Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(13):2783-2797. doi:10.7150/ijbs.35128 This issue Cite
Review
Cyclooxygenase-2 in Endometriosis
Zhen-Zhen Lai1, Hui-Li Yang1, Si-Yao Ha1, Kai-Kai Chang2, Jie Mei3, We-Jie Zhou4, Xue-Min Qiu1, Xiao-Qiu Wang1, Rui Zhu5, Da-Jin Li1, Ming-Qing Li1,6 ![]()
1. NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Fudan University, Shanghai 200080, People's Republic of China.
2. Department of Gynecology, Hospital of Obstetrics and Gynecology, Fudan University, Shanghai 200011, People's Republic of China.
3. Reproductive Medicine Center, Department of Obstetrics and Gynecology, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, Jiangsu 210008, People's Republic of China.
4. Clinical and Translational Research Center, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, People's Republic of China.
5. Center for Human Reproduction and Genetics, Suzhou Municipal Hospital, Suzhou 215008, People's Republic of China.
6. Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, Hospital of Obstetrics and Gynecology, Fudan University, Shanghai 200011, People's Republic of China.
Received 2019-3-22; Accepted 2019-7-28; Published 2019-10-23
Abstract

Endometriosis (EMS) is the most common gynecological disease in women of reproductive age, and it is associated with chronic pelvic pain, dyspareunia and infertility. As a consequence of genetic, immune and environmental factors, endometriotic lesions have high cyclooxygenase (COX)-2 and COX-2-derived prostaglandin E2 (PGE2) biosynthesis compared with the normal endometrium. The transcription of the PTGS2 gene for COX-2 is associated with multiple intracellular signals, which converge to cause the activation of mitogen-activated protein kinases (MAPKs). COX-2 expression can be regulated by several factors, such as estrogen, hypoxia, proinflammatory cytokines, environmental pollutants, metabolites and metabolic enzymes, and platelets. High concentrations of COX-2 lead to high cell proliferation, a low level of apoptosis, high invasion, angiogenesis, EMS-related pain and infertility. COX-2-derived PGE2 performs a crucial function in EMS development by binding to EP2 and EP4 receptors. These basic findings have contributed to COX-2-targeted treatment in EMS, including COX-2 inhibitors, hormone drugs and glycyrrhizin. In this review, we summarize the most recent basic research in detail and provide a short summary of COX-2-targeted treatment.
Keywords: COX-2, PGE2, endometriosis, pain, estrogen
Introduction
Endometriosis (EMS) is a chronic gynecological disease that can usually be seen in women of reproductive age, and is characterized by the presence, transfer and invasion of functional endometrial tissue outside of the uterine cavity [1]. Some hypotheses, such as retrograde menstrual reflux [2], ectopic presence of endometrial stem cells (ESCs) [3] and defects in the immune system [4], have been proposed to explain the migration, implantation and survival of the ectopic endometrial tissue and stroma. The incidence rate of EMS is 5-15% of all women of reproductive age and 20-50% of all infertile women [5-7], and the quality of life for endometriosis patients is significantly reduced, due to the increase in symptoms including chronic pelvic pain, dyspareunia, and infertility in comparison with women without EMS [8]. The economic impact of EMS is compounded by the latency in the diagnosis of EMS, especially in young women who delay seeking treatment. The diagnosis of EMS is typically delayed by 8-10 years, because of the common misdiagnoses of EMS-induced pelvic pain as menstrual-related abdominal pain [9]. EMS can be confirmed by direct visualization using laparoscopy and biopsy. In the past few years, the field of diagnostic biomarkers for EMS has gained increasing attention [10]. When considering the theory of retrograde menstrual reflux, a puzzle emerges in that only around one tenth of women develop EMS, whereas retrograde menstruation is observed in most women, suggesting that other factors may also trigger the formation of endometriotic lesions, such as hormones, inflammatory factors, growth factors, angiogenic factors and cancer-related molecules [11].
The cyclooxygenase-2 (COX-2) / prostaglandin E2 (PGE2) pathway is closely related to EMS. There has been a general realization that EMS is a chronic pelvic inflammatory state, characterized by rising numbers of activated peritoneal immune cells, such as macrophages, and pro-inflammatory factors [12-14]. COX-2 is thought to play a significant role in the origin and development of EMS [15]. In endometrial and endometriotic tissues of women with EMS, elevated expression of COX-2 has also been described [16]. COX-2 which is a rate-limiting enzyme in the PGE2 compound [17], is overexpressed in endometriotic tissues and contributes to increased concentrations of PGE2 in EMS patients, which have also been found in the peritoneal fluid (PF), as well as leukotrienes. COX-2/PGE2 signaling biologically activate oxygenated fatty acids, eicosanoids, and has been shown to be involved in various inflammatory pathological process [18]. In EMS, they appear to play an important role in disease-associated pain [19, 20], essentially being the target of non-steroidal anti-inflammatory drugs (NSAIDs) [16]. These inflammatory mediators, particularly COX-2/PGE2, may also be directly implicated in the pathogenesis of EMS [16], including the regulation of ectopic implantation and the growth of the endometrium, angiogenesis and immunosuppression [21]. PGE2 is a major regulator of the immune response and can exert two opposing functions, exerting inflammatory or anti-inflammatory effects [22]. Therefore, this paper systematically reviews the elements affecting the level and role of, and targeted drugs for COX-2 in EMS.
COX-2
The enzyme COX was first demonstrated to exist in 1976 and cloned in 1988 [23]. COX has three isoforms: COX-1, COX-2 and COX-3 [24-26]. Among these, the COX-1 and COX-2 isoforms are often studied, due to the fact that they are associated with physiological as well as pathological processes. In the gastrointestinal and cardiovascular system, COX-1, a constitutively expressed house-keeping isozyme, is responsible for the basal production of essential PGs [27] that mediate homoeostatic functions. COX-3 is encoded by the COX-1 gene with reserve intron 1 in its mRNA. COX-3 is only expressed in some specific parts of the cerebral cortex and heart, and its exact functions are still unclear [28]. The COX-2 isozyme, by contrast, is synthesized at very low levels under normal conditions and can be induced to become over-expressed under pathological conditions. The PTGS2, the gene for COX-2, is located on human chromosome 8 [29]. The promotor of the immediate-early gene PTGS2 contains a TATA box and binding sites for several transcription factors, including nuclear factor-κB (NF-κB), the cyclic AMP response element binding protein (CRE), and the nuclear factor for interleukin-6 expression (NF-IL-6) [30, 31]. COX-2 expression is associated with multiple transcriptional pathways. There is accumulating evidence for the critical involvement of COX-2 in various pathological processes that include inflammation [32, 33], cancer [34-36], neurodegenerative diseases [37, 38] and multidrug resistance [39].
The expression of COX-2 is rapidly upregulated in response to diverse pro-inflammatory and pathogenic stimuli. All signals converge upon the activation of mitogen-activated protein kinases (MAPKs) that regulate COX-2 expression at both the transcriptional and post-transcriptional levels [40]. Lipopolysaccharide (LPS) signaling, the most pro-inflammatory mediators, induces the expression of COX-2 in the periphery. Specifically, LPS and other Toll-like receptor (TLR) ligands bind to MyD88-associated receptors and activate MEK/ERK pathway to induce the transcription factor activator protein 1 (AP1). LPS also can induce gene PTGS2 transcription by activating the TRAF6/NIK/Tpl2/IKK/NF-κB pathway [41, 42]. Nitric oxide (NO) affects the transcription of PTGS2 in direct and indirect ways; directly, by increasing its catalytic activity, and indirectly, by triggering several signaling cascades that affect the gene transcription. NO and reactive oxygen species (ROS) increase PTGS2 expression [43] via β-catenin/TCF pathway-mediated activation of polyoma enhancer activator 3 (PEA3) [44]. Furthermore, several cytokines, including NO, several pro-inflammatory cytokines (e.g. IL-1, IFN-γ) and hypoxia inducible factor-1α (HIF-1α) can induce COX-2 expression through the cAMP/PKA/CREB and JNK/Jun/ATF2 signaling cascades [45-48]. Growth factors can induce COX-2 expression in both normal and cancer cells, including insulin-like growth factor (IGF), transforming growth factor-α (TGF-α) and epidermal growth factor (EGF). Notably, this regulatory effect of IGF is mediated by PI3K and Src/extracellular signal-regulated kinase (ERK), while the effects of TGF and EGF are achieved through p38MAPK, ERK1/2 and PI3K [49]. There are negative regulators for COX-2 expression. For example, glycogen synthase kinase 3 (GSK3) suppresses COX-2 expression through inhibition of the β-catenin/transcription factor-4 (TCF4) and PKCδ/ERK1/2 signaling pathways [50].
COX-2 expression in EMS
COX-2 is mainly expressed in the endometrial glandular epithelium in healthy women and varies during the menstrual cycle. The expression of COX-2 is at its lowest in the early proliferative phase and gradually increased thereafter, and it maintains a high level throughout the secretory phase [51]. In women with EMS, the expression of COX-2 in the endometrial glandular epithelium, endometrial stroma [4] and PF was higher than that in the control group [52], and it also varies throughout the menstrual cycle [53]. Cho et al. [54] demonstrated that in EMS patients, the expression of COX-2 was elevated significantly in the eutopic endometrium during the proliferative phase and in ovarian endometriotic tissue during the secretory phase compared with the control groups. In addition, ectopic lesions highly express COX-2 in endometriosis patients with chronic stress [54]. Notably, mRNA expression of PTGS2 in the endometrium and ovarian lesions significantly correlates with serum CA-125 and the diameter of endometriomas [54]. In recent research, Mei et al. [55]found that the number of COX-2+CD16- NK cells with impaired cytotoxic activity in the abdominal cavity fluid of patients with EMS was markedly higher than that of the control group.
Genetic variation in PTGS2 (COX-2) and the risk of EMS
Gene polymorphisms in PTGS2 are associated with a high risk of many diseases, such as EMS [56], cancer [57], and acute pancreatitis [58]. The cloning, sequencing and expression of human PTGS2 cDNA have been previously described [59]. There are 51 CpG sites in the promoter region of the COX-2 gene from -590 to +186. Three main transcription factors predominantly regulate COX-2 expression, including NF-κB, NF-IL6, and CRE [60, 61]. Moreover, in many cancers, aberrant methylation of promoter CpG island of the COX-2 has been regarded as an alternative mechanism of its abnormal expression and contributes to carcinogenesis [62, 63]. Genes associated with endometriosis have abnormal DNA methylation. Wang et al [64]and Zidan et al [56] reported that DNA hypomethylation of the NF-IL6 site within the promoter of the PTGS2 gene was highly correlated with the pathological process of EMS, suggesting that EMS may be an epigenetic disease. Wang et al. [64] found that PTGS2 genotypic frequencies of G to A at the -1195 locus in the promoter region in EMS were significantly different from those in normal women. Moreover, the allele frequency in EMS was markedly higher than that in normal women. The risk of EMS for those carrying two A alleles was 2.19 and 2.41 times greater than that for the to non-A genotype. In addition, Wang et al. [65] demonstrated that on the promoter region of the PTGS2 gene, the -1195 A/G may increase the risk of pain occurrence in women with EMS. The presence of the ancestral allele, -765G/C, of the PTGS2 gene is associated with an increased risk of pathological progression in moderate/severe EMS which is related to fertility, and the expression of COX-2 in the eutopic endometrium of women with EMS has shown a tendency to increase when compared to the control group [66, 67]. In a Korean study, the -765C allele was a protective agent against the development of the disease [68].
Regulation of COX-2 expression
Over the years, many epidemiological, pharmacological and laboratory studies have demonstrated that various factors are involved in the regulation of COX-2 expression in EMS (Table 1, Figure 1).
The factors that regulates COX-2 expression in EMS
| Classification | Regulatory factor | Function | Reference |
|---|---|---|---|
| Estrogen | hastens COX-2 expression by activated by NF-κB | Maia et al.2012 | |
| Proinflammatory Cytokine | IL-1β | stimulates the phosphorylation of ERK, p38 and JNK and results in high level of COX-2 | Tamura et al.2002 Huang et al.1998 |
| NGF | increases PTGS2/COX-2 mRNA and protein levels by binding to TrkA | Wang et al.2009 Peng et al.2018 | |
| Hypoxia | mediates DUSP2 down-regulation, activates ERKs and MAPK, and ultimately results in the hypersensitivity of COX-2 | Wu et al.2005 Wu et al.2011 Teague et al. 2010 Lin et al.2012 Pan et al.2007 Hsiao et al. 2015 | |
| Environmental pollutants | PCBs | plays a role in the development of endometriosis | Porpora et al. 2013 |
| HCB | activates of cytosolic AhR complex (AhR-dioxin-c-Src), triggers PTGS2 transcription | Smith et.al. 1993 Deger et al.2007 Chiappini et al. 2016 | |
| Metabolites and metabolic enzymes | omega-3 PUFA | inhibits the activation of NF-κB and decreases the production of pro-inflammatory cytokines to reduces COX-2 expression | Tomio et al. 2013 Attaman et al.2014 |
| IDO | up-regulates COX-2 expression via the activation of JNK signaling pathway | Mei et al.2013 Mei et al. 2012 | |
| LXA4 | inhibits COX-2 expression | Kumar et al. 2014 | |
| Platelets | increases IL-1β level and increases COX-2 expression | Ding et al. 2015 | |
| Others | COUP-TFII | binds to PTGS2 promoter to inhibit its transcription and IL-1β-induced COX-2 up-regulation | Li et al. 2013 Li et al. 2013 |
Multiple factors regulate COX-2 expression. Estrogen (E2), omega-3 PUFA and IL-1β promote COX-2 expression through the NF-κB signaling pathway. IL-1β stimulates the phosphorylation of MERK, p38 and JNK, then CRE and NF-κB p65 bind to sites on the COX-2 promoter to increase COX-2 expression. In hypoxic conditions, activated HIF-1α will suppress DUSP2 expression directly, and then result in the hypersensitivity of COX-2 in response to proinflammatory stimuli (e.g. IL-1β). Elevated NGF markedly upregulates the expression of PTGS2/COX-2 via the PI3K/AKT signaling pathway. Environmental pollutants, for example HCB and CB126, are known to act via the AhR. These organic compounds have some biological effects mediated by the activation of the cytosolic AhR complex (AhR-dioxin-c-Src), and regulate PTGS2 transcription indirectly. The combination of organic compounds and AhR induces HSD17B7 expression and results in the upregulation of E2, IL-6 and IL-8, which will further promote COX-2 expression.
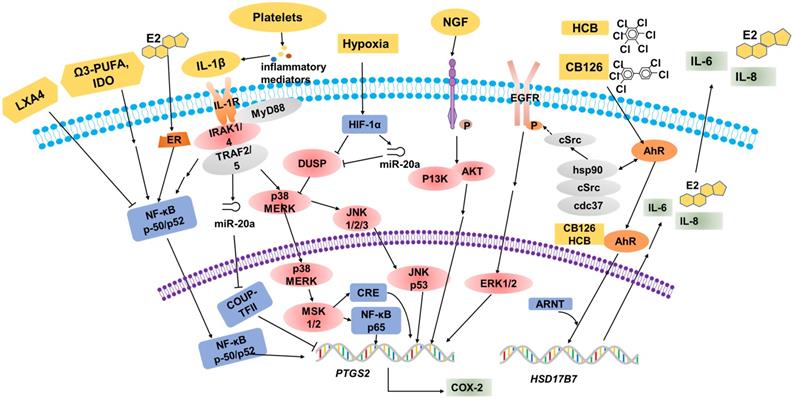
Estrogen
Estradiol and progesterone are core hormones regulating the function of endometrial tissue. In the course of different phases of the menstrual cycle, each steroid hormone is estimated to regulate the translation of hundreds of genes successively [15, 69]. Ectopic and eutopic endometrial tissues have apparently similar histological changes in response to estradiol and progesterone, and both tissues express immunoreactive estrogen and progesterone receptors (PRs). This locally produced estrogen in both the ectopic and eutopic endometrium is considered to exert a crucial role in the regulation of the immunological mechanisms responsible for controlling the development of EMS [15]. Local estrogen production hastens prostaglandin synthesis by stimulating COX-2 activity, thus creating a positive feedback loop of augmented estrogen formation and enhanced inflammation. The synthesis of proinflammatory PGs such as COX-2-derived PGE2, can be activated by NF-κB and increased by estrogen in the endometrium [70]. The synthesis of aromatase seems to play a pivotal role in the development of EMS, which is stimulated by PGs and other inflammatory mediators in endometrial cells but not in aromatase-negative endometrial cells [71]. Thus, a large amount of local estrogen production will further enhance PG synthesis by activating COX-2 expression.
Proinflammatory Cytokines
It has been reported that ectopic ESCs are hypersensitive to the stimulating effect of cytokines, such as interleukin-1β (IL-1β), in terms of overexpression of COX-2 [46]. IL-1β can accelerate the synthesis of COX-2 at the mRNA, protein, and enzyme activity levels in a model system of EMS. Notably, IL-1β can activate MAPK-dependent signaling by binding to the CRE site at -571/-564 of the COX-2 promoter to increase IL-1β-induced COX-2 expression [46]. COX-2 gene induction by IL-1β involves the ERK1/2 and NF-κB signaling pathway, because IL-1β stimulates the phosphorylation of ERK, p38 and JNK [72-74]. Nerve growth factor (NGF), a core endocrine regulator for the growth of neurons, plays crucial roles in the regulation of neuronal survival and maturation [75]. In inflamed tissues in numerous diseases, overexpressed NGF regulates immune responses; directly or indirectly: directly, by influencing innate and adaptive immune responses, and indirectly inducing the release of immune-active neuropeptides and neurotransmitters[76]. NGF is believed to contribute to pathological pain associated with various medical conditions, such as cancer and rheumatoid arthritis (RA) [77]. Elevated NGF levels markedly increase the expression of PTGS-2/COX-2 at the mRNA and protein levels as well as PGE2 secretion in women with EMS. This association may be regulated by enhanced nerve bundle density and by COX-2/PGE2 stimulation via the high-affinity Trk receptor [78-80].
Hypoxia
Hypoxia, which plays a key role in immunity and inflammation under both physiological and pathological conditions, arises when cellular oxygen demand exceeds supply [81]. Hypoxia triggers a profound change in gene transcription, and hypoxia-inducible factor (HIF) is a master regulator [82]. HIF-1α is one of the major transcriptionally active isoforms of HIF that have been described [83]. Dual-specificity phosphatase-2 (DUSP2) which is a nuclear phosphatase that can specifically dephosphorylate p38 MAPK and ERK [84], is markedly downregulated in stromal cells of ectopic endometriotic tissues, leading to prolonged activation of p38 MAPK and ERK and increased COX-2 expression [85]. HIF-1α suppresses DUSP2 expression directly, leads to sustained activation of p38 MAPK and ERK, and ultimately contributes to aberrant COX-2 synthesis in ectopic endometriotic stromal cells [86]. The ERK and p38 MAPK signaling pathways have been reported to play important roles in the modulation of PGE2 synthesis in ectopic endometrial cells, and abnormal phosphorylation of ERK and/or p38 MAPK may lead to over-expression of COX-2 in ectopic lesions [45, 87]. Down-regulation of hypoxia-mediated DUSP2 leads to more activated ERKs and p38 MAPK, and ultimately results in the hypersensitivity of COX-2 in response to proinflammatory stimuli. In addition, microRNAs (miRNAs) are related to tissue repair, hypoxia, inflammation, cell proliferation, extracellular matrix remodeling, apoptosis and angiogenesis in EMS [88]. It has been demonstrated that the expression of miR-20a induced by hypoxia is relatively high in ectopic endometrial tissues compared to that in eutopic endometrial tissues [86, 89]. Interestingly, DUSP2 is a target of miR-20a. A previous study suggested that hypoxia-induced miR-20a expression leads to downregulation of DUSP2 expression, and results in the overexpression of downstream ERK-regulated genes, such as angiogenic, and mitogenic factors, and COX-2 [87]. Taken together, these data strongly support the hypothesis that hypoxia is a vital factor that potentiates PTGS2 gene sensitivity in ESCs [90].
Environmental pollutants
During the last few years, increasing evidence has emerged in support of the relationship between exposure to chemicals with endocrine disruption potential and hormone-related gynecological diseases shows steadily increased [91]. Environmental organochlorine pollutants, particularly polychlorinated biphenyls (PCBs) and dioxins, are thought to be involved in the development of EMS [94]. Dioxin-like [92, 93] rather than non-dioxin-like PCB congeners [94] tend to be responsible for the pathological risk of EMS, according to current epidemiological evidences. Huang et al. [95] found that CB126 (a dioxin-like PCB) enhances estradiol (E2) biosynthesis and promotes the secretion of both IL-6 and IL-8. CB126 is known to act via the aromatic hydrocarbon receptor (AhR). Using DMF to inhibit this receptor can abolish the effects induced by CB126 [96]. The gene expression of HSD17B7, rather than aromatase (CYP19A) or HSD17B1, is up-regulated after exposure to CB126. For local E2 production in endometriotic lesions, CYP19A was previously thought to be significant [97, 98]. The expression of HSD17B7 can be enhanced by LPS and IL-1β which can be observed in ESCs. Thus, the development of EMS can be promoted by the interaction between the endocrine and immune systems and CB126 may provoke this process through stimulation of both E2 synthesis and the inflammatory response. This may support the idea that PCB-induced EMS is related to COX-2. Another type of organochlorinated pollutant, hexachlorobenzene (HCB), is a “dioxin-like” organic compound that binds to AhR [99], accumulating in lipid tissue and inducing the synthesis of xenobiotic metabolic enzymes. These organic compounds have some biological effects which are mediated by the activation of the cytosolic AhR complex (AhR-dioxin-c-Src), triggering membrane actions where c-Src activates growth factor receptors, and nuclear actions where AhR regulates gene transcription including for COX-2 [100, 101]. Chiappini et al [102] found that exposure to HCB enhanced COX-2, PGE2 and EP4 expression, and c-Src kinase activation in T-HESC, thereby contributing to EMS development through both hormonal regulation and immune function.
Metabolites and metabolic enzymes
In vivo and in vitro studies have demonstrated that omega-3 polyunsaturated fatty acids (omega-3 PUFAs) have potential antiapoptotic, anti-inflammatory, antiangiogenic, and antiproliferative effects [103]. Omega-3 PUFAs block the activation of NF-κB, cut down the production of pro-inflammatory cytokines such as IL-6, TNF-α and IL-1, and reduce COX-2 expression to protect against the development of EMS [104, 105]. In particular, the 12/15-LOX-pathway products of eicosatetraenoic acid (EPA) may be critical mediators in suppressing EMS[104]. In inflammatory bowel disease (IBD), PUFAs of the n-3 series have reported to exert an inhibitory action on PTGS2 gene expression in vivo using a genetically-modified mouse [106]; they compete with arachidonic acid (AA) for binding to the COX-2 catalytic site and finally obstructed prostaglandin formation [107]. Indoleamine 2,3-dioxygenase (IDO) has the capacity of tryptophan consumption and the generation of proapoptotic metabolites, thus it was confirmed to be an immune modulator [108] and to be highly expressed in EMS-derived eutopic and ectopic ESCs; it also upregulates COX-2 expression by means of the activation of the JNK signaling pathway [109, 110], along with the enhancement of cell survival, proliferation and invasion. In the canonical JNK pathway, activated JNK can lead to phosphorylation of the transcriptional activation domain of c-Jun; this phosphorylated domain constitutes AP-1, a kind of transcription factors which is acted on the human IDO gene promoter region [111], with c-Fos [112]. Subsequently, G protein-coupled receptors regulate MAPK signaling pathways that result in specific response gene expression, including the genes associated with cell proliferation, apoptosis and invasion [113]. Lipoxins are endogenous eicosanoids, generally produced via a transcellular biosynthetic pathway, the functions of which exhibit both pro-resolving and anti-inflammatory properties [114]. In vivo studies, Lipoxin A4 (LXA4) mediates anti-inflammatory activities through multiple receptors [115], and the best characterized lipoxin A4 receptors is (ALX)/formyl peptide receptor 2 (FPR2). LXA4 treatment significantly attenuated COX-2 and PGE2 levels in both endometriotic lesions and peritoneal fluid cells, which might be the result of downregulating CYP19a1 expression or via direct transcriptional repression [116].
Platelets
Inflammation and coagulation are intricately entwined: inflammation stimulates the coagulation cascade and coagulation modulates the inflammatory response in many ways [117, 118]. Platelets are aggregated in endometriotic lesions, concomitantly with the elevated levels of VEGF and microvessel density. A co-culture system of endometriotic stromal cells and platelets led to enhanced cellular proliferation, and increased COX-2 expression. Analysis of the underlying mechanisms demonstrated that platelet granules contain a variety of inflammatory mediators, such as, IL-1β, which induce the expression of COX-2 in a dose-dependent manner in both normal ESCs and ectopic ESCs [119].
Others
Chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII, also known as NR2F2) is an orphan nuclear receptor that has a pivotal impact in embryonic implantation and placentation, indicating that it is a key regulator in uterine physiology [120-122]. In normal and endometriotic stroma, the expression levels of COUP-TFII mRNA and protein have been dentified to be different, which highlights its potential functions in endometriotic pathogenesis. In normal endometrial tissue, COUP-TFII directly binds to the PTGS2 promoter to inhibit its transcription and diminish IL-1β-induced COX-2 over-expression [123]. In endometriotic stroma, cytokines IL-1β, TNF-α and TGF-β1 can repress COUP-TF II expression mediated by miR-302a, then suppress the binding of COUP-TFII to the COX-2 promoter [123]. Therefore, the decreased COUP-TFII results in the derepression of COX-2 in ESCs [124]. However, the detailed mechanism requires further research.
The role of COX-2 in EMS
Cell proliferation and apoptosis
The growth of endometriotic lesions is a process tightly regulated by a delicate balance between proliferation and apoptosis in endometrial cells. This abnormal survival ability has been associated with the concomitant overproduction of antiapoptotic factors and underproduction of proapoptotic factors [125]. As shown in Figure 2, COX-2-induced PGE2 is a significant antiapoptotic mediator; it can activate cell survival and antiapoptotic pathways to prevent cells from undergoing programmed cell death or apoptosis. The binding of PGE2 and its receptors, EP2 and EP4, regulates these complex molecular interactions and promotes the survival of human ESCs outside the uterus via multiple trans-activating complex signaling pathways (such as c-Src/β-arrestin 1/EGFR/ERK1/2, c-Src/βarrestin1/TNFαR1, IL-1βR1/IκB/NFκB or Gsα/axin/β-catenin)[128]. Selective inhibitors of EP2 and EP4 impair ESC survival pathways and facilitate interactions between antiapoptotic proteins (Bcl-2/Bcl-XL) and proapoptotic proteins (Bax/Bad) leading to an augmentation of the release of cytochrome c and activation of the caspase-3/PARP pathways [126]. The results indicated that administration of NS-398, a kind of selective COX-2-inhibitor, and siRNA can significantly reduce COX-2 concentration, PGE2 production, and endometriotic epithelial and stromal cell proliferation [127]. Laschke et al. [127] showed that in an EMS mouse model, treatment with NS-398 applied to endometrial grafts led to a tendency towards decreased cell proliferation, along with a sustained reduction in proliferating cell nuclear antigen (PCNA) expression; in addition, an increased number of apoptotic cells was observed, as indicated by an upregulation of activated caspase-3. Furthermore, epithelial cell lines stably transfected to overexpress the PTGS2 gene appear to have a higher proliferation rate and to inhibit apoptosis by means of reacting with cyclin D to elongate the G1 phase of the cell cycle [128, 129]. Therefore, the administration of selective COX-2 inhibitors to the ectopic and eutopic endometrium may contribute to an inhibition in proliferative potential and a growth rate in apoptosis [130].
The role of COX-2 in EMS. Overexpression of COX-2 has been demonstrated to be a master regulator in the progression of endometriosis. A high level of COX-2 can promote cell proliferation and suppress cell apoptosis via trans-activating multiple complex signaling pathways, which are triggered by PGE2 and its receptors, EP2 and EP4. In addition, MMP-2/9 activity regulated by PGE2 is be involved in angiogenesis, and ESC migration and invasion, via the intracellular MAPK, AKT and Wnt signaling pathways. COX-2 can induce COX-2+CD16-NK cell (Granzyme BlowPerforinlowIFN-γlowCD16-NK cell) differentiation in the peritoneal fluids of patients with endometriosis, which is beneficial to the immune escape of endometriotic lesions. The COX-2/PGE2/EP2-EP4 signaling decreases the threshold and enhances the excitability of nociceptor sensory fibers through TRPV1 and SCN11A, and contributes to EMS-associated pain. A high level of COX-2/PGE2 and COX-2-induced inflammatory mediators increase uterine tone and contractions and cause pain. TCs are important in maintaining the structural and reproductive functional normality of the oviduct, while overproduced COX-2 may damage the functions of TCs, which will lead to infertility. The low production of COX-2 in cumulus cells is regarded as a possible mechanism of EMS-related infertility.
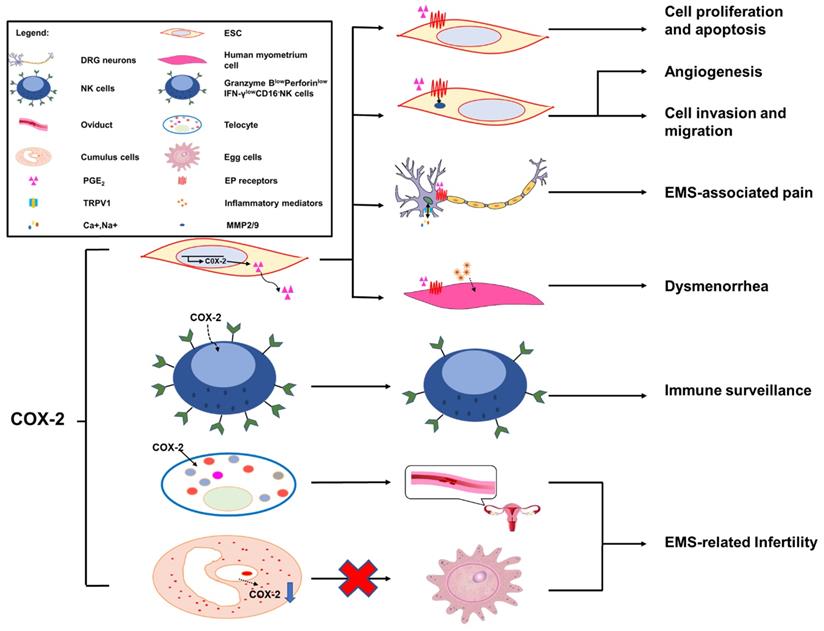
Cell invasion and migration
PGE2 exerts its biological effects through G protein-coupled receptors and by activating multiple cell signaling pathways. These G protein-coupled receptors are designated according to the four subtypes of the PGE receptor (EP1, EP2, EP3 and EP4) [131]. Previous studies have illustrated that EP receptors intracellularly trans-activate the MAPK, AKT and Wnt signaling pathways, resulting in the modulation of cell apoptosis, proliferation, invasion, migration, angiogenesis, pain and immunomodulation [132, 133]. Administration of COX-2 inhibitors decreases the survival, migration and invasion of endometriotic cells as a result of decreased production of PGE2 [127, 134]. Additionally, COX-2-associated migration and invasion are decreased when COX-2 is inhibited in endometriotic cells, and are mediated by matrix metalloproteinase (MMP)-2 and MMP-9 in humans [135]. In addition, there is an interesting observation that COX-2 inhibitors produce more detrimental effects on invasion compared with migration in endometriotic cells; however, the underlying molecular mechanisms of these selective effects are unknown [21].
Angiogenesis
In the pathological process of EMS, the development of new blood vessels represents a core factor, because the long-term survival and growth of the exfoliated endometrium requires an effective blood supply; this is a major prerequisite at ectopic lesions. The development of the ectopic endometrium relies on angiogenesis, which is a characterizing factor of EMS [48]. MMPs, a group of zinc-dependent proteolytic enzymes, are mainly involved in extracellular matrix degradation to promote cellular invasion, migration and angiogenesis [136, 137]. In vitro, some evidence suggests that PGE2 dramatically increases MMP-2 activity as well as tube formation [138]. Blocking the expression of COX-2 and/or a phosphorylated protein kinase (AKT) suppresses MMP-2 activity and endothelial tube formation, indicating that the MMP-2 activity modulated by PGE2 is potentially involved in angiogenesis. Moreover, treatment with a chemical inhibitor can specifically inhibit MMP-2 by significantly inhibiting cellular migration, invasion and tube formation. Furthermore, a notable decrease in endometrial lesion numbers was observed after applying inhibitors of MMP-2 and COX-2 to the mouse model of EMS. Collectively, COX-2 can promote angiogenesis indirectly via the involvement of MMP-2 activity during EMS progression [138]. In particular, COX-2 inhibitors could exert an anti-angiogenic effect on endometriotic lesions. On one hand, the angiogenic factor vascular endothelial growth factor (VEGF) plays an important role in the pathogenesis of EMS [48], and selective COX-2 inhibitors suppress the expression of VEGF in endometrial grafts initially [127] and in tumor researches [139]. On the other hand, in a study on hamsters, firm platelet adhesion to the endothelium of microvessels was increased when treated with a selective inhibitor of COX-2 [140].
EMS-associated pain: chronic pelvic pain and dysmenorrhea
COX-2 is inducible and is involved in pain- and inflammation-associated pathological pathways [141]. Increased expression levels of COX-2 in central nervous system (CNS) regions within the pain-processing pathway were found at the spinal [142], thalamic and cortical levels [143], and in dorsal root ganglion (DRG) neurons [144]. COX-2 expression is viewed as a sensitive and responsive biomarker of centralized inflammatory pain in the CNS [142]. In a rat EMS model, sympathetic and sensory C and Aδ fibers innervated endometriosis lesions, which expressed calcitonin gene related peptide (CGRP) and TRPV1 proteins, thereby contributing to the formation of the proinflammatory microenvironment of DRG neurons from L1-S1. Neurons from L1-S1 innervate the pelvis and pelvic organs and increase pelvic floor hyperalgesia [145]. Greaves et al [143] found that in an EMS mouse model, the COX-2/PGE2 signaling pathway was overexpressed. PGE2 plays a significant role in the pathophysiology of COX-2-induced EMS [143]. PGE2 acts on peripheral nociceptors, lowering the threshold and enhancing the excitability of nociceptor sensory fibers through TRPV1 and Nav1.9 voltage-gated sodium channels (SCN11A) [146], and induces chronic inflammatory pain through EP2 and EP4 [147, 148]. Localized peripheral inflammation increases the expression of EP4 protein in L5 DRG neurons. Inhibition of EP4 decreases the PGE2-induced sensitization of DRG neurons and the release of the neuropeptides SP and CGRP [147, 148]. At the level of the PTSG2 gene, the -1195 A/G on the promoter region of the COX-2 gene may increase the risk of pain occurrence in patients with EMS, possibly by affecting the rate of gene expression, especially in patients with the pain phenotype [66].
Dysmenorrhea, defined as painful cramps in the lower abdomen that occurs with menstruation, is one of three main characteristics of EMS [149]. Primary dysmenorrhea is one of two types of dysmenorrhea, caused by an increased or unbalanced level of endometrial prostaglandins, most importantly PGE2, during menstruation [150]. COX-2-derived PGE2 increases uterine tone and contractions, and causes pain [151]. COX-2 can induce the production of a large number of inflammatory mediators, including PGs [152], and contribute to dysmenorrhea in patients with EMS.
EMS-related infertility
Around 20-50% of the EMS population is estimated to be infertile [153]. Telocytes (TCs; previously considered to be interstitial Cajal-like cells, ICLC), a peculiar type of stromal cell, have been identified in many organs, including the endometrium, myometrium and fallopian tube [154], and have been reported to be decreased in women with EMS and tubal ectopic pregnancy [155]. Structural and reproductive functional abnormalities of the oviduct are observed as a result of TC damage [156]. In oviduct tissues, overproduced COX-2 may be responsible for the TC damage [157]. The pathologic niche of EMS is considered to have deleterious effect on oocyte quality. Cumulus cells are indirect biomarkers of this [158-160]. In eutopic and ectopic endometrial tissues from women with EMS, the transcription of PTGS2 is upregulated [15, 161, 162]. By contrast, the transcript levels of PTGS2 in cumulus cells of infertile women with EMS are decreased [163]. Reduced PTGS2/COX-2 expression may lead to an impairment of oocyte quality, which is regarded as a possible mechanism of EMS-related infertility [163].
Immune surveillance
The transcription of the aromatase gene is favored by epigenetic changes in the endometrium, allowing endometrial cells to survive in ectopic locations by producing enough estrogen to protect them from destruction by activated macrophages [70]. Local estrogen production accelerates PG synthesis by stimulating the activation of COX-2, thus creating a positive-feedback sequence of facilitated estrogen formation and enhanced inflammation [70]. Therefore, the increased inflammation in EMS may reflect the overexpression of estrogen, which alone activates COX-2 and NF-κB to increase inflammation and PG production. In a recent study, a high level of COX-2+CD16-NK cells was observed in the peritoneal fluid of patients with EMS [55]. COX-2 can induce the differentiation of low-cytotoxicity CD16-NK cells (with low levels of Granzyme B, Perforin and IFN-γ), and promote the immune escape of endometriotic lesions. In addition, these COX-2+CD16-NK cells promote the proliferation and restrict the apoptosis of ectopic lesions; however, the mechanism needs further study [23]. The population of Foxp3+ regulatory T (Treg) cells is upregulated in the PF of EMS patients, which contributes to the local dysfunctional immune microenvironment in EMS and the immune escape of ectopic endometrial tissue. The estrogen-IDO1-MRC2 axis is involved in regulating the differentiation and function of Treg cells [164]. It was reported that Treg cells upregulate the expression of MMP2 and COX-2 and promote the survival, migration and invasion of endometriotic cells [165]. In the gastric tumor microenvironment, COX-2 expression is also strongly correlated with Foxp3, a reliable marker of Treg cells [166]. Yuan X.Y et al [150] found that Treg cells could express high levels of COX-2, and produced a high level of PGE2. PGE2 binds to EP2 and EP4 and triggers the cAMP Csk inhibitory pathway to suppress T-cell immune responses. Foxp3high Treg cells suppress the proliferation of autologous CD4+CD25- T cells, which can be reversed by COX inhibitors and PGE2 receptor-specific antagonists. These data show that in the development of gastric cancer, tumor-infiltrating Treg cells can induce immune suppression via the COX-2/PGE2 axis [150,167].
Anti-EMS drugs targeting COX-2 (Figure 3)
COX-2 inhibitors
COX-2 is an essential therapeutic target for anti-inflammatory drugs, which are known as nonsteroidal anti-inflammatory drugs (NSAIDs), including naproxen and diclofenac, as well as newer COX-2 selective inhibitors such as Celebrex (celecoxib; Pfizer). A clinical trial recruited 28 women (age range 23-39 years) who were diagnosed with EMS by laparoscopy. They were treated with a placebo or a COX-2 specific inhibitor. It was found that the administration of NSAIDs was safe and effective in the management of EMS-related pain and might block angiogenesis in endometriotic foci, which was considered to be a long-term effect in that it may help prevent relapses of EMS [168]. In a rat model, a new selective COX-2 enzyme inhibitor, dexketoprofen trometamol, remarkably reduced the development of experimentally-induced endometriotic lesions, both macroscopically and microscopically [169, 170]. COX-2 induces the production of PGE2 and E2, which are known to increase VEGF expression [171]. The binding of VEGF to the Fms-like tyrosine kinase 1 (Flt-1) receptor [170] leads to an upregulation in mitogenesis, migration enhancement, and the release of various proteolytic enzymes. It has been demonstrated that treatment with parecoxib downregulates the expression of VEGF and Flk-1, and reinforces its antiangiogenic activity in rat endometriotic lesions [172]. It was reported that patients with EMS showed increased numbers of activated macrophages in the PF [14, 173], which are the primary source of VEGF produced in areas of inflammation [14]. Treatment with COX-2 inhibitors significantly decreases microvessel density and macrophage numbers, and is associated with decreased expression of VEGF and Flk-1 [172, 174]. In mouse model, the group administered with COX-2 inhibitors showed a low concentration of PGE2. Combined use of COX-2 inhibitors and telmisartan may be more effective in the treatment of endometriotic lesions. Combining the inhibition of COX-2 with the peroxisome proliferator-activated receptor (PPAR)-γ agonist telmisartan appears to be a promising strategy in EMS as it suppresses cell proliferation and induces apoptosis. Decreased expression of p-Akt/Akt and downstream p-eNOS/eNOS in parecoxib/telmisartan-treated lesions has also been shown experimentally [175]. However, COX-2 inhibitors may damage the gastrointestinal tract, and induce the development of erosions and ulcers, with potential complications of protein loss, stricture formation, bleeding and perforation [176]. The side effects of COX-2 inhibitors should be monitored.
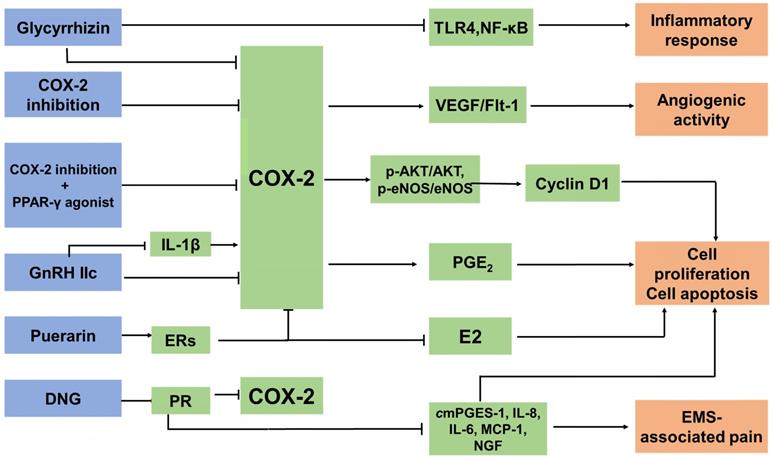
The anti-EMS strategy of targeted COX-2. There are three main types of anti-EMS drugs targeting COX-2: COX-2 inhibitors, hormone drugs and other drugs. They inhibit COX-2 expression in different ways. Treatment with COX‐2 inhibitors significantly decreases microvessel density and macrophage numbers, and is associated with decreased expression of VEGF and Flk-1. Combining the inhibition of COX-2 with peroxisome proliferator-activated receptor (PPAR)-γ agonists suppresses cell proliferation and induces apoptosis by decreasing the expression of p-Akt/Akt and p-eNOS/eNOS. GnRH-II decreases the COX-2 secretion of the ectopic, eutopic and normal ESC in EMS, and it can reverse the IL-1β-induced expression of COX-2 in ESCs. DNG, a selective PR agonist, downregulates the mRNA expression of CYP19A1, COX-2, mPGES-1, IL-8, IL-6, MCP-1, VEGF and NGF, and PGE2 production, as well as suppressing the development of endometriotic lesions and relieving EMS-associated pain. Glycyrrhizin is able to attenuate the expression of COX-2 and dramatically diminishes LPS-induced TLR4 expression and NF-κB activation in MEECs. As a result, it can inhibit the LPS-induced inflammatory response. Puerarin can inhibit the expression of P450arom and COX-2 in the ectopic endometrium, restrict the levels of E2 and PGE2, and block the positive feedback mechanism of E2 synthesis.
Hormone drugs
Type-II gonadotropin releasing hormone (GnRH II), a secondary form of GnRH, is distributed in discrete regions of the central and peripheral nervous systems and in nonneural tissues; GnRH-II functions in the nervous system and, notably, in areas associated with sexual behavior [177]. GnRH-II has the effect of promoting apoptosis, especially on the ectopic ESC, as a result of inhibiting the secretion of IL-8 protein and the level of COX-2 mRNA and IL-8 mRNA in endometriotic cells, and in the case of the downregulation of endogenous GnRH-II expression it can lead to the initiation and development of EMS [178]. In addition, GnRH-II decreases VEGF secretion in the ectopic, eutopic and normal ESC in EMS in vitro, which contributes to the downregulation of the number of newly-formed blood vessels [177]. The IL-1β-induced expression of COX-2 in ESC can be reversed by GnRH-II [179]. Dienogest (DNG) is a selective progesterone receptor (PR) agonist. One of the current clinical anti-EMS strategies is oral administration of DNG. However, PR has been reported to appear as two major isoforms, PR-A and PR-B, and they have mostly distinct physiological functions [180]. DNG exerts therapeutic efficacy against the pain and progression of EMS regardless of PR expression patterns. It was reported that DNG downregulates the mRNA expression of CYP19A1, COX-2, mPGES-1, IL-8, IL-6, monocyte chemoattractant protein (MCP)-1, VEGF and NGF, and PGE2 production in human endometriotic epithelial cell lines that specifically express either PR-A or PR-B [181, 182].
Other drugs
Glycyrrhizin, a triterpene isolated from the roots and rhizomes of licorice (Glycyrrhiza glabra), has been shown to have anti-inflammatory effects. Wang et al [182] found that glycyrrhizin was able to attenuate the expression of inducible nitric oxide synthase (iNOS) and COX-2 in mouse endometrial epithelial cells (MEECs). Furthermore, glycyrrhizin dramatically diminishes LPS-inducing TLR4 expression and NF-κB activation in MEECs. As a result, it can inhibit the LPS-induced inflammatory response. Glycyrrhizin may be used as a potential agent for the treatment of EMS, partly by targeting COX-2 [183]. Another traditional Chinese medicine, puerarin, extracted from Radix puerariae, is widely known as a natural conditioner of selective estrogen receptors (ERs) [184]. Puerarin can inhibit the expression of P450arom and COX-2 in the ectopic endometrium, restrict the levels of E2 and PGE2, and block the positive feedback mechanism of E2 synthesis. It could be a potential therapeutic agent for the treatment of EMS in clinic [185].
Conclusion and future perspectives
Under the regulation of hormone, hypoxia and so on, the increased COX-2 in the glandular epithelial cells and ESCs of ectopic lesions leads to the high proliferation, low level of apoptosis, high invasion and angiogenesis, and impaired cytotoxic NK cell differentiation, which further promotes the occurrence and development of EMS. By producing PGE2 to induce EMS-related pain, COX-2 in endometriotic cells can further accelerate the development of EMS. Many drugs and COX-2 inhibitors play an important role in the treatment of EMS by targeting COX-2, especially for EMS-related pain. However, further investigation of their actions, apart from analgesic functions, is needed, which will enlarge therapeutic horizon of these drugs in EMS. For example, considering the important role of COX-2 in the survival, invasion, angiogenesis and immune escape of ectopic lesions, COX-2 may be an important indicator for predicting the recurrence of EMS. Prophylactic drugs may become available in high-risk populations. COX-2-targeting treatments may inhibit the growth of the ectopic intima, relieve pain, reduce angiogenesis and remove residual lesions. By analyzing the expression level of COX-2 and the PGE2 concentration in the endometriotic microenvironment, there is potential to provide individualized and precise treatment for preventing the recurrence of EMS.
Abbreviations
AA: Arachidonic acid; CNS: Central nervous system; COX-2: Cyclooxygenase-2; EMS: Endometriosis; ESCs: Endometrial stem cells; GnRH II: II-type gonadotropin releasing hormone; IL-1β: Interleukin-1β; MAPK: Mitogen-activated protein kinases; MMP: Matrix metalloproteinase; PGE2: Prostaglandin E2; VEGF: Vascular endothelial growth factor.
Acknowledgements
This study was supported by the Major Research Program of National Natural Science Foundation of China (NSFC, No. 31970798, 91542108, 81471513, 31671200 and 81571509), the Shanghai Rising-Star Program 16QA1400800, and the Innovation-oriented Science and Technology Grant from NPFPC Key Laboratory of Reproduction Regulation (CX2017-2), and the Program for Zhuoxue of Fudan University.
Authors' contributions
ZZL performed the literature search, drafted the manuscript and prepared the figure. HLY, SYH, KKC, JM, WJZ, XUQ, XQW, RZ and DJL helped to perform revisions and critically discussed the completed manuscript. MQL designed, supervised and critically reviewed the complete manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Frackiewicz EJ. Endometriosis: an overview of the disease and its treatment. J Am Pharm Assoc (Wash). 2000;40(5):645-57 quiz 99-702
2. Griffith JS, Liu YG, Tekmal RR, Binkley PA, Holden AE, Schenken RS. Menstrual endometrial cells from women with endometriosis demonstrate increased adherence to peritoneal cells and increased expression of CD44 splice variants. Fertil Steril. 2010;93(6):1745-9
3. Figueira PG, Abrao MS, Krikun G, Taylor HS. Stem cells in endometrium and their role in the pathogenesis of endometriosis. Ann N Y Acad Sci. 2011;1221:10-7
4. Goumenou AG, Matalliotakis IM, Tzardi M, Fragouli YG, Mahutte NG, Arici A. Apoptosis and differential expression of apoptosis-related proteins in endometriotic glandular and stromal cells. J Soc Gynecol Investig. 2004;11(5):318-22
5. Eskenazi B, Warner ML. Epidemiology of endometriosis. Obstet Gynecol Clin North Am. 1997;24(2):235-58
6. Pritts EA, Taylor RN. An evidence-based evaluation of endometriosis-associated infertility. Endocrinol Metab Clin North Am. 2003;32(3):653-67
7. Meuleman C, Vandenabeele B, Fieuws S, Spiessens C, Timmerman D, D'Hooghe T. High prevalence of endometriosis in infertile women with normal ovulation and normospermic partners. Fertil Steril. 2009;92(1):68-74
8. Fuldeore MJ, Soliman AM. Prevalence and Symptomatic Burden of Diagnosed Endometriosis in the United States: National Estimates from a Cross-Sectional Survey of 59,411 Women. Gynecol Obstet Invest. 2017;82(5):453-61
9. Greene R, Stratton P, Cleary SD, Ballweg ML, Sinaii N. Diagnostic experience among 4,334 women reporting surgically diagnosed endometriosis. Fertil Steril. 2009;91(1):32-9
10. Ahn SH, Singh V, Tayade C. Biomarkers in endometriosis: challenges and opportunities. Fertil Steril. 2017;107(3):523-32
11. Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447):1789-99
12. Gazvani R, Templeton A. Peritoneal environment, cytokines and angiogenesis in the pathophysiology of endometriosis. Reproduction. 2002;123(2):217-26
13. Lousse JC, Van Langendonckt A, Gonzalez-Ramos R, Defrere S, Renkin E, Donnez J. Increased activation of nuclear factor-kappa B (NF-kappaB) in isolated peritoneal macrophages of patients with endometriosis. Fertil Steril. 2008;90(1):217-20
14. Ahn SH, Monsanto SP, Miller C, Singh SS, Thomas R, Tayade C. Pathophysiology and Immune Dysfunction in Endometriosis. Biomed Res Int. 2015;2015:795976
15. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E. et al. Role of estrogen receptor-beta in endometriosis. Semin Reprod Med. 2012;30(1):39-45
16. Lousse JC, Defrere S, Colette S, Van Langendonckt A, Donnez J. Expression of eicosanoid biosynthetic and catabolic enzymes in peritoneal endometriosis. Hum Reprod. 2010;25(3):734-41
17. Murakami M, Kudo I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog Lipid Res. 2004;43(1):3-35
18. Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423-8
19. Wu MH, Shoji Y, Chuang PC, Tsai SJ. Endometriosis: disease pathophysiology and the role of prostaglandins. Expert Rev Mol Med. 2007;9(2):1-20
20. Ray K, Fahrmann J, Mitchell B, Paul D, King H, Crain C. et al. Oxidation-sensitive nociception involved in endometriosis-associated pain. Pain. 2015;156(3):528-39
21. Banu SK, Lee J, Speights VJ, Starzinski-Powitz A, Arosh JA. Cyclooxygenase-2 regulates survival, migration, and invasion of human endometriotic cells through multiple mechanisms. Endocrinology. 2008;149(3):1180-9
22. Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L797-805
23. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97-120
24. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271(52):33157-60
25. Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY. et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384(6610):644-8
26. Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145-82
27. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56(3):387-437
28. Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS. et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926-31
29. Kraemer SA, Meade EA, DeWitt DL. Prostaglandin endoperoxide synthase gene structure: identification of the transcriptional start site and 5'-flanking regulatory sequences. Arch Biochem Biophys. 1992;293(2):391-400
30. Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T. Structure of the human cyclo-oxygenase-2 gene. Biochem J. 1994;302( Pt 3):723-7
31. Kang YJ, Mbonye UR, DeLong CJ, Wada M, Smith WL. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog Lipid Res. 2007;46(2):108-25
32. Mitchell JA, Warner TD. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol. 1999;128(6):1121-32
33. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986-1000
34. Benelli R, Vene R, Ferrari N. Prostaglandin-endoperoxide synthase 2 (cyclooxygenase-2), a complex target for colorectal cancer prevention and therapy. Transl Res. 2018;196:42-61
35. Gomes RN, Felipe DCS, Colquhoun A. Eicosanoids and cancer. Clinics (Sao Paulo). 2018;73(suppl 1):e530s
36. Yu T, Lao X, Zheng H. Influencing COX-2 Activity by COX Related Pathways in Inflammation and Cancer. Mini Rev Med Chem. 2016;16(15):1230-43
37. Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S. et al. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci U S A. 2003;100(9):5473-8
38. Mulet M, Blasco-Ibanez JM, Crespo C, Nacher J, Varea E. Early increased density of cyclooxygenase-2 (COX-2) immunoreactive neurons in Down syndrome. Folia Neuropathol. 2017;55(2):154-60
39. Li W, Cao Y, Xu J, Wang Y, Li W, Wang Q. et al. YAP transcriptionally regulates COX-2 expression and GCCSysm-4 (G-4), a dual YAP/COX-2 inhibitor, overcomes drug resistance in colorectal cancer. J Exp Clin Cancer Res. 2017;36(1):144
40. Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol. 2006;38(10):1654-61
41. Berthou F, Ceppo F, Dumas K, Massa F, Vergoni B, Alemany S. et al. The Tpl2 Kinase Regulates the COX-2/Prostaglandin E2 Axis in Adipocytes in Inflammatory Conditions. Mol Endocrinol. 2015;29(7):1025-36
42. Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN. Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 2002;21(18):4831-40
43. Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A. 1993;90(15):7240-4
44. Liu Y, Borchert GL, Phang JM. Polyoma enhancer activator 3, an ets transcription factor, mediates the induction of cyclooxygenase-2 by nitric oxide in colorectal cancer cells. J Biol Chem. 2004;279(18):18694-700
45. Wu MH, Lin SC, Hsiao KY, Tsai SJ. Hypoxia-inhibited dual-specificity phosphatase-2 expression in endometriotic cells regulates cyclooxygenase-2 expression. J Pathol. 2011;225(3):390-400
46. Wu MH, Wang CA, Lin CC, Chen LC, Chang WC, Tsai SJ. Distinct regulation of cyclooxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J Clin Endocrinol Metab. 2005;90(1):286-95
47. Endale M, Kim TH, Kwak YS, Kim NM, Kim SH, Cho JY. et al. Torilin Inhibits Inflammation by Limiting TAK1-Mediated MAP Kinase and NF-kappaB Activation. Mediators Inflamm. 2017;2017:7250968
48. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249-57
49. Chun KS, Surh YJ. Signal transduction pathways regulating cyclooxygenase-2 expression: potential molecular targets for chemoprevention. Biochem Pharmacol. 2004;68(6):1089-100
50. Wang Q, Zhou Y, Wang X, Evers BM. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene. 2006;25(1):43-50
51. Maia HJ, Maltez A, Studard E, Zausner B, Athayde C, Coutinho E. Effect of the menstrual cycle and oral contraceptives on cyclooxygenase-2 expression in the endometrium. Gynecol Endocrinol. 2005;21(1):57-61
52. Nandakishore R, Yalavarthi PR, Kiran YR, Rajapranathi M. Selective cyclooxygenase inhibitors: current status. Curr Drug Discov Technol. 2014;11(2):127-32
53. Ota H, Igarashi S, Sasaki M, Tanaka T. Distribution of cyclooxygenase-2 in eutopic and ectopic endometrium in endometriosis and adenomyosis. Hum Reprod. 2001;16(3):561-6
54. Cho S, Park SH, Choi YS, Seo SK, Kim HY, Park KH. et al. Expression of cyclooxygenase-2 in eutopic endometrium and ovarian endometriotic tissue in women with severe endometriosis. Gynecol Obstet Invest. 2010;69(2):93-100
55. Mei J, Zhou WJ, Zhu XY, Lu H, Wu K, Yang HL. et al. Suppression of autophagy and HCK signaling promotes PTGS2(high) FCGR3(-) NK cell differentiation triggered by ectopic endometrial stromal cells. Autophagy. 2018;14(8):1376-97
56. Zidan HE, Rezk NA, Alnemr AA, Abd EGA. COX-2 gene promoter DNA methylation status in eutopic and ectopic endometrium of Egyptian women with endometriosis. J Reprod Immunol. 2015;112:63-7
57. Luo MX, Long BB, Li F, Zhang C, Pan MT, Huang YQ. et al. Roles of Cyclooxygenase-2 gene -765G>C (rs20417) and -1195G>A (rs689466) polymorphisms in gastric cancer: A systematic review and meta-analysis. Gene. 2019;685:125-35
58. Barbeiro DF, Koike MK, Coelho AM, Da SF, Machado MC. Intestinal barrier dysfunction and increased COX-2 gene expression in the gut of elderly rats with acute pancreatitis. Pancreatology. 2016;16(1):52-6
59. Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89(16):7384-8
60. Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem. 1995;270(42):24965-71
61. Inoue H, Tanabe T. Transcriptional role of the nuclear factor kappa B site in the induction by lipopolysaccharide and suppression by dexamethasone of cyclooxygenase-2 in U937 cells. Biochem Biophys Res Commun. 1998;244(1):143-8
62. Jing F, Yuping W, Yong C, Jie L, Jun L, Xuanbing T. et al. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumour Biol. 2010;31(4):321-31
63. Li B, Liu W, Wang L, Li M, Wang J, Huang L. et al. CpG island methylator phenotype associated with tumor recurrence in tumor-node-metastasis stage I hepatocellular carcinoma. Ann Surg Oncol. 2010;17(7):1917-26
64. Wang D, Chen Q, Zhang C, Ren F, Li T. DNA hypomethylation of the COX-2 gene promoter is associated with up-regulation of its mRNA expression in eutopic endometrium of endometriosis. Eur J Med Res. 2012;17:12
65. Wang Y, Qu Y, Song W. Genetic variation in COX-2 -1195 and the risk of endometriosis and adenomyosis. Clin Exp Obstet Gynecol. 2015;42(2):168-72
66. Wang H, Sun L, Jiang M, Liu L, Wang G. -1195 A/G promoter variants of the cyclooxygenase-2 gene increases the risk of pain occurrence in endometriotic women. Clin Exp Obstet Gynecol. 2016;43(2):254-7
67. Cavalcanti V, Ponce TG, Mafra FA, Andre GM, Christofolini DM, Barbosa CP. et al. Evaluation of the frequency of G-765C polymorphism in the promoter region of the COX-2 gene and its correlation with the expression of this gene in the endometrium of women with endometriosis. Arch Gynecol Obstet. 2016;293(1):109-15
68. Kim HY, Cho S, Choi YS, Yang HI, Lee KE, Seo SK. et al. Cyclooxygenase-2 ( COX -2) gene-765G/C polymorphism and advanced-stage endometriosis in Korean women. Am J Reprod Immunol. 2012;68(3):238-43
69. Kao LC, Tulac S, Lobo S, Imani B, Yang JP, Germeyer A. et al. Global gene profiling in human endometrium during the window of implantation. Endocrinology. 2002;143(6):2119-38
70. Maia HJ, Haddad C, Coelho G, Casoy J. Role of inflammation and aromatase expression in the eutopic endometrium and its relationship with the development of endometriosis. Womens Health (Lond). 2012;8(6):647-58
71. Maia HJ, Casoy J, Valente FJ. Is aromatase expression in the endometrium the cause of endometriosis and related infertility? Gynecol Endocrinol. 2009;25(4):253-7
72. Tamura M, Sebastian S, Yang S, Gurates B, Fang Z, Bulun SE. Interleukin-1beta elevates cyclooxygenase-2 protein level and enzyme activity via increasing its mRNA stability in human endometrial stromal cells: an effect mediated by extracellularly regulated kinases 1 and 2. J Clin Endocrinol Metab. 2002;87(7):3263-73
73. Huang JC, Liu DY, Yadollahi S, Wu KK, Dawood MY. Interleukin-1 beta induces cyclooxygenase-2 gene expression in cultured endometrial stromal cells. J Clin Endocrinol Metab. 1998;83(2):538-41
74. Huang F, Cao J, Liu Q, Zou Y, Li H, Yin T. MAPK/ERK signal pathway involved expression of COX-2 and VEGF by IL-1beta induced in human endometriosis stromal cells in vitro. Int J Clin Exp Pathol. 2013;6(10):2129-36
75. Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217-81
76. Minnone G, De Benedetti F, Bracci-Laudiero L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int J Mol Sci. 2017:18 (5)
77. Kelleher JH, Tewari D, McMahon SB. Neurotrophic factors and their inhibitors in chronic pain treatment. Neurobiol Dis. 2017;97(Pt B):127-38
78. Wang G, Tokushige N, Markham R, Fraser IS. Rich innervation of deep infiltrating endometriosis. Hum Reprod. 2009;24(4):827-34
79. Kajitani T, Maruyama T, Asada H, Uchida H, Oda H, Uchida S. et al. Possible involvement of nerve growth factor in dysmenorrhea and dyspareunia associated with endometriosis. Endocr J. 2013;60(10):1155-64
80. Peng B, Zhan H, Alotaibi F, Alkusayer GM, Bedaiwy MA, Yong PJ. Nerve Growth Factor Is Associated With Sexual Pain in Women With Endometriosis. Reprod Sci. 2018;25(4):540-9
81. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017;17(12):774-85
82. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399-408
83. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J. et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284(25):16767-75
84. Simoens S, Hummelshoj L, D'Hooghe T. Endometriosis: cost estimates and methodological perspective. Hum Reprod Update. 2007;13(4):395-404
85. Filigheddu N, Gregnanin I, Porporato PE, Surico D, Perego B, Galli L. et al. Differential expression of microRNAs between eutopic and ectopic endometrium in ovarian endometriosis. J Biomed Biotechnol. 2010;2010:369549
86. Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26(22):3203-13
87. Lin SC, Wang CC, Wu MH, Yang SH, Li YH, Tsai SJ. Hypoxia-induced microRNA-20a expression increases ERK phosphorylation and angiogenic gene expression in endometriotic stromal cells. J Clin Endocrinol Metab. 2012;97(8):E1515-23
88. Teague EM, Print CG, Hull ML. The role of microRNAs in endometriosis and associated reproductive conditions. Hum Reprod Update. 2010;16(2):142-65
89. Pan Q, Luo X, Toloubeydokhti T, Chegini N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol Hum Reprod. 2007;13(11):797-806
90. Hsiao KY, Lin SC, Wu MH, Tsai SJ. Pathological functions of hypoxia in endometriosis. Front Biosci (Elite Ed). 2015;7:309-21
91. Smarr MM, Kannan K, Buck LG. Endocrine disrupting chemicals and endometriosis. Fertil Steril. 2016;106(4):959-66
92. Porpora MG, Resta S, Fuggetta E, Storelli P, Megiorni F, Manganaro L. et al. Role of environmental organochlorinated pollutants in the development of endometriosis. Clin Exp Obstet Gynecol. 2013;40(4):565-7
93. Martinez-Zamora MA, Mattioli L, Parera J, Abad E, Coloma JL, van Babel B. et al. Increased levels of dioxin-like substances in adipose tissue in patients with deep infiltrating endometriosis. Hum Reprod. 2015;30(5):1059-68
94. Trabert B, De Roos AJ, Schwartz SM, Peters U, Scholes D, Barr DB. et al. Non-dioxin-like polychlorinated biphenyls and risk of endometriosis. Environ Health Perspect. 2010;118(9):1280-5
95. Huang Q, Chen Y, Chen Q, Zhang H, Lin Y, Zhu M. et al. Dioxin-like rather than non-dioxin-like PCBs promote the development of endometriosis through stimulation of endocrine-inflammation interactions. Arch Toxicol. 2017;91(4):1915-24
96. Guo SW, Simsa P, Kyama CM, Mihalyi A, Fulop V, Othman EE. et al. Reassessing the evidence for the link between dioxin and endometriosis: from molecular biology to clinical epidemiology. Mol Hum Reprod. 2009;15(10):609-24
97. Bulun SE. Endometriosis. N Engl J Med. 2009;360(3):268-79
98. Colette S, Donnez J. Endometriosis. N Engl J Med. 2009;360(18):1911-2 author reply 2
99. Smith AG, Carthew P, Francis JE, Cabral JR, Manson MM. Enhancement by iron of hepatic neoplasia in rats caused by hexachlorobenzene. Carcinogenesis. 1993;14(7):1381-7
100. Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer. 2007;59(2):248-57
101. Haarmann-Stemmann T, Bothe H, Abel J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol. 2009;77(4):508-20
102. Chiappini F, Baston JI, Vaccarezza A, Singla JJ, Pontillo C, Miret N. et al. Enhanced cyclooxygenase-2 expression levels and metalloproteinase 2 and 9 activation by Hexachlorobenzene in human endometrial stromal cells. Biochem Pharmacol. 2016;109:91-104
103. Akyol A, Simsek M, Ilhan R, Can B, Baspinar M, Akyol H. et al. Efficacies of vitamin D and omega-3 polyunsaturated fatty acids on experimental endometriosis. Taiwan J Obstet Gynecol. 2016;55(6):835-9
104. Tomio K, Kawana K, Taguchi A, Isobe Y, Iwamoto R, Yamashita A. et al. Omega-3 polyunsaturated Fatty acids suppress the cystic lesion formation of peritoneal endometriosis in transgenic mouse models. PLoS One. 2013;8(9):e73085
105. Attaman JA, Stanic AK, Kim M, Lynch MP, Rueda BR, Styer AK. The anti-inflammatory impact of omega-3 polyunsaturated Fatty acids during the establishment of endometriosis-like lesions. Am J Reprod Immunol. 2014;72(4):392-402
106. Gravaghi C, La Perle KM, Ogrodwski P, Kang JX, Quimby F, Lipkin M. et al. Cox-2 expression, PGE(2) and cytokines production are inhibited by endogenously synthesized n-3 PUFAs in inflamed colon of fat-1 mice. J Nutr Biochem. 2011;22(4):360-5
107. Malkowski MG, Thuresson ED, Lakkides KM, Rieke CJ, Micielli R, Smith WL. et al. Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J Biol Chem. 2001;276(40):37547-55
108. Palafox D, Llorente L, Alberu J, Torres-Machorro A, Camorlinga N, Rodriguez C. et al. The role of indoleamine 2,3 dioxygenase in the induction of immune tolerance in organ transplantation. Transplant Rev (Orlando). 2010;24(3):160-5
109. Mei J, Li MQ, Ding D, Li DJ, Jin LP, Hu WG. et al. Indoleamine 2,3-dioxygenase-1 (IDO1) enhances survival and invasiveness of endometrial stromal cells via the activation of JNK signaling pathway. Int J Clin Exp Pathol. 2013;6(3):431-44
110. Mei J, Jin LP, Ding D, Li MQ, Li DJ, Zhu XY. Inhibition of IDO1 suppresses cyclooxygenase-2 and matrix metalloproteinase-9 expression and decreases proliferation, adhesion and invasion of endometrial stromal cells. Mol Hum Reprod. 2012;18(10):467-76
111. Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H. et al. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. 2006;139(4):655-62
112. Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B. et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15(11):2760-70
113. Gutkind JS. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci STKE. 2000;2000(40):re1
114. Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids. 2005;73(3-4):141-62
115. Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE. et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58(3):463-87
116. Kumar R, Clerc AC, Gori I, Russell R, Pellegrini C, Govender L. et al. Lipoxin A(4) prevents the progression of de novo and established endometriosis in a mouse model by attenuating prostaglandin E(2) production and estrogen signaling. PLoS One. 2014;9(2):e89742
117. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38(2 Suppl):S26-34
118. Lipinski S, Bremer L, Lammers T, Thieme F, Schreiber S, Rosenstiel P. Coagulation and inflammation. Molecular insights and diagnostic implications. Hamostaseologie. 2011;31(2):94-102 4
119. Ding D, Liu X, Duan J, Guo SW. Platelets are an unindicted culprit in the development of endometriosis: clinical and experimental evidence. Hum Reprod. 2015;30(4):812-32
120. Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d'Etat: an orphan takes control. Endocr Rev. 2011;32(3):404-21
121. Kurihara I, Lee DK, Petit FG, Jeong J, Lee K, Lydon JP. et al. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007;3(6):e102
122. Petit FG, Jamin SP, Kurihara I, Behringer RR, DeMayo FJ, Tsai MJ. et al. Deletion of the orphan nuclear receptor COUP-TFII in uterus leads to placental deficiency. Proc Natl Acad Sci U S A. 2007;104(15):6293-8
123. Lin SC, Li YH, Wu MH, Chang YF, Lee DK, Tsai SY. et al. Suppression of COUP-TFII by proinflammatory cytokines contributes to the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2014;99(3):E427-37
124. Li X, Large MJ, Creighton CJ, Lanz RB, Jeong JW, Young SL. et al. COUP-TFII regulates human endometrial stromal genes involved in inflammation. Mol Endocrinol. 2013;27(12):2041-54
125. Nasu K, Nishida M, Kawano Y, Tsuno A, Abe W, Yuge A. et al. Aberrant expression of apoptosis-related molecules in endometriosis: a possible mechanism underlying the pathogenesis of endometriosis. Reprod Sci. 2011;18(3):206-18
126. Banu SK, Lee J, Speights VJ, Starzinski-Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta-catenin pathways and activation of intrinsic apoptotic mechanisms. Mol Endocrinol. 2009;23(8):1291-305
127. Laschke MW, Elitzsch A, Scheuer C, Vollmar B, Menger MD. Selective cyclo-oxygenase-2 inhibition induces regression of autologous endometrial grafts by down-regulation of vascular endothelial growth factor-mediated angiogenesis and stimulation of caspase-3-dependent apoptosis. Fertil Steril. 2007;87(1):163-71
128. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83(3):493-501
129. DuBois RN, Shao J, Tsujii M, Sheng H, Beauchamp RD. G1 delay in cells overexpressing prostaglandin endoperoxide synthase-2. Cancer Res. 1996;56(4):733-7
130. Lee J, Banu SK, Subbarao T, Starzinski-Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 inhibits invasion of human immortalized endometriotic epithelial and stromal cells through suppression of metalloproteinases. Mol Cell Endocrinol. 2011;332(1-2):306-13
131. Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108(1):25-30
132. Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115-22
133. Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278(37):35451-7
134. Lucidi RS, Witz CA, Chrisco M, Binkley PA, Shain SA, Schenken RS. A novel in vitro model of the early endometriotic lesion demonstrates that attachment of endometrial cells to mesothelial cells is dependent on the source of endometrial cells. Fertil Steril. 2005;84(1):16-21
135. Yao M, Kargman S, Lam EC, Kelly CR, Zheng Y, Luk P. et al. Inhibition of cyclooxygenase-2 by rofecoxib attenuates the growth and metastatic potential of colorectal carcinoma in mice. Cancer Res. 2003;63(3):586-92
136. Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3(3):207-14
137. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221-33
138. Jana S, Chatterjee K, Ray AK, DasMahapatra P, Swarnakar S. Regulation of Matrix Metalloproteinase-2 Activity by COX-2-PGE2-pAKT Axis Promotes Angiogenesis in Endometriosis. PLoS One. 2016;11(10):e0163540
139. Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105(11):1589-94
140. Buerkle MA, Lehrer S, Sohn HY, Conzen P, Pohl U, Krotz F. Selective inhibition of cyclooxygenase-2 enhances platelet adhesion in hamster arterioles in vivo. Circulation. 2004;110(14):2053-9
141. Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J. et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci U S A. 1994;91(6):2046-50
142. Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ. et al. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119(2):287-94
143. Greaves E, Horne AW, Jerina H, Mikolajczak M, Hilferty L, Mitchell R. et al. EP2 receptor antagonism reduces peripheral and central hyperalgesia in a preclinical mouse model of endometriosis. Sci Rep. 2017;7:44169
144. Amaya F, Samad TA, Barrett L, Broom DC, Woolf CJ. Periganglionic inflammation elicits a distally radiating pain hypersensitivity by promoting COX-2 induction in the dorsal root ganglion. Pain. 2009;142(1-2):59-67
145. Berkley KJ, Dmitrieva N, Curtis KS, Papka RE. Innervation of ectopic endometrium in a rat model of endometriosis. Proc Natl Acad Sci U S A. 2004;101(30):11094-8
146. Rush AM, Waxman SG. PGE2 increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res. 2004;1023(2):264-71
147. Lin CR, Amaya F, Barrett L, Wang H, Takada J, Samad TA. et al. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319(3):1096-103
148. Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002;8(8):390-6
149. Koninckx PR, Ussia A, Adamyan L, Keckstein J, Wattiez A. Primary Dysmenorrhea. J Obstet Gynaecol Can. 2017;39(7):578-9
150. Yuan XL, Chen L, Li MX, Dong P, Xue J, Wang J. et al. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin Immunol. 2010;134(3):277-88
151. Soontornchaiboon W, Joo SS, Kim SM. Anti-inflammatory effects of violaxanthin isolated from microalga Chlorella ellipsoidea in RAW 264.7 macrophages. Biol Pharm Bull. 2012;35(7):1137-44
152. Haidari F, Homayouni F, Helli B, Haghighizadeh MH, Farahmandpour F. Effect of chlorella supplementation on systematic symptoms and serum levels of prostaglandins, inflammatory and oxidative markers in women with primary dysmenorrhea. Eur J Obstet Gynecol Reprod Biol. 2018;229:185-9
153. Holoch KJ, Lessey BA. Endometriosis and infertility. Clin Obstet Gynecol. 2010;53(2):429-38
154. Cretoiu SM, Cretoiu D, Suciu L, Popescu LM. Interstitial Cajal-like cells of human Fallopian tube express estrogen and progesterone receptors. J Mol Histol. 2009;40(5-6):387-94
155. Yang XJ, Xu JY, Shen ZJ, Zhao J. Immunohistochemical alterations of cajal-like type of tubal interstitial cells in women with endometriosis and tubal ectopic pregnancy. Arch Gynecol Obstet. 2013;288(6):1295-300
156. Yang J, Chi C, Liu Z, Yang G, Shen ZJ, Yang XJ. Ultrastructure damage of oviduct telocytes in rat model of acute salpingitis. J Cell Mol Med. 2015;19(7):1720-8
157. Yang XJ, Yang J, Liu Z, Yang G, Shen ZJ. Telocytes damage in endometriosis-affected rat oviduct and potential impact on fertility. J Cell Mol Med. 2015;19(2):452-62
158. Barcelos ID, Vieira RC, Ferreira EM, Martins WP, Ferriani RA, Navarro PA. Comparative analysis of the spindle and chromosome configurations of in vitro-matured oocytes from patients with endometriosis and from control subjects: a pilot study. Fertil Steril. 2009;92(5):1749-52
159. Barcelos ID, Donabella FC, Ribas CP, Meola J, Ferriani RA, de Paz CC. et al. Down-regulation of the CYP19A1 gene in cumulus cells of infertile women with endometriosis. Reprod Biomed Online. 2015;30(5):532-41
160. Donabela FC, Meola J, Padovan CC, de Paz CC, Navarro PA. Higher SOD1 Gene Expression in Cumulus Cells From Infertile Women With Moderate and Severe Endometriosis. Reprod Sci. 2015;22(11):1452-60
161. Chen Q, Zhang C, Chen Y, Lou J, Wang D. Identification of endometriosis-related genes by representational difference analysis of cDNA. Aust N Z J Obstet Gynaecol. 2012;52(2):140-5
162. Rakhila H, Carli C, Daris M, Lemyre M, Leboeuf M, Akoum A. Identification of multiple and distinct defects in prostaglandin biosynthetic pathways in eutopic and ectopic endometrium of women with endometriosis. Fertil Steril. 2013;100(6):1650-9.e1 -2
163. Da LC, Da BM, Donabela FC, Paro DPC, Meola J, Navarro PA. PTGS2 down-regulation in cumulus cells of infertile women with endometriosis. Reprod Biomed Online. 2017;35(4):379-86
164. Wei C, Mei J, Tang L, Liu Y, Li D, Li M. et al. 1-Methyl-tryptophan attenuates regulatory T cells differentiation due to the inhibition of estrogen-IDO1-MRC2 axis in endometriosis. Cell Death Dis. 2016;7(12):e2489
165. Li MQ, Wang Y, Chang KK, Meng YH, Liu LB, Mei J. et al. CD4+Foxp3+ regulatory T cell differentiation mediated by endometrial stromal cell-derived TECK promotes the growth and invasion of endometriotic lesions. Cell Death Dis. 2014;5:e1436
166. Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209-26
167. Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177(1):246-54
168. Cobellis L, Razzi S, De Simone S, Sartini A, Fava A, Danero S. et al. The treatment with a COX-2 specific inhibitor is effective in the management of pain related to endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;116(1):100-2
169. Nap AW, Griffioen AW, Dunselman GA, Bouma-Ter SJ, Thijssen VL, Evers JL. et al. Antiangiogenesis therapy for endometriosis. J Clin Endocrinol Metab. 2004;89(3):1089-95
170. Machado DE, Berardo PT, Landgraf RG, Fernandes PD, Palmero C, Alves LM. et al. A selective cyclooxygenase-2 inhibitor suppresses the growth of endometriosis with an antiangiogenic effect in a rat model. Fertil Steril. 2010;93(8):2674-9
171. Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M. et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59(2):455-67
172. Liang Y, Xie H, Wu J, Liu D, Yao S. Villainous role of estrogen in macrophage-nerve interaction in endometriosis. Reprod Biol Endocrinol. 2018;16(1):122
173. Kilico I, Kokcu A, Kefeli M, Kandemir B. Regression of experimentally induced endometriosis with a new selective cyclooxygenase-2 enzyme inhibitor. Gynecol Obstet Invest. 2014;77(1):35-9
174. Nenicu A, Gu Y, Korbel C, Menger MD, Laschke MW. Combination therapy with telmisartan and parecoxib induces regression of endometriotic lesions. Br J Pharmacol. 2017;174(16):2623-35
175. Millar RP. GnRH II and type II GnRH receptors. Trends Endocrinol Metab. 2003;14(1):35-43
176. Bjarnason I, Scarpignato C, Holmgren E, Olszewski M, Rainsford KD, Lanas A. Mechanisms of Damage to the Gastrointestinal Tract From Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology. 2018;154(3):500-14
177. Morimoto C, Osuga Y, Yano T, Takemura Y, Harada M, Hirata T. et al. GnRH II as a possible cytostatic regulator in the development of endometriosis. Hum Reprod. 2005;20(11):3212-8
178. Huang F, Wang H, Zou Y, Liu Q, Cao J, Yin T. Effect of GnRH-II on the ESC proliferation, apoptosis and VEGF secretion in patients with endometriosis in vitro. Int J Clin Exp Pathol. 2013;6(11):2487-96
179. Ichioka M, Mita S, Shimizu Y, Imada K, Kiyono T, Bono Y. et al. Dienogest, a synthetic progestin, down-regulates expression of CYP19A1 and inflammatory and neuroangiogenesis factors through progesterone receptor isoforms A and B in endometriotic cells. J Steroid Biochem Mol Biol. 2015;147:103-10
180. Grimm SL, Hartig SM, Edwards DP. Progesterone Receptor Signaling Mechanisms. J Mol Biol. 2016;428(19):3831-49
181. Grandi G, Mueller M, Bersinger NA, Cagnacci A, Volpe A, McKinnon B. Does dienogest influence the inflammatory response of endometriotic cells? A systematic review. Inflamm Res. 2016;65(3):183-92
182. Wang XR, Hao HG, Chu L. Glycyrrhizin inhibits LPS-induced inflammatory mediator production in endometrial epithelial cells. Microb Pathog. 2017;109:110-3
183. Namavar JB, Farrokhnia F, Tanideh N, Vijayananda KP, Parsanezhad ME, Alaee S. Comparing The Effects of Glycyrrhiza glabra Root Extract, A Cyclooxygenase-2 Inhibitor (Celecoxib) and A Gonadotropin-Releasing Hormone Analog (Diphereline) in A Rat Model of Endometriosis. Int J Fertil Steril. 2019;13(1):45-50
184. Atteritano M, Marini H, Minutoli L, Polito F, Bitto A, Altavilla D. et al. Effects of the phytoestrogen genistein on some predictors of cardiovascular risk in osteopenic, postmenopausal women: a two-year randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2007;92(8):3068-75
185. Yu J, Zhao L, Zhang D, Zhai D, Shen W, Bai L. et al. The effects and possible mechanisms of puerarin to treat endometriosis model rats. Evid Based Complement Alternat Med. 2015;2015:269138
Author contact
![]() Corresponding author: Ming-Qing Li, Email: mqliedu.cn; NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Fudan University Shanghai Medical College, Shanghai 200080, People's Republic of China. Tel/Fax: +86 21 33189900.
Corresponding author: Ming-Qing Li, Email: mqliedu.cn; NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Fudan University Shanghai Medical College, Shanghai 200080, People's Republic of China. Tel/Fax: +86 21 33189900.