Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Overview of Pyroptosis
Activators of Pyroptosis
Differential Inflammasome...
Temporal Pattern of Inflammasome...
Anti-Pyroptotic Therapies
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(12):2042-2050. doi:10.7150/ijbs.45467 This issue Cite
Review
Role of Pyroptosis in Traumatic Brain and Spinal Cord Injuries
Xinli Hu1,2*, Huanwen Chen3*, Hui Xu1,2, Yaosen Wu1,2, Chenyu Wu1,2, Chang Jia4, Yao Li1,2, Sunren Sheng1,2, Cong Xu1,2, Huazi Xu1,2, Wenfei Ni1,2 ![]() , Kailiang Zhou1,2
, Kailiang Zhou1,2 ![]()
1. Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, China
2. Zhejiang Provincial Key Laboratory of Orthopaedics, Wenzhou 325027, China
3. University of Maryland School of Medicine, Baltimore, MD 21201, USA
4. Pediatric Research Institute, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, China
*These authors contributed equally to this work.
Received 2020-2-29; Accepted 2020-4-5; Published 2020-4-27
Abstract

Central nervous system (CNS) trauma, including traumatic brain injury (TBI) and spinal cord injury (SCI), remains a leading cause for morbidity and mortality worldwide. Past research has shown that cell death plays a critical role in the pathophysiology of CNS injuries. More recently, pyroptosis has been identified as a form of programmed inflammatory cell death, and it is a unique form of cell death in various aspects. Mechanistically, pyroptosis can be categorized into canonical (mediated by caspase-1) and non-canonical (mediated by caspase-4/5/11). In canonical pyroptosis, Nod-like receptors (NLRs) inflammasomes play a critical role, and their activation promotes the maturation and secretion of the inflammatory cytokines interleukin-1β/18 (IL-1β/18), cleavage of gasdermin D (GSDMD), and ultimately pyroptotic cell death. Despite a plethora of new knowledge regarding pyroptosis, detailed understanding of how pyroptosis is involved in CNS injuries and possible ways to improve clinical outcomes following CNS injuries remain elusive. This review discusses the current knowledge on how pyroptosis is involved in CNS injuries, focusing on new discoveries regarding how pyroptosis activation occurs, differences between CNS cell types following injury, time-course of inflammatory responses, and key regulatory steps of pyroptosis. In addition, we highlight various investigational agents that are capable of regulating key steps in pyroptotic cell death, and we discuss how these agents may be used as therapies to improve outcomes following CNS trauma.
Keywords: Pyroptosis, Traumatic brain injury, Spinal cord injury, Inflammasomes, Cell death
Introduction
Neurotrauma is one of the most serious traumatic injuries and is a common cause of long-term disability and death among young adults. Despite clear clinical need, traumatic brain injury (TBI) and spinal cord injury (SCI) are still without adequate treatments [1]. These devastating injuries not only impair physical and psychological health, but also place financial burdens on families and society. In the United States alone, the incidences of TBI and SCI are 333 and 26 per 100 000 patients per year, respectively. Together, TBI and SCI incur an economic burden of approximately 23.7 billion on the American economy ($9.2 billion for TBI, and $14.5 billion for SCI) [1]. Clinically, the treatments of CNS trauma mainly include methylprednisolone, surgical decompression, supportive medical care, and rehabilitation. However, patient recovery is limited [2, 3]. While many experimental therapies (mainly involving growth factors, biomaterials, and cell transplantation etc.) are being explored in basic and translational research settings, results have been disappointing [4, 5]. Thus, new ideas and therapeutic targets are needed for the development of new and effective CNS trauma treatments.
While TBI and SCI share many pathophysiological features, the complex mechanism of CNS injury has been a major obstacle for developing treatments [6]. The time course of both TBI and SCI include two overall stages. The first stage features direct brain or spinal cord tissue damage by external force, which causes cell membrane rupture and results in irreversible cell injury and tissue necrosis [7, 8]. This is followed by the second stage, during which cell necrosis induces rapid release of intracellular and intra-axonal contents, such as glutamate, ROS, potassium, and cathepsin B. These substances are highly pro-inflammatory, and thus trigger a strong inflammatory response [9]. More specifically, pattern recognition receptors (PRRs) are activated by pro-inflammatory substances released from dead cells, thus inducing neuroinflammation [10]. Therefore, cell death, and subsequent inflammation, are central biological processes in CNS injury [11]. Classically, three different types of cell death have been widely researched: apoptosis [12], autophagic cell death [13], and necrosis [11]. These classifications are based largely on morphology, with apoptosis featuring cell shrinkage and chromatin condensation [14], autophagic cell death featuring cytoplasmic vacuolization, phagocytic uptake and lysosomal degradation [15], and necrosis featuring cell swelling, loss of cell membrane integrity, DNA degradation and release of cytoplasmic content [16]. More recently, researchers have identified detailed mechanisms regarding programmed necrotic cell death, which is characterized as mitochondrial permeability transition (MPT) dependent necrosis, necroptosis, ferroptosis, and pyroptosis [17]. Among these novel mechanisms, accumulating evidence suggests a critical role of pyroptosis, a process of programmed inflammatory cell death, in CNS trauma. In this review, we summarize recent studies of pyroptosis in CNS injuries, which have begun to elucidate how pyroptosis is involved in CNS trauma and how its regulation may be used as potential novel neuroprotective treatments.
Overview of Pyroptosis
Pyroptosis was first termed in 2002, and it was first observed in macrophages that underwent a unique caspase-1-programmed cell death following exposure to Salmonella. Pyroptosis is different from other forms of programmed cell death especially in morphological and biochemical characteristics [18]. Morphologically, classical findings include cell swelling induced by incoming water molecules, formation of pores 10-15 nm in diameter on plasma membranes, and release of pro-inflammatory cytokines (IL-1β and IL-18) from the cytoplasm [19]. Biochemically, while apoptotic caspases include initiating apoptotic caspases (caspase-2,8,9,10) and execution apoptotic caspases (caspase-3,6,7), pyroptosis caspases include caspases (caspase-1, -4, -5, -11) with both initiator and effector functions [17]. Pyroptosis has been extensively observed and well-studied in endothelial cells (EC), smooth muscle cells (SMC), phagocytes, macrophages, neurons and astrocytes, as well as various other cell types [20]. In CNS injuries, however, while researchers have reported evidence of pyroptosis in injured cells, the mechanisms of pyroptosis and the consequences of pyroptosis are less clear.
The molecular processes of pyroptosis have been described previously and is depicted in Figure 1. Mechanistically, pyroptosis involves two pathways: the canonical caspase-1 inflammasome pathway, and the non-canonical caspase-4/5/11 inflammasome pathway [21]. In canonical pyroptosis, which centers around caspase-1, the process is initiated in the “priming step,” where danger or pathogen-associated molecular patterns (DAMPs or PAMPs, respectively) are recognized by Toll-like receptors (TLRs) and Nod-like receptors (NLRs) [22]. Once this occurs, expression of inflammasome associated genes is increased, leading to the production of pro-IL-1β and pro-IL-18. Post-translational modifications can also occur during the priming step of canonical pyroptosis [23]. After the priming step is complete, the process progresses to the “activation step,” which is centered around inflammasome assembly and caspase activation [24]. Here, pro-caspase-1 and adaptor protein apoptosis-associated speck like proteins (ASC) are recruited (often by nod-like receptor family pyrin domain containing 3 (NLRP3)) to form inflammasomes. After that, pro-caspase-1 is cleaved to form caspase-1, which not only promotes cleavage of pro-IL-1β/18 but also cleaves GSDMD into two fragments [25]. The N-terminal fragment forms 10-15nm pores in the cell membrane, which eventually leads to discharge of inflammatory factors, cell swelling, membrane rupture [26].
Graphical depiction of the mechanism of pyroptosis in traumatic brain and spinal cord injuries.
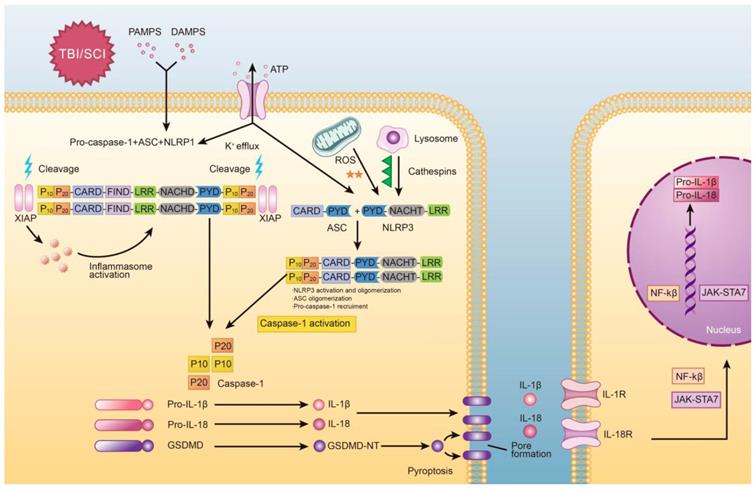
Overall, caspase-1 can be activated by various inflammasomes including nod-like receptors (NLRs), AIM2-like receptors (ALRs) [27], or tripartite motif family (TRIM). Among these, nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family have been extensively researched, especially NLRP1 [28], NLRP2 [10], NLRP3 [29], NLRP6 [30], NLRC4 [31], NLRP9b [32], Pyrin proteins [33] and IFI16 [34]. These molecules are depicted graphically in Figure 2. While the signaling mechanism may differ slightly depending on the molecules involved, all of these molecules ultimately lead to the activation of caspase-1.
Molecules that can trigger pyroptosis. The nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) have common NACHT, C-terminal leucine-rich repeat (LRR), and the N-terminal effector domains. It is reported that NLRs have either a pyrin domain or a caspase recruitment domain (CARD). The melanoma 2 (AIM2)-like receptors (AIM2 and IFI16) consist of a C-terminal dsDNA- binding one or two 200 amino acids (HIN200) domain and an N-terminal PYD. Pyrin is a human protein encoded by the MEFV gene, which carries a PYD, 2 B-boxes and a coiled-coil domain, and a SPRY domain.

In non-canonical pyroptosis, caspase-4/5 in humans and caspase-11 in mice are involved instead of caspase-1. It is believed that caspase 4/5 and 11 perform similar functions to caspase-1. In the lipopolysaccharide (LPS) induced non-canonical inflammasome pathway, Toll-like receptor 4 (TLR4) recognizes extracellular LPS and caspases recognize cytosolic LPS. Caspase-11/-4/-5 can be directly activated by LPS via binding with their CARD domains, however, it is unclear how LPS is sensed by the CARD domain of caspase-11 [35, 36]. After the caspases are activated, they then directly cleave GSDMD to initiate pyroptosis [37, 38].
Activators of Pyroptosis
When external force impacts brain or spinal tissue, immediate compression and contusion occur, accompanied by vascular injury which leads to the destruction of the blood-brain or blood-spinal-cord barrier [39]. These events are largely grouped together as “primary injury.” Subsequently, “secondary injury” occurs, which involves the local and systemic release of DAMPs, triggering an inflammatory response via binding to LRR (leucine- rich repeat) domains [40]. The mechanism by which NLRP3 inflammasome formation occurs remains unclear. Previous reports have suggested that overproduction of reactive oxygen species (ROS), potassium efflux, and leak of cathepsin are the three main activators of the NLRP3 inflammasome [41]. These signaling pathways may not be mutually exclusive, and it is likely that each signal may independently interact with NLRP3 to cause different conformational changes in the molecules involved in inflammasome assembly. Here, we discuss details of how potassium efflux, ROS, and CTSB activate the NLRP3 inflammasome.
Potassium Efflux
K+ efflux is linked with many known NLRP3 activators including ATP, nigericin and maitotoxin. ATP can bind to the P2X7 receptor, which facilitates an immediate K+ efflux. Subsequently, low intracellular potassium concentration causes changes in NLRP3 inflammasome components, eventually leading to the activation of NLRP3 [42-44]. Interestingly, NLRP3 is not affected by high extracellular K+ concentrations [45]. Importantly, Petrilli et al found that inhibiting K+ efflux is an effective way to block NLRP3 inflammasome activation, which may be a strategy that can be used to inhibit pyroptosis in CNS injuries [46]. Further work on modulating potassium efflux as a therapeutic approach in CNS injuries is eagerly awaited. NIMA-related kinase 7 (NEK7), a Ser/Thr mitotic kinase, is a modulator which can regulate the activity of NLRP3 inflammasomes and downstream neuroinflammatory responses to K+ efflux. The mechanism of NEK7 involves binding to NLRP3 and then recruiting pro-caspase-1, leading to activation of caspase-1 and inducing pyroptosis. NEK7 may be another mechanism to activate NLRP3 inflammasome [29, 47].
Reactive Oxygen Species (ROS)
Production of ROS can also trigger NLRP3 inflammasome activation [45]. There are many sources of ROS, such as mitochondria, NADPH oxidase, xanthine/xanthine oxidase (X/XO) and incomplete phagocytosis of macrophages. Among these pathways, ROS generated from mitochondria and NADPH oxidase are the most studied [48]. Many studies have shown that inhibition of ROS prevents caspase-1 activation and Interleukin 18/1β production. Bae et al found that NLRP3 has a disulfide bond that connects the PYD and nucleotide-binding site domains, and is sensitive to altered redox states, suggesting that ROS may trigger NLRP3 inflammasome activation via modifying this disulfide bond [49]. Furthermore, evidence suggests that ROS may play a key role in the priming step of pyroptosis and NLRP3 activation [50].
Release of Cathepsin B (CTSB)
A third trigger for NLRP3 inflammasome activation is the release of CTSB from lysosomes. Jin et al demonstrated that lysosomes cannot digest crystals after phagocytosis, and that phagocytosed crystals result in lysosomal swelling and damage, release of CTSB, and activation of NLRP3. Furthermore, this study found that NLRP3 inflammasome activation was not due to the presence of crystals, but rather lysosomal membrane rupture [51]. More recently, studies have shown that lack of CTSB markedly reduces NLRP3 activation [52]. However, the relationship between CTSB and CNS trauma remains largely unknown. In addition, it was reported that cytoplasmic phospholipase A2 (cPLA2) is activated after SCI and TBI, which can damage lysosome cellular membranes and then lead to leakage of CTSB [53, 54]. Thus, it is reasonable to hypothesize that inhibiting CTSB may also curtail pyroptosis in CNS injuries, and that activation of cPLA2 may contribute to CTSB mediated pyroptosis.
Differential Inflammasome Expression Among Cell Types in CNS Injury
Normal CNS physiology and response to injury involve numerous cell types including neurons, astrocytes, and microglia [55]. Following injury, neurons, astrocytes, and microglia generate neurotoxic molecules, such as ROS and CTSB. The formation of NLRP inflammasomes can be activated by these molecules via mechanisms described above. Past studies have shown that there is differential expression of inflammasome components in neurons, astrocytes, and microglia, suggesting that these cells may respond differently to activating signals [56]. Xu et al, using flow cytometry and immunofluorescence staining, showed that microglia are the main source of NLRP3 inflammasome expression [57], but do not express NLRP1 [58]. This is in stark contrast to neurons, which mainly express NLRP1-inflammasomes and AIM2, though NLRP3 positive neurons can also be found in rat models of TBI [59]. Overall, among known inflammasomes, NLRP1 and NLRP3 are the most commonly studied in TBI and SCI, and they have been shown to play key roles in the innate immune response [60, 61].
Interestingly, astrocytes mainly express the NLRP2 inflammasome, which operates in a unique fashion: ATP binds to P2X7 receptor, opening the pannexin-1 channel, allowing potassium efflux, activating NLRP2, leading to maturation of caspase-1 and secretion of IL-18 and IL-1β. Thus, NLRP2, which is activated by potassium efflux, has a critical role in the astrocyte innate immune response [10]. To this end, probenecid, which can inhibit the pannexin-1 channel, has been shown to reduce NLRP2 activation, and may be a potential therapeutic agent for CNS injuries.
Temporal Pattern of Inflammasome Following CNS Trauma
The formation of inflammasomes and secretion of cytokines following CNS injuries follow a complex temporal pattern. In a moderate parasagittal fluid-percussion injury rat model, maturation of caspase-1, secretion of IL-1β and formation of NLPR1 inflammasome complexes were detected 4 hours following TBI [62]. In a TBI rat model, NLRP3 mRNA levels gradually increased following TBI, peaked at 6 hours, gradually decreased until 24 h, and again increased until 7 day after injury; protein levels of NLRP3 were significantly increased from 24 hours to 7 days after injury [59]. Finally, in a mouse model of TBI, protein expression of NLRP3, ASC, and caspase-1 were shown to be increased at 1 day following TBI, peaked at 3 days, and gradually decreased over time while levels continued to be elevated compared to control animals [57]. Thus, determinants of the temporal pattern of inflammasome formation appears to be multifactorial, and further research is eagerly awaited.
Similarly, temporal patterns of cytokine release following CNS injuries are also highly variable among published results. Overall, TNFα is produced as part of the inflammatory process that occurs early after TBI; TNFα, IL-1β, IL-10 and IL-6 are increased as early as day 1 and decreased at days 2-4 after injury. TGFβ peaks at day 1, and gradually decreases over 21 days. CXCL8 (IL-8) peaks at day 1 and markedly declines at days 2-3, although remaining elevated for up to 108 hours after injury. Many triggers and brakes of inflammation also follow intricate temporal patterns. For example, adenosine increases within hours of injury, but rapidly declines over 12-24 hours. Complement proteins peak at 1 day after injury and decline on days 2-7. Similarly, glutamate peaks at 1 day and declines during days 2-3 [63]. Data regarding the temporal pattern of inflammasome formation and cytokine release in SCI remain largely unknown.
Anti-Pyroptotic Therapies
As NLRP3 is believed to be the most commonly involved inflammasome in pyroptosis following CNS injury, development of effective NLRP3 inflammasome inhibitors to treat SCI and TBI has generated significant interest in the scientific community. Various efforts have attempted to make use of the diverse range of candidate targets to inhibit NLRP3 inflammasome activation through the complex signaling pathway of NLRP3 inflammasomes. In particular, therapies may target inflammatory cytokines induced by the NLRP3 inflammasome, GSDMD cleavage to inhibit pore formation, upstream signals, or inflammasome assembly and caspase-1 activation. While there is currently no pyroptosis inhibiting drug approved to treat CNS injuries, many therapies are being developed and hold great promise (Table 1).
Effects of various therapeutic agents on inflammasome signal pathway after TBI and SCI.
| Therapeutic agents | Targets | Potential Mechanism |
|---|---|---|
| Canakinumab [64] [65] | IL-1β | Anti-IL-1β antibody |
| Ac-FLTD-CMK [66] | GSDMD | Inhibits GSDMD cleavage by directly binding to the catalytic region of caspase1, caspase4, caspase 5 and caspase11 |
| VX-740 and VX-765 [68, 69] and Parthenolide [70] | Caspase-1 | Compound act by covalent modification of the catalytic cysteine residue in the active site of caspase-1 |
| MCC950 [73]/ 3,4-methylenedioxy-β-nitrostyrene (MNS) [76]/ glyburide [77]/ OLT1177 [78]/ CY09 [79] | NLRP3 | Blocks the ATPase domain of NLRP3 Inhibits NLRP3 ATPase activity by cysteine modification |
| A438079 [82, 83] | P2X7R | Blocks potassium(K+) efflux |
| BHB [85] | Mitochondrial and ROS | Inhibits Drp1 mitochondrial translocation and Prevent ROS generation |
| CA-074Me [51, 86] | Cathepsin B | Inhibits cathepsin B |
Cytokine Inhibition
As discussed previously, IL-1β signaling is a key step in pyroptosis and is involved in CNS injuries, and thus it has been proposed as a therapeutic target for TBI and SCI. Canakinumab, a monoclonal antibody against IL-1β, inhibits IL-1β binding to the IL-1 receptor, preventing the propagation of downstream inflammatory signals. Past studies have shown that canakinumab can prevent cardiovascular events, however, its possible efficacy in TBI and SCI treatment remains unknown [64, 65]. Salidroside, another new experimental agent, has been shown to inhibit pyroptosis via inhibiting inflammatory cytokine expression and NF-κB and MAPK signaling pathways [66].
GSDMD inhibition
Cleavage of GSDMD after binding to caspase-1 is a critical step in pyroptosis, and it was shown that the cleavage occurs at the FLTD peptide site. Thus, an inhibiting therapy targeting Ac-FLTD-CMK has been designed to block GSDMD cleavage [67]. Studies also found that necrosulfonamide (NSA) can interact with cleaved GSDMD, inhibiting oligomerization of p30-GSDMD and preventing the formation of pyroptotic pores and thus pyroptosis cell death [68]. The efficacy of these agents in CNS injuries is currently unknown and awaits further investigation.
Caspase-1 inhibition
Past studies have shown that caspase-1 deficiency reduces neuroinflammation and neuronal damage in the acute phase of TBI and SCI, giving justification to develop caspase-1 inhibitors as therapeutic agents. Pralnacasan (VX-740), and its analog VX-765, are peptidomimetic caspase-1 inhibitors that act via covalent modification of the catalytic site of caspase-1, thereby blocking caspase-1 activation and cleavage of pro-IL-1β and pro-IL-18 [69, 70]. Parthenolide, an agent that inhibits caspase-1 protease activity and ATPase activity of NLRP3, is another potential therapy [71]. Unfortunately, the clinical effectiveness of these agents is not promising due to disappointing pre-clinical data [71, 72].
NLRP3 inflammasome inhibition
Inhibition of the NLRP3 inflammasome, which is a central molecule in many pyroptosis pathways, has also been explored for therapeutic use [46]. MCC950, a diarylsulfonylurea-containing compound, is a potent NLRP3 inhibitor. While studies have shown that MCC950 does not decrease the expression of NLRP3, pro-IL-1β, or pro-caspase-1 in early stages of TBI [73], MCC950 repressed all these molecules 24 hours post-injury, strongly inhibiting caspase-1 activity and IL-1β maturation for up to 72 hours post-TBI [74]. These data suggest that MCC950 may not have any effect during the priming stage of pyroptosis, instead targeting the subsequent assembly of NLRP3 inflammasomes. Mechanistically, MCC950 binds directly to the Walker B motif within the NACHT domain of NLRP3, thus blocking ATP hydrolysis and inhibit NLRP3 inflammasome formation [75]. Interestingly, MCC950 does not affect TNFα secretion and has no impact on the activation of other inflammasomes such as AIM2, NLRP1, and NLRC4 [73, 76]. 3,4-methylenedioxy-β-nitrostyrene (MNS), identified by He et al via screening a kinase inhibitor library, also inhibits the activation of NLRP3. MNS acts similarly to MCC950 and also suppresses ATPase activity, here via interacting with the NOD and LRR domains, and MNS also leaves AIM2 and NLRC4 inflammasomes unaffected [77]. Thus, MNS may be another potential therapeutic agent for CNS injuries awaiting further research. Of note, many other NLRP3 inhibitors are also under investigation including glyburide [78], OLT1177 [79] and CY-09 [80].
Suppression of upstream signals
Suppression of upstream activators of inflammasomes, such as K+ efflux, ROS, and CTSB, have also been explored as therapeutic options. Research has shown that preventing K+ efflux leads to a significant decrease in the ability of other inflammasome activators to function [51]. P2X7R, an extracellular ATP-gated receptor capable of activating NLPR3-inflammasomes and promoting IL-18 and IL-1β release, has been implicated in CNS injuries by possibly forming homo-trimer ion channels to allow K+ efflux (along with Na+ and Ca2+ influx) and subsequently lead to pyroptosis [81]. A438079 is an experimental agent that inhibits P2X7R activity, and Jiang et al found that P2X7R inhibition leads to a 50% decrease in caspase-1 activation and promotes functional recovery following SCI in mice [42, 82]. Furthermore, A438079 not only decreases the expression of NLRP3 and ASC, it also decreases expression of pro-IL-1β and pro-IL18 by inhibiting the NF-κB pathway [83, 84].
Inhibition of ROS-associated inflammasome activation has also been explored as potential treatments. In TBI, acute cerebral glucose hypometabolism occurs in the brain, and utilization of alternative energy substrates such as ketones is a common response [85]. The ketones BHB and acetoacetate are generated in the brain as alternative energy sources to enable neuron survival. Interestingly, BHB has been shown to inhibit Drp1 mitochondrial translocation and promote mitochondrial stability, thus inhibiting ROS-associated NLRP3 inflammasome activation [86].
Finally, CTSB inhibitors have also been studied for their ability to curtail pyroptosis. Recent data suggest that the CTSB inhibitor CA-074Me could inhibit NLRP3 inflammasome activation induced by silica and asbestos [51, 87]. However, whether CTSB inhibition is effective in treating CNS injuries is currently unknown. Furthermore, as specific mechanism of how CTSB induces pyroptosis is still unclear, further work is needed to develop effective therapies targeting this pathway.
Conclusion
The role of pyroptosis is very complicated in CNS injuries, and many unanswered questions remain. In this review, we discussed mechanisms involved in the canonical and non-canonical inflammasome pathways and progression of pyroptosis in TBI and SCI. We also highlighted novel agents that may be able to curtail this form of cell death. While canonical pyroptosis involving NLRP3 is thought to be the predominant pathway in CNS injuries, the roles of many other potential pathways such as other NLR inflammasomes and non-canonical pyroptosis caspases may also be significant. To accurately determine the function of pyroptosis after SCI and TBI, transgenic animals with pyroptosis defects, such as caspase-1-/- mice, should be used in future studies. To date, there are only a few studies that have shed light on the role of non-coding RNAs in pyroptosis of the field. Thus, investigating whether microRNA (miRNA) or long noncoding RNAs (lncRNAs) affect pyroptosis, particularly in TBI and SCI, is of great interest. Overall, while more work is needed to elucidate pyroptosis signaling pathways in CNS injuries, current experimental agents hold great promise, suggesting that novel effective therapies may soon become realities.
Acknowledgements
This work was supported by grants from Zhejiang Provincial Medicine and Health Technology Project (No. 2017KY472 to Kailiang Zhou; No.2015RCB022 to Hui Xu); Public Welfare Technology Research Project of Zhejiang Province (No. LGF20H150003 to Kailiang Zhou; No. LGF20H060013 to Yaosen Wu); Zhejiang Provincial Natural Science Foundation (No. LY17H060009 to Wenfei Ni; No. LQ18C010003 to Chang Jia); Natural Science Foundation of China (No. 81601705 to Kailiang Zhou, and No. 81873992 to Huazi Xu). Wenzhou Science and Technology Bureau Foundation (No. Y20190064 to Chang Jia).
Author Contributions
Xinli Hu and Huanwen Chen searched and reviewed literature, drafted manuscript and revision; Hui Xu, Yaosen Wu and Chenyu Wu discussed and revised the manuscript; Chang Jia, Yao Li, Sunren Sheng, Cong Xu and Huazi Xu provided critical comments; Wenfei Ni and Kailiang Zhou designed and formulated the review theme, revised and finalized the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Habtemariam S. Antioxidant and Anti-inflammatory Mechanisms of Neuroprotection by Ursolic Acid: Addressing Brain Injury, Cerebral Ischemia, Cognition Deficit, Anxiety, and Depression. Oxidative medicine and cellular longevity. 2019;2019:8512048
2. Ahuja CS, Martin AR, Fehlings M. Recent advances in managing a spinal cord injury secondary to trauma. F1000Research. 2016 5
3. Varma AK, Das A, Wallace Gt. et al. Spinal cord injury: a review of current therapy, future treatments, and basic science frontiers. Neurochemical research. 2013;38:895-905
4. Raspa A, Pugliese R, Maleki M. et al. Recent therapeutic approaches for spinal cord injury. Biotechnology and bioengineering. 2016;113:253-9
5. Fuhrmann T, Anandakumaran PN, Shoichet MS. Combinatorial Therapies After Spinal Cord Injury: How Can Biomaterials Help? Advanced healthcare materials. 2017 6
6. Lipinski MM, Wu J, Faden AI. et al. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid Redox Signal. 2015;23:565-77
7. Schoch KM, Madathil SK, Saatman KE. Genetic manipulation of cell death and neuroplasticity pathways in traumatic brain injury. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2012;9:323-37
8. Mortezaee K, Khanlarkhani N, Beyer C. et al. Inflammasome: Its role in traumatic brain and spinal cord injury. Journal of cellular physiology. 2018;233:5160-9
9. Dixon KJ. Pathophysiology of Traumatic Brain Injury. Physical medicine and rehabilitation clinics of North America. 2017;28:215-25
10. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61:1113-21
11. Bao Z, Fan L, Zhao L. et al. Silencing of A20 Aggravates Neuronal Death and Inflammation After Traumatic Brain Injury: A Potential Trigger of Necroptosis. Front Mol Neurosci. 2019;12:222
12. Li G, Shen F, Fan Z. et al. Dynasore Improves Motor Function Recovery via Inhibition of Neuronal Apoptosis and Astrocytic Proliferation after Spinal Cord Injury in Rats. Molecular neurobiology. 2017;54:7471-82
13. Tang P, Hou H, Zhang L. et al. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Molecular neurobiology. 2014;49:276-87
14. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
15. Galluzzi L, Vitale I, Aaronson SA. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation. 2018;25:486-541
16. Lee SY, Ju MK, Jeon HM. et al. Regulation of Tumor Progression by Programmed Necrosis. Oxidative medicine and cellular longevity. 2018;2018:3537471
17. Jia C, Chen H, Zhang J. et al. Role of pyroptosis in cardiovascular diseases. International immunopharmacology. 2019;67:311-8
18. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular microbiology. 2006;8:1812-25
19. Lu F, Lan Z, Xin Z. et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. Journal of cellular physiology. 2019
20. Liu W, Chen Y, Meng J. et al. Ablation of caspase-1 protects against TBI-induced pyroptosis in vitro and in vivo. J Neuroinflammation. 2018;15:48
21. Wang YY, Liu XL, Zhao R. Induction of Pyroptosis and Its Implications in Cancer Management. Frontiers in oncology. 2019;9:971
22. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clinical and experimental immunology. 2011;166:1-15
23. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews Immunology. 2016;16:407-20
24. Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432-41
25. Tsuchiya K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiology and immunology. 2020
26. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. International journal of biological sciences. 2019;15:1345-57
27. Adamczak SE, de Rivero Vaccari JP, Dale G. et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:621-9
28. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002;10:417-26
29. Chen Y, Meng J, Bi F. et al. EK7 Regulates NLRP3 Inflammasome Activation and Neuroinflammation Post-traumatic Brain Injury. Front Mol Neurosci. 2019;12:202
30. Levy M, Thaiss CA, Zeevi D. et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163:1428-43
31. Broz P, Newton K, Lamkanfi M. et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. The Journal of experimental medicine. 2010;207:1745-55
32. Zhu S, Ding S, Wang P. et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature. 2017;546:667-70
33. Xu H, Yang J, Gao W. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237-41
34. Kerur N, Veettil MV, Sharma-Walia N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell host & microbe. 2011;9:363-75
35. Kayagaki N, Wong MT, Stowe IB. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (New York, NY). 2013;341:1246-9
36. Hagar JA, Powell DA, Aachoui Y. et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (New York, NY). 2013;341:1250-3
37. Kayagaki N, Warming S, Lamkanfi M. et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117-21
38. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61-75
39. Zhou K, Sansur CA, Xu H. et al. The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury. International journal of molecular sciences. 2017 18
40. Hirsiger S, Simmen HP, Werner CM. et al. Danger signals activating the immune response after trauma. Mediators of inflammation. 2012;2012:315941
41. Gao L, Dong Q, Song Z. et al. NLRP3 inflammasome: a promising target in ischemic stroke. Inflammation research: official journal of the European Histamine Research Society [et al]. 2017;66:17-24
42. Wang S, Zhao J, Wang H. et al. Blockage of P2X7 attenuates acute lung injury in mice by inhibiting NLRP3 inflammasome. International immunopharmacology. 2015;27:38-45
43. Jiang S, Zhang Y, Zheng JH. et al. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacological research. 2017;117:82-93
44. Lorden G, Sanjuan-Garcia I, de Pablo N. et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. The Journal of experimental medicine. 2017;214:511-28
45. Abais JM, Xia M, Zhang Y. et al. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111-29
46. Petrilli V, Papin S, Dostert C. et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell death and differentiation. 2007;14:1583-9
47. Mathur A, Hayward JA, Man SM. Molecular mechanisms of inflammasome signaling. Journal of leukocyte biology. 2018;103:233-57
48. Brown DI, Griendling KK. Nox proteins in signal transduction. Free radical biology & medicine. 2009;47:1239-53
49. Bae JY, Park HH. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. The Journal of biological chemistry. 2011;286:39528-36
50. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nature reviews Immunology. 2010;10:210-5
51. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. Journal of clinical immunology. 2010;30:628-31
52. Wang Y, Jia L, Shen J. et al. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLoS pathogens. 2018;14:e1006872
53. Liu NK, Deng LX, Zhang YP. et al. Cytosolic phospholipase A2 protein as a novel therapeutic target for spinal cord injury. Annals of neurology. 2014;75:644-58
54. Li Y, Jones JW, H MCC. et al. cPLA2 activation contributes to lysosomal defects leading to impairment of autophagy after spinal cord injury. Cell death & disease. 2019;10:531
55. Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253-63
56. Kaushal V, Dye R, Pakavathkumar P. et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell death and differentiation. 2015;22:1676-86
57. Xu X, Yin D, Ren H. et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiology of disease. 2018;117:15-27
58. de Rivero Vaccari JP, Lotocki G, Marcillo AE. et al. A molecular platform in neurons regulates inflammation after spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:3404-14
59. Liu HD, Li W, Chen ZR. et al. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochemical research. 2013;38:2072-83
60. Wallisch JS, Simon DW, Bayir H. et al. Cerebrospinal Fluid NLRP3 is Increased After Severe Traumatic Brain Injury in Infants and Children. Neurocritical care. 2017;27:44-50
61. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nature reviews Neuroscience. 2014;15:84-97
62. de Rivero Vaccari JP, Lotocki G, Alonso OF. et al. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:1251-61
63. Simon DW, McGeachy MJ, Bayir H. et al. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171-91
64. Anders HJ, Muruve DA. The inflammasomes in kidney disease. Journal of the American Society of Nephrology: JASN. 2011;22:1007-18
65. Newell EA, Todd BP, Mahoney J. et al. Combined Blockade of Interleukin-1alpha and -1beta Signaling Protects Mice from Cognitive Dysfunction after Traumatic Brain Injury. eNeuro. 2018 5
66. Su Y, Zong S, Wei C. et al. Salidroside promotes rat spinal cord injury recovery by inhibiting inflammatory cytokine expression and NF-kappaB and MAPK signaling pathways. Journal of cellular physiology. 2019;234:14259-69
67. Yang J, Liu Z, Wang C. et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:6792-7
68. Rathkey JK, Zhao J, Liu Z. et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Science immunology. 2018 3
69. Cornelis S, Kersse K, Festjens N. et al. Inflammatory caspases: targets for novel therapies. Current pharmaceutical design. 2007;13:367-85
70. Linton SD. Caspase inhibitors: a pharmaceutical industry perspective. Current topics in medicinal chemistry. 2005;5:1697-717
71. Juliana C, Fernandes-Alnemri T, Wu J. et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. The Journal of biological chemistry. 2010;285:9792-802
72. Wannamaker W, Davies R, Namchuk M. et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. The Journal of pharmacology and experimental therapeutics. 2007;321:509-16
73. Coll RC, Robertson AA, Chae JJ. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine. 2015;21:248-55
74. Ismael S, Nasoohi S, Ishrat T. MCC950, the Selective Inhibitor of Nucleotide Oligomerization Domain-Like Receptor Protein-3 Inflammasome, Protects Mice against Traumatic Brain Injury. J Neurotrauma. 2018;35:1294-303
75. Coll RC, Hill JR, Day CJ. et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nature chemical biology. 2019;15:556-9
76. Daniels MJ, Rivers-Auty J, Schilling T. et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nature communications. 2016;7:12504
77. He Y, Varadarajan S, Munoz-Planillo R. et al. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. The Journal of biological chemistry. 2014;289:1142-50
78. Chen H, Ding Y, Chen W. et al. Glibenclamide alleviates inflammation in oleic acid model of acute lung injury through NLRP3 inflammasome signaling pathway. Drug design, development and therapy. 2019;13:1545-54
79. Marchetti C, Swartzwelter B, Gamboni F. et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E1530-e9
80. Jiang H, He H, Chen Y. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. The Journal of experimental medicine. 2017;214:3219-38
81. Furini F, Giuliani AL, Parlati ME. et al. P2X7 Receptor Expression in Patients With Serositis Related to Systemic Lupus Erythematosus. Frontiers in pharmacology. 2019;10:435
82. Jiang W, Li M, He F. et al. Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J Neuroinflammation. 2017;14:207
83. Yan Y, Bai J, Zhou X. et al. P2X7 receptor inhibition protects against ischemic acute kidney injury in mice. American journal of physiology Cell physiology. 2015;308:C463-72
84. Bauernfeind FG, Horvath G, Stutz A. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. Journal of immunology (Baltimore, Md: 1950). 2009;183:787-91
85. Eiden M, Christinat N, Chakrabarti A. et al. Discovery and validation of temporal patterns involved in human brain ketometabolism in cerebral microdialysis fluids of traumatic brain injury patients. EBioMedicine. 2019;44:607-17
86. Guo M, Wang X, Zhao Y. et al. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front Mol Neurosci. 2018;11:86
87. Hentze H, Lin XY, Choi MS. et al. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell death and differentiation. 2003;10:956-68
Author contact
![]() Corresponding author: Dr. Wenfei Ni, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002814; E-mail: wenfeininet. #Dr. Kailiang Zhou, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002814; E-mail: zhoukailiangedu.cn
Corresponding author: Dr. Wenfei Ni, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002814; E-mail: wenfeininet. #Dr. Kailiang Zhou, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002814; E-mail: zhoukailiangedu.cn