Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Influence of TME and mechanical...
How cells sense mechanical...
Pathways regulating metastasis...
Future perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(12):2014-2028. doi:10.7150/ijbs.44943 This issue Cite
Review
Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis
Xingchen Li1, Jianliu Wang1,2 ![]()
1. Department of Obstetrics and Gynecology, Peking University People's Hospital, Beijing, 100044, China
2. Beijing Key Laboratory of Female Pelvic Floor Disorders Diseases, Beijing, 100044, China
Received 2020-2-14; Accepted 2020-4-15; Published 2020-4-27
Abstract

Metastasis is a complicated, multistep process that is responsible for over 90% of cancer-related death. Metastatic disease or the movement of cancer cells from one site to another requires dramatic remodeling of the cytoskeleton. The regulation of cancer cell migration is determined not only by biochemical factors in the microenvironment but also by the biomechanical contextual information provided by the extracellular matrix (ECM). The responses of the cytoskeleton to chemical signals are well characterized and understood. However, the mechanisms of response to mechanical signals in the form of externally applied force and forces generated by the ECM are still poorly understood. Furthermore, understanding the way cellular mechanosensors interact with the physical properties of the microenvironment and transmit the signals to activate the cytoskeletal movements may help identify an effective strategy for the treatment of cancer. Here, we will discuss the role of tumor microenvironment during cancer metastasis and how physical forces remodel the cytoskeleton through mechanosensing and transduction.
Keywords: metastasis, cytoskeleton, mechanical force, TRP channel, mechanotransduction
Introduction
Metastasis is the process by which cancers migrate to a distant organ and develop into a metastatic lesion[1]. However, cancer cells are unable to accomplish this process alone. The tumor microenvironment (TME) also is known to play an essential role in tumor metastasis [2]. Reciprocal biochemical and biophysical interactions among tumor cells, stromal cells and the extracellular matrix (ECM) result in a unique TME that determines disease outcome. The cellular component of the TME contributes to tumor growth by providing nutrients, assisting in the infiltration of immune cells, and regulating the production and remodeling of the ECM [3]. The TME consists of surrounding blood vessels, the extracellular matrix, secreted soluble factors, and other stromal cells [4, 5].
Mechanical forces within the TME play a pivotal role in driving physiological and pathological processes of cancers [6]. These forces have been identified as critical components of the TME and coordinate their behaviors during various biological processes, including cell division, survival, differentiation and migration [7, 8]. In solid tumor, mechanical force is caused by an elevation in the structural constitutions, particularly in the amount of cancer cells, stromal cells, and EMC components. With the increasing number of the cancer and noncancerous cells, the pressure inside the tumor rises and the signals of mechanical forces transfer to cancer cells, leading to mechanotransduction and cancer progression [9]. There are many types of stresses from TME could be loaded to cancer cells including substrate rigidity, fluid shear stress, hydrostatic pressure, and tensile and compressive forces [10].
Mechanosensing describes a cell's ability to sense mechanical cues from its microenvironment, including not only force, stress and strain, but also substrate stiffness, topography and adhesiveness. This ability is critical for cells to react to the surrounding mechanical cues and adapt to the varying environment [11]. Various mechanical signals are detected by and transmitted to the cells through activation of superficial mechanosensors such as integrins, G protein-coupled receptors (GPCR), transient receptor potential (TRP) ion channels, Piezo channels and YAP/TAZ [12-16]. The TME provides changing mechanical cues to the mechanoreceptors of cancer cells, which convey the signals to their internal machinery and affect the cellular behaviors. This communication process is called mechanotransduction and taking place in a continuous feedback cycle [17]. Mechanotransduction translates mechanical stimuli into biochemical signals, changing gene expression or regulating the cytoskeleton and membrane traffic, to ultimately alter cellular functions [18].
In response to mechanosensors, the cytoskeleton, an intracellular architecture composed of microtubules, microfilaments, and intermediate filaments that together determine the mechanical properties of cells, undergoes dramatic changes [19]. Cells are intricately connected to the external environment through their cytoskeleton, which receives external signals that guide complex behaviors such as lamellipodia formation, invasion and migration [20]. Whereas the contribution of chemical signals in the TME has long been understood, mechanical signals have only recently been widely recognized to be pervasive and powerful [21]. The cytoskeletal structure plays an integral role in transducing external mechanical signals to internal responses [22].
Physical forces mediate the cytoskeleton through mechanosensors by activating various pathways, such as GTP-binding protein RhoA [23], the Hippo pathway, the focal adhesion kinases (FAK), JAK/STAT, and PI3K-AKT pathways et al. Knowing the pathological mechanical force and signaling pathways is critical for selecting therapeutic strategies for metastatic cancers.
In this review, we will discuss recent progress towards an integrated understanding of the mechanical TME and its physical influence on cancers. Furthermore, we especially focus on how these mechanical signals transmitted by mechanosensors influence metastasis through cytoskeletal structures.
Influence of TME and mechanical properties of TME on tumor progression
Solid tumor is consisted of a complicated mixture of cancer cells and noncancerous cells. Overall, these noncancerous cells together with factors including the extracellular matrix, cytokines, growth factors, and hormones, make up the tumor microenvironment [24]. The major constitutions of TME include vascular, CAFs, immune cells, TAMs, tumor-associated endothelial cells, and ECM [25]. TME has an influence on the entire process of tumors from initiation to metastasis. What's more, tumor cells in turn influence the biochemical and biophysical properties of the TME to make TME conductive to the growth of tumor [26]. Variations in physical aspects, such as matrix stiffness, geometry, gradients of soluble factors, and electromagnetic fields are also features of the tumor microenvironment [27].
Within the last decade of cancer research, it has been shown that mechanical stimuli in the TME affect cells as profoundly as chemical signals do. Multiple substrates in TME produce or transmit mechanical signals to cancer cells, thus leading to the cancer cells acquiring features that are primarily focused on invasion and metastasis [28]. In this review, we concentrate on three components that play essential roles in the TME: ECM, TGF-β and CAFs.
ECM regulates cytoskeleton and metastasis through substrate stiffness in TME
ECM provides biochemical signals and mechanical support, which can both sustain cellular constituents. Fundamentally, the ECM is composed of proteoglycans (PGs), glycoproteins (GAGs) and fibrous proteins such as collagen, elastin, fibronectin (FN) and laminin, which are controlled by ECM and provide cancer cells with mechanical support [29]. Rather than serving simply as a physical support, the ECM is a physiologically active component of living tissue, responsible for cell-cell communication, cell proliferation and metastasis [30].
It has previously been proposed that tumors are stiffer than their surrounding normal tissue, and tumor stiffness is mainly determined by the amount of ECM, particularly collagen and hyaluronan [31]. ECM stiffness may cause intracellular contractions and a subsequent increase in the stiffness of an actin cytoskeleton that favors cancer migration [32]. Increasing ECM stiffness also induces malignant phenotypes, characterized by Rho-dependent cytoskeletal tension that leads to enhanced cell-ECM adhesions, disruption of cell-cell junctions and increased growth [33]. ECM stiffening can also enhance the connection between the ECM and the cytoskeleton through local adhesion proteins, and increase cytoskeletal tension by Rho/ROCK signaling activation [34]. The ECM also regulates cytoskeletal tension in hepatocellular carcinomas [35].
In addition, stiffening of the ECM is accompanied by an incremental increase in collagen deposition and a progressive linearization and thickening of interstitial collagen, which induces tumor aggression and causes immune cells to infiltrate tumor cells [36]. As the collagen receptor and the transmembrane connector between cellular cytoskeleton and ECM, integrin allows the transmission of forces, which the cells generate with their actomyosin onto the ECM [37, 38].
Matrix stiffening can also activate TGF-β signaling, which mediates epithelial-mesenchymal transition (EMT), leading to the acquisition of a more aggressive phenotype that promotes cancer metastasis [39]. EMT contributes pathologically to cancer progression through signaling pathways and among these, the actin cytoskeleton has played a predominant role [40]. Cells that undergo EMT reorganize their cortical actin cytoskeleton into one that enables dynamic cell elongation and directional motility [41]. Therefore, dysregulation of ECM composition and stiffness contributes to changes in cytoskeletal structure and enables metastasis [42].
TGF-β regulation of the cytoskeleton in Epithelial-Mesenchymal Transition
Furthermore, the TME also sequesters and locally releases growth factors including epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF), and other signaling molecules, such as WNTs and transforming growth factor-β (TGF-β) [43]. ECM components release these factors through ECM cleavage, and these factors also regulate ECM architecture and influence cell behaviors [44]. Mechanical stimuli can result in the local release or activation of the above-mentioned factors stored in the ECM [45]. Among all these factors, TGF-β is also stored in the matrix as part of a large latent complex and can be activated by cell contractile force. Matrix straining and stiffening lower the threshold for TGF-β activation by increasing the mechanical resistance to cell pulling [46].
TGF-β regulates the actin cytoskeleton in cancer cells, mainly by promoting EMT, an event of reorganization of cytoskeleton architecture and dissolution of the epithelial cell-cell junctions [41]. TGF-β-induced EMT is characterized by dramatic changes in cytoskeletal structure mediated in part by changes in the expression and organization of cytoskeleton proteins, including intermediate filaments, microtubules and microfilaments [47]. In carcinoma, increased expression and activation of TGF-β promotes the epithelial plasticity response, which leads to cancer cell invasion and dissemination [48]. Binding of TGF-β family proteins in cancer cells activated SMAD signaling pathway [49], which controls disruption and rearrangement of the actin-cytoskeleton [50]. In response to TGF-β, SMAD signaling not only activates the expression of EMT transcription factors such as SNAIL, MRTF and ZEB, but also increases their activity [51-53]. TGF-β also induces the activation of SMAD-independent pathways like Rho GTPases, p38MAPK and ERK1/2 [54], and drives actin reorganization and formation of lamellipodia and filopodia [55-57]. For example, one study showed that TGF-β induced activation of the RhoA-LIMK2-cofilin-1 pathway to modulate the actin cytoskeleton by increasing actin polymerization in colorectal cancer cells [58]. In non-small cell lung cancer, TGF-β induced EMT and migration by activating cytoskeleton microtubules and the functions can be attenuated by RCCD1 depletion [59]. It has been shown that TGF-β also causes changes in the cytoskeletons of gastrointestinal and prostate cancer cells [60, 61]. Tight junctions connect adjacent cells and associate with the intracellular actin cytoskeleton. TGF-β dissolves tight junctions and down-regulates the potential proteins including claudins, occludins and ZO1 [62].
In TGFβ-induced cytoskeletal rearrangement, miRNA participates in the process. For example, miR-155, which is expressed in response to TGF-β, targets the mRNA encoding RhoA, resulting in the dissolution of tight junctions [63]. TGF-β also induces the expression of miR-24, which targets neuroepithelial cell-transforming 1A (NET1A), a RHO-GEF that activates RhoA, therefore promoting EMT through the disruption of adhering and tight junctions [64].
CAF produces growth factors, chemokines and extracellular matrix to facilitate metastasis
By dynamically interacting with tumor cells, stromal cells participate in all stages of tumor initiation, progression, metastasis, recurrence and drug response, and consequently, affect the fate of patients. During the processes of tumor evolution and metastasis initiation, stromal cells in TME also experience some changes and play roles in both the suppression and promotion of metastasis. However, the overall function of stromal cells is beneficial for cancer cell survival and movement [65]. Stromal cells in the TME include endothelial cells, smooth muscle cells, pericytes, fibroblast cells and immune cells. Fibroblasts that are found in the TME are called Cancer-Associated Fibroblasts (CAFs) and can be identified by expression of certain markers, including α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP) and fibroblast-specific protein-1 (FSP1) [66]. A large percentage of tissue fibroblasts are transformed to CAFs that contain high levels of α-SMA. Therefore, it is proposed that TGF-β activates fibroblasts to become CAFs, which in turn produce more ECM fibers leading to desmoplasia [67].
During metastasis, CAFs are recruited by cancer cells and lead the way ahead of cancer cells to promote proliferation and migration by alignment of the actin cytoskeleton [68, 69]. They generate force to reorganize the matrix through a Rho-mediated myosin function, allowing them to clear the path for the cancer cells. Induction of the YAP transcription factor is required for the ability of CAFs to remodel EMT to support tumorigenesis. YAP induction in turn regulates multiple factors that modulate the cytoskeleton and matrix stiffness [70]. Expression of multiple cytoskeletal regulators is activated by YAP, which enables the fibroblasts to stiffen the surrounding matrix and promote cancer cell invasion [28]. FXR activation inhibits the tumor stimulatory activities of CAFs by governing cytoskeleton organization, stress fiber formation and contractility [71].
CAFs become activated by several tumor-derived growth factors such as TGF-β, which increases stiffness of CAFs by reorganizing their cytoskeletons to increase their elongation, cell spreading, lamellipodia formation and spheroid invasion [72]. TGF-β also enhances CAF formation, which is regulated by the rate of microtubule polymerization, depending on β-tubulin composition [73, 74].
CAFs are also the main contributors to ECM stiffness. CAFs interact with almost all cells within the TME and regulate cancer cell cytoskeletons indirectly through mediating EMC stiffness [75]. Hypoxia and TGF-β are the key inducers for CAFs in regulating TME stiffness and they strongly influence tumor and stromal cell properties such as proliferation and motility [32, 76].
CAFs respond differently to diverse levels of substrate stiffness, mainly by secreting the α-SMA and thus causing cytoskeleton remodeling and tumor invasion [77]. Mechanical stimuli also activate CAFs through MRTF-SRF and YAP-TEAD pathways interacting indirectly to control cytoskeletal dynamics [78]. In another study, Cdc42EP3 also responds to mechanical stimulation and plays a role in CAFs through tight regulation by Cdc42, which is a key regulator of cytoskeletal organization through its effects on actin assembly, actomyosin contractility and microtubules [79].
CAFs are known to possess the ability to reorganize the stromal cells by secreting ECM and enzymes that covalently cross-link collagen fibers, and by pulling the collagen network [80]. What's more, the contribution of secretions from tumor-associated macrophage (TAM)-like cells to the stimulation of mechanical property changes in TME is also an important factor for stiffness of the ECM in cancer cells [81] (Figure 1).
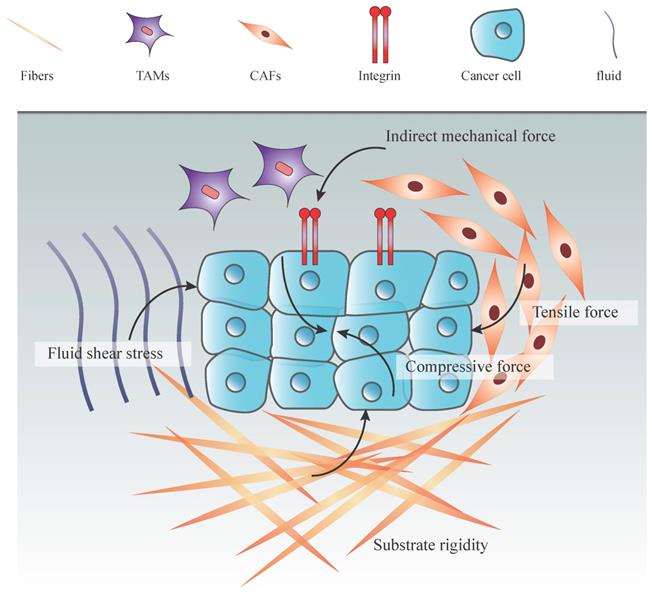
Types of mechanical forces in tumor microenvironment. An increasing number of cancer cells increas the solid stress among tumor cells, including tensile and compressive forces. Together with the ECM stiffening and proliferation of stromal cells, these forces promote the interstitial pressure. Moreover, fluid from leakage of blood vessels and secretion of stromal cells increase both fluid pressure and hydrostatic pressure. Indirect mechanical forces transduced by CAFs and TAMs to the mechanosensors (integrin) also play an essential role in the mechanical tumor microenvironment.
How cells sense mechanical signals and mediate the cytoskeleton
As we stated above, biomechanical elements, biochemical elements and stromal cells work together to control cancer cell fate during progression and are crucial for the maintenance of TME homeostasis. Loss of mechanical homeostasis in the TME accompanies tumorigenesis and also contributes to invasion and metastasis [82]. Nevertheless, the biomechanical nature of TME is influenced by both biochemical cues and stromal cells indirectly. For example, the secretion of biochemical factors such as TGF-β and MMPs activates alterations in the biomechanical properties of the TME and remodeling of the ECM [83]. Stromal cells can then regulate matrix alignment by releasing increasing amounts of proteases and auxiliary growth factors that trigger mechanical changes to the ECM [84], and CAFs are able to remodel the tumor matrix within the TME and provide the nutrients and chemicals to promote cancer cell invasion [85] and migration [86].
Progression of a tumor is characterized by increasing ECM stiffness. With stiffening of the ECM, the external force and plasticity of cancer cells increase [87]. Meanwhile, the ECM is a source of biochemical and biomechanical signals that promote tumor progression, and it is in turn strongly influenced by the cancer in a reciprocal relationship that is driven by the cytoskeletons of cancer cells [34]. Cells are equipped with several different mechanisms to sense the physical properties of the microenvironment and the mechanical forces arising within it [88]. Mechanical forces in the TME can mediate the cytoskeleton and promote cancer cell migration in two ways: by directly transducing mechanical force via the cytoskeleton, or indirectly by mechanosensors [89]. These mechanosensors translate mechanical forces into biochemical signals that trigger changes in the structure of cancer cell cytoskeletons [90]. Then, at the cellular level, cancer cells actively respond to externally applied forces and then couple to intracellular signaling pathways and effectors [91]. For instance, integrin-mediated adhesion of cells to matrix stimulates the activity of Rho GTPases and actin remodeling to regulate cell contractility and modify cellular behaviors such as survival and migration [92].
Mechanical force remodels the cytoskeleton directly
The cytoskeleton senses and transduces mechanical stress directly and the extracellular force mainly produced by the ECM conversely regulates cytoskeletal formation and structure [93, 94]. The principal components of the cytoskeleton include the actin filaments, microtubules and intermediate filaments [95]. Mechanical forces impact all three components of the cytoskeleton.
Previous reports have indicated that when a mechanical force is applied to cancer cells, the actin filaments act as a mechanosensor that senses the mechanical forces [96, 97]. They play a crucial role both in generating contractile forces by combining with the motor protein myosin II and by polymerization, which pushes the plasma membrane forward [98]. The pitch length of helical actin filaments was increased by the tensile force, thus reducing the affinity of cofilin and increasing the affinity of myosin II to the actin filaments [99]. Cancer cells interact with extracellular tension through this mechanism, which also helps them regulate cell proliferation and gene expression [100]. The extent of actin filament alignment and the direction of the applied force relative to this alignment are key determinants of mechanotransduction efficiency [101].
Microtubules (MTs) are highly dynamic structures involved in cellular growth, vesicular formation and especially mitosis. MTs are critical for mediating mechanical force-directed spindle organization and chromosome alignment in mitosis [102]. Tensional force at the surface of MTs is crucial for the mitotic spindle assembly checkpoint and the regulation of orientation and positioning [103]. Cell migration requires the involvement of MTs in the formation of pseudopodia responding to mechanical cues from TME, and IncRNA may participate in that progress [104]. Although these studies reveal the role of MTs in mechanoresponse, there is no direct evidence that MTs act independently as mechanosensors.
Compared with the mentioned actin filaments and MTs, intermediate filaments (IFs) are relatively stable filaments as the third component of the cytoskeleton, and are also flexible with the ability to be stretched 2.6-fold by tensile force [105]. Due to the stable and resilient nature of IFs, they serve as an essential component in sensing the strength and direction of mechanical forces endured by cancer cells [89]. It remains unclear whether IFs act independently as mechanosensors, but they are believed to be involved in mechanical response. Further studies are required to confirm the mechanisms of IF mechanical response.
Mechanical force-induced cytoskeleton modification indirectly by cellular mechanosensors
Mechanical force must be transduced to an intracellular signaling pathway in order to influence cell behavior. Cancer cells contain several mechanosensing components that jointly connect the ECM with the cytoskeleton, thus transducing mechanical signals into biochemical cascades [106]. Cellular mechanosensing is based on force-induced conformational changes in mechanosensitive proteins subjected to molecular forces. These changes result in opening of transmembrane channels or altered affinities to binding partners, thereby activating signaling pathways (Figure 2).
Components and roles of mechanical microenvironment in regulating cytoskeleton. Potential roles of biomechanical and biochemical factors in tumor microenvironment (TME). The TME embeds tumor cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), and their derivatives. The mechanical environment is mainly composed of extracellular matrix and its components such as proteoglycans (PGs), glycoproteins (GAGs) and collagenous fibers. The increased ECM deposition elevates its stiffness and rigidity. With the increasing rigidity of the ECM, mechanical force applied on tumor cells also increases. Mechanical force activates tumor cell progression directly by exerting pressure on the cell membrane or indirectly by putting force on integrins. The Rho/ROCK pathway is subsequently activated and contributes to the reorganization of F-actin. In addition, TGF-β and other growth factors are stored within the ECM and released in a tension-dependent manner. TGF-β combines with its ligands TβR-I/TβR-II. TGF-β regulates the actin cytoskeleton by promoting EMT, which is characterized by dramatic changes in cytoskeletal structure through the SMAD pathway. TGFβ-SMAD signaling activates the expression of EMT transcription factors, and SMAD complexes cooperate with these transcription factors to increase their transcriptional activities. In another way, TGF-β and hypoxia activate the CAFs which release α-SMA. α-SMA is transduced to the intracellular plasma and accelerates the formation and organization of the cytoskeleton. These biomechanical and biochemical parameters act together and lead to the reorganization of the cytoskeleton and the formation of lamellipodia, which act as the motile force and stimulate tumor cell invasion and metastasis.

Integrins
Cell-ECM interactions in both normal and pathological conditions are principally mediated via integrins. The ability of cells to sense ECM stiffness can be attributed to the integrins [107]. Considering that the communication between cancer cells and ECM takes place mainly through the ECM, the involvement of integrins in the mechanotransduction is significant [108]. Integrin-mediated adhesions interact with the ECM and sense its rigidity, which in turn regulates cellular behaviors such as motility and migration [109]. One critical mechanosensory response that underlies migration is the strengthening of ligand-integrin-cytoskeleton linkages under mechanical forces [110]. Integrins regulate cytoskeletal organization and activate intracellular signaling pathways, conveying both mechanical and chemical signaling.
The binding of integrins with specific components of the ECM initiates outside-in signaling that eventually triggers the regulation of the cytoskeleton, while the mechanical forces generated by the cytoskeleton can be transmitted to the integrin-ECM interaction causing cancer metastasis [111]. Integrin-mediated mechanotransduction also modulates gene expression via the nucleoskeleton [112]. Recent study revealed that physical deformation of the membrane, either by mechanical force or curvature, can induce integrin activation [113].
YAP/TAZ
Recently, the transcriptional regulators YAP and TAZ were found to be the key mediators of cell growth and differentiation activated by matrix rigidity [114]. These proteins can localize to the nucleus where they interact with transcription factors and promote expression of a number of genes involved in cell growth and differentiation [115]. Corruption of cell-environment interplay leads to aberrant YAP/TAZ activation, which is instrumental for multiple cell behaviors including cancer proliferation, metastasis and stemness essential for tissue regeneration [116].
YAP/TAZ localized in the cell membrane is directly regulated by ECM stiffness, cell shape and cytoskeleton tension, which are required for YAP/TAZ nuclear localization [114]. For example, YAP/TAZ is mechanically activated when they are cultured on the stiff ECM [117]. Several different mechanisms and molecules have been implicated to explain mechanotransduction, such as membrane dynamics, nuclear mechanics, Hippo signaling and the Rho pathway [118]. Integrins also have been reported to be the central transducers of mechanical cues from the ECM [119]. Tumorigenesis requires increased force transmission between oncogene-expressing cells and their surrounding extracellular matrix, and these regulations rely on YAP/TAZ mechanotransduction [120].
Given the critical connection between cell mechanics and YAP/TAZ activity, it is not surprising that YAP/TAZmay also function to reinforce the cytoskeleton and the mechanical properties of ECM responses to mechanical stress [121]. Expression of several regulators and components of the actomyosin cytoskeleton, including myosin IIB, myosin regulatory light chain 2 and filamin A, also is enhanced by YAP [122, 123]. Furthermore, YAP/TAZ activity is tightly coupled to actin cytoskeleton architecture and enhances the membrane-cytoskeleton integrity resulting in the viability of cancer cells during metastasis [124].
The reciprocal regulation between YAP/TAZ and cytoskeletons allows us to understand how cells translate and influence mechanical stimuli during the promotion of survival, migration and metastasis.
Transient Receptor Potential (TRP) ion channels
Transient receptor potential (TRP) cation channels represent a large and diverse family of ion channels that is sensitive to multiple environmental factors such as temperature, light and mechanical force both at organismal and cellular levels [125-127]. Discovery of these channels has greatly increased our understanding of mechanotransduction pathways in a variety of fundamental cell processes, and they are recognized as potential biomarkers for many types of cancer [14, 128, 129]. The majority of TRP channels are non-selective and mainly calcium-permeable. Dysregulation of intracellular calcium gating or expression of the TRP family causes deregulation of downstream effectors, thus promoting proliferation and metastasis of cancer cells.
Several TRP channels are gated from opening by the application of certain mechanical forces, and thus function as stretch-activated channels [130]. A mechanical stimulus could exert its influence on a TRP channel directly or alter the membrane tension that in turn indirectly opens the channel. For instance, mechanical tension elevates cAMP and activate PKA, thereby enhancing the mechanosensitive activation of some TRP channels associated with cell migration [131]. TRPM7 functions as a part of mechanosensory complex driving metastasis, formation and invasiveness of breast cancer cells, involving the kinase domain of the channel and phosphorylation of MAPK [132].
Mechanical force transmitted through adhesion sites can be translated into biochemical signals by TRP channels, which may serve to localize signal transduction pathways as well as intracellular cytoskeletal dynamics [133]. TRP channels are engaged in a reciprocal interplay with the cytoskeleton and control the TME for cytoskeletal dynamics [134]. However, they can also be regulated by the cytoskeleton. For instance, TRPM7 is involved in regulating myosin II-based cellular tension and modifying focal adhesion (FA) through F-actin and paxillin [135]. The interaction between TRPC1 and the calcium sensor STIM1 depends on an intact actomyosin cytoskeleton [136]. The bidirectional regulation between TRP channels and the cytoskeleton occurs in multiple signaling pathways. TRPM7 controls actomyosin dynamics by phosphorylation of cytoskeleton components in a kinase-dependent manner [137]. TRPV4 modifies the cytoskeletons in breast cancer cells, allowing them to migrate. This occurs via Ca2+-dependent activation of AKT and down-regulation of E-cadherin cell cortex proteins [138]. Other TRP subfamilies also play important roles in cancers through various mechanisms [139]. These findings provide new insight into the role of TRP channels in tumor progression and cytoskeletal structure.
Piezo1/2
Piezo channels are key force sensors, which sense the mechanical signals and transduce mechanical stimuli into intracellular signals typically by increasing cytosolic Ca2+ concentration [15, 140]. Piezo channels have two main variants, Piezo1 and Piezo2. Piezo1 is shown to be a cation-selective channel that does not require ancillary proteins for its activity, and senses changes in the rigidity of the environment [141]. Piezo2 is a critical regulator in tumor angiogenesis and vascular permeability [142]. Piezo channels are regulated by cellular signaling pathways, typically by calcium influx [143]. Piezo1 triggers a variety of intracellular processes, mainly related to cytoskeleton modulation. The actomyosin cytoskeleton exerts complex effects on Piezo1 activity and it in turn modulates cytoskeletal dynamics. Emerging data in the field support the idea for such a feedback loop [21]. For Piezo2, it generates Ca2+ influx that triggers downstream activation of the RhoA-mDia pathway, which is necessary for regulation of the actin cytoskeleton [144]. In conclusion, Piezo channels are closely associated with cytoskeletons and regulate their organization through calcium influx or interaction.
Pathways regulating metastasis through the cytoskeleton
The cytoskeleton has mechanical functions that involve supporting the structural integrity of the cell (such as cellular shape, adhesion and motility), and non-mechanical functions that may include the regulation of cellular architecture, cell growth, and stress pathways. Cytoskeleton reorganization is induced by the activation of multiple signaling pathways to modulate diverse physiological and pathological processes including angiogenesis, proliferation and migration. Many signaling pathways have been reported to have an impact on actin cytoskeleton regulation through metastasis. The molecules in the pathways are major drivers of many of the steps required for metastatic success. In this section, we will focus on some key regulators in the signaling pathways controlling the cytoskeletal organization processes important for metastasis.
RhoA/ROCK
One of the main cellular signaling pathways involved in regulating actin and cytoskeletons is the Rho family GTPase signaling pathway. This pathway has been identified as a fundamental contributor controlling several biochemical pathways underlying migration, such as cytoskeletal dynamics, directional sensing, cell-cell junction assembly/disassembly and integrin-matrix adhesion [20]. To date, approximately 20 Rho GTPases have been reported in humans (of which Rho, Rac, and Cdc42 are the best studied), which orchestrate the remodeling of actin-containing cytoskeletal structures and regulate the cell contractile machinery to control many cellular processes [145].
Ras homolog gene family member A (RhoA) plays a key role in the regulation of actin polymerization, basement membrane disassembly and cortical contractility [146]. Several proteins, including Rho-associated protein kinase (ROCK), formins, and other scaffolding molecules, have been identified as downstream targets of RhoA [147]. ROCK, the major target of RhoA, directly phosphorylates Lim kinase (LIMK), an actin cytoskeleton regulator [148]. LIMK also acts as an important downstream effector of Rho directly [149]. Cofilin is a key regulator of actin severing, nucleation and capping within protrusive machinery. The phosphorylation of cofilin at Ser3 blocks the actin binding interface, preventing its actin binding function and promoting F-actin stability and elongation [150].
Ras-related C3 botulinum toxin substrate 1 (Rac1) reorganizes the actin cytoskeleton to promote formation of large membrane protrusions, called lamellipodia, which drive motility in many cell types [151]. Cell division control protein 42 homolog (Cdc42) promotes the formation of actin-rich microspikes to sense extracellular chemotactic gradients and initiates directed cell movement [152]. Cdc42 signaling also can generate actomyosin contraction through p21 protein (Cdc42/Rac)-activated kinase 2 (PAK2) and Cdc42-binding kinase (MRCK) kinases, which are related to myotonic dystrophy kinases [153]. Shear stress and Cdc42 activation are also sufficient to promote filopodia formation by adjusting cytoskeleton in cancer [154]. Rho family GTPase play a crucial role in a range of human diseases and is now considered as a potential target for the treatment of several malignancies including gastric cancer [155], breast cancer [156] and prostate cancer [157].
Rho-ROCK signaling is a key regulator of actomyosin contractility and regulates cell shape, cytoskeletal arrangement and thereby cellular functions such as cell proliferation, differentiation, motility and adhesion. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness [158]. Rho-ROCK signaling has been shown to promote cancer cell growth, migration and invasion [148]. Pre-clinical evidence supports a role for Rho-ROCK signaling in enhancing the malignancy of cancers. Pharmacological inhibition of Rho-ROCK using either Fasudil or Y-27632 decreased the invasion, stress fiber organization and migration in pancreatic adenocarcinoma cells and breast cancer cells in vitro [159], suggesting a cell-autonomous role for Rho-ROCK signaling in tumor progression.
G protein-coupled receptors (GPCRs)
G protein-coupled receptors (GPCRs) are a family of proteins able to promote changes in cell shape. To date, GPCRs transduce signals into rapid changes on the actin-cytoskeleton via the activation of small GTP-binding proteins of the Rho GTPases family, such as Rho, Rac and Cdc42. GPCRs also modulate cAMP levels in order to exert diverse effects on cytoskeleton remodeling [160]. As the effector of cAMP, PKA induces stabilization of the microtubule cytoskeleton, resulting in the inhibition of RhoA activity, myosin light chain (MLC) phosphorylation, and actomyosin contractility [161]. Activated myosin connects the actin filaments to form stress fibers that generate actomyosin force to facilitate cell movement.
Actin polymerization in response to GPCR activation may imply the recruitment of proteins capable of modulating the activation of RhoA-mediated pathways, which lead to the formation of actin stress fibers [162]. Chemokine signals participate in the recruitment of cells during metastasis, promoting spreading of different types of tumors. Chemokine receptors coupled to GPCR proteins induce cell motility and actin reorganization, acting through activation of Rho, PI3K and MAPK signaling [163]. GPCRs can also activate the Hippo mechanical pathway by pressure-controlled YAP regulation [164].
As a steroid-acting GPCR, G-protein estrogen receptor (GPER) has important transcription-dependent outcomes in the regulation of cell growth and programmed cell death secondary to GPER-regulated second-messenger pathways [165]. GPER is expressed ubiquitously and has diverse biological effects, including regulation of proliferation and migration [166]. Non-genomic transcriptional effects induced by estrogen regulate F-actin cytoskeleton assembly and breast cancer cell metastasis through GPER acting on the Rho/ROCK-LIMK-cofilin pathway [167].
PI3K-mTOR-RhoA/Rac
Phosphatidylinositol 3-kinase (PI3K) plays a central role in a complex, multi-armed signaling network that orchestrates cell responses including cell proliferation, migration and glucose metabolism [168]. PI3K is presumed to activate most of its downstream targets via the Akt protein, which phosphorylates diversified downstream substrates including the mammalian target of rapamycin (mTOR), a master regulator of protein translation [168].
mTOR signaling is activated in conditions of migration deregulation in many cancer types including breast, ovarian, renal and glioblastoma [169]. mTOR is a downstream effector of the PI3K/AKT pathway and forms two distinct multiprotein complexes: mTORC1 and mTORC2. mTORC2 functions to regulate spatial aspects of yeast cell growth, by controlling the actin cytoskeleton [170]. It is activated by cancer hallmarks, phosphorylates PKC-α, PI3K/AKT and regulates the activity of Rho GTPase, which is related to the regulation of the actin cytoskeleton. mTORC2 signals to the actin cytoskeleton by activating a RhoA GTPase switch. Upon activation, RhoA interacts and activates PKC1, which in turn signals to the actin cytoskeleton via the MAPK pathway [171]. mTORC2 also signals to the actin cytoskeleton, and although the direct targets of mTORC2 are unknown, this signaling may involve PKCα and the small GTPases Rho and Rac [172]. PI3K/AKT can also inhibit the mechanical and mechanosensing properties of tumor cells [173].
Hence, the mTOR pathway is a central coordinator of fundamental biological events, playing a key role in cell growth and regulation of the actin cytoskeleton and cell survival [174].
The Hippo pathway
The Hippo signaling pathway regulates diverse physiological processes, and genetic deletion or aberrant expression of some Hippo pathway genes leads to enhanced cell proliferation, tumorigenesis and cancer metastasis [175]. There are many upstream signals that regulate the Hippo pathway, among which rearrangement of the cytoskeleton exerts the strongest effects on the pathway by the action of cell-cell and cell-matrix junction components and by the mechanical properties of the ECM [176-178]. The actin cytoskeleton is believed to play a critical role in relaying mechanical forces to Hippo signaling, but the exact biochemical mechanisms by which the actin cytoskeleton impacts the core kinase cascade remain poorly understood.
The Hippo kinase cascade converges on its nuclear effector Yki/YAP/TAZ to regulate gene expression programs. Phosphorylation of Yki/YAP/TAZ by Hippo signaling inactivates these transcriptional co-activators by excluding them from the nucleus, and additionally for YAP/TAZ, by promoting their degradation [179]. When Hippo signaling is low, Yki/YAP/TAZ enters the nucleus to drive gene expression. Other identified regulators of the Hippo pathway include mechanical cues, ligands of GPCRs, cell polarity, energy status, and hormonal signals [180].
Ca2+ acts as the secondary messenger
Calcium is one of the most important elements for human beings and usually acts as a secondary messenger. Proper control of Ca2+ signaling through the actin cytoskeleton is mandatory and critical for cancer cell metastasis. At the cellular level, increases in Ca2+ trigger a wide variety of physiological and pathological processes. Thus, it is not surprising that aberrant Ca2+ signaling can induce malignancy for a broad spectrum of diseases.
Local Ca2+ pulses are generated from two sources, the internal Ca2+ storage and the external Ca2+ influx. Most internal Ca2+ signals originate from the endoplasmic reticulum (ER) through inositol triphosphate (IP3) receptors and extracellular calcium influx is induced by calcium channels of the TRP family, Piezo1/2, and Cav family, for example. Some of the cellular calcium signaling is triggered by mechanical force [181]. Increased Ca2+ concentration in the cytoplasm is regulated locally and globally for effective cytoskeletal remodeling, cell migration and cancer metastasis. Ca2+ pulses and spikes occurring at particular locations and times activate numerous downstream structural and signaling targets [182].
Ca2+ signaling regulates the Rho GTPases [183], which are mandatory for the formation of actin bundles in lamellipodia, focal adhesion complexes, and filopodia, which are the major components for cell migration. Although the present data reveals no evidence of direct binding between Ca2+ and Rho GTPases, it is reasonable to expect their mutual interactions considering their perfect coordination during cell migration [184]. It has been shown that constitutively active Rac1 fully counteracted the effects of SOC influx inhibition in migrating breast cancer cells [185], indicating the regulatory role of Ca2+ on Rho GTPases. Knockdown of the calcium channel reduces calcium influx, thus decreasing RhoA activity [144]. There are many other Ca2+-related pathways that regulate cytoskeletal proteins in cancer, such as ERKs [186] and lipid signaling [187].
As we previously described, the oscillation of local Ca2+ pulses activate the Rho GTPases in the front of migrating cells and synchronizes with the retraction phases of lamellipodia [188]. STIM1-ORAI1 also plays a key role in the control of Ca2+ entry at the leading edge of migrating cells [189], where this Ca2+ mobilization enhances the reorganization of the cortical cytoskeleton required for the formation of filopodia and lamellipodia [190].
In addition, the F-actin severing protein cofilin also depends on the cytosolic Ca2+ for its proper activity. Moreover, myosin, as one of the major actin regulators, is totally dependent on Ca2+ for proper activity [191]. Therefore, though not a direct regulator, Ca2+ modulates actin dynamics through multiple signaling pathways and structural molecules (Figure 3).
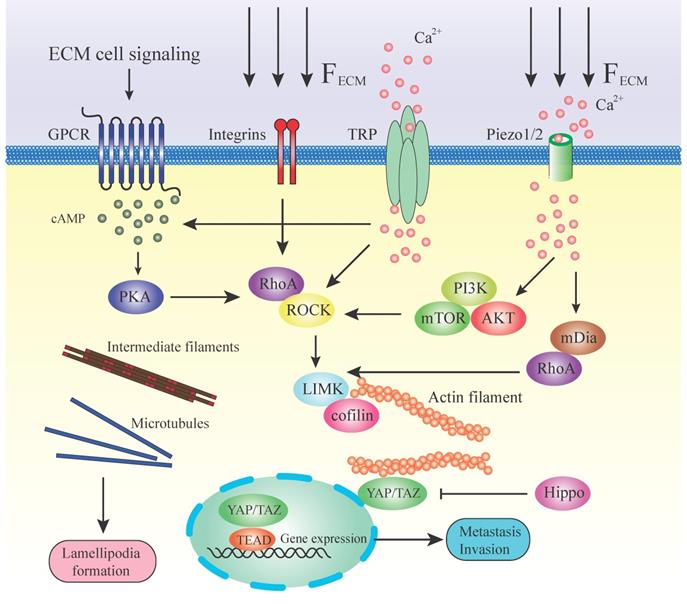
Mechanotransduction and pathways regulating cytoskeleton during metastasis. This illustration summarizes some of the mechanisms by which a stiff ECM or extracellular signaling alters cell fate through mechanotransducers. A stiffened matrix prompts cell tension and generates mechanical force, activating different mechanosensors. RhoA/ROCK is located in the center position among all these molecules in the pathways. RhoA is facilitated by numerous effector proteins. GPCR, via activation of cAMP and PKA, enhances the expression of RhoA. Integrins activate the RhoA directly by transducing the mechanical ECM force. TRP channels and Piezo 1/2 can sense the ECM force and lead to the release of Ca2+. Ca2+ influx stimulates RhoA or via PI3K/AKT/mTOR and mDia. Downstream of RhoA/ROCK lies the key regulators of cytoskeleton LIMK/cofilin. ROCK activates LIMK, which subsequently inhibits cofilin via phosphorylation. Cofilin facilitates actin filament severing and depolymerization; therefore, its inhibition results in elevated polymerized actin stability. LIMK also directly enhances actin polymerization and myosin contractility. This, in turn, initiates a paracrine signaling mechanism that causes increased production of ECM components in the tumor microenvironment. High expression of actin contractility and inhibition of the Hippo pathway activates YAP/TAZ function. YAP/TAZ accumulates in the nucleus and regulates gene transcription together with DNA‑binding transcription factors such as TEAD. ECM directly transduces the mechanical force to intermediate filaments and microtubules and assists in the formation of lamellipodia.
Future perspectives
In this review, we reported on the critical role of the mechanical TME in cancer cell migration and the actin cytoskeleton. We focused on essential mechanisms by which mechanical signals produced by ECM and stromal cells in the TME promote cancer metastasis through the regulation of cytoskeletal dynamics and biomechanical and biochemical transduction occurring during this regulation. Under normal conditions, cell motility and cytoskeletal structure are tightly controlled in order to maintain cell physiological activity and tissue homeostasis. If either the mechanical microenvironment or the expression of mechanotransduction molecules is disturbed, the balance of the mechanical microenvironment and cell will have to be reorganized to restore equilibrium.
Often, structure, mechanotransduction, and cellular behavior are tightly linked. The current challenge that remains is to distinguish the multiple target markers in the process of the TME-cellular function relationship. In particular, there is still a lack of understanding of how the stromal components generate the physical force and transduce to modify the cytoskeletal reorganization and cellular functions such as cancer metastasis. What's more, another great challenge is lacking of methods to gauge the specific mechanical pressure during the whole progression of tumor. Understanding these processes may provide us with new clues in addressing the question of how cells manage to sense their physical environment and how we could intervene during the metastasis.
Understanding the basic mechanisms of mechanical properties in TME contributes to the progress of tumor treatment and this will bring hope to thousands of patients with cancer. Studying the mechanisms that underlie these processes and identifying key molecular targets will also lead us to new therapeutic strategies and further exploration of new therapeutic approaches based on the mechanical signals generated by tumors. It will also provide us with innovative treatment and potential curable strategy from another new point of view.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No 81874108 and 81802607).
Author Contributions
X.L. contributed to conceptualization and writing of the manuscript. J.W. helped finalized the presented idea and supervised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559-64
2. Yuan Y, Jiang YC, Sun CK, Chen QM. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncology reports. 2016;35:2499-515
3. Johan MZ, Samuel MS. Rho-ROCK signaling regulates tumor-microenvironment interactions. Biochemical Society transactions. 2019;47:101-8
4. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423-37
5. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
6. Moraes C, Sun Y, Simmons CA. (Micro)managing the mechanical microenvironment. Integrative biology: quantitative biosciences from nano to macro. 2011;3:959-71
7. Ladoux B, Mege RM. Mechanobiology of collective cell behaviours. Nature reviews Molecular cell biology. 2017;18:743-57
8. Subramanian A, Kanzaki LF, Galloway JL, Schilling TF. Mechanical force regulates tendon extracellular matrix organization and tenocyte morphogenesis through TGFbeta signaling. eLife. 2018 7
9. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annual review of biomedical engineering. 2014;16:321-46
10. Jang I, Beningo KA. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers. 2019 11
11. Chen Y, Ju L, Rushdi M, Ge C, Zhu C. Receptor-mediated cell mechanosensing. Molecular biology of the cell. 2017;28:3134-55
12. Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. The Journal of cell biology. 2016;215:445-56
13. Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15463-8
14. Shapovalov G, Ritaine A, Skryma R, Prevarskaya N. Role of TRP ion channels in cancer and tumorigenesis. Seminars in immunopathology. 2016;38:357-69
15. He L, Si G, Huang J, Samuel ADT, Perrimon N. Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature. 2018;555:103-6
16. Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M. et al. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS letters. 2014;588:2663-70
17. Chandorkar Y, Castro Nava A, Schweizerhof S, Van Dongen M, Haraszti T. et al. Cellular responses to beating hydrogels to investigate mechanotransduction. Nature communications. 2019;10:4027
18. Iskratsch T, Wolfenson H, Sheetz MP. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nature reviews Molecular cell biology. 2014;15:825-33
19. Pachenari M, Seyedpour SM, Janmaleki M, Babazadeh Shayan S, Taranejoo S. et al. Mechanical properties of cancer cytoskeleton depend on actin filaments to microtubules content: investigating different grades of colon cancer cell lines. Journal of biomechanics. 2014;47:373-9
20. Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. British journal of pharmacology. 2014;171:5507-23
21. Nourse JL, Pathak MM. How cells channel their stress: Interplay between Piezo1 and the cytoskeleton. Seminars in cell & developmental biology. 2017;71:3-12
22. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485-92
23. Marjoram RJ, Lessey EC, Burridge K. Regulation of RhoA activity by adhesion molecules and mechanotransduction. Current molecular medicine. 2014;14:199-208
24. Hirata E, Sahai E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harbor perspectives in medicine. 2017 7
25. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer letters. 2017;387:61-8
26. Meng W, Xue S, Chen Y. The role of CXCL12 in tumor microenvironment. Gene. 2018;641:105-10
27. White FM, Gatenby RA, Fischbach C. The Physics of Cancer. Cancer research. 2019;79:2107-10
28. Nagelkerke A, Bussink J, Rowan AE, Span PN. The mechanical microenvironment in cancer: How physics affects tumours. Seminars in cancer biology. 2015;35:62-70
29. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Advanced drug delivery reviews. 2016;97:4-27
30. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. Journal of cell science. 2010;123:4195-200
31. Gkretsi V, Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Frontiers in oncology. 2018;8:145
32. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nature reviews Cancer. 2014;14:430-9
33. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI. et al. Tensional homeostasis and the malignant phenotype. Cancer cell. 2005;8:241-54
34. Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D. et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer cell. 2011;19:776-91
35. Pang M, Teng Y, Huang J, Yuan Y, Lin F. et al. Substrate stiffness promotes latent TGF-beta1 activation in hepatocellular carcinoma. Biochemical and biophysical research communications. 2017;483:553-8
36. Acerbi I, Cassereau L, Dean I, Shi Q, Au A. et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative biology: quantitative biosciences from nano to macro. 2015;7:1120-34
37. Kim SN, Jeibmann A, Halama K, Witte HT, Walte M. et al. ECM stiffness regulates glial migration in Drosophila and mammalian glioma models. Development. 2014;141:3233-42
38. Eble JA, Niland S. The extracellular matrix in tumor progression and metastasis. Clinical & experimental metastasis. 2019;36:171-98
39. Mortezaee K. Human hepatocellular carcinoma: Protection by melatonin. Journal of cellular physiology. 2018;233:6486-508
40. Kim HY, Nelson CM. Extracellular matrix and cytoskeletal dynamics during branching morphogenesis. Organogenesis. 2012;8:56-64
41. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178-96
42. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nature reviews Molecular cell biology. 2014;15:786-801
43. Kanematsu A, Marui A, Yamamoto S, Ozeki M, Hirano Y. et al. Type I collagen can function as a reservoir of basic fibroblast growth factor. Journal of controlled release: official journal of the Controlled Release Society. 2004;99:281-92
44. Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216-9
45. Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Current rheumatology reports. 2009;11:120-6
46. Hinz B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix biology: journal of the International Society for Matrix Biology. 2015;47:54-65
47. Nalluri SM, O'Connor JW, Gomez EW. Cytoskeletal signaling in TGFbeta-induced epithelial-mesenchymal transition. Cytoskeleton. 2015;72:557-69
48. Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Current opinion in oncology. 2013;25:76-84
49. Moustakas A, Heldin CH. Dynamic control of TGF-beta signaling and its links to the cytoskeleton. FEBS letters. 2008;582:2051-65
50. Caligaris C, Vazquez-Victorio G, Sosa-Garrocho M, Rios-Lopez DG, Marin-Hernandez A. et al. Actin-cytoskeleton polymerization differentially controls the stability of Ski and SnoN co-repressors in normal but not in transformed hepatocytes. Biochimica et biophysica acta. 2015;1850:1832-41
51. Hoot KE, Lighthall J, Han G, Lu SL, Li A. et al. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. The Journal of clinical investigation. 2008;118:2722-32
52. Morita T, Mayanagi T, Sobue K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. The Journal of cell biology. 2007;179:1027-42
53. Nishimura G, Manabe I, Tsushima K, Fujiu K, Oishi Y. et al. DeltaEF1 mediates TGF-beta signaling in vascular smooth muscle cell differentiation. Developmental cell. 2006;11:93-104
54. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME. et al. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603-10
55. Ridley AJ. Life at the leading edge. Cell. 2011;145:1012-22
56. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
57. Moustakas A, Heldin CH. Non-Smad TGF-beta signals. Journal of cell science. 2005;118:3573-84
58. Sousa-Squiavinato ACM, Rocha MR, Barcellos-de-Souza P, de Souza WF, Morgado-Diaz JA. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochimica et biophysica acta Molecular cell research. 2019;1866:418-29
59. Wu J, He Z, Yang XM, Li KL, Wang DL. et al. RCCD1 depletion attenuates TGF-beta-induced EMT and cell migration by stabilizing cytoskeletal microtubules in NSCLC cells. Cancer letters. 2017;400:18-29
60. Collazo J, Zhu B, Larkin S, Martin SK, Pu H. et al. Cofilin drives cell-invasive and metastatic responses to TGF-beta in prostate cancer. Cancer research. 2014;74:2362-73
61. Luo J, Chen XQ, Li P. The Role of TGF-beta and Its Receptors in Gastrointestinal Cancers. Translational oncology. 2019;12:475-84
62. Gruvel J. [Activities of Glossina tachinoides W. (V)]. Revue d'elevage et de medecine veterinaire des pays tropicaux. 1975;28:173-93
63. Kong W, Yang H, He L, Zhao JJ, Coppola D. et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Molecular and cellular biology. 2008;28:6773-84
64. Papadimitriou E, Vasilaki E, Vorvis C, Iliopoulos D, Moustakas A. et al. Differential regulation of the two RhoA-specific GEF isoforms Net1/Net1A by TGF-beta and miR-24: role in epithelial-to-mesenchymal transition. Oncogene. 2012;31:2862-75
65. Guo S, Deng CX. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. International journal of biological sciences. 2018;14:2083-93
66. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews Cancer. 2006;6:392-401
67. Papageorgis P, Stylianopoulos T. Role of TGFbeta in regulation of the tumor microenvironment and drug delivery (review). International journal of oncology. 2015;46:933-43
68. Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF. et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nature cell biology. 2007;9:1392-400
69. Garcia-Palmero I, Torres S, Bartolome RA, Pelaez-Garcia A, Larriba MJ. et al. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen alpha1(VI). Oncogene. 2016;35:5224-36
70. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature cell biology. 2013;15:637-46
71. Barone I, Vircillo V, Giordano C, Gelsomino L, Gyorffy B. et al. Activation of Farnesoid X Receptor impairs the tumor-promoting function of breast cancer-associated fibroblasts. Cancer letters. 2018;437:89-99
72. Stylianou A, Gkretsi V, Stylianopoulos T. Transforming growth factor-beta modulates pancreatic cancer associated fibroblasts cell shape, stiffness and invasion. Biochimica et biophysica acta General subjects. 2018;1862:1537-46
73. Wawro ME, Chojnacka K, Wieczorek-Szukala K, Sobierajska K, Niewiarowska J. Invasive Colon Cancer Cells Induce Transdifferentiation of Endothelium to Cancer-Associated Fibroblasts through Microtubules Enriched in Tubulin-beta3. International journal of molecular sciences. 2018 20
74. Wawro ME, Sobierajska K, Ciszewski WM, Niewiarowska J. Nonsteroidal Anti-Inflammatory Drugs Prevent Vincristine-Dependent Cancer-Associated Fibroblasts Formation. International journal of molecular sciences. 2019 20
75. Najafi M, Farhood B, Mortezaee K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. Journal of cellular biochemistry. 2019;120:2782-90
76. Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Seminars in cancer biology. 2010;20:161-8
77. Stylianou A, Gkretsi V, Louca M, Zacharia LC, Stylianopoulos T. Collagen content and extracellular matrix cause cytoskeletal remodelling in pancreatic fibroblasts. Journal of the Royal Society, Interface. 2019;16:20190226
78. Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes & development. 2017;31:2361-75
79. Farrugia AJ, Calvo F. Cdc42 regulates Cdc42EP3 function in cancer-associated fibroblasts. Small GTPases. 2017;8:49-57
80. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Current opinion in cell biology. 2010;22:697-706
81. Kim H, Ishibashi K, Okada T, Nakamura C. Mechanical Property Changes in Breast Cancer Cells Induced by Stimulation with Macrophage Secretions in Vitro. Micromachines. 2019 10
82. Northey JJ, Przybyla L, Weaver VM. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer discovery. 2017;7:1224-37
83. Claudio-Rizo JA, Rangel-Argote M, Castellano LE, Delgado J, Mata-Mata JL. et al. Influence of residual composition on the structure and properties of extracellular matrix derived hydrogels. Materials science & engineering C, Materials for biological applications. 2017;79:793-801
84. Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R. et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148-63
85. Attieh Y, Vignjevic DM. The hallmarks of CAFs in cancer invasion. European journal of cell biology. 2016;95:493-502
86. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochemical Society transactions. 2017;45:229-36
87. Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Frontiers in oncology. 2018;8:431
88. Bukoreshtliev NV, Haase K, Pelling AE. Mechanical cues in cellular signalling and communication. Cell and tissue research. 2013;352:77-94
89. Ohashi K, Fujiwara S, Mizuno K. Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction. Journal of biochemistry. 2017;161:245-54
90. Martino F, Perestrelo AR, Vinarsky V, Pagliari S, Forte G. Cellular Mechanotransduction: From Tension to Function. Frontiers in physiology. 2018;9:824
91. Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nature reviews Molecular cell biology. 2009;10:63-73
92. Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nature reviews Cancer. 2009;9:108-22
93. Bertocchi C, Wang Y, Ravasio A, Hara Y, Wu Y. et al. Nanoscale architecture of cadherin-based cell adhesions. Nature cell biology. 2017;19:28-37
94. Sanyour HJ, Li N, Rickel AP, Torres HM, Anderson RH. et al. Statin-mediated cholesterol depletion exerts coordinated effects on the alterations in rat vascular smooth muscle cell biomechanics and migration. The Journal of physiology. 2020
95. Kapitein LC, Hoogenraad CC. Building the Neuronal Microtubule Cytoskeleton. Neuron. 2015;87:492-506
96. Jegou A, Carlier MF, Romet-Lemonne G. Formin mDia1 senses and generates mechanical forces on actin filaments. Nature communications. 2013;4:1883
97. Krieg M, Dunn AR, Goodman MB. Mechanical control of the sense of touch by beta-spectrin. Nature cell biology. 2014;16:224-33
98. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453-65
99. Hayakawa K, Tatsumi H, Sokabe M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. The Journal of cell biology. 2011;195:721-7
100. Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nature reviews Molecular cell biology. 2010;11:353-65
101. Gouget CL, Hwang Y, Barakat AI. Model of cellular mechanotransduction via actin stress fibers. Biomechanics and modeling in mechanobiology. 2016;15:331-44
102. Civelekoglu-Scholey G, Scholey JM. Mitotic force generators and chromosome segregation. Cellular and molecular life sciences: CMLS. 2010;67:2231-50
103. Maresca TJ, Salmon ED. Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. Journal of cell science. 2010;123:825-35
104. Tang Y, He Y, Zhang P, Wang J, Fan C. et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Molecular cancer. 2018;17:77
105. Kreplak L, Bar H, Leterrier JF, Herrmann H, Aebi U. Exploring the mechanical behavior of single intermediate filaments. Journal of molecular biology. 2005;354:569-77
106. Jahed Z, Shams H, Mehrbod M, Mofrad MR. Mechanotransduction pathways linking the extracellular matrix to the nucleus. International review of cell and molecular biology. 2014;310:171-220
107. Jiang G, Huang AH, Cai Y, Tanase M, Sheetz MP. Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophysical journal. 2006;90:1804-9
108. Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K. et al. Integrins in mechanotransduction. Current opinion in cell biology. 2013;25:613-8
109. Seetharaman S, Etienne-Manneville S. Integrin diversity brings specificity in mechanotransduction. Biology of the cell. 2018;110:49-64
110. Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. The Journal of cell biology. 2013;200:9-19
111. Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Current opinion in cell biology. 2012;24:600-6
112. Elbediwy A, Vincent-Mistiaen ZI, Spencer-Dene B, Stone RK, Boeing S. et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143:1674-87
113. Kim J, Lee J, Jang J, Ye F, Hong SJ. et al. Topological Adaptation of Transmembrane Domains to the Force-Modulated Lipid Bilayer Is a Basis of Sensing Mechanical Force. Current biology: CB. 2020
114. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S. et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179-83
115. Choi W, Kim J, Park J, Lee DH, Hwang D. et al. YAP/TAZ Initiates Gastric Tumorigenesis via Upregulation of MYC. Cancer research. 2018;78:3306-20
116. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nature cell biology. 2018;20:888-99
117. Hino N, Ichikawa T, Kimura Y, Matsuda M, Ueda K. et al. An amphipathic helix of vinexin alpha is necessary for a substrate stiffness-dependent conformational change in vinculin. Journal of cell science. 2019 132
118. Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Experimental cell research. 2016;343:42-53
119. Wang L, Luo JY, Li B, Tian XY, Chen LJ. et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540:579-82
120. Panciera T, Citron A, Di Biagio D, Battilana G, Gandin A. et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nature materials. 2020
121. Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. The Journal of biological chemistry. 2019;294:17693-706
122. Lin C, Yao E, Zhang K, Jiang X, Croll S. et al. YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. eLife. 2017 6
123. Bai H, Zhu Q, Surcel A, Luo T, Ren Y. et al. Yes-associated protein impacts adherens junction assembly through regulating actin cytoskeleton organization. American journal of physiology Gastrointestinal and liver physiology. 2016;311:G396-411
124. Qiao Y, Chen J, Lim YB, Finch-Edmondson ML, Seshachalam VP. et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell reports. 2017;19:1495-502
125. Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517-24
126. Visser D, Middelbeek J, van Leeuwen FN, Jalink K. Function and regulation of the channel-kinase TRPM7 in health and disease. European journal of cell biology. 2014;93:455-65
127. Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF. et al. Mammalian TRP ion channels are insensitive to membrane stretch. Journal of cell science. 2019 132
128. Kaneko Y, Szallasi A. Transient receptor potential (TRP) channels: a clinical perspective. British journal of pharmacology. 2014;171:2474-507
129. Chen J, Luan Y, Yu R, Zhang Z, Zhang J. et al. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Bioscience trends. 2014;8:1-10
130. Liu C, Montell C. Forcing open TRP channels: Mechanical gating as a unifying activation mechanism. Biochemical and biophysical research communications. 2015;460:22-5
131. Howe AK. Cross-talk between calcium and protein kinase A in the regulation of cell migration. Current opinion in cell biology. 2011;23:554-61
132. Meng X, Cai C, Wu J, Cai S, Ye C. et al. TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer letters. 2013;333:96-102
133. Kuipers AJ, Middelbeek J, van Leeuwen FN. Mechanoregulation of cytoskeletal dynamics by TRP channels. European journal of cell biology. 2012;91:834-46
134. Goswami C, Hucho T. Submembraneous microtubule cytoskeleton: biochemical and functional interplay of TRP channels with the cytoskeleton. The FEBS journal. 2008;275:4684-99
135. Middelbeek J, Kuipers AJ, Henneman L, Visser D, Eidhof I. et al. TRPM7 is required for breast tumor cell metastasis. Cancer research. 2012;72:4250-61
136. Lopez JJ, Salido GM, Pariente JA, Rosado JA. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. The Journal of biological chemistry. 2006;281:28254-64
137. Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG. et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. The EMBO journal. 2006;25:290-301
138. Lee WH, Choong LY, Mon NN, Lu S, Lin Q. et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Scientific reports. 2016;6:27903
139. Hantute-Ghesquier A, Haustrate A, Prevarskaya N, Lehen'kyi V. TRPM Family Channels in Cancer. Pharmaceuticals. 2018 11
140. Zhou T, Gao B, Fan Y, Liu Y, Feng S. et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-beta-catenin. eLife. 2020 9
141. Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J. et al. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:16148-53
142. Yang H, Liu C, Zhou RM, Yao J, Li XM. et al. Piezo2 protein: A novel regulator of tumor angiogenesis and hyperpermeability. Oncotarget. 2016;7:44630-43
143. Borbiro I, Rohacs T. Regulation of Piezo Channels by Cellular Signaling Pathways. Current topics in membranes. 2017;79:245-61
144. Pardo-Pastor C, Rubio-Moscardo F, Vogel-Gonzalez M, Serra SA, Afthinos A. et al. Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:1925-30
145. Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nature reviews Molecular cell biology. 2016;17:496-510
146. Haga RB, Ridley AJ. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases. 2016;7:207-21
147. Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell adhesion & migration. 2011;5:170-80
148. Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67:545-54
149. Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167-79
150. Islam SU, Ahmed MB, Lee SJ, Shehzad A, Sonn JK. et al. PRP4 kinase induces actin rearrangement and epithelial-mesenchymal transition through modulation of the actin-binding protein cofilin. Experimental cell research. 2018;369:158-65
151. Bottcher RT, Veelders M, Rombaut P, Faix J, Theodosiou M. et al. Kindlin-2 recruits paxillin and Arp2/3 to promote membrane protrusions during initial cell spreading. The Journal of cell biology. 2017;216:3785-98
152. Dayma K, Radha V. Cytoskeletal remodeling by C3G to induce neurite-like extensions and inhibit motility in highly invasive breast carcinoma cells. Biochimica et biophysica acta. 2011;1813:456-65
153. Gadea G, Sanz-Moreno V, Self A, Godi A, Marshall CJ. DOCK10-mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Current biology: CB. 2008;18:1456-65
154. Liu L, Jiang H, Zhao W, Meng Y, Li J. et al. Cdc42-mediated supracellular cytoskeleton induced cancer cell migration under low shear stress. Biochemical and biophysical research communications. 2019;519:134-40
155. Matsuoka T, Yashiro M. Rho/ROCK signaling in motility and metastasis of gastric cancer. World journal of gastroenterology. 2014;20:13756-66
156. Matsubara M, Bissell MJ. Inhibitors of Rho kinase (ROCK) signaling revert the malignant phenotype of breast cancer cells in 3D context. Oncotarget. 2016;7:31602-22
157. Wen S, Shang Z, Zhu S, Chang C, Niu Y. Androgen receptor enhances entosis, a non-apoptotic cell death, through modulation of Rho/ROCK pathway in prostate cancer cells. The Prostate. 2013;73:1306-15
158. Peng Y, Chen Z, Chen Y, Li S, Jiang Y. et al. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta biomaterialia. 2019;88:86-101
159. Timpson P, McGhee EJ, Morton JP, von Kriegsheim A, Schwarz JP. et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer research. 2011;71:747-57
160. Nager AR, Goldstein JS, Herranz-Perez V, Portran D, Ye F. et al. An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell. 2017;168:252-63 e14
161. Bachmann VA, Mayrhofer JE, Ilouz R, Tschaikner P, Raffeiner P. et al. Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:7786-91
162. Vazquez-Victorio G, Gonzalez-Espinosa C, Espinosa-Riquer ZP, Macias-Silva M. GPCRs and actin-cytoskeleton dynamics. Methods in cell biology. 2016;132:165-88
163. Salvi V, Sozio F, Sozzani S, Del Prete A. Role of Atypical Chemokine Receptors in Microglial Activation and Polarization. Frontiers in aging neuroscience. 2017;9:148
164. Gao J, He L, Zhou L, Jing Y, Wang F. et al. Mechanical force regulation of YAP by F-actin and GPCR revealed by super-resolution imaging. Nanoscale. 2020;12:2703-14
165. Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M. et al. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. The Journal of steroid biochemistry and molecular biology. 2018;176:4-15
166. Hao J, Bao X, Jin B, Wang X, Mao Z. et al. Ca2+ channel subunit alpha 1D promotes proliferation and migration of endometrial cancer cells mediated by 17beta-estradiol via the G protein-coupled estrogen receptor. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:2883-93
167. Wang Z, Sun L, Liang S, Liu ZC, Zhao ZY. et al. GPER stabilizes F-actin cytoskeleton and activates TAZ via PLCbeta-PKC and Rho/ROCK-LIMK-Cofilin pathway. Biochemical and biophysical research communications. 2019;516:976-82
168. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7
169. Costa RLB, Han HS, Gradishar WJ. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast cancer research and treatment. 2018;169:397-406
170. Diz-Munoz A, Thurley K, Chintamen S, Altschuler SJ, Wu LF. et al. Membrane Tension Acts Through PLD2 and mTORC2 to Limit Actin Network Assembly During Neutrophil Migration. PLoS biology. 2016;14:e1002474
171. Fadri M, Daquinag A, Wang S, Xue T, Kunz J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Molecular biology of the cell. 2005;16:1883-900
172. Morrison Joly M, Williams MM, Hicks DJ, Jones B, Sanchez V. et al. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast cancer research: BCR. 2017;19:74
173. Lee T. Mechanical and Mechanosensing Properties of Tumor Affected Bone Cells Were Inhibited via PI3K/Akt Pathway. Journal of bone metabolism. 2019;26:179-91
174. Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. International journal of molecular sciences. 2012;13:1886-918
175. Zheng Y, Pan D. The Hippo Signaling Pathway in Development and Disease. Developmental cell. 2019;50:264-82
176. Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811-28
177. Sun S, Irvine KD. Cellular Organization and Cytoskeletal Regulation of the Hippo Signaling Network. Trends in cell biology. 2016;26:694-704
178. Chang YC, Wu JW, Wang CW, Jang AC. Hippo Signaling-Mediated Mechanotransduction in Cell Movement and Cancer Metastasis. Frontiers in molecular biosciences. 2019;6:157
179. Dong J, Feldmann G, Huang J, Wu S, Zhang N. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120-33
180. Zhang C, Wang F, Gao Z, Zhang P, Gao J. et al. Regulation of Hippo Signaling by Mechanical Signals and the Cytoskeleton. DNA and cell biology. 2019
181. Huang B, Ling Y, Lin J, Du X, Fang Y. et al. Force-dependent calcium signaling and its pathway of human neutrophils on P-selectin in flow. Protein & cell. 2017;8:103-13
182. Zhang QY, Bai JD, Wu XA, Liu XN, Zhang M. et al. Microniche geometry modulates the mechanical properties and calcium signaling of chondrocytes. Journal of biomechanics. 2020: 109729.
183. Evans JH, Falke JJ. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16176-81
184. Tkachenko E, Sabouri-Ghomi M, Pertz O, Kim C, Gutierrez E. et al. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nature cell biology. 2011;13:660-7
185. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer cell. 2009;15:124-34
186. Lopez-Guerrero AM, Pascual-Caro C, Martin-Romero FJ, Pozo-Guisado E. Store-operated calcium entry is dispensable for the activation of ERK1/2 pathway in prostate cancer cells. Cellular signalling. 2017;40:44-52
187. Tsai FC, Seki A, Yang HW, Hayer A, Carrasco S. et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nature cell biology. 2014;16:133-44
188. Tsai FC, Kuo GH, Chang SW, Tsai PJ. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. BioMed research international. 2015;2015:409245
189. Mo P, Yang S. The store-operated calcium channels in cancer metastasis: from cell migration, invasion to metastatic colonization. Frontiers in bioscience. 2018;23:1241-56
190. Lopez-Guerrero AM, Tomas-Martin P, Pascual-Caro C, Macartney T, Rojas-Fernandez A. et al. Regulation of membrane ruffling by polarized STIM1 and ORAI1 in cortactin-rich domains. Scientific reports. 2017;7:383
191. Tsai FC, Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Current biology: CB. 2012;22:837-42
Author contact
![]() Corresponding author: Jianliu Wang. Department of Obstetrics and Gynecology, Peking University People's Hospital, No. 11, Xizhimen South Street, Xicheng District, Beijing 100044, China. Tel +86 10 8832 4474; Email: wangjianliuedu.cn
Corresponding author: Jianliu Wang. Department of Obstetrics and Gynecology, Peking University People's Hospital, No. 11, Xizhimen South Street, Xicheng District, Beijing 100044, China. Tel +86 10 8832 4474; Email: wangjianliuedu.cn