete assembly of COPII [21]. After that, the COPII-containing vesicles leave the ER membrane and SEC23 functions as GAP for SAR1 dissociation from COPII [22], leading to the formation of the ER-Golgi intermediate compartment, along which, the vesicles enter the Golgi apparatus [23]. This observation indicates that SEC23 is a core component of COPII and is involved in the formation of the inner coat by acting as a GAP and participating in cargo selection [7].
It is well established that the SEC23/SEC24 complex is recruited to the ER membrane by SAR1, a small G protein, while SEC23A cycling can be altered by ER stress, which will reduce the stability of SEC23 proteins and their binding to the ER membrane [24]. After being recruited to the ER membrane, SEC23 will act as a GAP for SAR1, while SEC24 will select and capture most of the cargo [25]. Indeed, a previous structural analysis study demonstrated that the size of the COPII vesicles was usually between 60 and 100 nm and that the size of the COPII vesicles bound to the concave surface of SEC23/SEC24 and SAR1 was approximately 60 nm [11]. Moreover, SEC23, residing at the inner and outer coats, will interact with SEC31, residing at the outer COPII coat, to form a SEC23-SEC31 interface in order to recognize and capture various cargo molecules [26]. Molecularly, the Phe380 residue of the SEC23 protein forms the binding groove to accommodate SEC31 interaction and promotion of SEC23 GAP activity and COPII disassembly [27]. In addition, SEC23 can also interact with the p150 dynactin subunit and the dynein motor in order to facilitate microtubule-driven traffic from the ER-Golgi intermediate compartment [28, 29]. However, other previous studies showed that Rab1b, localized at the ER-Golgi interface and the Golgi [30, 31], was able to interact with SEC23 and change the kinetics of COPII association/dissociation at the ER exit site [32].
Overall, SEC23 and the COPII-vesicle play an important role in the transportation of newly translated proteins or lipids that are destined for secretion in cells. Indeed, during cancer development and progression, a great number of proteins are produced and transported in cancer cells, and SEC23B may play an important role in this process.
SEC23 Regulation of Embryo Development
Previous studies demonstrated that SEC23B played an essential role in mouse embryo development, especially in the development of the secretory tissues, like the pancreas [33, 34]. SEC23B-knockout mice died shortly after birth, due to the degeneration of the secretory tissues [34]. SEC23B knockout not only led to an accumulation of proteins in the ER lumen but also activated the apoptosis pathway in response to unfolded proteins (i.e., the ER stress response) [34]. The mice with SEC23B deficiency had smaller pancreas than the wild-type mice, and their pancreas structures also showed abnormalities, such as a lack of zymogen granules in exocrine cells [33, 34].
In addition, a previous study showed that MIA SH3 Domain ER Export Factor 2 (MIA2) also participated in brain development [35, 36] and protein secretion in embryos and loss of MIA2 expression affected the interaction between SAR1 and SEC23, leading to the persistent SAR1 activation to interrupt COPII vesicle formation and transportation from the ER to the Golgi in neurons [37]. Similarly, knockout of SEC23A expression resulted in death of mice during embryonic stages with defects in embryo and neural tube development [38].
Furthermore, it was reported that SEC23B was able to regulate cell growth. For example, epidermal growth factor (EGF), functioning in cell growth and differentiation, can upregulate SEC23B expression through the transcriptional regulator RNF11 [39]. EGF treatment was able to induce the transportation efficiency of newly synthesized EGFRs from the ER to the cell membrane by upregulation of SEC23B, SEC24B, and SEC24D [39]. This was an important process for maintaining physiological levels of EGFR in cells, otherwise it could lead to the proliferation of human cancer cells. It was also reported that SEC23 could directly interact with the Trk-fused gene (TFG) through the TFG C-terminus in order to induce outer coat dissociation [40]. TFG was implicated in multiple neurodegenerative diseases and oncogenesis [40]; thus, SEC23 interaction with TFG could lead to tumor cell growth.
Thus, SEC23 could regulate embryo development, especially the secretory tissues and the neural system. It could also promote cancer cell growth and inhibit cell apoptosis, indicating that SEC23 may accelerate cancer development and progression.
SEC23 Mutations in Human Diseases
To date, various mutations associated with SEC23A and SEC23B were reported in human diseases [10], although there was no two null SEC23B alleles occurring in patients, indicating that complete SEC23B deficiency could be lethal for cells [41]. Previous studies revealed SEC23A mutations in CLSD, which is an autosomal recessive disease [5, 42]. CLSD patients have craniofacial and skeletal malformations, associated with defects in collagen secretion [5, 42]. The germline SEC23A deletion in mice showed the same phenotype of human CLSD [38], while SEC23A mutations in zebra fish jeopardized cartilage development [43].
On the other hand, SEC23B mutations could cause congenital CDAII in human beings, which is an autosomal recessive disease. This disease is characterized by moderate anemia and a lack of erythropoiesis [4, 44]. However, mice with complete SEC23B deficiency did not show visible anemia or any of the characteristics of CDAII [34], although the mice would be unable to survive after birth, probably because of hypoglycemia [34]. In contrast, insertion of the SEC23A coding sequences into the murine SEC23B locus was able to rescue the lethal SEC23B-deficient pancreatic phenotype completely in mice [1]. Furthermore, an increase in SEC23A expression could also compensate for the function of mutated SEC23B in CDAII patients, a potential therapeutic strategy for CDAII patients [45, 46]. In addition, SEC23B mutations showed completely different phenotypes among humans, mice, and zebra fish [1]. In human tissues, SEC23B was mainly expressed in the bone marrow, whereas SEC23B was strongly expressed in the mouse pancreas [13, 14] and disruption of SEC23B expression showed defects in the secretion of extracellular matrix protein in zebra fish, which mimics human CLSD [4].
Furthermore, approximately 3-9% of all thyroid cancer cases are a familial non-medullary thyroid cancer (FNMTC), which may be due to Cowden syndrome [47], a rare autosomal dominant familial cancer syndrome with a high risk of developing breast cancer, metrocarcinoma, and non-medullary thyroid cancer [48]. A previous study of the whole exome sequences in FNMTC revealed that SEC23B was a novel susceptibility gene in Cowden syndrome and identified a heterozygous missense SEC23B variant (c. 1781T>G [p.Val594Gly]) in a multi-generation Cowden syndrome family [49].
Therefore, both SEC23A and SEC23B have vital functions in human beings, loss of which would cause serious diseases; particularly, lost SEC23B closely associated with Cowden syndrome, which has potentially cancer predisposing.
SEC23 Regulation of Autophagy in Cells
The transport protein particle (TRAPP) III complex could be recruited to the phagophore assembly site of COPII during macroautophagy and bind to SEC23, providing the membrane components to form the autophagosome and autophagy [50]. Moreover, SEC23 protein could be phosphorylated by a serine/threonine protein kinase Hrr25 to trigger autophagy-related pathways; however, if there is a loss of SEC23 expression, starvation-induced autophagy could be impaired [51]. A previous study has demonstrated that autophagy was controlled by a novel mechanism based on ULK-FBXW5-SEC23B action, showing that F-Box and WD Repeat Domain Containing 5 (FBXW5) could inhibit biogenesis of the COPII-mediated autophagosome by targeting and promoting SEC23B degradation in the presence of nutrients, whereas ULK1 phosphorylated SEC23B at S186 and prevented SEC23B and FBXW5 interaction in order to inhibit SEC23B degradation during cell starvation, which in turn led to cell autophagy [52]. The UNC51-like kinase 1 (ULK1) is an important enzyme in the regulation of autophagy in mammalian cells [53, 54] and is activated after nutrient deprivation through several upstream signals to initiate autophagy processes [55]. Interestingly, a mutation at the SEC23B S186 site, a cancer-related mutation occurring in human melanoma, could abolish the interaction between SEC23B and FBXW5, resulting in an increase in autophagy and promoting the survival of cancer cells [52].
Similarly, ULK1 can phosphorylate of SEC23A to decrease the interaction between SEC23A and SEC31A and suppress the traffic from the ER to Golgi apparatus. Again, ULK1 can also phosphorylate SEC23A at the residues serine 207 and threonine 405 to induce cell autophagy [56]. Interestingly, SEC23A and SEC23B are associated with autophagy, which is involved in cancer development and progression.
SEC23 Alterations in Human Cancers
Thus far, we discussed the function of SEC23 in cells and its alterations and associated mutations in human disease. This section will discuss the role of SEC23 in human cancer development and progression, although previous studies revealed that SEC23A and SEC23B might have been opposing roles in cancer. In Table 1, we summarized the known miRNAs that may directly affect SEC23 expression.
SEC23 as the target gene of miRNAs
| miR | Target | Cancer type | Ref. |
|---|---|---|---|
| miR-200b | SEC23A | Prostate | [58] |
| miR-200c | SEC23A | Prostate | [57] |
| miR-200s | SEC23A | Breast | [59] |
| miR-21 | SEC23A | Colorectal | [67] |
| miR-375 | SEC23A | BreastProstate | [57][60] |
| miR-130a | SEC23B | Prostate | [79] |
SEC23A Alterations in Human Cancer
To date, there is no report in PubMed showing SEC23A alterations in human cancer, but the role of SEC23A in human cancer has been reported as a target of other genes, like miRNAs (see below for details); thus, it remains to be determined how SEC23A regulates the development of human cancer.
miRNAs are a class of small non-coding RNA molecules up to 24 nucleotides in length and function to regulate cell proliferation, differentiation, apoptosis, and tumor metastasis by downregulation of protein-coding gene expression [57]. A previous study reported that SEC23A was the target gene of miR-200b, miR-200c, or miR-200s, miR-21, miR-375 [58-62]. For example, miR-200 played an important role in the regulation of embryonic and cancer stem cells and sensitivity of cancer cells to chemotherapy [63-67]. SEC23A is one of the miR-200b targeting genes, which was inversely associated with prostate cancer tissues [59]. Moreover, when miR-200c and miR-200s were overexpressed, SEC23A expression was reduced [58, 60]. In addition, a recent study revealed that miR-21 expression enhanced colorectal cancer cell proliferation and metastasis by targeting and inhibiting SEC23A expression [68]. Furthermore, miR-375 possesses tumor-suppressive activity in different human cancers [69]; for example, miR-375 was downregulated in invasive breast cancer, leading to tumor cell migration and invasion [58, 70]. Another previous study reported that miR-375 could target the expression of the SEC23A protein by binding to the 3'-untranslatable region (3'-UTR) of SEC23A cDNA, while SEC23A expression was reduced in primary prostate carcinoma tissues [61]. Furthermore, RNA binding motif 5 (RBM5) functions to regulate gene transcription and mRNA splicing in cells and after knockdown of RBM5 expression in neurons, the level of SEC23A mRNA was upregulated, although there was no significant change in the level of the SEC23A protein [71].
Knockdown of SEC23A expression enhanced tumor cell proliferation and metastatic colonization in prostate and colorectal cancer [58, 59, 61, 68]. It was possibly because SEC23A could mediate secretion of Insulin-Like Growth Factor Binding Protein 4 (IGFBP4), a metastasis-suppressive protein [58]. Interestingly, SEC23A has a clear inhibitory role in breast cancer metastasis, especially the step of colonization during tumor cell metastasis but not at the step of tumor cell migration. Knockout of SEC23A could increase breast cancer colonization and reduce tumor cell migration, where these two effects of SEC23A may cancel each other out. In fact, endogenous SEC23A was lower in highly metastatic breast cancer cells than in lowly metastatic cells. Consistently, SEC23A was also expressed at a lower level in metastatic than in the primary tumors [60]. Taken together, both clinical and animal data demonstrates that SEC23A plays a key role in the inhibition of breast cancer metastasis.
Furthermore, detection of miRNA-375 vs. SEC23A expression in thyroid carcinoma cells was a useful biomarker to assess the in vitro efficacy of vandetanib, a selective kinase inhibitor of the vascular endothelial growth factor receptor (VEGFR), the epidermal growth factor receptor (EGFR), and the RET-tyrosine kinase [72]. SEC23A expression was also reported to be associated with the resistance of prostate cancer to docetaxel treatment [73]. In addition, SEC23A was used as a biomarker of pain flare in patients with painful bone metastases [74].
SEC23B Alterations in Human Cancer
Previous studies revealed that germline heterozygous SEC23B variants were potentially cancer predisposing, even though the mutant SEC23B (associated with ER stress-mediated tumorigenesis) did not cause an overall decrease in SEC23B expression [49, 75]. Indeed, ER stress is a hallmark of cancer [76, 77]. Other previous studies showed that alteration of SEC23B was associated with the development of thyroid cancer [78], hepatocellular cancer [79], and prostate cancer [80].
As a novel susceptibility gene in Cowden syndrome, the SEC23B variant (c. 1781T>G [p.Val594Gly]) was able to promote proliferation, colony formation, survival, and invasion of normal thyroid cells, which was associated with cancer predisposition induced by ER stress [49]. Furthermore, another study demonstrated that SEC23B contributed to cancer predisposition through the ribosome biogenesis pathway, independent of SEC23B-mediated COPII functions [75]. In addition, tandem mass spectrometry [79] compared the transitional endoplasmic reticulum (tER) in the liver tumor nodules vs. the control rat liver and showed that SEC23B expression was upregulated in tumor ER, and this differential SEC23B expression suggests that SEC23B could be used as a potential novel tumor marker for hepatocellular carcinoma [79].
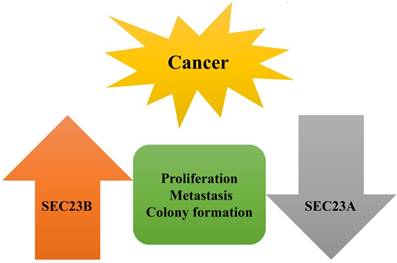
In addition, SEC23B is shown to be a target of miR-130a and the latter was downregulated in prostate cancer [80], whereas miR-130a overexpression in prostate cancer cells reduced tumor cell proliferation and invasion capacity but induced apoptosis [80]. However, knockdown of SEC23B expression mimicked the effect of miR-130a overexpression in prostate cancer cells [80]. This study implies that SEC23B could be an oncogene in prostate cancer. Based on these few studies, we concluded that SEC23B expression is frequently upregulated in these types of human cancer and functions as an oncogene or possesses oncogenic activity in these cancers. However, more studies are needed to clarify the role of SEC23B in human cancer (Fig. 4).
Opposite functions of SEC23A and SEC23B in human cancer. SEC23A was expressed at a lower level in cancer tissues and cells, whereas SEC23B expression was often higher in tumor than in normal tissues, indicating their opposite functions in cancer cells, which effects were confirmed in tumor cell proliferation, metastasis, and colony formation.
Summary and Future Direction of Research on SEC23
To date, different studies have shown the importance of SEC23 participation and function in COPII, particularly the SEC23/SEC24 complex, in order to facilitate newly translated secretion proteins transferred from the ER to the Golgi apparatus for secretion. SEC23 interacts with SAR1 and SEC31 to select cargo and induce the GTPase activity of SAR1 during vesicle formation, leading to the secretion of proteins and lipids. Thus, as an important member of COPII, SEC23B is involved in collagen secretion [81]. SEC23 is involved in embryo development and SEC23B plays an essential role in mouse secretory tissue development, whereas SEC23B mutations could lead to the death of the mice shortly after birth. The main reason for this could be that the loss of SEC23 expression enhanced apoptosis of the secretory tissues, like the pancreas, while SEC23 also plays a role in brain development and function in mice. In addition, SEC23 affects on the transportation of EGFR from the ER to the Golgi for cell proliferation.
Interestingly, the loss of SEC23B mainly affects the exocrine glands, not the endocrine tissues. The SEC23B-deficient mice also shows apoptosis in salivary gland and gastric glands, which may be caused by ER stress pathways, such as PERK or other two ways. The loss of SEC23A could also cause the death of mice embryos. And about half of the live SEC23A-deficient mice embryos would appear neural tube opening at midbrain, which could be found in all of the surviving and dead embryos. Besides, the deletion of SEC23A could lead to the apoptosis in extraembryonic membranes, which may be also caused by ER stress, just like SEC23B-deficient mice. Therefore, both SEC23A and SEC23B may have effect on ER stress, especially the PERK pathway.
Furthermore, SEC23B can also help in the regulation of autophagy in cells because SEC23B can be phosphorylated by ULK1, Hrr25, and TRAPPIII, all of which are in the autophagy pathway. ULK1 can also phosphorylate of SEC23A and inhibit the traffic from ER to Golgi. SEC23 was shown to be a master regulator of budding and fusion through the phosphorylation-dephosphorylation cycle in order to facilitate the formation of the autophagosome [f]. Autophagy is an evolutionarily conserved cellular process and its alteration is involved in cancer development and progression [83, 84]. In vitro SEC23A expression suppressed proliferation and metastasis of breast, prostate, and colorectal cancer cells. In contrast, downregulated SEC23A expression was associated with poor disease relapse-free survival (RFS) of breast cancer patients, indicating that SEC23A might be a potential tumor metastasis suppressor. In addition, reduced SEC23A expression increased sensitivity of medullary thyroid carcinoma to vandetanib treatment, all of which suggest that SEC23A could be a tumor suppressor gene, indicating opposing roles for SEC23A and SEC23B in cancer, although their role in cells is interchangeable in vivo.
However, additional research is needed to understand the precise function and role of SEC23A and SEC23B in human disease and cancer development. Future study will analyze their expression using online databases, like TCGA data, to find an association between expression and different human cancers. Functionally, further study will investigate the gain and loss of these proteins' function in cells in order to better understand their role in individual human cancers. Human cell lines remain one of the better models to study and understand SEC23 activity in the context of human disease, while animal experiments could help us to assess the functional role of SEC23.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (#81872272).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khoriaty R, Hesketh GG, Bernard A. et al. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc Natl Acad Sci U S A. 2018;115:E7748-e57
2. Lee MC, Orci L, Hamamoto S. et al. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605-17
3. Antonny B, Madden D, Hamamoto S. et al. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531-7
4. Schwarz K, Iolascon A, Verissimo F. et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936-40
5. Boyadjiev SA, Fromme JC, Ben J. et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38:1192-7
6. Pagano A, Letourneur F, Garcia-Estefania D. et al. Sec24 proteins and sorting at the endoplasmic reticulum. J Biol Chem. 1999;274:7823-40
7. Aridor M, Weissman J, Bannykh S. et al. Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol. 1998;141:61-70
8. Aridor M, Bannykh SI, Rowe T. et al. Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J Biol Chem. 1999;274:4389-99
9. Paccaud JP, Reith W, Carpentier JL. et al. Cloning and functional characterization of mammalian homologues of the COPII component Sec23. Mol Biol Cell. 1996;7:1535-46
10. Russo R, Esposito MR, Asci R. et al. Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am J Hematol. 2010;85:915-20
11. Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271-7
12. Russell C, Stagg SM. New insights into the structural mechanisms of the COPII coat. Traffic. 2010;11:303-10
13. Supek F, Madden DT, Hamamoto S. et al. Sec16p potentiates the action of COPII proteins to bud transport vesicles. J Cell Biol. 2002;158:1029-38
14. Ma W, Goldberg J. TANGO1/cTAGE5 receptor as a polyvalent template for assembly of large COPII coats. Proc Natl Acad Sci U S A. 2016;113:10061-6
15. Hutchings J, Stancheva V, Miller EA. et al. Subtomogram averaging of COPII assemblies reveals how coat organization dictates membrane shape. Nat Commun. 2018;9:4154
16. Alberts B, Johnson A, Lewis J. et al. The Endoplasmic Reticulum. Molecular Biology of the Cell. 4th ed. New York: Garland Science. 2002
17. Alberts B, Johnson A, Lewis J. et al. Transport from the ER through the Golgi Apparatus. Molecular Biology of the Cell. 4th ed. New York: Garland Science. 2002
18. Gillon AD, Latham CF, Miller EA. Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta. 2012;1821:1040-9
19. Jensen D, Schekman R. COPII-mediated vesicle formation at a glance. J Cell Sci. 2011;124:1-4
20. Lee MC, Miller EA, Goldberg J. et al. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87-123
21. Lee MC, Miller EA. Molecular mechanisms of COPII vesicle formation. Semin Cell Dev Biol. 2007;18:424-34
22. Yoshihisa T, Barlowe C, Schekman R. Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science. 1993;259:1466-8
23. Palmer KJ, Watson P, Stephens DJ. The role of microtubules in transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Biochem Soc Symp. 2005:1-13
24. Amodio G, Venditti R, De Matteis MA. et al. Endoplasmic reticulum stress reduces COPII vesicle formation and modifies Sec23a cycling at ERESs. FEBS Lett. 2013;587:3261-6
25. Zanetti G, Pahuja KB, Studer S. et al. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2011;14:20-8
26. Kim SD, Pahuja KB, Ravazzola M. et al. The SEC23-SEC31 interface plays critical role for export of procollagen from the endoplasmic reticulum. J Biol Chem. 2012;287:10134-44
27. Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13:635-45
28. Presley JF, Cole NB, Schroer TA. et al. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81-5
29. Watson P, Forster R, Palmer KJ. et al. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol. 2005;7:48-55
30. Plutner H, Cox AD, Pind S. et al. Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J Cell Biol. 1991;115:31-43
31. Saraste J, Lahtinen U, Goud B. Localization of the small GTP-binding protein rab1p to early compartments of the secretory pathway. J Cell Sci. 1995;108( Pt 4):1541-52
32. Slavin I, Garcia IA, Monetta P. et al. Role of Rab1b in COPII dynamics and function. Eur J Cell Biol. 2011;90:301-11
33. Khoriaty R, Everett L, Chase J. et al. Pancreatic SEC23B deficiency is sufficient to explain the perinatal lethality of germline SEC23B deficiency in mice. Sci Rep. 2016;6:27802
34. Tao J, Zhu M, Wang H. et al. SEC23B is required for the maintenance of murine professional secretory tissues. Proc Natl Acad Sci U S A. 2012;109:E2001-9
35. Fan J, Wang Y, Liu L. et al. cTAGE5 deletion in pancreatic beta cells impairs proinsulin trafficking and insulin biogenesis in mice. J Cell Biol. 2017;216:4153-64
36. Wang Y, Liu L, Zhang H. et al. Mea6 controls VLDL transport through the coordinated regulation of COPII assembly. Cell Res. 2016;26:787-804
37. Zhang F, Wang Y, Wang T. et al. cTAGE5/MEA6 plays a critical role in neuronal cellular components trafficking and brain development. Proc Natl Acad Sci U S A. 2018;115:E9449-E58
38. Zhu M, Tao J, Vasievich MP. et al. Neural tube opening and abnormal extraembryonic membrane development in SEC23A deficient mice. Sci Rep. 2015;5:15471
39. Scharaw S, Iskar M, Ori A. et al. The endosomal transcriptional regulator RNF11 integrates degradation and transport of EGFR. J Cell Biol. 2016;215:543-58
40. Hanna MGt, Block S, Frankel EB. et al. TFG facilitates outer coat disassembly on COPII transport carriers to promote tethering and fusion with ER-Golgi intermediate compartments. Proc Natl Acad Sci U S A. 2017;114:E7707-E16
41. Khoriaty R, Vasievich MP, Ginsburg D. The COPII pathway and hematologic disease. Blood. 2012;120:31-8
42. Boyadjiev SA, Kim SD, Hata A. et al. Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin Genet. 2011;80:169-76
43. Lang MR, Lapierre LA, Frotscher M. et al. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet. 2006;38:1198-203
44. Bianchi P, Fermo E, Vercellati C. et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30:1292-8
45. Russo R, Langella C, Esposito MR. et al. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol Dis. 2013;51:17-21
46. Pellegrin S, Haydn-Smith KL, Hampton-O'Neil LA. et al. Transduction with BBF2H7/CREB3L2 upregulates SEC23A protein in erythroblasts and partially corrects the hypo-glycosylation phenotype associated with CDAII. Br J Haematol. 2019;184:876-81
47. Nelen MR, Padberg GW, Peeters EA. et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13:114-6
48. Liaw D, Marsh DJ, Li J. et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64-7
49. Yehia L, Niazi F, Ni Y. et al. Germline Heterozygous Variants in SEC23B Are Associated with Cowden Syndrome and Enriched in Apparently Sporadic Thyroid Cancer. Am J Hum Genet. 2015;97:661-76
50. Tan D, Cai Y, Wang J. et al. The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc Natl Acad Sci U S A. 2013;110:19432-7
51. Tanaka C, Tan LJ, Mochida K. et al. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J Cell Biol. 2014;207:91-105
52. Jeong YT, Simoneschi D, Keegan S. et al. The ULK1-FBXW5-SEC23B nexus controls autophagy. Elife. 2018:7
53. Hurley JH, Young LN. Mechanisms of Autophagy Initiation. Annu Rev Biochem. 2017;86:225-44
54. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132-9
55. Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2015;10:257-61
56. Gan W, Zhang C, Siu KY. et al. ULK1 phosphorylates Sec23A and mediates autophagy-induced inhibition of ER-to-Golgi traffic. BMC Cell Biol. 2017;18:22
57. Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586-91
58. Luo D, Wilson JM, Harvel N. et al. A systematic evaluation of miRNA:mRNA interactions involved in the migration and invasion of breast cancer cells. J Transl Med. 2013;11:57
59. Hart M, Nolte E, Wach S. et al. Comparative microRNA profiling of prostate carcinomas with increasing tumor stage by deep sequencing. Mol Cancer Res. 2014;12:250-63
60. Korpal M, Ell BJ, Buffa FM. et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101-8
61. Szczyrba J, Nolte E, Wach S. et al. Downregulation of Sec23A protein by miRNA-375 in prostate carcinoma. Mol Cancer Res. 2011;9:791-800
62. Hurteau GJ, Spivack SD, Brock GJ. Potential mRNA degradation targets of hsa-miR-200c, identified using informatics and qRT-PCR. Cell Cycle. 2006;5:1951-6
63. Iliopoulos D, Lindahl-Allen M, Polytarchou C. et al. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761-72
64. Samavarchi-Tehrani P, Golipour A, David L. et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64-77
65. Shimono Y, Zabala M, Cho RW. et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592-603
66. Wellner U, Schubert J, Burk UC. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487-95
67. Cochrane DR, Howe EN, Spoelstra NS. et al. Loss of miR-200c: A Marker of Aggressiveness and Chemoresistance in Female Reproductive Cancers. J Oncol. 2010;2010:821717
68. Li C, Zhao L, Chen Y. et al. MicroRNA-21 promotes proliferation, migration, and invasion of colorectal cancer, and tumor growth associated with down-regulation of sec23a expression. BMC Cancer. 2016;16:605
69. Yan JW, Lin JS, He XX. The emerging role of miR-375 in cancer. Int J Cancer. 2014;135:1011-8
70. Wang F, Li Y, Zhou J. et al. miR-375 is down-regulated in squamous cervical cancer and inhibits cell migration and invasion via targeting transcription factor SP1. Am J Pathol. 2011;179:2580-8
71. Jackson TC, Kotermanski SE, Kochanek PM. Whole-transcriptome microarray analysis reveals regulation of Rab4 by RBM5 in neurons. Neuroscience. 2017;361:93-107
72. Lassalle S, Zangari J, Popa A. et al. MicroRNA-375/SEC23A as biomarkers of the in vitro efficacy of vandetanib. Oncotarget. 2016;7:30461-78
73. Wang Y, Lieberman R, Pan J. et al. miR-375 induces docetaxel resistance in prostate cancer by targeting SEC23A and YAP1. Mol Cancer. 2016;15:70
74. Furfari A, Wan BA, Ding K. et al. Genetic biomarkers associated with pain flare and dexamethasone response following palliative radiotherapy in patients with painful bone metastases. Ann Palliat Med. 2017;6:S240-S7
75. Yehia L, Jindal S, Komar AA. et al. Non-canonical role of cancer-associated mutant SEC23B in the ribosome biogenesis pathway. Hum Mol Genet. 2018;27:3154-64
76. Urra H, Dufey E, Avril T. et al. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer. 2016;2:252-62
77. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89-102
78. Peiling Yang S, Ngeow J. Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr Relat Cancer. 2016;23:R577-R95
79. Roy L, Laboissiere S, Abdou E. et al. Proteomic analysis of the transitional endoplasmic reticulum in hepatocellular carcinoma: an organelle perspective on cancer. Biochim Biophys Acta. 2010;1804:1869-81
80. Ramalho-Carvalho J, Martins JB, Cekaite L. et al. Epigenetic disruption of miR-130a promotes prostate cancer by targeting SEC23B and DEPDC1. Cancer Lett. 2017;385:150-9
81. Malhotra V, Erlmann P. The pathway of collagen secretion. Annu Rev Cell Dev Biol. 2015;31:109-24
82. Conibear E. Vesicle transport: springing the TRAPP. Curr Biol. 2011;21:R506-8
83. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69-79
84. Rybstein MD, Bravo-San Pedro JM, Kroemer G. et al. The autophagic network and cancer. Nat Cell Biol. 2018;20:243-51
85. Ren J, Wen L, Gao X. et al. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271-3
Author contact
![]() Corresponding author: Dr. Peijun Liu, Center for Translational Medicine, The First Affiliated Hospital, Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, China. E‑mail: liupeijunxjtu.edu.cn
Corresponding author: Dr. Peijun Liu, Center for Translational Medicine, The First Affiliated Hospital, Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, China. E‑mail: liupeijunxjtu.edu.cn