Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(2):284-297. doi:10.7150/ijbs.38487 This issue Cite
Research Paper
Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1G93A Mice
Qi Zhou1*, Lei Zhu1*, Weiwen Qiu1*, Yue Liu2, Fang Yang2, Wenzhi Chen2, Renshi Xu ![]()
1. Department of Neurology, Jiangxi Provincial People's Hospital, Affiliated People's Hospital of Nanchang University, Nanchang 330006, Jiangxi, China
2. Department of Neurology, First Affiliated Hospital of Nanchang University, Nanchang 330006, Jiangxi, China
*These authors contributed equally to this work.
Received 2019-7-17; Accepted 2019-9-28; Published 2020-1-1
Abstract

Amyotrophic lateral sclerosis (ALS) is caused by the progressive degeneration of motor neurons in the spinal cord, the brain stem, and the motor cortex. So far, there is still a lack of effective drugs. Nicotinamide adenine dinucleotide (NAD+) takes part in redox reactions and the NAD-dependent signaling pathway. The NAD+ decline is related with many neurological diseases, leading to the accumulation of neurotoxic protein in the central nervous system. Moreover, the NAD+ supplementation is shown to promote neural stem cells/neuronal precursor cells (NSCs/NPCs) pool maintenance. Regulatory mechanisms and functions of NAD+ metabolism in ALS are still unknown. Thus, we hypothesized the aggregation of human SOD1 toxic protein and the fate of NSCs/NPCs in the ALS disease could be improved by the administration of nicotinamide riboside (NR), an NAD+ precursor. In this study, we treated SOD1G93A transgenic and wild-type mice by the oral administration of 20 mg/ml NR starting at 50 days of age. Effects of NR on the body weight, the motor function, the onset and the survival were assessed during the experiment. The expression of mutant hSOD1 protein, mitochondrial unfolded protein response (UPRmt) related protein, mitophagy markers and NAD+ metabolism related protein were detected by immunoblotting. Effects of NR on the NSCs/NPCs in neurogenic niches of brain were identified by the immunofluorescence staining. Our investigation elucidated that the NR treatment exhibited better hanging wire endurance but did not postpone the onset or extend the life span of SOD1G93A mice. Besides, we observed that the NR repletion promoted the clearance of mitochondrial hSOD1 neurotoxic protein. Meanwhile, the mitochondrial function pathway was disrupted in the brain of SOD1G93A mice. What's more, we demonstrated that the inadequate function of NAD+ salvage synthesis pathway was the primary explanation behind the decline of NAD+, and the NR treatment enhanced the proliferation and migration of NSCs/NPCs in the brain of SOD1G93A mice. At last, we found that levels of UPRmt related protein were significantly increased in the brain of SOD1G93A mice after the NR treatment. In summary, these findings reveal that the administration of NR activates UPRmt signaling, modulates mitochondrial proteostasis and improves the adult neurogenesis in the brain of SOD1G93A mice.
Keywords: Nicotinamide riboside, Amyotrophic lateral sclerosis, Mitochondrial unfolded protein response related protein, Neural stem cells, Neuronal precursor cells
Introduction
NAD+, known as active metabolite types of vitamin B3, is a fundamental little molecule co-factor in metabolic redox responses [1], conveying high vitality electrons to help oxidative phosphorylation by reversibly oxidizing or lessening NAD+ [2, 3], and filling in as a substrate for NAD-subordinate compounds that connect cell metabolism with the epigenetic guideline and the DNA damage repair [3]. Mammalian cells make NAD+ by three distinct methods: (1) De novo synthesis from the tryptophan; (2) Generation from the nicotinic acid using the Preiss-Handler (PH) pathway; or (3) Synthesis from nicotinamide (NAM) or NR via the salvage pathway [1, 4].
The NAD+ biosynthesis mediated by the nicotinamide monophosphoribosyl transferase (NAMPT) and the NAD+ utilization by NAD+-consuming enzymes are in a sensitive balance [5]. The decrease of NAD+ indicates the dysfunction of basic physiological system of whole body [6]. The ongoing development of understanding that the NAD+ homeostasis is vulnerable to aging and disease processes [7, 8], has invigorated tests to determine whether the replenishment of cellular or tissue NAD+ improves disease phenotypes in neurodegeneration-related diseases.
Mitochondria, the primary site of cellular energy acquisition, are gotten from proteobacteria that developed within our cells in endosymbiosis. The significant capacity of mitochondria is the creation of adenosine triphosphate (ATP) through the oxidative phosphorylation system (OXPHOS) [9]. During the cell respiration, electrons are exchanged to oxygen atoms and produce superoxide anions. Since they are exceptionally poisonous, superoxide anions are generally neutralized by antioxidant enzymes. However, the mitochondrial dysfunction prompts to the ATP depletion, the superoxide anion over-burden and release of proapoptotic molecules, such as cytochrome c in pathological conditions [9]. Cells have nevertheless adapted a mitochondrial quality control (MQC) framework, which including the mitochondrial biogenesis, mitochondrial derived vesicles, mitochondrial dynamics, mitophagy and UPRmt in the mammalian, to defeat mitochondrial defects [10, 11]. Thus, MQC is especially crucial for neurons which are long living cells and relatively easily lead to the accumulate damage in mitochondria in a state of stress [12].
The loss of mitochondrial proteostasis has been proposed to assume an important role in the age-related decline [13, 14]. Recent studies have implicated a mitochondrial stress reaction, the UPRm, as a connection between mitochondrial proteostasis and aging in different organisms [15, 16]. The UPRmt is a vigorous transcriptional reaction that has been proposed to alleviate the proteostatic stress in mitochondria by promoting folding, restricting import, and diminishing the translation of mitochondrial proteins [17]. In addition, a related basic pathway of mitochondrial damage in many situations is the consumption of NAD+, which occur by the initiation of pathways that utilize up cellular and mitochondrial NAD+ pools, such as the actuation of poly (ADP-ribose) polymerases (PARPs) and the cyclic ADP-ribose hydrolase (CD38) [18, 19]. And it replenishes the NAD+ pool to counteract carbon stress by improving the activity of sirtuins, or forestalling the oxidative damage with antioxidants, helping to keep up the mitochondria proteostasis [20].
Several lines of proof support the role of NAD+ supplementation for reestablishing function of NSCs/NPCs during ageing and with regards to neurodegenerative diseases. NAD+ levels decrease with age in the hippocampus of mice, alongside the expression of NAMPT, an enzyme associated with the NAD+ salvage pathway [21]. The deletion of NAMPT in NSCs diminishes the proliferation and self-renewal ability in the adult hippocampus, which could be protected with the nicotinamide mononucleotide (NMN) supplementation [21]. The NAD+ repletion with the NR supplementation advances the NSC proliferation and salvages neurogenesis in the ageing subventricular zone (SVZ) and the hippocampal dentate gyrus (SGZ) [20]. The NR supplementation in a 3xTg-AD mouse that contains the insufficient POLG displayed the less phosphorylated tau pathology, diminished the DNA damage, expanded the survival of hippocampal neurons and improved the cognitive function [22]. All above proofs shows that NAD+ metabolic homeostasis are central regulators of NSCs/NPCs destiny decision, which stimulate us to interest in potential advantages enhancing the tissue NAD+ as a way to treat neurodegenerative diseases.
ALS is caused by the progressive degeneration of motor neurons in the spinal cord, the brain stem and the motor cortex. The motor neuron death prompts the muscle weakness and paralysis, causing death in 1-5 years from the time of symptom onset. Most ALS cases are sporadic ALS (SALS), and the introduction to yet unidentified natural toxicants might be responsible for SALS [23]. About 5-10% of cases are the inherited familial ALS (FALS), and the primary identified ALS-linked gene was the superoxide dismutase 1 (SOD1) [24]. Mutations in the SOD1 gene represent up to 20% of FALS and 1-2% of obviously SALS cases. Mutations in several other genes have now been recognized in many FALS pedigrees [23, 25, 26]. Each mutated gene has its own genetic and molecular signature, FALS and SALS are phenotypically unclear yet, and a noteworthy share of our understanding originates from the investigation of rodents overexpressing the ALS-linked mutant SOD1 that develops an ALS-like phenotype [27]. Various molecular pathways have been involved in the neuronal death in ALS. Only two disease-modifying therapies currently approved by US Food and Drug Administration (FDA) for ALS are riluzole and edaravone, but the exact neuroprotective action of drugs is obscure and has no obvious improvement on the quality of life [28-30]. The absence of alternative drugs for the treatment of ALS requests the improvement of new remedial methodologies. Recently, it is exhibited the SOD1 protein misplacing and mitochondrial dysfunction in the brain of ALS mice [31]. Besides, several lines of evidences suggest the NSCs/NPCs niche is modified in ALS mice [32, 33]. Notably, the enhancing NAD+ salvage pathway returns the lethality of primary astrocytes expressing ALS-linked mutant superoxide dismutase 1 [34]. In line with this observation, NAD+ salvage pathway proteins suppress the proteotoxicity in yeast models of neurodegeneration by advancing the clearance of misfolded proteins [35]. On the contrary, the deletion of NAMPT in projection neurons coming about of NAD+ decrease of adult mice prompts the motor dysfunction, neurodegeneration, and death [36]. Collectively, these discoveries strongly suggest that the improving NAD+ biosynthesis by administering NAD+ precursors, might be a candidate therapeutic target against the ALS disease related to the mitochondrial dysfunction and the adult neurogenesis.
In this study, our investigation elucidated the NR treatment exhibited better hanging wire endurance but did not postpone the onset or extend the life span of SOD1G93A mice. Besides, we demonstrated that the replenishment of intracellular NAD+ by providing NR could reduce neurotoxic protein aggregates of mitochondria in the brain of SOD1G93A mice, and the mitochondrial function pathway was disrupted in the brain of SOD1G93A mice. Meanwhile, we suggested that the NAMPT-mediated NAD+ biosynthesis was the main reason for the NAD+ levels decline and the de novo biosynthesis of NAD+ might act as an adaptive response of body in the ALS. As we predicted, we demonstrated that the NR treatment enhanced the proliferation and migration of NPCs/NSCs in the brain of SOD1G93A mice. What's more, we found that levels of UPRmt related protein were significantly increased in the brain of SOD1G93A mice after the NR treatment. At last, we concluded that the NR might modulate the mitochondrial proteostasis and improve the adult neurogenesis through activating the mitochondrial UPR signaling in the brain of SOD1G93A mice.
Materials and methods
Animals, treatments and sample
Transgenic mice of SOD1 (G93A) mutation on a C57BL/6 foundation (Jackson laboratory, Bar Harbour, Maine) that harbor the high copy number of mutant allele hSOD1 were kept up as a hemizygous line in an SPF-reproducing facility (The neurological lab of first affiliated hospital of Nanchang university). The hemizygous line was kept up by mating transgenic males with C57BL/6 WT females. All creatures were housed in a room with controlled photoperiod (08:00-20:00 light) and temperature (22±1ºC) with free access to the standard nourishment and water. Wild-type (WT) and transgenic (TG) mice were recognized by numbering toe marks. Preceding the begin of different experiments, they were randomly apportioned to different treatment groups.
NR was specially synthesized as the previously described [37], and purified by the reverse-phase HPLC. The mother liquor of NR was obtained by adding 10 ml 0.9% saline to 6 g NR powder with purity > 98%. The concentration of NR mother liquor was 600 mg/ml. After the NR powder completely dissolved, 290 ml distilled water was added to the 10 ml mother liquor to set up a 20 mg/ml NR working fluid. For the drug treatment, NR was administered to mice from postnatal 50 days through drinking water, provided in light-protected bottles. Based on drinking water measurement taken in hSOD1G93A mice, NR dosages for every one equated to 400 mg/kg/day.
For animal grouping, mice were randomly divided into 4 groups: (1) SOD1-G93A TG mice receiving NR treatment (G93A-NR); (2) Litter-matched SOD1-G93A TG mice receiving NR-free drinking water (G93A-Vehicle); (3) WT control mice receiving NR treatment (WT-NR); (4) Litter-matched WT mice receiving NR-free drinking water (WT-Vehicle). For the immunohistochemistry analysis, n=3 mice were for each group. For the western blot analysis, n=3 mice were for each group. For the animal behavior test, n=4 mice were for each group, and the drug treatment was continued daily until the end-stage of disease (i.e. point of euthanasia). To evade any potential bias, all drug and vehicle treatments were coded and afterward administered and subsequently investigated by a researcher blinded to treatment groups. Decoding only happened after each animal experiment was finished. All animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals of China. All experiments involving mice were checked and affirmed by the ethics committee for animal care and utilization of the first affiliated hospital of Nanchang University, China.
Behavioral recording
Every three days, mice underwent the hanging wire test and the weight measure. Mice were trained one week before treated, and data were gathered from 70 days of age. For the hanging wire test, the latency of mice to fall from a wire cage top, which was gradually inverted and suspended at roughly 30 cm to the floor, was utilized as a record of motor weakness. The test was repeated three times to obtain the mean estimation of three trials. The disease onset was characterized by age at which hind limb tremors were apparent while suspending the mouse in the air by its tail. The survival was determined by the failure of mice to right itself inside 15-30 seconds if laid on either side. This is a broadly accepted end point for life expectancy studies in ALS mice [38, 39], and ensures that mice are killed before they are unfit to reach food or water. Mice were executed by the cervical disengagement following anesthesia.
Immunofluorescent staining
Mice were anesthetized and perfused utilizing 20 ml of 0.9% saline and 40 ml of 4% paraformaldehyde in 1xPBS (pH 7.5) at the room temperature. Brain was extracted and set in 4% paraformaldehyde buffer overnight, hatched in 20% sucrose in 1xPBS (pH 7.5), embedded utilizing the optimum cutting temperature (OCT). Slices were coronally and progressively cut into 12 μm sections from the rostral to the caudal on a leica cryostat and gathered on superfrost plus slides. In the fluorescent immunohistochemical stain, areas of SVZ, SGZ and olfactory bulb (OB) tissues were permeabilized utilizing 0.2% TritonX-100 and blocked utilizing 10% goat serum in 1xPBS after rehydrated in 1xPBS (pH7.4), incubated utilizing primary antibodies: Vimentin 1:100, Doublecortin (DCX) 1:200 (Abcam, Hong Kong Ltd.) at 4°C overnight, trailed by washing 6 times with 0.2% Triton X-100 in 1xPBS, incubating utilizing secondary antibodies (Donkey anti rabbit, 1:200) conjugated to the fluorescence rhodamine (Red) for 2 hours at the room temperature, and the DAPI recolor (Blue), widely washing for 5 times, each for 5 minutes, mounting utilizing the antifade medium, examining in a Nikon E800 fluorescent magnifying instrument outfitted with a spot digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) and Photoshop software (Adobe Systems, San Jose, CA, USA), and taking pictures. Multiple labeled histochemical stains conjugated to Vimentin, DCX and DAPI were utilized to watch and analyze the cell proliferation and migration. The analysis of immunohistochemical positive cells was performed by counting amount of positive cells in sections of SGZ, SVZ and OB at 200 magnifications in 3 sections and computing positive cells sum of all 3 sections, then the sum was divided by the section number, 3 mice per group were utilized, the averaged amount was utilized for the quantitative investigation.
Protein preparation
Total protein fractions
For the absolute protein extraction, brain tissues acquired from WT-Vehicle vs. WT-NR or G93A-Vehicle vs. G93A-NR mice were washed in PBS (0.1 mmol/L), and homogenized with the buffer [Tris-HCl pH 7.5, 20 mmol/L; NP-40, 1%; EDTA, 2 mmol/L; PMSF, 2 mmol/L; Triton-100, 1%; aprotinin, 25 g/ml; pesptatin A, 10 ug/ml; leupeptin, 50 μg/ml; and dithiothreitol (DTT), 2 mmol/L] with the protein extraction reagent enhanced with a protease inhibitor cocktail (Animal tissue whole protein extraction kit, Solarbio, Beijing, China). Homogenates (Total fractions) were then solidified/thawed three times in the fluid nitrogen and centrifuged at 20,800×g for 10 minutes, at 4°C, in order to remove cell debris, the subsequent supernatant was gathered and stored at -80 °C for the later use.
Mitochondrial-enriched fractions
Fresh brain tissues acquired from WT-Vehicle vs. WT-NR or G93A-Vehicle vs. G93A-NR mice were washed with PBS, and the blood was washed out. The filter paper was sucked dry. Fragments were cut by scissors and put into a little volume glass homogenizer. Adding 1.0 ml ice pre-cooled lysis buffer (Mitochondrial extraction kit, Solarbio, Beijing, China). After homogenization, were centrifuged at 1,000×g for 12 minutes for three times (4°C) to pellet the nuclei and the cell flotsam and jetsam. The supernatant was additionally centrifuged at 12,000×g for 10 minutes (4°C) and the subsequent pellet (Mitochondrial-enriched fraction) resuspended in the supplemented 100 ml store buffer and stored at -80°C for the later use.
Western blot analysis
Subsequent to mixing with the loading buffer and boiling at 100°C for 5 minutes, equivalent amounts of proteins were isolated by 8-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Polyvinylidene fluoride (PVDF) membranes were utilized for the protein transfer and afterward incubated with relating primary antibodies overnight at 4°C after obstructing by 5% bovine serum or skim milk. Primary rabbit-derived antibodies included SOD1 (1:500, Proteintech, 10269-1-AP), LC3 (1:1000, Proteintech, 14600-1-AP), P62 (1:1000, Proteintech, 18420-1-AP), CLPP (1:3000, Proteintech, 15698-1-AP), HSP60 (1:1000, Proteintech, 15282-1-AP), LONP1 (1:3000, Proteintech, 15440-1-AP), NAMPT (1:5000, Proteintech, 11776-1-AP), NMNAT3 (1:1000, Proteintech, 13236-1-AP), and β-actin (1:10000, Proteintech, 20536-1-AP). After washing with the Tris-buffered saline-Tween (TBST) for 3 times (10 minutes each time), anti-rabbit HRP-linked secondary antibodies were added. Membranes were incubated at the room temperature for 1 hour and after washed with TBST for 3 times (10 minutes each time). The chemical luminescence was evaluated using the BeyoECL Plus Kit (Beyotime, Shanghai, China).
Statistical analysis
Data were analyzed with the GraphPad Prism software (GraphPad Software, San Diego, CA). The Kaplan-Meier survival curve was analyzed using a Log-rank (Mantel-Cox) test. The comparison between two groups was performed by the Student's t test. All other data were analyzed with two-way ANOVA, followed by the Tukey's multiple comparisons post hoc test when appropriate. Data are expressed as mean + SEM as indicated. P-values below 0.05 were considered as significantly.
Results
The mutant hSOD1 protein incorrectly aggregated in the mitochondria of ALS SOD1G93A mice brain tissue
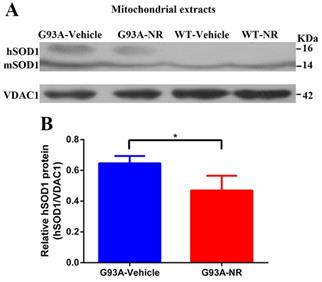
As previously shown, the WT SOD1 protein is ordinarily restricted in both the cytoplasm and the nucleus of human cells [40, 41]. And the mutant hSOD1 has been shown to associate with the mitochondria [42-46], exclusively in tissues from the central nervous system (CNS). To characterize the distribution of hSOD1 protein in the G93A-Vehicle and WT-Vehicle mice at the age of 120 days, hSOD1 protein levels of mitochondria-enriched portions were measured by the western blot analysis using mice brain tissues. As expected, our data demonstrated that there was obvious hSOD1 protein expression in the mitochondria of G93A-Vehicle group; however, we observed no mitochondrial hSOD1 protein expression in the WT-Vehicle group (Fig. 1A).
Mutant hSOD1 protein incorrectly aggregated in the mitochondria, and NR repletion promoted the clearance of mutant hSOD1 neurotoxic protein in ALS SOD1G93A mice. (A) Representative western blots showed hSOD1 levels of mitochondria enriched fractions in four groups mice (G93A-Vehicle, G93A-NR, WT-Vehicle, WT-NR). (B) Bar graphs showed the quantification of relative protein density of hSOD1 in the mitochondria fraction. VDAC1 was shown as the loading control. Data were expressed as mean ± SEM of n=3 mice/group; *P<0.05.
The mitochondrial proteostasis was disturbed in the brain of ALS SOD1G93A mice
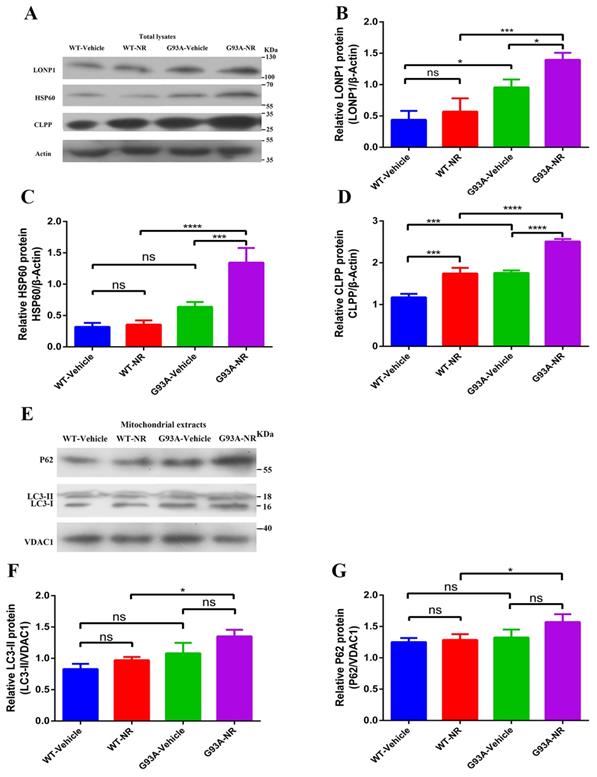
Mitochondrial anomalies in ALS incorporate to decrease the mitochondrial respiration activity and change in the mitochondrial morphology [47, 48]. However, the relevance of other aspects of mitochondrial homeostasis, such as the mitochondrial proteostasis, to the pathogenesis of ALS is still mostly unknown. Because the UPRmt and mitophagy, two major MQC pathways, are connected with the mitochondrial proteostasis, we analyzed cortex samples of G93A-Vehicle and WT-Vehicle mice of ALS at the age of 120 days. The immunoblotting of total lysates from the brain cortex of G93A-Vehicle group showed significantly up-regulated expression of mitochondrial LONP1, HSP60 and ClPP protein, the representative of UPRmt markers in vivo, relative to WT-Vehicle mice (Fig. 2B, C, D). However, we observed no statistical changes in the mitochondrial protein expression level of LC3-II, P62, the representative of mitophagy markers in vivo, between two groups (Fig. 2F, G).
Mitochondrial proteostasis were disturbed in the brain of ALS SOD1G93A mice. Representative immunoblots showed the expression of ClPP, HSP60, LONP1 (UPRmt pathways) in total lysates fraction (A) and P62, LC3-II (Mitophagy pathways) in the mitochondria fraction (E). Bar graphs showed the quantification of relative protein density of CLPP (B), HSP60 (C), LONP1 (D) in total lysates. Bar graphs showed the quantification of relative protein density of LC3-II (F) and P62 (G) in the mitochondria fraction. The protein density of CLPP, HSP60, LONP1 proteins was normalized with β-actin in total lysates. The protein density of LC3-II, P62 was normalized with VDAC1 in the mitochondria fraction. Data was showed as mean ± SEM of n=3 mice/group; *P<0.05, **P<0.01, ***P<0.001****P<0.0001.
The inadequate NAMPT-mediated NAD+ salvage synthesis was the main explanation behind the decline of NAD+ in the ALS SOD1G93A mice
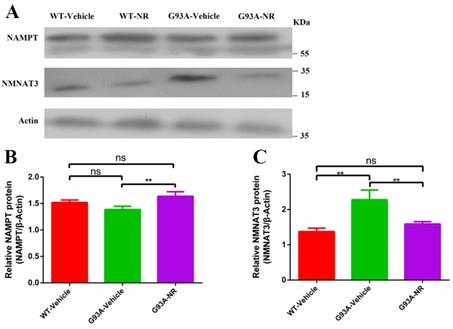
To investigate the reason for NAD+ decline in the development and progression of ALS, we firstly evaluated protein expression levels related to the NAD+ homeostasis in G93A-Vehicle and litter-matched WT-Vehicle mice at the age of 120 days. Protein levels of key enzymes NAMPT (Representative of key rate-limiting enzymes of salvage pathway for NAD+) and NMNAT3 (Representative of key rate-constraining enzymes of de novo biosynthesis pathway for NAD+) in the process of NAD+ synthesis were compared in two groups. Protein levels of NAMPT in the G93A-Vehicle group were significantly lower than that in the WT-Vehicle group (Fig. 3B). However, reverse changes were observed in levels of NMNAT3 protein. NMNAT3 levels in brain cortex tissues of G93A-Vehicle group were significantly higher than that in the WT-Vehicle group (Fig. 3C). These above data implied that the NAD+ decline was related to susceptibility factors in the ALS, and suggested that the NAMPT-regulated NAD+ salvage synthesis pathway may be the primary explanation behind the NAD+ decline, while the protein of NMNAT that regulates the NAD+ denovo biosynthesis may increase as an adaptive response in the ALS.
Inadequate NAMPT-mediated NAD+ salvage synthesis was the main reason for the decline of NAD+ in ALS SOD1G93A mice. (A) Representative western blots showed NAMPT and NMNAT3 levels in three groups. Bar graphs showed the quantification of relative protein levels of (B) NAMPT, (C) NMNAT3 in total lysates. Protein levels of NAMPT and NMNAT3 protein were normalized with β-actin. Data were showed as mean ± SEM of n=3 mice/group; (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
The NR treatment exhibited the better hanging wire endurance but did not delay the disease onset or extend the life span of ALS SOD1G93A mice
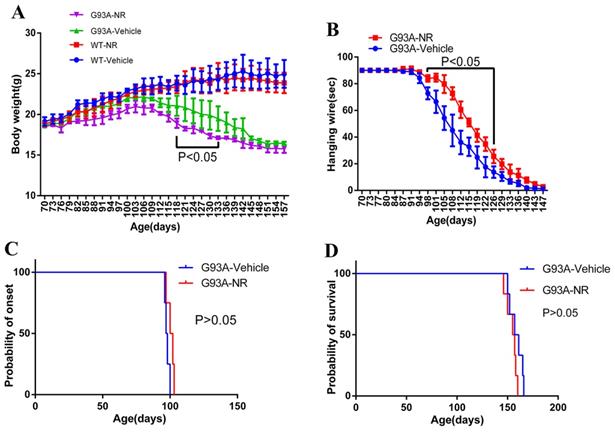
Starting from the age of 70 days, mice were weighted the bodyweight every three days in successive 87 days. As a result, G93A-NR group mice showed significantly lower bodyweight than the G93A-Vehicle group from 118 days to 133 days (Fig. 4A). Motor functions were assessed using the hanging wire test. The G93A-NR treatment significantly exhibited better hanging wire endurance in the disease from 98 and 126 days of age when compared to G93A-Vehicle mice (Fig. 4B). G93A-NR mice treated with NR from 50 days of age had no significant extension in the disease onset when compared with litter-matched G93A-Vehicle mice (Fig. 4C). There was also no difference in the survival time between G93A-NR and G93A-Vehicle groups (Fig. 4D).
Effects of NR treatment on body weight, hanging wire, disease onset or life expectancy of ALS SOD1G93A mice. (A) Body weight was different between G93A-NR and G93A-Vehicle groups treated from 50 days of age with significant values from 118 to 133 days of age. (B) Hanging wire was significantly different between G93A-NR and G93A-Vehicle groups from 98 to 126 days of age. (C) Disease onset (Defined by age at which hind limb tremors were apparent when suspending the mice in the air by its tail) in the G93A-NR group were not significantly different from G93A-Vehicle group mice. (D) G93A-NR mice showed no significant effect of survival time (The age when mice achieve the complete hind limb paralysis and have a failure to right itself once placed on its back) when compared with G93A-Vehicle mice. Data are expressed as mean ± SEM of n=4 mice/group; *P<0.05, **P<0.01, ***P<0.001, ****P< 0.0001.
The NR repletion promoted the clearance of mutant hSOD1 neurotoxic protein
NR is a naturally occurring precursor of NAD+ initially isolated from the fresh milk [49]. The exogenous treatment with NR has been shown to increase intracellular NAD+ levels in an assortment of cell lines [50]. Thus, we sought to determine in vivo the effect of NR administration on the hSOD1 aggregation in the brain cortex of SOD1G93A ALS mice model. As expected, expression levels of hSOD1 protein were significantly reduced in the G93A-NR group compared with the G93A-Vehicle group (Fig. 1B). Interestingly, when compared protein levels of NAMPT and NMNAT3 between G93A-Vehicle and G93A-NR groups, we found that these were significantly increased the expression of NAMPT protein in the G93A-NR group (Fig. 3B). On the contrary, protein levels of NMNAT3 in the G93A-NR group were significantly reduced when compared with the G93A-Vehicle group (Fig. 3C).
The NR treatment improved the proliferation, migration of NSCs/NPCs in the brain of ALS SOD1G93A mice
Restoring the NAD+ content is not exclusively protecting neurons, since it also has been reported to prevent the stem cells exhaustion in vivo [51, 52]. Data from several studies recommend that the NR treatment restores muscle, neuronal and melanocyte stem cell pools through the enlistment of UPRmt and the synthesis of preclusion proteins, prompts the oxidative respiration and ATP levels and the higher mitochondrial layer potential [51-53]. It also has been reported that the restoration of NAD+ in aged somatic cells improves the reprogramming proficiency and prolongs the life expectancy of mesenchymal stem cell by delaying senescence [53].
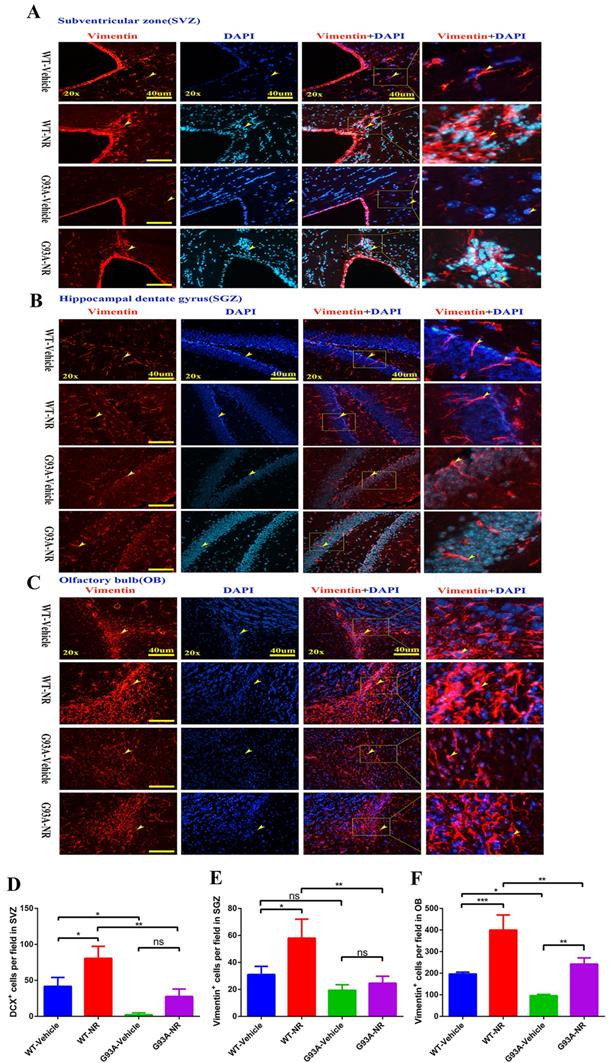
Thus, we initially determined the impact of NR on the NSCs/NPCs proliferation in the SVZ, SGZ and OB of SOD1G93A ALS mice brain at the age of 120 days. We performed the immunostaining of vimentin, a marker of NSCs/NPCs indicating neurogenesis. We found that the number of vimentin+ cells in the SVZ and OB, were significantly reduced in the G93A-Vehicle as compared with the WT-Vehicle group (Fig. 5D, F). And the number of vimentin+ cells in the SGZ of G93A-Vehicle had marginally reduction compared with WT-Vehicle mice (P=0.0518, Fig. 5E). Strikingly, the NR treatment in G93A-NR mice, significantly enhanced the number of vimentin+ proliferating cells in the SVZ, OB, as compared with G93A-Vehicle mice (Fig. 5D, F). However, the NR treatment to G93A-NR mice had no significantly effects on vimentin+ cells in the SGZ, when compared with G93A-Vehicle mice (Fig. 5E).
NR treatment increased the proliferation of NSCs/NPCs in the brain of ALS SOD1G93A mice. Representative photomicrographs showed the immunostaining of vimentin (A cell proliferation marker; red) in the SVZ (A), SGZ (B), OB (C) at 120 days of four groups mice. Cell nuclei were counterstained with DAPI (blue). Yellow arrows indicated the colocalization of vimentin+ cell and DAPI. Bar graphs showed the analysis of vimentin+ cells in SVZ (D), SGZ (E), OB (F) regions. Scale bar: 40μm. Data were expressed as mean ± SEM of n=3 mice/group; *P<0.05, **P<0.01, ***P<0.001, **** P<0.0001.
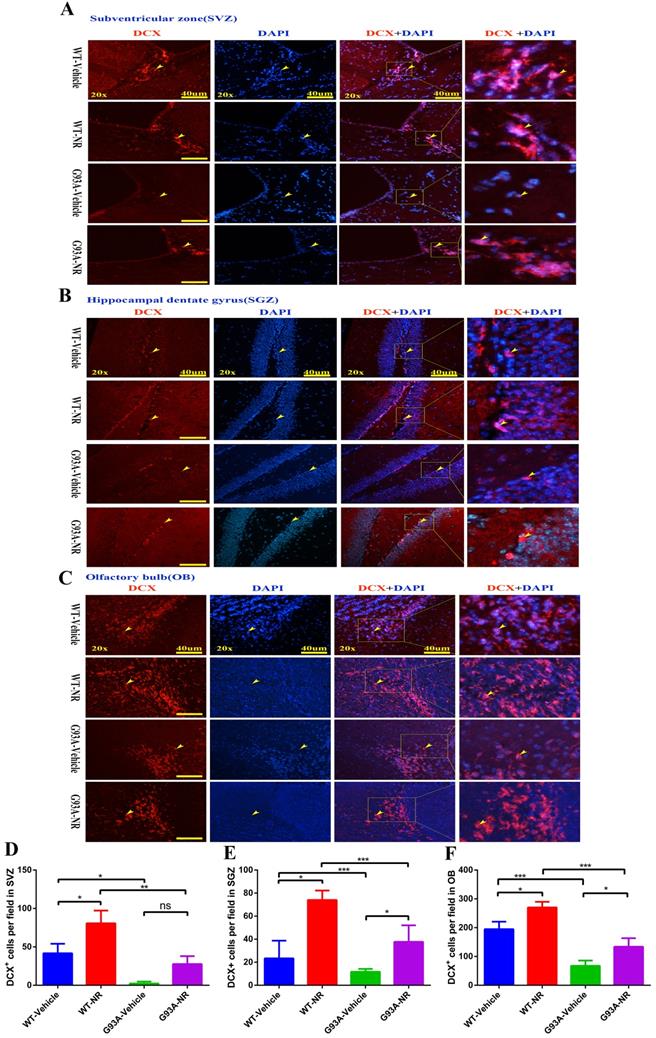
Further, we performed the immunostaining of DCX, a marker of neuroblasts to inspect the impact of NR on immature neurons in ALS mice at the age of 120 days. Accordingly, the number of DCX+ cells was significantly reduced in the SVZ, OB of G93A-Vehicle group mice, as compared to the WT-Vehicle group (Fig. 6D, F). However, the number of DCX+ cells in the SGZ between G93A-Vehicle and WT-Vehicle mice had no statistical differences (Fig. 6E). Interestingly, the NR treatment to G93A-NR mice, significantly increased the number of DCX+ cells in the SVZ, SGZ and OB as compared to G93A-Vehicle mice (Fig. 6D, E, F). It is well definitely recommended that DCX+ cells migrate from SVZ to OB rostral migratory stream (RMS) means migration of immature neurons [54]. Thus, the NR treatment tended to improve the migration of NPCs in the brain of SOD1G93A mice.
NR treatment extended the survival of newborn neurons in the brain of ALS SOD1G93A mice. Representative photomicrographs showed the immunostaining of DCX (A maker of newborn neuron; red) and DAPI (blue) in SVZ (A), SGZ (B), OB (C) regions at 120 days from four groups mice. Cell nuclei were counterstained with DAPI. Yellow arrows indicated the colocalization of DCX+ cell and DAPI. Bar graphs showed the quantitative analysis of DCX+ cells in SVZ (D), SGZ (E), OB (F) regions. Scale bar: 40μm. Data were expressed as mean ± SEM of n=3 mice/group; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To investigate whether NR mediated its effects via the mitochondrial UPR or the mitophagy pathway in the brain of SOD1G93A ALS mice model, we investigated the effect of NR on LONP1, HSP60, and CLPP proteins, downstream target molecules of NAD+ metabolites. We found that levels of LONP1, HSP60 and CLPP proteins were significantly increased in the brain cortex of G93A-NR mice as compared to G93A-Vehicle mice (Fig. 2B, C, D). Nonetheless, we did not observe that mitophagy-related proteins (LC3-II, P62) have significantly differences between G93A-NR and G93A-Vehicle groups (Fig. 2F, G).
Discussion
We obtained following several findings from our studied results: 1) The NR treatment exhibited the better hanging wire perseverance but did not postpone the disease onset or extended the life span of ALS SOD1G93A mice. 2) The mutant hSOD1 protein mistakenly aggregated in the mitochondria of ALS SOD1G93A mice cerebrum tissue, and the NR repletion promoted the clearance of mutant hSOD1 neurotoxic protein. 3) The mitochondrial function pathway was disrupted in the brain of ALS SOD1G93A mice. 4) The inadequate function of NAD+ salvage synthesis pathway was the primary explanation behind the decline of NAD+ in the ALS SOD1G93A mice. 5) The NR treatment enhanced the proliferation and migration of NSCs/NPCs in the brain of ALS SOD1G93A mice. 6) Levels of UPRmt related proteins were significantly increased in the brain of SOD1G93A mice after the NR treatment.
Numerous molecular pathways have been ensnared in the neuronal death in ALS. Pathogenic mechanisms such as the oxidative stress, the glutamate excitotoxicity, the protein aggregation, the inflammation, apoptosis, the proteasomal and mitochondrial dysfunction happen during the disease progression [55-57]. However, it is as yet unclear which of these occasions is the essential trigger for the beginning of ALS and which events occur as a result of initial activation of ALS disease. A substantial number of promising drugs had been preclinically found and indicated successful therapeutic effects in different ALS animal models, yet a large portion of them failed to demonstrate clinical viability. Two main approved drugs for the ALS disease treatment by FDA are riluzole and edaravone which just have marginal effects on clinic manifestations and the patient survival [28, 29, 58, 59]. Accordingly, it is necessary to grow progressively effective and prudent methodologies for the ALS treatment.
The misfolded protein accumulation is a noteworthy pathological feature of ALS, which is distinguished in the brain and the spinal cord even at the presymptomatic stage, and may spread through the cell-to-cell transmission. The previous study proposed that NAD+ salvage pathway proteins could smother proteotoxicity in yeast models of neurodegeneration by advancing the clearance of misfolded proteins [35]. In accordance with this research, our study showed the NR treatment reduced aggregated mutant hSOD1 proteins in the brain of SOD1G93A mice, which indicted that NAD+ salvage pathway was a potential therapeutic target suppressing the collection of misfolded SOD1 in the ALS. More specifically, the mutant hSOD1 has been shown to associate with the mitochondria. It has been discovered that the mutant hSOD1 deposited on the cytoplasmic substance of spinal cord mitochondrial [45], accompanied by the modified mitochondria shape and the distribution in ALS [60]. Similarly, our data demonstrated that obvious expression levels of hSOD1 protein in the mitochondria of SOD1G93A mice, and the hSOD1 protein tended to be partly eliminated by the NR administration. Taken together, our observations provided evidences that the recharging of intracellular NAD+ by providing NR might decrease neurotoxic impacts of protein aggregates of mitochondria in the brain of ALS SOD1G93A mice.
NAD+ is delivered in all eukaryotic cells. The basic intracellular NAD+ concentration can be up to 800 mM in yeast [61], 100-400 mM in human HEK293 cells [62, 63], and roughly 0.2 mmol/kg in the mouse tibialis foremost muscle [64]. New strategies have been developed to empower the detection of subcellular NAD+ levels [62, 63, 65, 66]. In our present study, we measured protein levels of NAMPT and NMNAT3 to indirectly assess the basal intracellular level of NAD+, for the reason they are key rate-limiting enzymes of salvage pathway and the de novo biosynthesis pathway for NAD+, respectively. It has been recently revealed that NAD+ levels decrease normally with age and involve in the support of mitochondrial health in the aged tissue [67, 68]. Moreover, NAD+ levels decline with age in the hippocampus of mice, alongside the reduced expression of NAMPT [69]. To examine whether the NAD+ decline was actually involved in the pathogenic mechanism of ALS disease, we measured expression levels of key proteins, NAMPT and NMNAT3 in SOD1G93A mice and litter-matched WT mice, respectively. As a result, we observed protein levels of NAMPT in the cortical brain of SOD1G93A mice were significantly reduced compared with WT mice. On the contrary, the expression level of NMNAT3 protein of cortical brain in the SOD1G93A group was significantly upgraded compared with the WT group. Curiously, when compared protein levels of NAMPT and NMNAT3 between NR-treated and NR-untreated SOD1G93A groups, we observed that these were significantly increased levels of NAMPT protein in G93A-NR mice compared with the G93A-Vehicle group. On the contrary, protein levels of NMNAT3 in the G93A-NR group were significantly decreased compared with G93A-Vehicle mice. In summary, our research confirmed that the NAD+ decline was related to susceptibility factors in the ALS, suggested that the insufficient NAMPT-mediated NAD+ salvage synthesis was the main reason for the NAD+ levels decline and the de novo biosynthesis of NAD+ might act as an adaptive response in the ALS.
Recent years have witnessed a resurgence of enthusiasm for the NAD+ biology. This has been driven to some degree by disclosing the intermediate of NAD+ biosynthesis, NR, effectively increases the NAD+ concentration in a variety of tissues, much of time with beneficial or therapeutic effects. The direct commitment of NR to the NAD+ metabolism was first perceived by Bieganowski and Brenner in 2004 [70]. This report described a class of enzymes known as NRKs (NR kinases) that could directly change over NR to NMN, bypassing the requirement for NAMPT in the salvage pathway. NR has additionally been reported to have a number of intriguing advantages in the CNS. One of studies to examine impacts of NR in vivo uncovered a striking improvement in the progression of Alzheimer's disease pathology in the Tg2576 model [71]. NR also has been shown to prevent the noise-induced hearing loss and the neuritis withdrawal from hair cells in the internal ear through a SIRT3-dependent mechanism [72]. NR additionally secures against the diabetic and chemotherapy-instigated neuropathy in mice [73], the raising expectations may have a role in the administration of chronic pain. In concordance with these outcomes, our data uncovered that the NR treatment displayed the better hanging wire endurance. One clarification for the better hanging wire test in the present investigation was that the morphological damage of muscle might be mitigated by the NR administration which was suggested by researches [20, 74, 75]. Notably, it has been demonstrated that the NAD+/ sirtuin pathway adjusts the life span through the enactment of mitochondrial UPR and FOXO signaling [51]. However, we observed no effects of NR administration on the lifespan in our study, suggesting the mechanism of longevity in the state of ALS disease may be different from that in the healthy. Besides, it is proposed that the maintenance of efficient DNA repair by NAD+ recharging in cells, c.elegans, and mice may delay the onset of aging and age-related diseases [76, 77]. In contrary, our results indicated that the NR treatment did not postpone the disease onset of SOD1G93A mice, which indicted that the difference might be the consequence of insufficient NAD+ supplementation or the course of treatment in our study.
There are two neurogenic niches in the mammalian adult brain: the SGZ of hippocampus and the SVZ, delivering new neurons that utilize the RMS to achieve OB. Cells in two discrete locales hold the ability to generate multiple lines composed of various cells, including NSCs, NPCs, newborn immature neurons and mature neurons. It has been recently reported that the NSCs and NPCs niche is altered in the ALS mice brain [32, 33, 78]. Besides, the NR treatment has been shown to improve the stem cell function and somewhat relieve the muscle wasting phenotype in the mdx mice [20, 75]. Furthermore, the NAD+ supplementation reestablished the mitochondrial function in NSCs during ageing and in the context of neurodegenerative disease [20, 22]. Thus, we determined whether the NAD+ repletion with the NR supplementation have potential effects to promote the NSCs/NPCs proliferation or the immature neurons migration in the SVZ, SGZ and OB in SOD1G93A mice. Intriguingly, in the present study, we observed that the NR treatment significantly attenuated the ALS-induced loss of NSCs/NPCs in SVZ, SGZ and OB. Meantime, the NR administration also continually renewed the pool of immature neurons, jointly suggesting the promotion to the proliferation and migration of NPCs/NSCs was improved by NR in the brain of SOD1G93A mice. This may be clarified by that the NR treatment could reduce the stem cell exhaustion through the induction of UPRmt in ALS SOD1G93A mice.
Conclusion
In summary, we conclude that the NR modulates the mitochondrial proteostasis and improves the adult neurogenesis through the activation of mitochondrial UPR signaling in the brain of SOD1G93A mice, and NR may as a viable clinical therapeutic scheme of translational medicine for ALS and other neurodegenerative diseases. While many studies on NAD+ precursors are progressing, major issues remain. The pleotropic role of NAD+ in the human physiology is unpredictable and requires further mechanistic insight. For instance, a previous research proposed the NAD+ metabolism governs the proinflammatory senescence-related secretome and may animate the development of tumor cells [79]. It is likewise conceivable that some unanticipated side effects may exhibit in certain human populations. Hence, profoundly stringent and carefully designed clinical trials are necessary to guarantee safety.
Abbreviations
ALS: amyotrophic laterale sclerosis; NAD+: nicotinamide adenine dinucleotide; UPRmt: mitochondrial unfolded protein response; hSOD1: human SOD1; NSCs/NPCs: neural stem cells/neuronal precursor cells; NR: nicotinamide riboside; PH: Preiss-Handler; NAM: nicotinamide; NAMPT: nicotinamide monophosphoribosyl transferase; ATP: adenosine triphosphate; OXPHOS: oxidative phosphorylation system; SVZ: subventricular zone; SGZ: hippocampal dentate gyrus; OB: olfactory bulb; FDA: Food and Drug Administration; WT: wild-type; TG: transgenic; DCX: doublecortin; DTT: dithiothreitol; CNS: central nervous system; MQC: mitochondrial quality control; NMN: nicotinamide mononucleotide; SOD1: superoxide dismutase 1; OCT: optimum cutting temperature; SDS-PAGE: sulfate-polyacrylamide gel electrophoresis; PVDF: polyvinylidene fluoride; TBST: tris-buffered saline-Tween; RMS: rostral migratory stream; NRKs: NR kinases.
Acknowledgements
Funding
This work was supported by grants from the National Natural Science Foundation of China (30560042, 81160161 and 81360198), Education Department of Jiangxi Province (GJJ13198, GJJ170021), Jiangxi Provincial Department of Science and Technology ([2014]-47, 20142BBG70062, 20171BAB215022), Health and Family Planning Commission of Jiangxi province (20181019), Jiangxi Provincial Department of Science and Technology Gan Po Elite 555 (Jiangxi Finance Elite Education Refers to [2015] 108) and the Innovation Fund Designated for Graduate Students of Jiangxi Province (YC2016-B027, YC2015-S097).
Authorship
All authors contributed significantly to this research and preparation of the manuscript. RX and QZ conceived and designed the experiments and wrote the manuscript. QZ, LZ, WQ, YL, FY and WC performed the experiments and analyzed the data. QZ is the first author to the work. QZ, LZ and WQ contributed equally to this work. All authors have been involved in the drafting, critical revision and final approval of the manuscript for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval
All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of China. All experiments involving animal were reviewed and approved by the ethics committee for animal care and use of First Affiliated Hospital of Nanchang University, China.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31-53
2. Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW. et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018;27:1067-80 e5
3. Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Kraus WL. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science. 2018;360:eaan 5780
4. Kaelin WG Jr McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56-69
5. Katsyuba E, Auwerx J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017;36:2670-2683
6. Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208-1213
7. Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y. et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24:795-806
8. Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. P Natl Acad Sci U S A. 2015;112:2876-2881
9. Kadenbach B. Introduction to mitochondrial oxidative phosphorylation. Adv Exp Med Biol. 2012;748:1-11
10. Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci Ther. 2017;23:5-22
11. Durand F, Hoogenraad N. Assessing Mitochondrial Unfolded Protein Response in Mammalian Cells. Methods Mol Biol. 2017;1567:363-378
12. Dawson TM, Dawson VL. Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:437-454
13. de Castro IP, Martins LM, Loh SH. Mitochondrial quality control and Parkinson's disease: a pathway unfolds. Mol Neurobiol. 2011;43:80-86
14. Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D. et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187-193
15. Callegari S, Dennerlein S. Sensing the Stress: A Role for the UPR(mt) and UPR(am) in the Quality Control of Mitochondria. Front Cell Dev Biol. 2018;6:31
16. Moehle EA, Shen K, Dillin A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J Biol Chem. 2019;294:5396-5407
17. Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20:214-225
18. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM. et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23:1127-1139
19. Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A. et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016;17:69-85
20. Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P. et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436-1443
21. Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321-1340
22. Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E. et al. NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018;115:E1876-E1885
23. Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17-23
24. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59-62
25. Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124:339-352
26. Ravits J, Appel S, Baloh RH, Barohn R, Brooks BR, Elman L. et al. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:S5-18
27. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772-1775
28. Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. An outcome study of riluzole in amyotrophic lateral sclerosis-a population-based study in Ireland, 1996-2000. J Neurol. 2003;250:473-479
29. Rothstein JD. Edaravone: A new drug approved for ALS. Cell. 2017;171:725
30. Jawaid A, Khan R, Polymenidou M, Schulz PE. Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol Neurodegener. 2018;13:63
31. Tafuri F, Ronchi D, Magri F, Comi GP, Corti S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci. 2015;9:336
32. Liu Z, Martin LJ. The adult neural stem and progenitor cell niche is altered in amyotrophic lateral sclerosis mouse brain. J Comp Neurol. 2006;497:468-488
33. Chi L, Gan L, Luo C, Lien L, Liu R. Temporal response of neural progenitor cells to disease onset and progression in amyotrophic lateral sclerosis-like transgenic mice. Stem Cells Dev. 2007;16:579-588
34. Harlan BA, Pehar M, Sharma DR, Beeson G, Beeson CC, Vargas MR. Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1). J Biol Chem. 2016;291:10836-10846
35. Ocampo A, Liu J, Barrientos A. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum Mol Genet. 2013;22:1699-1708
36. Wang X, Zhang Q, Bao R, Zhang N, Wang Y, Polo-Parada L. et al. Deletion of Nampt in Projection Neurons of Adult Mice Leads to Motor Dysfunction, Neurodegeneration, and Death. Cell Rep. 2017;20:2184-200
37. Yang T, Chan NY, Sauve AA. Syntheses of nicotinamide riboside and derivatives: effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J Med Chem. 2007;50:6458-6461
38. Ludolph AC, Bendotti C, Blaugrund E, Hengerer B, Löffler JP, Martin J. et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler. 2007;8:217-223
39. Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N. et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4-15
40. Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc Natl Acad Sci U S A. 1992;89:10405-10409
41. Zhong Y, Wang J, Henderson MJ, Yang P, Hagen BM, Siddique T. et al. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. eLife. 2017;6:e23759
42. Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF. et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626-29633
43. Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM. et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5-17
44. Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E. et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142-7147
45. Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022-4027
46. Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V. et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575-587
47. Palomo GM, Manfredi G. Exploring new pathways of neurodegeneration in ALS: the role of mitochondria quality control. Brain Res. 2015;1607:36-46
48. Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2017 S0304-3940(17)30544-X
49. Trammell SA, Yu L, Redpath P, Migaud ME, Brenner C. Nicotinamide Riboside Is a Major NAD+ Precursor Vitamin in Cow Milk. J Nutr. 2016;146:957-963
50. Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP. et al. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol Med. 2017;23:899-916
51. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C. et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430-441
52. Kim HJ, Oh GS, Shen A, Lee SB, Choe SK, Kwon KB. et al. Augmentation of NAD(+) by NQO1 attenuates cisplatin-mediated hearing impairment. Cell Death Dis. 2014;5:e1292
53. Son MJ, Kwon Y, Son T, Cho YS. Restoration of Mitochondrial NAD(+) Levels Delays Stem Cell Senescence and Facilitates Reprogramming of Aged Somatic Cells. Stem Cells. 2016;34:2840-2851
54. Bond AM, Ming GL, Song H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell. 2015;17:385-395
55. Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol. 2001;54:386-392
56. Pandya RS, Mao LL, Zhou EW, Bowser R, Zhu Z, Zhu Y. et al. Neuroprotection for amyotrophic lateral sclerosis: role of stem cells, growth factors, and gene therapy. Cent Nerv Syst Agents Med Chem. 2012;12:15-27
57. Zhu Y, Fotinos A, Mao LL, Atassi N, Zhou EW, Ahmad S. et al. Neuroprotective agents target molecular mechanisms of disease in ALS. Drug Discov Today. 2015;20:65-75
58. Schultz J. Disease-modifying treatment of amyotrophic lateral sclerosis. Am J Manag Care. 2018;24:S327-S35
59. Jaiswal MK. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39:733-748
60. Vande Velde C, McDonald KK, Boukhedimi Y, McAlonis-Downes M, Lobsiger CS, Bel Hadj S. et al. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PloS one. 2011;6:e22031
61. Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell. 2007;129:473-484
62. Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ. et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095-1107
63. Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL. et al. Biosensor reveals multiple sources for mitochondrial NAD(+). Science. 2016;352:1474-1477
64. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056-1060
65. Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y. et al. SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab. 2015;21:777-789
66. Frederick DW, Loro E, Liu L, Davila A Jr, Chellappa K, Silverman IM. et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016;24:269-282
67. Srivastava S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin Transl Med. 2016;5:25
68. Yoshino J, Baur JA, Imai SI. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018;27:513-528
69. Zhang C, Yang Y, Liang W, Wang T, Wang S, Wang X. et al. Neuroprotection by urate on the mutant hSOD1-related cellular and Drosophila models of amyotrophic lateral sclerosis: Implication for GSH synthesis via activating Akt/GSK3beta/Nrf2/GCLC pathways. Brain Res Bull. 2019;146:287-301
70. Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495-502
71. Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L. et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging. 2013;34:1581-1588
72. Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W. et al. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059-1068
73. Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats. Pain. 2017;158:962-972
74. Chaturvedi P, Tyagi SC. NAD(+): A big player in cardiac and skeletal muscle remodeling and aging. J Cell Physiol. 2018;233:1895-1896
75. Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L. et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. 2016;8:361ra139
76. Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb Perspect Med. 2015;5:a025130
77. White RR, Vijg J. Do DNA Double-Strand Breaks Drive Aging? Mol Cell. 2016;63:729-238
78. Galan L, Gomez-Pinedo U, Guerrero A, Garcia-Verdugo JM, Matias-Guiu J. Amyotrophic lateral sclerosis modifies progenitor neural proliferation in adult classic neurogenic brain niches. BMC Neurol. 2017;17:173
79. Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S. et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 2019;21:397-407
Author contact
![]() Corresponding author: Prof. Renshi Xu, Email: xurenshiedu.cn or 13767015770com, Department of Neurology, Jiangxi Provincial People's Hospital, Affiliated People's Hospital of Nanchang University, Nanchang 330006, Jiangxi, China. Tel: +86 0791-88603798.
Corresponding author: Prof. Renshi Xu, Email: xurenshiedu.cn or 13767015770com, Department of Neurology, Jiangxi Provincial People's Hospital, Affiliated People's Hospital of Nanchang University, Nanchang 330006, Jiangxi, China. Tel: +86 0791-88603798.