Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The expression of...
The type and structure of...
PHGDH expression and cancer
PHGDH plays a non-metabolic role...
The factors controlling PHGDH...
The inhibitor of PHGDH against...
PHGDH and tumor resistance to...
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(9):1495-1506. doi:10.7150/ijbs.41051 This issue Cite
Review
The Role of D-3-Phosphoglycerate Dehydrogenase in Cancer
Xiaoya Zhao1, Jianfei Fu2, Jinlin Du3 ![]() , Wenxia Xu1
, Wenxia Xu1 ![]()
1. Central Laboratory, Jinhua Hospital of Zhejiang University, Jinhua 321000, Zhejiang Province, China
2. Department of Medical Oncology, Jinhua Hospital of Zhejiang University, Jinhua 321000, Zhejiang Province, China
3. Department of Colorectal Surgery, Jinhua Hospital of Zhejiang University, Jinhua 321000, Zhejiang Province, China
Received 2019-10-10; Accepted 2020-2-17; Published 2020-3-5
Abstract

Serine, a non-essential amino acid, can be imported from the extracellular environment by transporters and de novo synthesized from glycolytic 3-phosphoglycerate (3-PG) in the serine biosynthetic pathway (SSP). It has been reported that active serine synthesis might be needed for the synthesis of proteins, lipids, and nucleotides and the balance of folate metabolism and redox homeostasis, which are necessary for cancer cell proliferation. Human D-3-phosphoglycerate dehydrogenase (PHGDH), the first and only rate-limiting enzyme in the de novo serine biosynthetic pathway, catalyzes the oxidation of 3-PG derived from glycolysis to 3-phosphohydroxypyruvate (3-PHP). PHGDH is highly expressed in tumors as a result of amplification, transcription, or its degradation and stability alteration, which dysregulates the serine biosynthesis pathway via metabolic enzyme activity to nourish tumors. And some recent researches reported that PHGDH promoted some tumors growth via non-metabolic way by upregulating target cancer-promoting genes. In this article, we reviewed the type, structure, expression and inhibitors of PHGDH, as well as the role it plays in cancer and tumor resistance to chemotherapy.
Keywords: serine metabolism, SSP, structure of PHGDH, expression of PHGDH, cancer, non-metabolic activity, inhibitor of PHGDH, tumor resistance
Introduction
Metabolic reprogramming plays an important role in the development of tumors [1]. Glucose is one of the most primary nutrients utilized for mitochondrial oxidative phosphorylation to generate energy supporting cellular behaviors in normal differentiated cells. In contrast, most cancer cells exploit glucose for aerobic glycolysis to support the proliferation of cancer cells [2], which is called the “Warburg effect” [3]. Despite the universal alterations in glucose metabolism observed in tumors [2-4], diversion of glycolytic flux into amino acid synthesis is also necessary during tumor development [5, 6]. Serine, an unessential amino acid, is required for certain anabolic processes [7]. It is a critically important component of proteins and provides precursors for the synthesis of lipids, such as sphingolipids and phosphatidylserine (PS) [8], a major component of cellular membranes [7, 9]. Serine is a vital source of “one-carbon” units carried by tetrahydrofolate supplying methyl groups for the modification of lipids, nucleic acids, and proteins [9]. Thus, serine serves multiple essential roles in cellular metabolism.
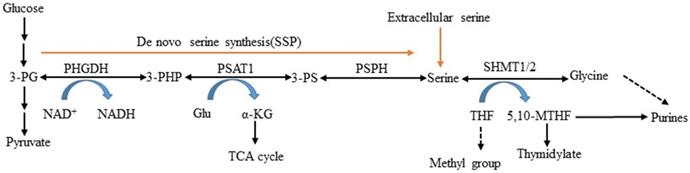
Serine is obtained exogenously via amino acid transporters and also can be synthesized from a branch of glycolysis [5]. NAD+-dependent enzyme 3-phosphoglycerate dehydrogenase (PHGDH) is the first and only rate-limiting enzyme in the serine and glycine de novo biosynthetic pathway [10]. It catalyzes glycolytic intermediate 3-phosphoglycerate (3-PG) transformed into 3-phosphohydroxypyruvate (3- PHP) [11, 12]. Subsequent enzymatic reactions in the pathway convert 3-PHP to serine via transamination (PSAT1) and phosphate ester hydrolysis (PSPH). Serine can be converted to glycine by serine hydroxymethyltransferase (SHMT) and then synthesize glutathione (GSH) to protect against oxidative stress [13, 14]. This process involves two important factors: PSAT1 converts glutamate to alpha-ketoglutarate (α- KG) serving as fuels for the TCA cycle, and SHMT1/2 catalyzes serine that is cleaved into CH2-THF in either the cytosol or mitochondria, which is necessary for the biosynthesis of thymidine and purine (Figure 1). In this study, we reviewed PHGDH's structures and functions, described its role in serine metabolism, and explored its association with diseases.
L-serine synthesis pathway. PHGDH first catalyzes the oxidation of 3-phosphoglycerate (3-PG) to 3-phosphohydroxypyruvate (3-PHP), with the coinstantaneous reduction of the cofactor NAD+ to NADH. The subsequent transamination reaction is catalyzed by phosphoserine aminotransferase (PSAT), which uses glutamate (Glu) as a nitrogen donor and thereby converts 3-phosphoserine (3-PS) and α-ketoglutarate (α-KG) into tricarboxylic acid (TCA) cycle. Dephosphorylation of phosphoserine via phosphoserine phosphatase (PSPH) produces serine, and then serine hydroxymethyltransferase (SHMT) converts serine into glycine and 5,10-methylenetetrahydrofolate (5,10-MTHF) via tetrahydrofolate (THF) supplying methyl.
The expression of 3-phosphoglycerate dehydrogenase
The nucleotide sequence of human PHGDH gene located at 1p12, which has 533 amino acid open reading frames (ORFs) sharing 88% and 94.0% homology with those of rat and mouse PHGDH, respectively [15], encodes a 56.6 kDa protein. However, the sequence similarity of the proximal promoter region (700 bp) of human PHGDH is 42% and 40% identical to its rat and mouse counterparts, respectively [16, 17]. This means that the regulation mechanism of human PHGDH gene expression differs from that of rats and mice. Similar to mouse PHGDH promoter, which previously demonstrated multiple transcription initiation sites at -136, -83, -81, -79, and -74 bases upstream from the first ATG codon, the human PHGDH promoter has Sp1 and NF-Y- binding sites in the absence of a TATA-box motif and thus showed multiplicity of transcriptional initiation sites [18, 19]. Two different transcripts of 3-PHGDH mRNA were detected in normal human tissues. A primary 2.1 kb transcript was markedly expressed in the testes, kidneys, ovaries, prostate, brain, liver, and pancreas and expressed at low levels in the colon, thymus, and heart. A 710bp transcript was also found at low levels predominantly in the heart and skeletal muscle [15]. Gromova et al. discovered that PHGDH has two major protein variants called α and β differing by approximately 3kDa in size and with each showing multiple modifications [20]. However, they did not prove that their sizes were consistent with the expected sizes of ENSEMBL transcripts ENST00000369409 and ENST00000369407 of PHGDH. Both translate into 533aa and 499aa proteins at 53.1 kDa and 56.6 kDa, respectively. Gromova et al. also found that the expression of PHGDH β variants could be caused by malignancy or deterioration of a malignant phenotype, but their relationship is unclear.
The type and structure of 3-phosphoglycerate dehydrogenase
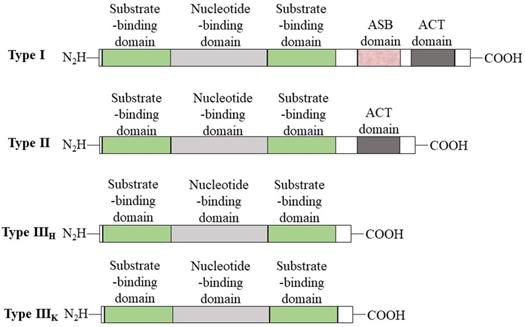
PHGDH is universally expressed in all organisms in at least three different basic structural forms, called types I, II, and III (Figure 2) [21]. These forms do not appear to be strictly specific for organism type, such as human, M. tuberculosis. rat and synechocystis have similar type I structures. PHGDH type III contains only substrate-binding and nucleotide-binding domains, which are structurally distinct and joined by two polypeptide chain segments at the active cleft site (Figure 3). Pyrococcus, Rhodopseudomonas and Clostridium have similar type III H, while Entamoeba hystolytica, Baterioides fragilis and Porphyromonas gingivalis have similar type III K. Type II has an aspartate kinase-chorismate mutase- tyrA perrhenate dehydrogenase (ACT) domain, a regulatory domain consisting of approximately 60-70 amino residues, and a βαββαβ structure. The ACT domain has been reported to function as a binding site for L-serine to provide feedback inhibition in E. coli and M. tuberculosis. However, this regulatory mechanism could not be confirmed for human PHGDH [22, 23]. The type I enzyme has an additional regulatory domain at the carboxyl terminal extremity, allosteric substrate-binding (ASB) domain, composed of approximately 150 amino acid residues with an αβααββ motif. The ASB domain is found between the substrate-binding domain and ACT domain and has been studied only in M. tuberculosis [24]. The function of the ASB domain is to provide additional levels of allosteric control. To date, no other protein containing the ASB domain has been found, except for L-serine dehydratase from Legionella pneumophila [25].
Basic domain structure of PHGDH. Basic domain structure found within the three enzyme types of PHGDH. Additional amino acids at the N-terminus change depending on the species. Two forms of the type III enzyme exist depending on whether histidine (type H) or lysine (type K) is present at the active site.
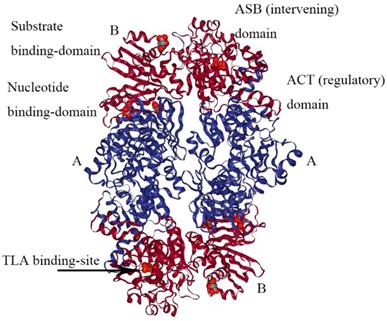
The structure of type III PHGDH. Each subunit is colored differently. NAD+ and D-malate (MLT) binding-site are designated with arrows.
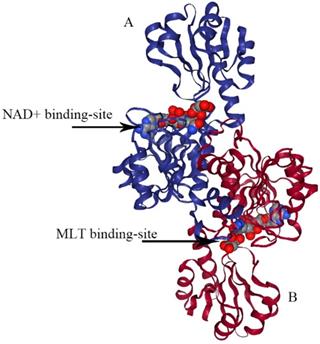
E.coli PHGDH is a tetramer consisting of selfsame subunits in regard to amino acid sequences. It contains three different structural domains as shown in Figure 4. The crystal structure of E.coli PHGDH was discovered by binding NAD+ plus a substrate analog (a-ketoglutarate) to the active site [26] and binding L-serine and NADH to the effector site and catalytic site, respectively [27]. When L-serine binds to the effector site, the conformation of the active site changes, resulting in the loss of activity. Binding research clarified that all four sites of the two ACT domains can be occupied with L-serine via a bridge formed by hydrogen bonds. The binding of serine to one site causes the other site to open and the last two sites have a negative cooperative relationship with the binding of the crystal structure [22].
The structure of E. coli PHGDH. The tetrameric structure of E. coli PHGDH is shown with NADH bound at the active sites and L-serine bound at the effector sites, which are designated with arrows. The subunits (A and B) are different colors for clarity and the structural domains of one subunit are indicated. Each subunit has the same amino acid sequence.
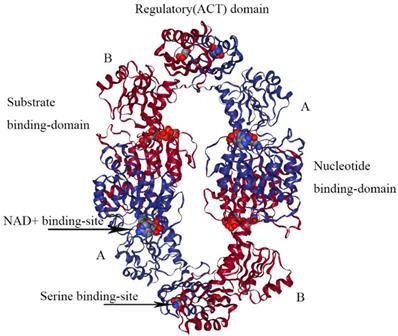
The crystal structure of M. tuberculosis PHGDH (PDB 1YGY) is dramatically different from that of E.coli PHGDH, although the catalytic domains adopt the same conformation in all four subunits. The structure of PHGDH has a distinct asymmetry as a result of the existence of the fourth domain, the ASB domain (Figure 5) [28]. To date, the full-length crystal structure of mammalian PHGDH remains unknown. Only the catalytic subunit of human PHGDH (PDB 2G76) has been solved as a dimer, probably because the crystallized truncated protein lacks the regulatory domains that influence inter-subunit interfaces. In human PHGDH, the active site consists of the substrate-binding domain and nucleotide domain. It is lined by several loop regions of the first monomers Arg54-Val59, Ala76-Val83, Asn97-Gly101, Gly152- Leu153, Asp175-Ile178, His206-Leu216, Cys234- Val240, Asp260-Asp269, and Cys281-Ser287 and one loop of the second monomer Trp133-Lys136 [29].
The structure of M. tuberculosis PHGDH. The structure of M. tuberculosis PHGDH (PDB code: 1YGY) is shown with each subunit (A and B) depicted in a different color for clarity. The active site histidine residue is shown with arrows.
PHGDH expression and cancer
The link between cancer and serine biosynthesis was first suggested by observations that PHGDH activity was greater in rat hepatoma cell lines than in normal liver cells, and the highest PHGDH activity led to the greatest growth rate [30]. Subsequent studies showed that PHGDH activity was most consistently increased and correlated with tumor growth in many other tumors [31]. To date, it has been reported that the level of PHGDH protein is increased in 16% of all cancers [32], 40% of melanoma samples [4], 70% of estrogen receptor (ER)-negative and triple-negative breast cancer samples, and even associated with subtypes of breast cancers[33]. This renewed interest in the serine synthesis relative with tumor development (Table 1).
The role of overexpression PHGDH in cancer
| Tumor types | Characteristics |
|---|---|
| Melanoma[4] | glucose metabolism improved, luminal organization and polarity disrupted, the viability of the inner, matrix-deprived cells to survive in an anchorage-independent fashion preserved, promoted oncogenesis |
| Estrogen receptor- negative breast [33] | poor prognosis, decreased overall survival, higher tumor grade, and high expression of the proliferative markers proliferating cell nuclear antigen and Ki-67 |
| Cervical[36] | large tumor, poor prognosis |
| Pancreatic[37] | advanced TNM stage, large tumor, higher tumor grade, rapid proliferation, migration and invasion |
| Colorectal[38] | advanced TNM stage, large tumor, tumor differentiation, and shorter overall survival time |
| Gastric[39] | histological type, tumor stage, and preoperative carcinoembryonic antigen |
| Lung adenocarcinoma[10] | high incidence, poor prognosis, rapid proliferation and migration |
| Leukemia[2] | high cell survival |
| Glioma[35] | PHGDH interacted with and stabilized FOXM1 at the protein level, promoting the proliferation, invasion and tumorigenicity of glioma cells |
| Breast cancer[49, 50] | high level of oncometabolite D-2-hydroxyglutarate (D-2HG) |
In metastatic variants of estrogen receptor-negative breast cancer cells, PHGDH and PSAT protein are highly expressed and related to poor prognosis, decreased overall survival, higher tumor grade, and high expression of the proliferative markers proliferating cell nuclear antigen and Ki-67 [33]. PHGDH regulates the oxidation of 3-PG, potentially influencing the NAD+⁄NADH ratio and cellular redox balance. Certain breast cancers depend on genomically amplified PHGDH, which diverts glucose carbons away from glycolysis into oxidative stress and biosynthetic pathways [4, 34]. In gliomas [35] and cervical [36], pancreatic [37], and colorectal cancer [38], the overexpression of PHGDH is associated with advanced TNM stage, large tumor, higher tumor grade, and shorter overall survival time, respectively. In the human pancreatic cancer cell lines BxPC-3 and SW1990, knockdown of PHGDH inhibits the cell proliferation, migration, and invasion abilities by downregulating the expression of cyclin B1, cyclin D1, MMP-2, and MMP-9. In gastric cancer, PHGDH was identified as an independent prognostic factor for outcomes, and the high expression of PHGDH was markedly related to histological type, tumor stage, and preoperative carcinoembryonic antigen [39]. Compared to the normal adjacent tissues, PHGDH expression is markedly and significantly elevated in lung adenocarcinoma and positively with poor prognosis. Accordingly, models with high levels of PHGDH display rapid proliferation and migration [10]. Reprogramming of tumor metabolism also involves leukemia cells. In leukemia cells (HL-60, K-562, and THP-1), inhibiting Gln metabolism leads to the upregulation of PHGDH, which maintains cell survival. Thus, it is necessary to combine the inhibition of Gln metabolism and serine metabolism in the treatment of hematological malignancies [2].
In most cases, tumor cell proliferation is promoted by increased level of PHGDH and supressed when PHGDH is knocked out or mutated in specific site. [40]. In some cases, extracellular serine is sufficient to promote tumor cell proliferation, whereas in other cases, extracellular serine is unable to support cell proliferation when PHGDH is absent. For example, adding excess serine to the growth medium of PHGDH knockdown PANC-1 cells was unable to rescue cell proliferation [41]. This suggests that PHGDH can promote cell proliferation not just by providing serine.
PHGDH plays a non-metabolic role or leads to additional enzymatic activity except supplying serine in cancer
PHGDH knockdown or inhibition inhibits the proliferation of some cancer cells in cultures in a manner that cannot be fully rescued by exogenous serine [4, 33, 42, 43].This demonstrated that PHGDH not only possesses metabolic enzyme activity by supplying serine but also plays a non-metabolic role or leads to additional enzymatic activity in tumorigenesis.
In glioma, PHGDH identified as directly upstream of FOXM1 prevents FOXM1, a proverbial oncogene, from proteasome degradation due to ubiquitination by interacting with its N-terminal, which facilitates tumor proliferation, invasion, and tumorigenicity [35]. In the translation initiation process, eIF4E recognizes m7 GTP on mRNA [44] and eIF4A unwinds the 5'mRNA structure to compose pre-initiation complexes [45]. In PANC-1 cells, PHGDH was found to interact with the translation initiation factors eIF4A1 and eIF4E and promote the assembly of the complex eIF4F on 5' mRNA structures to contribute to the expression of ZO-1 and E-cadherin involved in tight junctions [41, 46]. PHGDH possesses the dinucleotide-binding domain, which is close to its substrate-binding pockets, serving for RNA binding [47, 48]. It was speculated that the exact interactions between PHGDH and eIF4A1/eIF4E are related to its dinucleotide-binding domain and might also be close to its substrate-binding pockets. Therefore, reducing PHGDH expression and blocking the binding of PHGDH to eIF4A1/eIF4E provides a new anti-tumor therapeutic design.
PHGDH is a member of the isomer-specific 2- hydroxyacid dehydrogenase family, and its substrate is 3-phosphohydroxypyruvate, which is similar to α-ketoglutarate (α-KG) in structure. Evidence has demonstrated that PHGDH has additional enzymatic activity by converting NADH-dependent reduction of α-KG to the oncometabolite D-2-hydroxyglutarate (D-2HG) in breast cancer [49, 50]. D-2HG can competitively restrain α-KG-dependent dioxygenase enzymes, including DNA and histone demethylases [51-55]. Large amounts of D-2HG produced by IDH active site mutants in glioma and acute myeloid leukemia (AML) promotes the formation and malignant progression of gliomas and the transformation of leukemic cells [56-58].
The mechanisms of PHGDH amplification promoting tumor growth include supplying serine for protein synthesis and one-carbon metabolism, promoting the TCA cycle, and non-enzymatic functions including FOXM1 binding. Even overproducing the oncometabolite D-2HG impacts cell physiology. In the future, it is essential to explore the functional significance of PHGDH's multiple activities in different physiological and pathological settings.
The factors controlling PHGDH expression in cancer
PHGDH copy number gain is a reason for PHGDH high expression. PHGDH amplifies more quickly in triple-negative breast cancer than in other breast cancer subtypes, and PHGDH is more highly expressed in estrogen receptor-negative tumors than estrogen receptor-positive tumors [33]. PHGDH gene copy number gain is observed at a higher frequency in melanoma than other cancers [4, 33] and PHGDH expression can accelerate melanoma progression in mice [59].
In addition to amplification, PHGDH expression can be upregulated transcriptionally by Sp1/NF-Y [17], activating transcription factor 4 (ATF4)[60], protooncogene c-Myc[61], and TGF-β[62] and epigenetically by lysine methyltransferase G9A[63]. It has been clarified that Sp1 binds to the proximal GC-motif (-194 to -185) and NF-Y binds to the CCAAT-motif (165 to -154), dramatically regulating the promoter activity of the human PHGDH gene in HeLa cells by a ChIP assay. However, the positive regulatory region necessary for transcription was contained within the -196/+4 proximal sequence of the mouse PHGDH promoter, in which one Sp1-binding site was critical for the promoter activity. ATF4 has been reported as both a direct transcriptional target [64, 65] and heterodimerization partner of transcription factor nuclear factor erythroid-2- related factor 2 (NRF2). Human non-small cell lung cancer with high NRF2 protein expression displayed significantly higher expression of ATF4, PHGDH, PSAT1, and SHMT2, which was significantly related to poorer prognosis and higher tumor grade. It has also been verified that in NSCLC, NRF2 induced the expression of PHGDH, PSAT1, and SHMT2 by regulating ATF4 transcriptional activity to support glutathione and nucleotide production through serine biosynthetic metabolism. The indirect regulation mechanisms of ATF4 transcriptional activity by NRF2 are unknown, and it is speculated that NRF2 regulates serine biosynthesis genes combinatorially through ATF4 and additional factors [66]. Ben-Sahra reported that ATF4 can be activated by mTORC1 independent of its canonical induction downstream of eIF2α phosphorylation, stimulating nucleic acid synthesis, and helping cells adapt to amino acid deprivation and endoplasmic reticulum (ER) stress [67]. Methyltransferases play an essential role in the regulation of transcription by controlling the methylation of histone and DNA. G9A, an H3K9 methyltransferase, has a central role in catalyzing transcriptional activation of the serine-glycine biosynthetic pathway by upregulating the expression of PHGDH, PSAT1, PSPH, and SHMT with H3K9me1, an epigenetic marker associated with active chromatin [68, 69]. Moreover, higher G9A expression significantly increased serine and glycine biosynthesis in the cells depending on the upregulation of ATF4 [63]. In lung adenocarcinomas, differential PHGDH protein levels were defined by their degradation and stability, which were regulated by deubiquitinating enzyme JOSD2 [10].
The inhibitor of PHGDH against cancer
It is critical to find the inhibitor of PHGDH to treat patients with high PHGDH expression or PHGDH gene amplification. Many recent studies have reported compounds with activities against PHGDH.
Orthosteric inhibitors
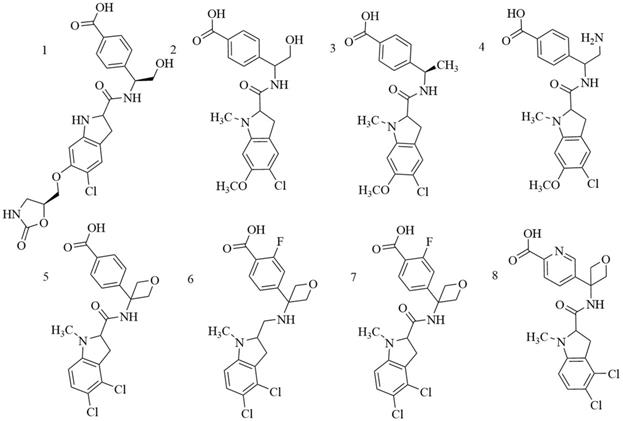
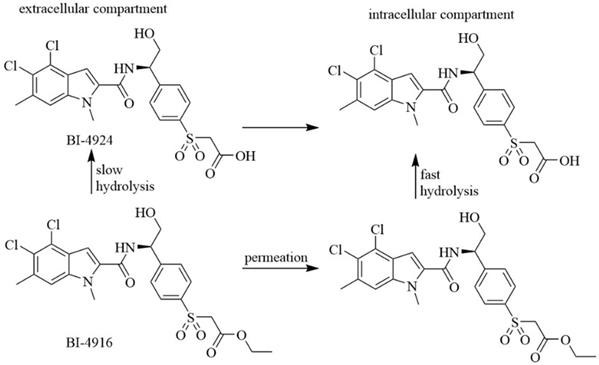
Indole derivatives. Mullarky et al. found a series of that bind the NAD+ pocket of PHGDH (Figure 6), inhibiting its activity and arresting the proliferation of cancer cells in serine-free media with low nanomolar affinities [70]. Weinstabl discovered a cofactor nicotinamide adenine dinucleotide (NADH/NAD+)- competitive PHGDH inhibitor, BI-4916, a prodrug of BI-4942. BI-4916 has shown high selectivity against the high cytosolic levels of the competitive cofactors NADH/NAD+ with high selectivity via an intracellular ester cleavage mechanism of the ester prodrug to achieve intracellular enrichment of the actual carboxylic acid-based drug (Figure 7) [71].
Chemical structures of indole derivatives.
BI-4916, a prodrug of the cofactor nicotinamide adenine dinucleotide (NADH/NAD+)-competitive PHGDH inhibitor BI-4924, has shown high selectivity against the majority of other dehydrogenase targets. An intracellular ester cleavage mechanism of the ester prodrug was utilized to achieve intracellular enrichment of the actual carboxylic acid-based drug to overcome high cytosolic levels of the competitive cofactors NADH/NAD+.
Allosteric inhibitors
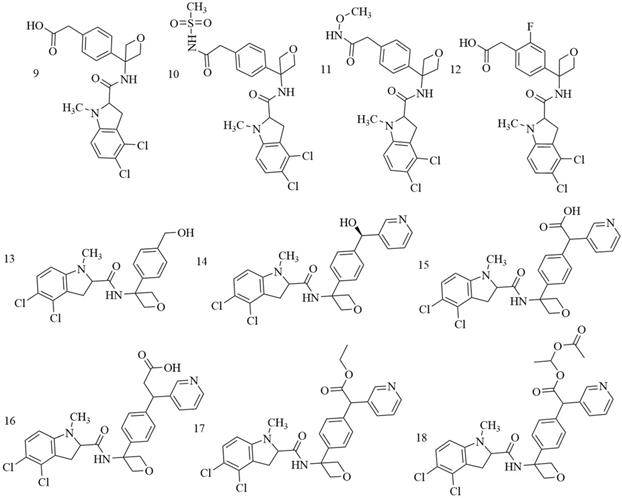
Mullarky et al. discovered PHGDH inhibitor by screening a library of 800,000 drug-like compounds and identified Compound 9 (CBR-5884) (Figure 8A) as superior [42]. CBR-5884 inhibited PHGDH enzymatic activity in a time-dependent manner with an IC50 of 33 ± 12 μM. CBR-5884 was identified as a covalent inhibitor that binds to a Cys in the non-active site and disrupts its oligomeric state. CBR-5884 inhibited the growth of cells by 35% to 60% in serine-abundant media, and by 80% to 90% in serine-depleted media at 30 μM. The substrate-binding pocket of PHGDH is rather small, approximately 100-200 Å3, and the physiological concentration of its cofactor NAD+ is as high as 0.3 mM [72]. These properties likely increase the difficulties of designing substrate-competitive inhibitors. Because NAD+ or NADH is a widely used cofactor, many studies focused on designing allosteric inhibitors for PHGDH that do not compete with the native ligand. Allosteric regulation can be achieved by various effectors, ranging from micromolecules to macromolecules and have high specificity, as allosteric-binding sites are not usually evolutionarily conserved [73]. Neither a direct binding test nor a postulated binding site was reported. CBR-5884 was unstable in mouse plasma and could not be used for in vivo testing. Spillier et al. found that disulfiram (DSF) (Figure 8B), an anti-alcohol drug, also inhibited PHGDH activity via converting PHGDH tetramer into either an inactive dimer covalently linked by a disulfide bridge involving Cys116 on adjacent monomers or, to a lesser extent, an inactive intermediate[74].
Chemical structures of PHGDH inhibitors. (A) CBR-5884. (B) DSF. (C) NCT-503. (D)α-ketothioamides. (E) PKUMDL-WQ-2101. (F) PKUMDL-WQ-2202. (G) Azacoccone E. (H) Ixocarpalactone A.
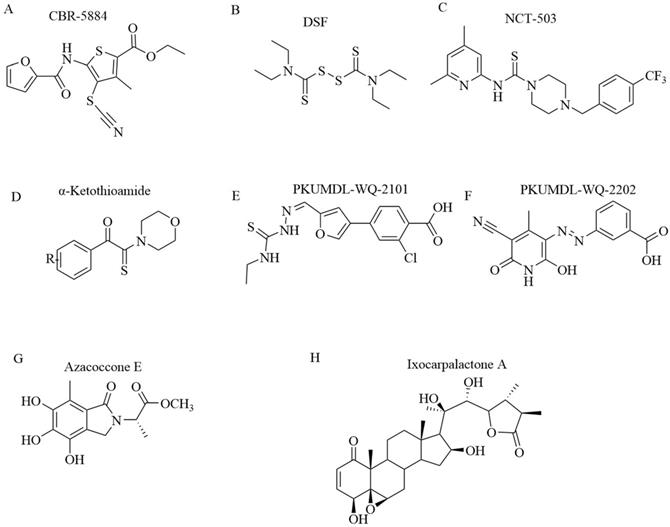
Similar to Mullarky et al., Pacold et al. found three PHGDH inhibitors by first screening a 400,000-compound NIH Molecular Libraries Small Molecule Repository (MLSMR) library and then optimizing the lead compounds [75]. The best compound, NCT-503 (Figure 8C), exhibited an IC50 value of 2.5 ± 0.6 μM and showed some selectivity in PHGDH-amplified breast cancer cell lines and had bioactivities in a xenograft model. NCT-503, also a non-competitive inhibitor in regards to substrates and cofactors, closely binds to the active site as a mutation of C234 in the protein's active site to reduce the inhibitory effect of PHGDH.
α-ketothioamide derivatives. Primary screening was implemented on a compound library of 336 molecules from a fragment library and an in-house compound collection at a high concentration (100 μM). Of 336 samples initially screened, 29 (8.6%) exhibited an inhibitory effect, and among them, 15 molecules (4.5%) decreased PHGDH activity by greater than 50%. A convergent pharmacophore approach contributed to the synthesis of α-ketothioamides by merging the arylketo moiety of 12-14 and the highlighted thioamide template after structural analyses (Figure 8D). Moreover, α-ketothioamides could selectively strain the proliferation of cancer cells with elevated PHGDH expression [76].
Qian Wang discovered novel non-NAD+ competing allosteric inhibitors for PHGDH using a structure-based design approach with the best IC50 of 28.1 ± 1.3 μM for enzyme inhibition. PKUMDL-WQ- 2101(Figure 8E) and PKUMDL-WQ-2201 (Figure 8F) were confirmed to specifically bind to PHGDH in PHGDH-amplified breast cancer cells with EC50 values less than 10 μM in serine-replete media. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 inhibited PHGDH activity mainly by forming hydrogen-bond networks with R134, K57, and T59 of site I and T59, T56, and K57 of site II, respectively, which limits the movement of the rigid domains and prevents the active sites from closing, thus stabilizing PHGDH in the inactive conformation [77]. Site I is close to the active site and the NAD+/NADH- cofactor-binding site, with a volume of 847 Å3 and a predicted maximal pKd of 8.71. It shares residues Gly 78, Val 79, Asp 80, Asn 81, and Val 82 with the active site. Site II is located in the substrate-binding domain, with a volume of 463 Å3 and a predicted maximal pKd of 7.79 [78, 79]. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 also suppressed tumor growth in mice.
Natural compounds. Guo J et al. found that natural compounds inhibit the activity of PHGDH. Azacoccone E, the first natural PHGDH inhibitor discovered by screening an in-house database of NPs, had an optimal IC50 of 9.8 ± 4.3 μM (Figure 8G). Furthermore, azacoccone E was a non-competitive inhibitor in a time-dependent manner as identified by the observed enzyme inhibition kinetics. Molecular docking demonstrated that azacoccone E fit at the allosteric site of PHGDH, which is essential for diminishing enzyme activity. A ligand-binding pocket with two protruding arginines and dramatically hydrophilic hydrogen bindings of Arg118 with two phenol groups and Arg113 with lactam carbonyl was predicted. Azacoccone E selectively inhibited PHGDH-expressing cancer cell proliferation and showed dose-dependent proapoptotic activity in HeLa cells [80]. However, the anti-tumor mechanism of azacoccone E remains unknown.
Zheng et al. extracted Ixocarpalactone A (Iox A) from dietary tomatillos (Physalis ixocarpa) and reported markedly inhibited PHGDH activity with an IC50 value of 1.66 ± 0.28 μM (Figure 8H). An MST assay and molecular docking experiments demonstrated that lox A directly coordinated at the allosteric site in the back side of the active site of PHGDH [81]. The ligand-binding pocket was dramatically hydrophobic, and hydrophobic interactions between Val301 and ring A, Pro331 and ring D, and Ile333 and ring B were predicted. Several hydrogen bonds were predicted between Gln302 with the carbonyl group of ring A, Asp305 and Thr313 with the 3-hydroxyl group, and Pro327 with the 22-hydroxyl group. Iox A was a non-competitive inhibitor relative to the substrate of NAD co-enzyme, which decreased the potential toxicities. Iox A selectively suppressed the proliferation of high PHGDH-amplified cancer cell lines and dose-dependently promoted the apoptosis of cells in a micromolar concentration. Iox A markedly restrained the tumor growth in a xenograft mouse model with low toxic effects. Iox A was identified as a novel natural PHGDH inhibitor with high specificity and low toxicity for the treatment of pancreatic cancer.
PHGDH and tumor resistance to chemotherapy drugs
Metabolic rewiring plays an essential role in the development of drug resistance [82-85]. In triple-negative breast cancer (TNBC) cells exposed to doxorubicin, the glucose flux for serine synthesis was increased by upregulating PHGDH. Serine was then transformed into GSH, which antagonized the doxorubicin-induced formation of ROS. Consequently, inhibition of PHGDH via shRNA caused doxorubicin-induced oxidative stress and increased doxorubicin sensitivity. The enhancement of doxorubicin efficacy through simultaneous suppression of PHGDH was demonstrated in a mouse tumor model [86]. In ER+ breast cancer, PHGDH may be a novel therapeutic target to reverse recurrence/resistance to tamoxifen therapy [87]. In multiple myeloma (MM) cells, the higher activity of SSP contributed to bortezomib resistance caused by upregulated PHGDH and increased anti-oxidant capacity. Interestingly, PHGDH is upregulated in different BTZ-resistant MM cells and serine starvation improves the efficacy of BTZ [88].
High PHGDH also contributes to tumor resistance to targeted therapy. In response to vemurafenib treatments of a vemurafenib-resistant melanoma cell line, SK-MEL-28VR1 established from parental BRAF V600E SK-MEL-28 cells, PHGDH was elevated to support cell proliferation by supplying nucleotides. Knockdown of PHGDH or using methotrexate to inhibit nucleotide metabolism by impeding the folate cycle downstream of serine biosynthesis can sensitize SK-MEL-28VR1 cells to vemurafenib [89]. The PHGDH level was also significantly increased in EGFR inhibitor erlotinib-resistant lung adenocarcinoma PC9ER4 and HCC827ER9 cells, which regulate the transcriptions of genes associated with DNA damage repair and nucleotide metabolism. Disturbing PHGDH by siRNA or NCT-503, an inhibitor of PHGDH, the tumoricidal effect was improved and sensitivity to erlotinib was restored in cell lines and xenografts [90, 91]. Additionally, suppression of PHGDH can eradicate advanced or metastatic clear cell renal cell carcinoma (ccRCC) resistant to HIF2α antagonists by inducing apoptosis [92]. PHGDH knocked down with RNAi and knocked out by CRISPR/Cas9 or inactivated by inhibitor can overcome tyrosine kinase inhibitor (TKI) drug resistance, including sorafenib, regorafenib, or lenvatinib, in hepatocellular carcinoma (HCC) [93]. Taken together, the high expression of PHGDH is dramatically related to tumor resistance to chemotherapies, and treatment with PHGDH inhibitor works synergistically with chemotherapy drugs and may be an effective approach to improve overall patient survival.
Conclusions
Targeting unique features of tumor metabolism is increasingly being deliberated as a successful approach. The strong dependence of cancer cells on certain nutrients or metabolites provides a therapeutic window and potentially has prognostic value. Although it has been more than 30 years since serine metabolism was proved to associated with tumorigenesis [31], the mechanism for this observation remains unclear. PHGDH is a rate-limiting enzyme in the serine biosynthetic pathway that consists of three enzymatic steps branching off the glycolytic pathway. Cells with high levels of PHGDH were connected with rapid proliferation, migration, and glycolytic metabolism supplying serine by SSP flux even when serine was available [33]. It is important to identify at what time point PHGDH expression is high during tumorigenesis, how PHGDH promotes tumorigenesis and metastasis, and the relationship between PHGDH and tumor resistance.
Due to the physiology of the blood-brain barrier, the de novo synthesis of serine in the central nervous system (CNS) is necessary to supply amino acids required in the brain [94]. To avoid potential neurological side effects, PHGDH inhibitor (or SSP enzyme inhibitor) must not cross the blood-brain barrier, obstructing serine homeostasis in the CNS. Many PHGDH inhibitors in the plasma are unstable and the anti-tumor mechanism of most parts of PHGDH remains unknown. Moreover, it is difficult to set thresholds to determine PHGDH-dependent and non-dependent tumors using existing techniques. Not only can serine be imported from the extracellular compartment via amino acid transporters and synthesized from glucose flux, but it can also be converted from glycine and glutamine. It may lead to resistance to PHGDH inhibitors by supplementing serine in other ways. Considering these factors, the targeted inhibition of PHGDH in tumor patients with high PHGDH expression will face significant challenges.
Abbreviations
3-PG: 3-phosphoglycerate; SSP: serine biosynthetic pathway; PHGDH: D-3-phosphoglycerate dehydrogenase; NADH: nicotinamide adenine dinucleotide; 3-PHP: 3-phosphohydroxypyruvate; PSAT: phosphoserine amino transferase; TCA: tricarboxylic acid cycle; 3-PS: 3-phosphoserine aminotransferase; PSPH: phosphoserine phosphatase; SHMT: serine hydroxymethyltransferase; THF: tetrahydrofolate; 5,10-MTHF: 5,10-methylenetetrahydrofolate; GSH: glutathione; α-KG: α-ketoglutarate; Glu: glutamate; ORF: open reading frame; NF-Y: nuclear factor Y; ACT: aspartate kinase-chorismate mutase-tyrA; ASB: allosteric substrate binding; ER: estrogen receptor; TGF-β: transforming growth factor-β; ATF4: activating transcription factor 4; NRF2: nuclear factor erythroid-2-related factor 2; NSCLC: non-small cell lung cancer; mTORC: mammalian target of rapamycincomplex; SAM: S-adenosylmethionine; G9A: histone H3 lysine 9 methyltransferase; BTZ: bortezomib; EGFR: epidermal growth factor receptor; eIF4F: eukaryotic translation initiation factor 4F; FOXM1: forkhead box protein M1; MMP-2: matrix metalloproteinase 2; D-2HG: D-2-hydroxyglutarate; AML: acute myeloid leukemia; TNBC: triple-negative breast cancer; ROS: reactive oxygen species; MM: multiple myeloma; ccRCC: clear cell renal cell carcinoma; TKIs: tyrosine kinase inhibitors; HCC: hepatocellular carcinoma; CNS: central nervous system.
Acknowledgements
This work was supported by Zhejiang Public Welfare Technology Research Program (LGF18H160029), Natural Science Foundation of Zhejiang province (LY18H160003), Doctor Foundation of Jinhua hospital (JY2019-3-001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
2. Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V. et al. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget. 2016;7:1765-76
3. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
4. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869-74
5. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11-20
6. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040-4
7. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191-8
8. Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. The Journal of cell biology. 2016;214:249-57
9. Newman AC, Maddocks ODK. Serine and Functional Metabolites in Cancer. Trends Cell Biol. 2017;27:645-57
10. Zhang B, Zheng A, Hydbring P, Ambroise G, Ouchida AT, Goiny M. et al. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017;19:2289-303
11. Liu S, Sun Y, Jiang M, Li Y, Tian Y, Xue W. et al. Glyceraldehyde-3-phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology. 2017;66:631-45
12. Pan S, Jang SY, Liew SS, Fu J, Wang D, Lee JS. et al. A Vinyl Sulfone-Based Fluorogenic Probe Capable of Selective Labeling of PHGDH in Live Mammalian Cells. Angew Chem Int Ed Engl. 2018;57:579-83
13. Blakley RL. The interconversion of serine and glycine: participation of pyridoxal phosphate. Biochem J. 1955;61:315-23
14. Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR. et al. Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J Biol Chem. 1993;268:11910-6
15. Cho HM, Jun DY, Bae MA, Ahn JD, Kim YH. Nucleotide sequence and differential expression of the human 3-phosphoglycerate dehydrogenase gene. Gene. 2000;245:193-201
16. Robbi M, Achouri Y, Szpirer C, Van Schaftingen E. The gene encoding rat 3-phosphoglycerate dehydrogenase. Mamm Genome. 2000;11:1034-6
17. Mitoma J, Furuya S, Shimizu M, Shinoda Y, Yoshida K, Azuma N. et al. Mouse 3-phosphoglycerate dehydrogenase gene: genomic organization, chromosomal localization, and promoter analysis. Gene. 2004;334:15-22
18. Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem. 2015;290:457-66
19. Jun DY, Park HS, Lee JY, Baek JY, Park HK, Fukui K. et al. Positive regulation of promoter activity of human 3-phosphoglycerate dehydrogenase (PHGDH) gene is mediated by transcription factors Sp1 and NF-Y. Gene. 2008;414:106-14
20. Gromova I, Gromov P, Honma N, Kumar S, Rimm D, Talman ML. et al. High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol Oncol. 2015;9:1636-54
21. Dey S, Hu Z, Xu XL, Sacchettini JC, Grant GA. D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J Biol Chem. 2005;280:14884-91
22. Grant GA. Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch Biochem Biophys. 2012;519:175-85
23. Grant GA. The ACT domain: a small molecule binding domain and its role as a common regulatory element. J Biol Chem. 2006;281:33825-9
24. Unterlass JE, Wood RJ, Basle A, Tucker J, Cano C, Noble MME. et al. Structural insights into the enzymatic activity and potential substrate promiscuity of human 3-phosphoglycerate dehydrogenase (PHGDH). Oncotarget. 2017;8:104478-91
25. Xu XL, Chen S, Grant GA. Kinetic, mutagenic, and structural homology analysis of L-serine dehydratase from Legionella pneumophila. Arch Biochem Biophys. 2011;515:28-36
26. Thompson JR, Bell JK, Bratt J, Grant GA, Banaszak LJ. Vmax regulation through domain and subunit changes. The active form of phosphoglycerate dehydrogenase. Biochemistry. 2005;44:5763-73
27. Schuller DJ, Grant GA, Banaszak LJ. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nature structural biology. 1995;2:69-76
28. Dey S, Grant GA, Sacchettini JC. Crystal structure of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase: extreme asymmetry in a tetramer of identical subunits. J Biol Chem. 2005;280:14892-9
29. Ravez S, Spillier Q, Marteau R, Feron O, Frederick R. Challenges and Opportunities in the Development of Serine Synthetic Pathway Inhibitors for Cancer Therapy. J Med Chem. 2017;60:1227-37
30. Davis JL, Fallon HJ, Morris HP. Two enzymes of serine metabolism in rat liver and hepatomas. Cancer Res. 1970;30:2917-20
31. Snell K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv Enzyme Regul. 1984;22:325-400
32. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J. et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905
33. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346-50
34. Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP. et al. The NAD(+) Salvage Pathway Supports PHGDH-Driven Serine Biosynthesis. Cell Rep. 2018;24:2381-91 e5
35. Liu J, Guo S, Li Q, Yang L, Xia Z, Zhang L. et al. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J Neurooncol. 2013;111:245-55
36. Jing Z, Heng W, Xia L, Ning W, Yafei Q, Yao Z. et al. Downregulation of phosphoglycerate dehydrogenase inhibits proliferation and enhances cisplatin sensitivity in cervical adenocarcinoma cells by regulating Bcl-2 and caspase-3. Cancer Biol Ther. 2015;16:541-8
37. Song Z, Feng C, Lu Y, Lin Y, Dong C. PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene. 2018;642:43-50
38. Jia XQ, Zhang S, Zhu HJ, Wang W, Zhu JH, Wang XD. et al. Increased Expression of PHGDH and Prognostic Significance in Colorectal Cancer. Transl Oncol. 2016;9:191-6
39. Xian Y, Zhang S, Wang X, Qin J, Wang W, Wu H. Phosphoglycerate dehydrogenase is a novel predictor for poor prognosis in gastric cancer. Onco Targets Ther. 2016;9:5553-60
40. Mattaini KR, Brignole EJ, Kini M, Davidson SM, Fiske BP, Drennan CL. et al. An epitope tag alters phosphoglycerate dehydrogenase structure and impairs ability to support cell proliferation. Cancer Metab. 2015;3:5
41. Ma X, Li B, Liu J, Fu Y, Luo Y. Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. Journal of Experimental & Clinical Cancer Research. 2019:38
42. Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN. et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113:1778-83
43. Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ. et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452-8
44. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF. et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422-37
45. Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol. 2004;5:232-41
46. Matter K, Balda MS. SnapShot: Epithelial tight junctions. Cell. 2014;157:992-e1
47. Castello A, Fischer B, Frese Christian K, Horos R, Alleaume A-M, Foehr S. et al. Comprehensive Identification of RNA-Binding Domains in Human Cells. Molecular Cell. 2016;63:696-710
48. Liao Y, Castello A, Fischer B, Leicht S, Foehr S, Frese CK. et al. The Cardiomyocyte RNA-Binding Proteome: Links to Intermediary Metabolism and Heart Disease. Cell Rep. 2016;16:1456-69
49. Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG. et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol. 2015;10:510-6
50. Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M. et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. The Journal of clinical investigation. 2014;124:398-412
51. Evans B, Griner E. Registered report: Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Elife. 2015;4:e07420
52. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484-8
53. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474-8
54. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463-9
55. Ye D, Guan KL, Xiong Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer. 2018;4:151-65
56. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44
57. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta neuropathologica. 2008;116:597-602
58. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine. 2009;361:1058-66
59. Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF. et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019;29:1410-21 e4
60. Adams CM. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem. 2007;282:16744-53
61. Nilsson LM, Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW. et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012;8:e1002573
62. Li Q, Gao H, Zhou S, Liao Y. LncRNA PlncRNA-1 overexpression inhibits the growth of breast cancer by upregulating TGF-beta1 and downregulating PHGDH. Breast Cancer. 2018;25:619-25
63. Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L. et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013;18:896-907
64. Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR. et al. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol. 2010;30:1007-13
65. Miyamoto N, Izumi H, Miyamoto R, Bin H, Kondo H, Tawara A. et al. Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE-19/HPV-16 cells. Investigative ophthalmology & visual science. 2011;52:1226-34
66. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475-81
67. Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728-33
68. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781-8
69. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491-507
70. Mullarky E, Xu J, Robin AD, Huggins DJ, Jennings A, Noguchi N. et al. Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorganic & medicinal chemistry letters. 2019;29:2503-10
71. Weinstabl H, Treu M, Rinnenthal J, Zahn SK, Ettmayer P, Bader G. et al. Intracellular Trapping of the Selective Phosphoglycerate Dehydrogenase (PHGDH) Inhibitor BI-4924 Disrupts Serine Biosynthesis. J Med Chem. 2019;62:7976-97
72. Yamada K, Hara N, Shibata T, Osago H, Tsuchiya M. The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Analytical biochemistry. 2006;352:282-5
73. Merdanovic M, Monig T, Ehrmann M, Kaiser M. Diversity of allosteric regulation in proteases. ACS Chem Biol. 2013;8:19-26
74. Spillier Q, Vertommen D, Ravez S, Marteau R, Themans Q, Corbet C. et al. Anti-alcohol abuse drug disulfiram inhibits human PHGDH via disruption of its active tetrameric form through a specific cysteine oxidation. Sci Rep. 2019;9:4737
75. Rohde JM, Brimacombe KR, Liu L, Pacold ME, Yasgar A, Cheff DM. et al. Discovery and optimization of piperazine-1-thiourea-based human phosphoglycerate dehydrogenase inhibitors. Bioorganic & medicinal chemistry. 2018;26:1727-39
76. Ravez S, Corbet C, Spillier Q, Dutu A, Robin AD, Mullarky E. et al. alpha-Ketothioamide Derivatives: A Promising Tool to Interrogate Phosphoglycerate Dehydrogenase (PHGDH). J Med Chem. 2017;60:1591-7
77. Wang Q, Liberti MV, Liu P, Deng X, Liu Y, Locasale JW. et al. Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem Biol. 2017;24:55-65
78. Yuan Y, Pei J, Lai L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Current pharmaceutical design. 2013;19:2326-33
79. Yuan Y, Pei J, Lai L. LigBuilder 2: a practical de novo drug design approach. Journal of chemical information and modeling. 2011;51:1083-91
80. Guo J, Gu X, Zheng M, Zhang Y, Chen L, Li H. Azacoccone E inhibits cancer cell growth by targeting 3-phosphoglycerate dehydrogenase. Bioorg Chem. 2019;87:16-22
81. Zheng M, Guo J, Xu J, Yang K, Tang R, Gu X. et al. Ixocarpalactone A from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct. 2019;10:3386-95
82. Liu H, Liu Y, Zhang JT. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther. 2008;7:263-70
83. Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL. et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71:4585-97
84. Staubert C, Bhuiyan H, Lindahl A, Broom OJ, Zhu Y, Islam S. et al. Rewired metabolism in drug-resistant leukemia cells: a metabolic switch hallmarked by reduced dependence on exogenous glutamine. J Biol Chem. 2015;290:8348-59
85. Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H. et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. 2016;10:73-84
86. Zhang X, Bai W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother Pharmacol. 2016;78:655-9
87. Gokmen-Polar Y, Neelamraju Y, Goswami CP, Gu Y, Gu X, Nallamothu G. et al. Splicing factor ESRP1 controls ER-positive breast cancer by altering metabolic pathways. EMBO Rep. 2019:20
88. Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017;5:7
89. Ross KC, Andrews AJ, Marion CD, Yen TJ, Bhattacharjee V. Identification of the Serine Biosynthesis Pathway as a Critical Component of BRAF Inhibitor Resistance of Melanoma, Pancreatic, and Non-Small Cell Lung Cancer Cells. Mol Cancer Ther. 2017;16:1596-609
90. Dong JK, Lei HM, Liang Q, Tang YB, Zhou Y, Wang Y. et al. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics. 2018;8:1808-23
91. Queiroz AL, Vakifahmetoglu-Norberg H, Norberg E. Resistant to Targeted Therapy - Aim for Metabolic Liabilities. Theranostics. 2018;8:2061-3
92. Yoshino H, Nohata N, Miyamoto K, Yonemori M, Sakaguchi T, Sugita S. et al. PHGDH as a Key Enzyme for Serine Biosynthesis in HIF2alpha-Targeting Therapy for Renal Cell Carcinoma. Cancer Res. 2017;77:6321-9
93. Wei L, Lee D, Law C-T, Zhang MS, Shen J, Chin DW-C. et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nature Communications. 2019:10
94. Tabatabaie L, Klomp LW, Berger R, de Koning TJ. L-serine synthesis in the central nervous system: a review on serine deficiency disorders. Molecular genetics and metabolism. 2010;99:256-62
Author contact
![]() Corresponding authors: djl9090com and xuwenxiaedu.cn
Corresponding authors: djl9090com and xuwenxiaedu.cn