Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. H2S plays...
3. H2S influences...
4. H2S influences...
5. H2S influences...
6. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(2):264-271. doi:10.7150/ijbs.38143 This issue Cite
Review
Hydrogen Sulfide Plays an Important Protective Role through Influencing Endoplasmic Reticulum Stress in Diseases
Honggang Wang1 ![]() , Xingzhuo Shi2, Mengyuan Qiu1, Shuangyu Lv1, Huiyang Liu1
, Xingzhuo Shi2, Mengyuan Qiu1, Shuangyu Lv1, Huiyang Liu1
1. Institute of Biomedical Informatics, Bioinformatics Center, School of Basic Medical Sciences, Henan University, Kaifeng, Henan, 475000, China.
2. School of Life Science, Henan University, Kaifeng, Henan, 475000, China.
Received 2019-7-4; Accepted 2019-9-10; Published 2020-1-1
Abstract

The endoplasmic reticulum is an important organelle responsible for protein synthesis, modification, folding, assembly and transport of new peptide chains. When the endoplasmic reticulum protein folding ability is impaired, the unfolded or misfolded proteins accumulate to lead to endoplasmic reticulum stress. Hydrogen sulfide is an important signaling molecule that regulates many physiological and pathological processes. Recent studies indicate that H2S plays an important protective role in many diseases through influencing endoplasmic reticulum stress, but its mechanism is not fully understood. This article reviewed the progress about the effect of H2S on endoplasmic reticulum stress and its mechanisms involved in diseases in recent years to provide theoretical basis for in-depth study.
Keywords: Hydrogen sulfide, endoplasmic reticulum stress, cardiomyopathy, neurological diseases, respiratory diseases, vascular diseases
1. Introduction
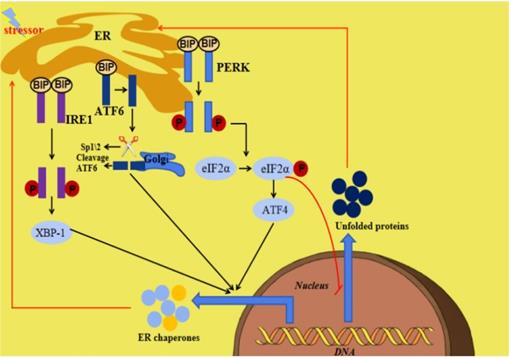
The endoplasmic reticulum (ER) is an important organelle responsible for protein synthesis, modification, folding, assembly and transport of new peptide chains [1-5]. In addition, it regulates the cholesterol and lipid-membrane biosynthesis and the signaling mechanisms of cell surviving and death [6, 7]. Under stress conditions including glucose deficiency, environmental toxins, viral infection, changes in Ca2+ levels, hypoxia, inflammation and oxidative stress, ER homeostasis can be interrupted, which is termed ER stress (ERS). ERS is defined as the disturbance of ER function, which interferes with protein folding, post-translational modification and secretion. Finally, the accumulation of unfolded proteins in ER initiates a homeostatic signaling network called as the unfolded proteins reaction (UPR) [8, 9]. When the perturbation is moderate, UPR activation will promote a homeostatic recovery of ER and help cells adapt to changes. However, if the interference is intense and prolonged, ERS and UPR will initiate the death signaling pathway, which will lead to the onset of various diseases [10]. The ERS and UPR are mediated by three transmembrane ER signaling proteins: pancreatic endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6), which mediate three parallel signal branches respectively [11, 12]. Under non-pressure conditions, the binding immunoglobulin (BIP) binds to PERK, IRE1 and ATF6 to stabilize and prevent their activation. The stressors and unfolded proteins promote the isolation of BIP from PERK, IRE1 and ATF6, thereby activating these three molecules. Subsequently, the autophosphorylated PERK phosphorylates eIF2a to inhibit mRNA translation and global protein synthesis, and increase ATF4 expression, the activated IRE1 cleaves Xbp1 mRNA and the isolated ATF6 is cleaved by 1-site protease (sp1) and 2-site protease (sp2) proteins in Golgi complex. At last, the cleaved Xbp1, the ATF4 and the spliced ATF6 promote the expression of ER chaperone genes, which are further involved in eliminating unfolded proteins and restoring homeostasis in normal cells (Figure 1) [10]. Many diseases have been reported to be related with ERS [13, 14].
Summary of ERS and the UPR. When ERS is activated, there are three parallel signaling branches in UPR. ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; BIP, binding immunoglobulin protein; ER, endoplasmic reticulum; ERS, endoplasmic reticulum stress; IRE1,inositol-requiring protein 1; PERK, PRKR-like ER kinase; SP1, site-1 protease; UPR, unfolded protein response; XBP1, X-box binding protein 1.
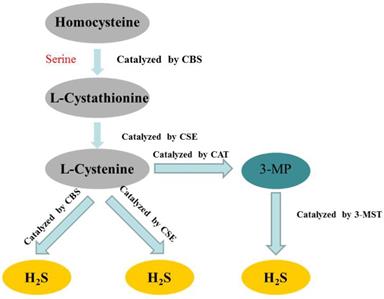
Hydrogen sulfide (H2S) has long been considered as a flammable, water-soluble, colorless and toxic gas. However, since the 1990s, more and more studies have confirmed that H2S belonged to a class of gasotransmitters, together with nitric oxide (NO) and carbon monoxide (CO) [15-17]. In mammalian cells, H2S is produced by endogenous enzymatic and non-enzymatic pathways. The enzymatic generation of H2S, which may be important for the regulation in given cells under special conditions, is the focus of the research. Several different mammalian enzymatic systems for H2S production have been described in detail. Most commonly, three typical H2S-producing enzymes are identified: cystathionine-gamma-lyase (CSE), cystathionine-beta-synthase (CBS) and 3-mercaptopyruvate thiotransferase (3-MST) [18-20]. Cystathionine is produced by β-substitution reaction of homocysteine with serine catalyzed by CBS. CSE catalyzes the elimination of α, γ-cysteine of cystathionine to produce cystenine. Under the catalysis of CBS and CSE, cysteine can form H2S through β elimination reaction. 3-mercaptopyruvate (3-MP) is produced by transferring amines from cystine to α-ketoglutarate via cysteine aminotransferase (CAT). 3-MST catalyzes the sulphur of 3-MP to convert into H2S [21] (Figure 2). The biological function of H2S does not depend on H2S itself, but on the formation of new molecules, such as S-nitrosothiols, whose possible mechanisms include reversible protein sulfidation [22]. H2S has many physiological functions, such as relaxing blood vessels, lowering blood pressure [23, 24], anti-apoptotic [25], anti-inflammatory [26], anti-oxidative stress and regulation of ER stress [27]. The role of H2S in the regulation of ERS has been one of the focuses of attention in recent years [28].
Summary of the production process of endogenous H2S. CBS: cystathionine-beta-synthase; CSE: cystathionine-gamma-lyase; 3-MST: 3-mercaptopyruvate thiotransferase; 3-MP: 3-mercaptopyruvate; CAT: cysteine aminotransferase.
In this review, we summarize the progress about the effects of H2S on ERS and the mechanism involved in recent years to provide ideas for relevant basic research in the future.
2. H2S plays cardioprotective role by influencing endoplasmic reticulum stress
Diabetic cardiomyopathy (DCM) is one of the major cardiac complications independent of coronary artery disease and hypertension in diabetic patients [29]. In recent years, many studies have shown that ERS plays a crucial role in the occurrence and development of DCM [30, 31]. Hyperglycemia induces cardiomyocyte apoptosis by activating ERS through caspase-12 dependent pathway and C/EBP-homologous protein (CHOP) dependent pathway [32-34]. Fang Li and her coworkers reported that in streptozotocin (STZ)-induced diabetic rats, ERS was increased by hyperglycemia, leading to myocardial fibrosis and cardiomyocyte apoptosis. While treatment with H2S reduced ERS to inhibit myocardial apoptosis and improve myocardial fibrosis, suggesting that H2S had a potential role in the treatment of DCM [35]. In this experiment, since the intervention of H2S is simultaneous with the establishment of DC model, not after the establishment of DC model, thus, the protective effect of H2S on DC cannot be fully demonstrated. Whether H2S regulated ERS to play a protective effect on DC remained to be studied. Moreover, long-term hyperglycemia can cause excessive production of reactive oxygen species (ROS) in mitochondria of cardiomyocyte [36, 37] and excessive ROS induces ERS which leads to cardiomyocyte apoptosis [38]. H2S can inhibit the production of ROS, indicating that H2S can regulate ERS through ROS. ER and mitochondria are spatially close organelles which are joined together by ER-mitochondrial associated membranes [39]. ROS regulate ER-mitochondrial crosstalk during ERS-induced apoptosis [40]. In streptozotocin (STZ)-induced diabetic rats, H2S reduces ROS in mitochondria and ERS-induced myocardial apoptosis through regulating ER-mitochondrial crosstalk [41]. ROS associates the antioxidant effect of H2S with its inhibition of ERS. Researches showed that the excessive lipid deposition and ERS might play a role in the pathogenesis of DCM [42-44]. In the hearts of STZ-induced rats or in AC16 cardiac cells treated with palmitic acid (PA), endogenous H2S decreased, ERS, apoptosis and lipid accumulation increased, suggesting that endogenous H2S, ERS and lipotoxicity are involved in the pathological process of DCM. The further experiment showed that exogenous H2S alleviated myocardial lipotoxicity and ER stress. The similar results can be obtained by using ERS inhibitors (4-PBA), suggesting that exogenous H2S inhibits lipid accumulation and myocardial toxicity through suppressing ERS [45]. This is consistent with previous reports that H2S mitigated high fat diet-induced cardiac dysfunction through suppression of ERS [46]. The mechanism of the effect of ERS on myocardial lipotoxicity of DCM remains to be studied. Myocardial ischemia reperfusion (I/R) injury is an important cause of myocardial injury [47]. Recently, it has been proved that ERS is related to myocardial I/R injury [48]. Myocardial I/R decreased endogenous H2S, increased ERS and ERS-induced cardiomyocyte apoptosis. H2S preconditioning could reverse these above changes. Moreover, pretreatment with ERS inhibitors yielded similar results as H2S. Collectively, these results indicated that H2S ameliorated myocardial I/R injury by attenuating excessive ERS induced by myocardial I/R [49].This added a new mechanism, which remains to be studied, to the myocardial protection of H2S. Several studies suggest that chronic intermittent hypoxia (CIH) may induce ER stress and lead to myocardial injury [50-52]. Zhou, et al. reported that CIH induced myocardial injury by activating ERS, while the treatment with the inhibitor of cystathionine γ-lyase (DL-propargylglycine, PAG) alleviated myocardial injury induced by CIH [53]. This result is inconsistent with previous study that H2S could alleviate myocardial injury in ischemia-reperfusion model [54]. The underlying mechanisms of the contradiction remain to be studied. It has been reported that post-conditioning (PC) inhibits apoptosis induced by I/R, however, its myocardial protection is lost in the elderly heart [55, 56]. Sun et al. reported that H2S restored the cardioprotective effect of PC and reduced I/R-induced ERS, the similar effects were obtained by using 4-PBA, which indicated that exogenous H2S restores PC-induced cardioprotection by inhibition of ERS in the aged cardiomyocytes [57] (Table 1). Although there are many studies about the protective effect of H2S on myocardium by influencing ER stress, the exact mechanism is not fully understood. Further researches are needed to provide a new way for the treatment of myocardial injury.
H2S playes cardioprotective role by influencing endoplasmic reticulum stress (ERS)
| Experimental models | Effects | Proposed mechanisms | References |
|---|---|---|---|
| Streptozotocin(STZ)-induced diabetic rats | Intraperitoneally administation of NaHS at 30 µmol/kg or 100 µmol/kg for 8 weeks could improve myocardial hypertrophy and myocardial collagen deposition in hearts of diabetic rats | Suppressing STZ‑induced ERS | [35] |
| AC16 cardiac cells treated with palmitic acid(PA) | Pretreatment AC16 cells with 100 μmol/L of NaHS could suppress the PA-induced myocardial injury | Suppressing PA‑induced ERS | [45] |
| Murine model of high fat diet (HFD)-induced cardiomyopathy | H2S therapy mitigated HFD-induced cardiac dysfunction | Suppressing cardiac ERS induced by HFD feeding | [46] |
| Model of hypoxia/reoxygenation in rat H9c2 cardiac myocytes. | H2S preconditioning significantly reduced myocardial infarct size, preserved left ventricular function, and inhibited I/R-induced cardiomyocyte apoptosis in vivo. | Attenuating I/R-induced ERS | [49] |
| Chronic intermittent hypoxia (CIH) model in rats | Inhibiting the production of endogenous H2S by PAG alleviated myocardial injury induced by CIH. | Reducing ERS induced by CIH | [53] |
| H2O2-induced H9C2 cells senescence model | Exogenous H2S restores PC-induced cardioprotection | Inhibition of ERS via down-regulating PERK-eIF 2α-ATF4, IRE 1α-XBP-1 and ATF 6 pathways | [57] |
PAG:DL-propargylglycine; I/R:ischemia reperfusion; PC:post-conditioning; PERK:pancreatic endoplasmic reticulum kinase; IRE1α:inositol-requiring enzyme1α; ATF6(4): activating transcription factor 6(4); XBP-1:X-box binding protein 1; eIF2α:Eukaryotic initiation factor 2α.
3. H2S influences endoplasmic reticulum stress in neurological diseases
In recent years, there have been many reports that H2S regulates ERS to inhibit neurological diseases. Homocysteine (Hcy), produced by demethylation of methionine [58], can induce ERS to lead the apoptosis of many types of neurons, including hippocampal and cortical neurons [59]. The research by Li et al. demonstrated that intraventricular injection of Hcy impaired learning and memory function, reduced the production of endogenous H2S and increased the ERS of hippocampal cells, which suggested that Hcy-induced learning and memory loss was associated with reduced endogenous H2S production and increased hippocampal ERS [60]. Similarly, the neurotoxicity to PC12 cells induced by arecoline is also involved with reduced endogenous H2S production and increased hippocampal ERS [61]. Wei et al. reported that H2S downregulated Hcy-induced neuronal ERS and upregulated the expression of brain-derived neurotrophic factor (BDNF) in the hippocampus of rats. In addition, blocking BDNF-TrkB pathway by inhibitor could reverse the abovementioned effect of H2S. Overall, these findings suggested that H2S alleviated Hcy-induced neurotoxicity through reducing ERS by upregulating the BDNF-TrkB pathway [62]. In PC12 cells, H2S also markedly inhibited homocysteine‑induced ERS and increased the protein level of silent mating type information regulation 2 homolog 1 (SIRT‑1) in the presence or absence of homocysteine treatment. Sirtinol, an inhibitor of SIRT‑1, eliminated the inhibitory effect of H2S on homocysteine‑induced ERS, which indicated that H2S protected PC12 cells against homocysteine‑induced ERS by upregulating SIRT‑1 [63]. The similar results were obtained in vivo [64] Increasing evidences suggest that diabetes can cause cognitive impairment and memory loss [65, 66]. ERS-induced apoptosis in the hippocampus is the mechanism of diabetic cognitive impairment [67]. Wei et al. reported that H2S improved cognitive impairment in diabetes mellitus by inhibiting ERS induced by hyperglycemia in hippocampus. Furthermore, the hippocampal endogenous H2S generation of diabetic rats was decreased, while this downregulation is reversed by exogenous H2S [68]. These results suggested that the neuroprotective effect of H2S might be related to its promotion of endogenous H2S production in hippocampal cells. However, it has been reported that the endogenous H2S production in pancreas and liver of STZ-induced diabetic rats increased significantly [69]. This conflict is probably due to that the concentration of endogenous H2S is diverse in different tissue. The mechanisms of H2S regulating ERS to protect nerve injury need to be further studied. ERS will be a new target of treatment for neurological diseases.
Depression is a chronic and recurrent serious mental disorder characterized by loss of happiness, emotional disorders and suicidal tendencies. It affects more than 10% of the world's population and causes a huge social burden [70, 71]. It has been reported that rats exposed to chronic unpredictable mild stress (CUMS) exhibited many behavioral and neurobiological changes in depression [72]. The research demonstrated that CUMS induced depression-like behavior, caused hippocampal ERS in rats and suppressed the production of endogenous H2S, while exogenous H2S alleviated the above depression-like behavior suggesting that H2S production disorder and ER stress in hippocampus played an important role in depressive behavior induced by CUMS [73]. The further studies showed that exogenous H2S attenuated CUMS-induced depression-like behaviors by suppressing hippocampal ERS and increased the SIRT‑1 expression in rats. Moreover, the inhibition of SIRT‑1 by inhibitor reversed the protective effect of H2S and promoted CUMS-induced ERS. Collectively, these indicated that H2S inhibited CUMS-induced depressive-like behavior through suppressing CUMS-induced ERS by upregulating SIRT‑1 pathway [74]. H2S also exerts its protection against the neurotoxicity of formaldehyde through overcoming ERS via upregulation of SIRT-1 [75]. In addition to the SIRT‑1 pathway, the BDNF/TrkB pathway is also related with the antidepressant effect of H2S. BDNF is an important endogenous neurotrophic factor, mainly expressed in hippocampus and cortex [76]. H2S mitigates CUMS-induced depressive-like behaviors, induces the expressions of BDNF and p-TrkB proteins and inhibits ERS in the hippocampus of CUMS-induced rats [77]. The inhibition of BDNF-TrkB pathway with K252a, an inhibitor of BDNF, reverses the protective role of H2S in CUMS-induced depressive-like behaviors and hippocampal ERS, which indicates that H2S plays an antidepressant-like effect through suppressing ERS via BDNF-TrkB pathway in CUMS-exposed rats [78] (Table 2). At present, the existing treatment of depression is often ineffective and cannot completely solve the symptoms. With the in-depth study of the mechanism of H2S antidepressant effect, the new H2S-related drugs will be provided for the treatment of depression. ERS will also become a new target for the treatment of depression.
H2S influences endoplasmic reticulum stress (ERS) in neurological diseases
| Experimental models | Effects | Proposed mechanisms | References |
|---|---|---|---|
| Adult male Sprague-Dawley rats were intracerebroventricularly injected with Hcy | H2S alleviated Hcy-induced neurotoxicity | Inhibiting ERS by upregulating the BDNF -TrkB pathway | [62] |
| Homocysteine-treated PC12 cells | Exogenous H2S significantly attenuated the homocysteine‑induced ERS response in hippocampal. | Inhibiting homocysteine‑induced ERS by upregulating SIRT‑1 | [63] |
| Streptozotocin-induced diabetic rats | H2S improved cognitive impairment in diabetes mellitus | Suppressing hippocampal endoplasmic reticulum stress induced by hyperglycemia | [68] |
| Rat model of chronic unpredictable mild stress | H2S inhibited CUMS-induced depressive-like behavior. | Suppressing CUMS-induced ERS by upregulating SIRT‑1 pathway | [74] |
| Formaldehyde (FA)-induced PC12 cells | H2S exerts its protection against the neurotoxicity of FA. | Through overcoming ERS via upregulating SIRT‑1 pathway | [75] |
| Rat model of chronic unpredictable mild stress | H2S inhibited CUMS-induced depressive-like behavior. | Suppressing ERS via BDNF-TrkB pathway. | [77,78] |
BDNF: brain-derived neurotrophic factor; TrkB: tyrosine protein kinase B; SIRT-1: silent mating type information regulation 2 homolog 1; CUMS: chronic unpredictable mild stress.
4. H2S influences endoplasmic reticulum stress in respiratory diseases
Chronic obstructive pulmonary disease (COPD) can be defined as a disease characterized by exposure to harmful substances, leading to irreversible airflow restriction and shortness of breath [79, 80]. Evidences suggest that ERS may play an important role in the development or pathology of COPD [81, 82]. Cigarette smoke (CS) induces ERS and ERS-mediated apoptosis and suppresses the production of endogenous H2S to lead COPD, which is reversed by exogenous H2S [83]. Intraperitoneal injection of endogenous H2S inhibitor in rat model of passive inhalation of CS aggravates these effects caused by CS; however, the ERS inhibitor suppresses CS-induced effects, which suggests that H2S may inhibit CS-induced bronchial epithelial cell apoptosis through suppressing ERS [84]. Artery endothelial dysfunction induced by apoptosis of arterial endothelial cells is associated with the severity of COPD [85]. Exogenous H2S reduces the apoptosis of pulmonary artery endothelial cells by suppressing ERS in a rat model of COPD [86]. The specific signaling pathways involved in the above effect need to be further studied. The decrease of ERS and endogenous H2S are involved in the pathogenesis of acute lung injury (ALI). Exogenous H2S can play protectice role during the early stage of ALI by increasing ERS, which is contrary to the former statement [87]. The reason needs to be studied (Table 3).
H2S influences endoplasmic reticulum stress (ERS) in respiratory diseases and vascular diseases
| Experimental models | Effects | Proposed mechanisms | References |
|---|---|---|---|
| Sprague-Dawley rats exposed to cigarette smoke (CS) generated from 20 commercial unfiltered cigarettes for 4 h/day, 7 days/week for 4 months | NaHS significantly inhibited CS-induced bronchial epithelial cell apoptosis in rat lungs | Inhibiting ERS | [84] |
| Rat model of chronic obstructive pulmonary disease established by means of passive smoke exposure and intratracheal injection with lipopolysaccharide (LPS) | Exogenous H2S reduced the apoptosis of pulmonary artery endothelial cells | Suppressing ERS | [86] |
| Rats with acute lung injury (ALI) induced by oleic acid (OA) | H2S could promote alveolar epithelial cell endoplasmic reticulum stress in rats with ALI. | [87] | |
| Vitamin D3 plus nicotine (VDN) model of rats | H2S alleviated vascular calcification (VC) and phenotype transformation of vascular smooth muscle cells. | Inhibiting ERS via activation of the Akt signaling pathway | [89] |
| 10-6 M AngII treated human umbilical vein endothelial cells (HUVECs) | H2S protected human umbilical vein endothelial cells (HUVECs) against AngII‑stimulated ET‑1 generation and subsequent cytotoxicity‑induced endoplasmic reticulum stress | Via inhibiting NF-кB signaling pathway | [93] |
Akt:serine threonine kinase; NF-кB:nuclear factor kappa-B
5. H2S influences endoplasmic reticulum stress in vascular disease
Vascular calcification (VC) refers to the abnormal deposition of calcium and phosphorus minerals on the wall of blood vessels; ERS-induced apoptosis plays a vital role in the development of VC [88], so inhibiting apoptosis is an effective treatment for VC. Yang et al. reported that H2S could inhibit VC and ERS of calcified aortic tissues. Furthermore, the ERS inducer Tm could block the ameliorated effect of H2S on VC, while the effect of the ER stress inhibitor PBA on VC in rats treated with vitamin D3 plus nicotine was similar as that of H2S.These indicated that H2S ameliorated VC by suppressing ERS. Moreover, the protein levels of phosphorylated AKT and Akt were both upregulated by H2S, suggesting that activation of the Akt signaling pathway is involved with the above effect of H2S [89]. With the development of research, it will provide a new strategy and target for the prevention and treatment of VC. Endothelial dysfunction (ED) caused by inflammation is very important in the development of atherosclerosis (AS). Angiotensin II (AngII) is involved in the progression of ED, leading to atherosclerosis [90]. There is evidence that ERS and ED are the key factors leading to AngII-induced cytotoxicity [91, 92]. The results of Hu et al. revealed that AngII markedly induced cytotoxicity by promoting ERS and ED in human umbilical vein endothelial cells (HUVECs), which are reversed by H2S supplementation via inhibiting NF-кB signaling pathway [93] (Table 3). Similar results were obtained in other studies [94, 95]. Whether ERS can directly induce ED and the molecular mechanism of interaction between ER stress and ED need to be studied. With the deepening of the research, it will certainly provide a new prevention and treatment of AS.
6. Conclusion
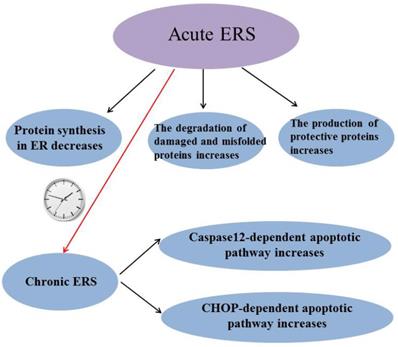
ERS has been reported to be involved in many diseases and it is a “double-edged sword”: Acute ERS can reduce protein synthesis in ER, increase degradation of damaged and misfolded proteins and induce the production of protective proteins to alleviate stress-induced injury, while chronic ERS can induce caspase-12 dependent apoptotic pathway and C/EBP-homologous protein (CHOP) dependent apoptotic pathway to lead some diseases (Figure 3). So it is very important to study how to maintain ERS at an appropriate level. Although the mechanism of how prolonged ERS leads to disease is not fully understood, it is now clear that abnormal ERS can cause disease by inducing excessive reactive oxygen species (ROS).The suppression of excessive ERS will provide a way to prevent and treat many diseases. H2S has been shown to play a protective role in many diseases by inhibiting ERS, but individual research has reported that H2S inhibits diseases by promoting ERS; perhaps the reason is that the basic level of ERS varies in different tissues or different diseases have different effects on ERS. The mechanism of H2S regulating ERS in diseases needs further study. In conclusion, ERS may be a potential target for H2S therapy with the in-depth study of the effect of H2S on ERS.
Summary of the role of endoplasmic reticulum stress. ERS: endoplasmic reticulum stress; CHOP:C/EBP-homologous protein.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant number 81600974), the key scientific research project of colleges and universities in Henan province, China (Grant number 16A310001) and Innovation and Entrepreneurship Training Program for Henan University Students in 2019 (Grant number 2019101907).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460-70
2. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. 2016;57:1329-38
3. Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203-17
4. Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1-13
5. King AL, Lefer DJ. Cytoprotective actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp Physiol. 2011;96:840-6
6. Yang F, Luo J. Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules. 2015;5:2538-53
7. Krebs J, Groenendyk J, Michalak M. Ca2+-signaling, alternative splicing and endoplasmic reticulum stress responses. Neurochem Res. 2011;36:1198-211
8. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581-97
9. Lan B, He Y, Sun H, Zheng X, Gao Y, Li N. The roles of mitochondria-associated membranes in mitochondrial quality control under endoplasmic reticulum stress. Life Sci. 2019;231:116587
10. Ji T, Han Y, Yang W, Xu B, Sun M, Jiang S. et al. Endoplasmic reticulum stress and NLRP3 inflammasome: Crosstalk in cardiovascular and metabolic disorders. J Cell Physiol. 2019
11. Ariyasu D, Yoshida H, Hasegawa Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. Int J Mol Sci. 2017:18
12. Fernandez A, Ordonez R, Reiter RJ, Gonzalez-Gallego J, Mauriz JL. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res. 2015;59:292-307
13. Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM. et al. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. 2016;124:687-702
14. Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. 2018;128:64-73
15. Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169-87
16. Olson KR. A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal. 2012;17:32-44
17. Kolluru GK, Shen X, Bir SC, Kevil CG. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide. 2013;35:5-20
18. Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791-896
19. Szabo C, Papapetropoulos A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol Rev. 2017;69:497-564
20. Rose P, Moore PK, Zhu YZ. H2S biosynthesis and catabolism: new insights from molecular studies. Cell Mol Life Sci. 2017;74:1391-412
21. Wang J, Wu D, Wang H. Hydrogen sulfide plays an important protective role by influencing autophagy in diseases. Physiol Res. 2019;68:335-45
22. Dongo E, Beliczai-Marosi G, Dybvig AS, Kiss L. The mechanism of action and role of hydrogen sulfide in the control of vascular tone. Nitric Oxide. 2018;81:75-87
23. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K. et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587-90
24. Sun Y, Huang Y, Zhang R, Chen Q, Chen J, Zong Y. et al. Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J Mol Med (Berl). 2015;93:439-55
25. Guo C, Liang F, Shah Masood W, Yan X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-kappaB dependent anti-inflammation pathway. Eur J Pharmacol. 2014;725:70-8
26. Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M. et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J Biol Chem. 2014;289:9741-53
27. Zheng J, Zhao T, Yuan Y, Hu N, Tang X. Hydrogen sulfide (H2S) attenuates uranium-induced acute nephrotoxicity through oxidative stress and inflammatory response via Nrf2-NF-kappaB pathways. Chem Biol Interact. 2015;242:353-62
28. Panthi S, Chung HJ, Jung J, Jeong NY. Physiological Importance of Hydrogen Sulfide: Emerging Potent Neuroprotector and Neuromodulator. Oxid Med Cell Longev. 2016;2016:9049782
29. Joshi M, Kotha SR, Malireddy S, Selvaraju V, Satoskar AR, Palesty A. et al. Conundrum of pathogenesis of diabetic cardiomyopathy: role of vascular endothelial dysfunction, reactive oxygen species, and mitochondria. Mol Cell Biochem. 2014;386:233-49
30. Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T. et al. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. 2013;45:438-47
31. Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y. et al. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. 2009;13:1499-512
32. Cicek FA, Toy A, Tuncay E, Can B, Turan B. Beta-blocker timolol alleviates hyperglycemia-induced cardiac damage via inhibition of endoplasmic reticulum stress. J Bioenerg Biomembr. 2014;46:377-87
33. Cao Y, Hao Y, Li H, Liu Q, Gao F, Liu W. et al. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33:809-16
34. Xu K, Wang X, Shi Q, Chen C, Tian C, Li XL. et al. Human prion protein mutants with deleted and inserted octarepeats undergo different pathways to trigger cell apoptosis. J Mol Neurosci. 2011;43:225-34
35. Li F, Luo J, Wu Z, Xiao T, Zeng O, Li L. et al. Hydrogen sulfide exhibits cardioprotective effects by decreasing endoplasmic reticulum stress in a diabetic cardiomyopathy rat model. Mol Med Rep. 2016;14:865-73
36. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058-70
37. Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of 'autoxidative glycosylation' in diabetes. Biochem J. 1987;245:243-50
38. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP. et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880-91
39. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656-67
40. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880-5
41. Yang F, Yu X, Li T, Wu J, Zhao Y, Liu J. et al. Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am J Physiol Endocrinol Metab. 2017;312:E190-E203
42. Cong XQ, Piao MH, Li Y, Xie L, Liu Y. Bis(maltolato)oxovanadium(IV) (BMOV) Attenuates Apoptosis in High Glucose-Treated Cardiac Cells and Diabetic Rat Hearts by Regulating the Unfolded Protein Responses (UPRs). Biol Trace Elem Res. 2016;173:390-8
43. Zhu M, Guo M, Fei L, Pan XQ, Liu QQ. 4-phenylbutyric acid attenuates endoplasmic reticulum stress-mediated pancreatic beta-cell apoptosis in rats with streptozotocin-induced diabetes. Endocrine. 2014;47:129-37
44. Yan X, Chen J, Zhang C, Zhou S, Zhang Z, Chen J. et al. FGF21 deletion exacerbates diabetic cardiomyopathy by aggravating cardiac lipid accumulation. J Cell Mol Med. 2015;19:1557-68
45. Guo R, Wu Z, Jiang J, Liu C, Wu B, Li X. et al. New mechanism of lipotoxicity in diabetic cardiomyopathy: Deficiency of Endogenous H2S Production and ER stress. Mech Ageing Dev. 2017;162:46-52
46. Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145-56
47. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121-35
48. Wang XB, Huang XM, Ochs T, Li XY, Jin HF, Tang CS. et al. Effect of sulfur dioxide preconditioning on rat myocardial ischemia/reperfusion injury by inducing endoplasmic reticulum stress. Basic Res Cardiol. 2011;106:865-78
49. Li C, Hu M, Wang Y, Lu H, Deng J, Yan X. Hydrogen sulfide preconditioning protects against myocardial ischemia/reperfusion injury in rats through inhibition of endo/sarcoplasmic reticulum stress. Int J Clin Exp Pathol. 2015;8:7740-51
50. Ding W, Zhang X, Huang H, Ding N, Zhang S, Hutchinson SZ. et al. Adiponectin protects rat myocardium against chronic intermittent hypoxia-induced injury via inhibition of endoplasmic reticulum stress. PLoS One. 2014;9:e94545
51. Bourdier G, Flore P, Sanchez H, Pepin JL, Belaidi E, Arnaud C. High-intensity training reduces intermittent hypoxia-induced ER stress and myocardial infarct size. Am J Physiol Heart Circ Physiol. 2016;310:H279-89
52. Belaidi E, Thomas A, Bourdier G, Moulin S, Lemarie E, Levy P. et al. Endoplasmic reticulum stress as a novel inducer of hypoxia inducible factor-1 activity: its role in the susceptibility to myocardial ischemia-reperfusion induced by chronic intermittent hypoxia. Int J Cardiol. 2016;210:45-53
53. Zhou X, Tang S, Hu K, Zhang Z, Liu P, Luo Y. et al. DL-Propargylglycine protects against myocardial injury induced by chronic intermittent hypoxia through inhibition of endoplasmic reticulum stress. Sleep Breath. 2018;22:853-63
54. Sun X, Wang W, Dai J, Jin S, Huang J, Guo C. et al. A Long-Term and Slow-Releasing Hydrogen Sulfide Donor Protects against Myocardial Ischemia/Reperfusion Injury. Sci Rep. 2017;7:3541
55. Li L, Li M, Li Y, Sun W, Wang Y, Bai S. et al. Exogenous H2S contributes to recovery of ischemic post-conditioning-induced cardioprotection by decrease of ROS level via down-regulation of NF-kappaB and JAK2-STAT3 pathways in the aging cardiomyocytes. Cell Biosci. 2016;6:26
56. Li H, Zhang C, Sun W, Li L, Wu B, Bai S. et al. Exogenous hydrogen sulfide restores cardioprotection of ischemic post-conditioning via inhibition of mPTP opening in the aging cardiomyocytes. Cell Biosci. 2015;5:43
57. Sun W, Yang J, Zhang Y, Xi Y, Wen X, Yuan D. et al. Exogenous H2S restores ischemic post-conditioning-induced cardioprotection through inhibiting endoplasmic reticulum stress in the aged cardiomyocytes. Cell Biosci. 2017;7:67
58. Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci U S A. 2006;103:6489-94
59. Ho YS, Yu MS, Yang XF, So KF, Yuen WH, Chang RC. Neuroprotective effects of polysaccharides from wolfberry, the fruits of Lycium barbarum, against homocysteine-induced toxicity in rat cortical neurons. J Alzheimers Dis. 2010;19:813-27
60. Li MH, Tang JP, Zhang P, Li X, Wang CY, Wei HJ. et al. Disturbance of endogenous hydrogen sulfide generation and endoplasmic reticulum stress in hippocampus are involved in homocysteine-induced defect in learning and memory of rats. Behav Brain Res. 2014;262:35-41
61. Jiang JM, Wang L, Gu HF, Wu K, Xiao F, Chen Y. et al. Arecoline Induces Neurotoxicity to PC12 Cells: Involvement in ER Stress and Disturbance of Endogenous H2S Generation. Neurochem Res. 2016;41:2140-8
62. Wei HJ, Xu JH, Li MH, Tang JP, Zou W, Zhang P. et al. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol Sin. 2014;35:707-15
63. Wang CY, Zou W, Liang XY, Jiang ZS, Li X, Wei HJ. et al. Hydrogen sulfide prevents homocysteineinduced endoplasmic reticulum stress in PC12 cells by upregulating SIRT1. Mol Med Rep. 2017;16:3587-93
64. Tang YY, Wang AP, Wei HJ, Li MH, Zou W, Li X. et al. Role of silent information regulator 1 in the protective effect of hydrogen sulfide on homocysteine-induced cognitive dysfunction: Involving reduction of hippocampal ER stress. Behav Brain Res. 2018;342:35-42
65. Gaspar JM, Baptista FI, Galvao J, Castilho AF, Cunha RA, Ambrosio AF. Diabetes differentially affects the content of exocytotic proteins in hippocampal and retinal nerve terminals. Neuroscience. 2010;169:1589-600
66. Zhao CH, Liu HQ, Cao R, Ji AL, Zhang L, Wang F. et al. Effects of dietary fish oil on learning function and apoptosis of hippocampal pyramidal neurons in streptozotocin-diabetic rats. Brain Res. 2012;1457:33-43
67. Zhang X, Xu L, He D, Ling S. Endoplasmic reticulum stress-mediated hippocampal neuron apoptosis involved in diabetic cognitive impairment. Biomed Res Int. 2013;2013:924327
68. Zou W, Yuan J, Tang ZJ, Wei HJ, Zhu WW, Zhang P. et al. Hydrogen sulfide ameliorates cognitive dysfunction in streptozotocin-induced diabetic rats: involving suppression in hippocampal endoplasmic reticulum stress. Oncotarget. 2017;8:64203-16
69. Yusuf M, Kwong Huat BT, Hsu A, Whiteman M, Bhatia M, Moore PK. Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis. Biochem Biophys Res Commun. 2005;333:1146-52
70. Tanti A, Belzung C. Open questions in current models of antidepressant action. Br J Pharmacol. 2010;159:1187-200
71. Ferrari AJ, Norman RE, Freedman G, Baxter AJ, Pirkis JE, Harris MG. et al. The burden attributable to mental and substance use disorders as risk factors for suicide: findings from the Global Burden of Disease Study 2010. PLoS One. 2014;9:e91936
72. Hill MN, Hellemans KG, Verma P, Gorzalka BB, Weinberg J. Neurobiology of chronic mild stress: parallels to major depression. Neurosci Biobehav Rev. 2012;36:2085-117
73. Tan H, Zou W, Jiang J, Tian Y, Xiao Z, Bi L. et al. Disturbance of hippocampal H2S generation contributes to CUMS-induced depression-like behavior: involvement in endoplasmic reticulum stress of hippocampus. Acta Biochim Biophys Sin (Shanghai). 2015;47:285-91
74. Liu SY, Li D, Zeng HY, Kan LY, Zou W, Zhang P. et al. Hydrogen Sulfide Inhibits Chronic Unpredictable Mild Stress-Induced Depressive-Like Behavior by Upregulation of Sirt-1: Involvement in Suppression of Hippocampal Endoplasmic Reticulum Stress. Int J Neuropsychopharmacol. 2017;20:867-76
75. Li X, Zhang KY, Zhang P, Chen LX, Wang L, Xie M. et al. Hydrogen sulfide inhibits formaldehyde-induced endoplasmic reticulum stress in PC12 cells by upregulation of SIRT-1. PLoS One. 2014;9:e89856
76. Ivanova T, Beyer C. Pre- and postnatal expression of brain-derived neurotrophic factor mRNA/protein and tyrosine protein kinase receptor B mRNA in the mouse hippocampus. Neurosci Lett. 2001;307:21-4
77. Hu M, Zou W, Wang CY, Chen X, Tan HY, Zeng HY. et al. Hydrogen Sulfide Protects against Chronic Unpredictable Mild Stress-Induced Oxidative Stress in Hippocampus by Upregulation of BDNF-TrkB Pathway. Oxid Med Cell Longev. 2016;2016:2153745
78. Wei L, Kan LY, Zeng HY, Tang YY, Huang HL, Xie M. et al. BDNF/TrkB Pathway Mediates the Antidepressant-Like Role of H2S in CUMS-Exposed Rats by Inhibition of Hippocampal ER Stress. Neuromolecular Med. 2018;20:252-61
79. Vijayan VK. Chronic obstructive pulmonary disease. Indian J Med Res. 2013;137:251-69
80. Wang YJ, Jiang YL, Tang HF, Zhao CZ, Chen JQ. Zl-n-91, a selective phosphodiesterase 4 inhibitor, suppresses inflammatory response in a COPD-like rat model. Int Immunopharmacol. 2010;10:252-8
81. Wei J, Rahman S, Ayaub EA, Dickhout JG, Ask K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease. Chest. 2013;143:1098-105
82. Zhang Y, Gao J, Luo Y. The effect of various durations of cigarette smoke exposure on muscle fibre remodeling in rat diaphragms. Biomed Pharmacother. 2019;117:109053
83. Somborac-Bacura A, van der Toorn M, Franciosi L, Slebos DJ, Zanic-Grubisic T, Bischoff R. et al. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp Physiol. 2013;98:316-25
84. Lin F, Liao C, Sun Y, Zhang J, Lu W, Bai Y. et al. Hydrogen Sulfide Inhibits Cigarette Smoke-Induced Endoplasmic Reticulum Stress and Apoptosis in Bronchial Epithelial Cells. Front Pharmacol. 2017;8:675
85. Bei Y, Duong-Quy S, Hua-Huy T, Dao P, Le-Dong NN, Dinh-Xuan AT. Activation of RhoA/Rho-kinase pathway accounts for pulmonary endothelial dysfunction in patients with chronic obstructive pulmonary disease. Physiol Rep. 2013;1:e00105
86. Ding HB, Liu KX, Huang JF, Wu DW, Chen JY, Chen QS. Protective effect of exogenous hydrogen sulfide on pulmonary artery endothelial cells by suppressing endoplasmic reticulum stress in a rat model of chronic obstructive pulmonary disease. Biomed Pharmacother. 2018;105:734-41
87. Liu ZW, Wang HY, Guan L, Zhao B. Regulatory effects of hydrogen sulfide on alveolar epithelial cell endoplasmic reticulum stress in rats with acute lung injury. World J Emerg Med. 2015;6:67-73
88. Shi Y, Wang S, Peng H, Lv Y, Li W, Cheng S. et al. Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats. Int J Biol Sci. 2019;15:138-47
89. Yang R, Teng X, Li H, Xue HM, Guo Q, Xiao L. et al. Hydrogen Sulfide Improves Vascular Calcification in Rats by Inhibiting Endoplasmic Reticulum Stress. Oxid Med Cell Longev. 2016;2016:9095242
90. Marampon F, Gravina GL, Scarsella L, Festuccia C, Lovat F, Ciccarelli C. et al. Angiotensin-converting-enzyme inhibition counteracts angiotensin II-mediated endothelial cell dysfunction by modulating the p38/SirT1 axis. J Hypertens. 2013;31:1972-83
91. Uhal BD, Nguyen H, Dang M, Gopallawa I, Jiang J, Dang V. et al. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1-7. Am J Physiol Lung Cell Mol Physiol. 2013;305:L33-41
92. Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ. et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734-41
93. Hu HJ, Jiang ZS, Zhou SH, Liu QM. Hydrogen sulfide suppresses angiotensin II-stimulated endothelin-1 generation and subsequent cytotoxicity-induced endoplasmic reticulum stress in endothelial cells via NF-kappaB. Mol Med Rep. 2016;14:4729-40
94. Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M. et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652-61
95. Hu HJ, Jiang ZS, Qiu J, Zhou SH, Liu QM. Protective effects of hydrogen sulfide against angiotensin II-induced endoplasmic reticulum stress in HUVECs. Mol Med Rep. 2017;15:2213-22
Author contact
![]() Corresponding author: Honggang Wang, E-mail: whg197167henu.edu.cn, Tel: 13663781967.
Corresponding author: Honggang Wang, E-mail: whg197167henu.edu.cn, Tel: 13663781967.