Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(12):2116-2130. doi:10.7150/ijbs.43557 This issue Cite
Research Paper
Inflammation and DNA methylation coregulate the CtBP-PCAF-c-MYC transcriptional complex to activate the expression of a long non-coding RNA CASC2 in acute pancreatitis
Jun Zeng1, Jian-Yong Chen1, Jun Meng1, Zhi Chen2,3 ![]()
1. Department of Gastroenterology, Jiangxi Provincial People's Hospital Affiliated to Nanchang University, Nanchang, Jiangxi, China
2. Department of critical care medicine, Jiangxi Provincial People's Hospital Affiliated to Nanchang University, Nanchang, Jiangxi, China
3. Department of Pulmonary and Critical Care Medicine, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
Received 2020-1-1; Accepted 2020-4-20; Published 2020-5-18
Abstract

Long non-coding RNAs (lncRNAs) are emerging as important regulators involved in the pathogenesis of many diseases. However, it is still unknown if they contribute to the occurrence of acute pancreatitis (AP). Here, we identified a lncRNA CASC2 (Cancer Susceptibility Candidate 2) was significantly upregulated in the pancreatic tissues from AP patients. Knockdown or overexpression of CASC2 in vitro could specifically repress or induce the expression of two proinflammatory cytokines including IL6 (Interleukin 6) and IL17, respectively. Changing the expression levels of several transcription factors that were predicted to bind to the promoter of CASC2, we found c-MYC could specifically regulate the expression of CASC2. Using immunoprecipitation, mass spectrometry, and co-immunoprecipitation assays, we proved that c-MYC assembled a transcriptional complex with PCAF (p300/CBP-associated Factor) and CtBP1/2 (C-terminal Binding Protein 1 and 2), terming as the CtBP-PCAF-c-MYC (CPM) complex. Further investigation revealed that CtBPs were amplified in the pancreatic tissues from AP patients and they functioned as coactivators to induce the expression of CASC2 and thus led to the upregulation of IL6 and IL17. Moreover, we identified that decreased DNA methylation levels in the promoters of CtBPs and inflammatory stimuli coactivated the expression of CtBPs. Collectively, we identified a new signaling pathway in which DNA methylation and inflammatory stimuli coregulate the CPM complex to activate CASC2 expression, whose induction further activates the expression of IL6 and IL17, eventually aggravating inflammation response and causing the pathology of AP.
Keywords: acute pancreatitis, CASC2, CtBP, PCAF, c-MYC
Introduction
Acute pancreatitis (AP) is an inflammatory disorder that occurs in the pancreas and is clinically characterized by severe abdominal pain and increased levels of circulating pancreatic enzymes [1, 2]. Current views recognize that gallstones and alcohol are the two major factors that contribute to the occurrence of AP [1, 2]. AP is often accompanied by other complications, including low blood pressure, fever, organ damage and pancreatic infection [1, 2]. The mortality of AP is still more than 10% even in developed countries [1, 2], which reminds us to pay more attention to investigate the molecular pathogenesis of AP and to identify more therapeutic targets.
Inflammation is a leading cause of AP [3, 4]. The destruction of acinar cells in the pancreas can activate macrophages and granulocytes to secrete proinflammatory cytokines, i.e., tumor necrosis factor (TNF)-α, interleukin-1 beta (IL-1β), IL6, IL17 and IL18 [5-7]. In addition, the activation of multiple signaling pathways has been observed in the pathological process of AP. Of them, the signaling cascades of TLR4/NF-κB (Toll-like receptor 4/Nuclear Transcription Factor-κB) [8], TGF-β/SMAD (Transforming Growth Factor-β/Mothers Against Decapentaplegic Homolog) [9], MAPK (Mitogen-activated Protein Kinase) [10], and JAK/STAT (Janus Kinase/Signal Transducers and Activator of Transcription) pathways have been well characterized [11]. Besides, microRNAs (miRNAs) are also involved in the pathogenesis of AP [12]. A variety of miRNAs such as miR-7, miR-10, miR-92b, miR-126-5p, miR-551b-5p, miR-24-3p, miR-222-3p, miR-361-5p, and miR-1246 are aberrantly expressed in AP patients [13-18]. However, it is still unknown if long non-coding RNAs (lncRNAs) also contribute to the pathogenesis of AP at present. In recent years, emerging evidence indicates that lncRNAs are critical regulators in the process of inflammation response through transcriptionally mediating the expression of inflammatory genes [19]. For instance, Zhao and colleagues found that MALAT1 (Metastasis-associated Lung Adenocarcinoma Transcript 1) could interact with NF-κB to control the expression of TNFA and IL6 [20]. Du and colleagues identified that Mirt2 (Myocardial Infarction-associated Transcript 2) associated with TRAF6 (TNF-associated Factor 6) to prevent the activation of NF-κB and MAPK (Mitogen-Activated Protein Kinase) pathways, thus repressing the expression of proinflammatory cytokines [21]. Recently, Zhang and colleagues revealed that ROCK1 (Regulator of Cytokines and Inflammation) induced by multiple TLR stimuli assembled a ribonucleoprotein complex to control inflammation response [22]. Huang and colleagues found that CASC2 (Cancer Susceptibility 2) was overexpressed in osteoarthritis patients and it could regulate IL17 expression and contribute to chondrocyte proliferation and apoptosis [23]. Except for this unique role in inflammation response, CASC2 also functions as a tumor suppressor and it is downregulated in multiple cancer types including lung cancer [24], gastric cancer [25], colorectal cancer [26], bladder cancer [27], melanoma [28], and glioma [29]. Mechanically, CASC2 exerts its suppressive role mainly through inhibiting the expression of oncogenic miRNAs and genes. For instance, CASC2 is able to repress the expression of miR-18a-5p, thereby inducing PIAS3 (Protein Inhibitor of Activated STAT 3) expression in colorectal cancer cells [26]. CASC2 can interact with miR-181a to control glioma cell growth through upregulating PTEN (Phosphatase and Tensin Homolog) pathway [29]. In gastric cancer cells, CASC2 inhibits the phosphorylation levels of ERK1/2 (Extracellular signal-regulated Kinase 1 and 2) and JNK1 (JUN-N-terminal Kinase 1) to decrease cell proliferation [25]. In bladder cancer cells, overexpression of CASC2 in vitro can decrease the expression of β‐catenin and its downstream target genes c‐MYC and CCND1 (cyclin D1) [27]. However, it is still unknown for the mechanism of CASC2 dysregulation in these biological processes.
Although a large number of lncRNAs have been found to be abnormally expressed in various diseases, the molecular mechanisms that cause their aberrant expression are still poorly understood [16-18]. A few studies reveal that DNA methylation and the transcriptional complex can mediate the aberrant expression of lncRNAs [30-32]. DNA methylation is a fundamental mechanism that regulates the expression of genes and non-coding RNAs and it occurs in the CpG dinucleotides [33]. In mammals, DNA methylation is controlled by three DNA methyltransferases (DNMTs) including DNMT1, DNMT3a and DNMT3b [34]. Transcriptional complexes are often composed of transcription factors [e.g., NF-κB, c-MYC, AP1 (Activator Protein 1), and SP1 (Specificity Protein 1)], coactivators [e.g., p300 (E1A Binding Protein 300), CBP (CREB Binding Protein) and PCAF (p300 and CBP-associated Factor)], and corepressors [e.g., CtBPs (C-terminal binding proteins) and RB1 (Retinoblastoma 1)] [35-37]. Recently, Zhang and colleagues found that IRF1 (Interferon Regulatory Factor 1) could recruit HDAC1/2 (Histone Deacetylase 1 and 2) and CtBP1 to assemble a complex in the promoter of a lncRNA GAS5 (Growth Arrest Specific 5) to repress its expression [38].
To identify aberrantly expressed lncRNAs that control the pathogenesis of AP, we carried out a microarray analysis using the pancreatic tissues from AP patients and totally found 21 differentially expressed lncRNAs. We focused the current study on revealing the downstream targets and the upstream signaling of CASC2, the mostly upregulated lncRNA in our microarray result. Our results demonstrated that CASC2 could control the expression of two proinflammatory cytokines including IL6 and IL17. The transcription factor c-MYC recruited PCAF and CtBP1/2 to the promoter of CASC2 and activated its expression. Both DNA methylation and inflammatory stimuli could trigger the activation of CtBP-associated transcriptional complex, inducing the expression of CASC2 and its downstream targets.
Materials and Methods
Blood sample collection and measurement of serum cytokines
Blood samples were collected from the veins of pancreatic cancer patients (stage 0, setting as controls) (n=48) and AP patients (n=48). The basic information about pancreatic cancer patients and AP patients is summarized in Supplementary Table-1. All participants were aware of the purpose of this study and signed a consent form reviewed and approved by the ethical board of Jiangxi Provincial People's Hospital in China. Blood samples were immediately stored in plastic whole blood tubes with spray-coated K2EDTA (BD, Franklin Lakes, NJ, USA, #367835). The concentrations of cytokines including TNF-α, IL-1β, IL4, IL6, IL8, IL10, IL13, IL15, and IL17 were examined using their corresponding enzyme-linked immunosorbent assay (ELISA) kits purchased from the Thermo Fisher Scientific company (Waltham, MA, USA). The catalog numbers of these kits were as follows: #KHC3011 (TNF-α), #KAC1211 (IL-1β), #KAC1281 (IL4), #KHC0061 (IL6), #KHC0081 (IL8), #KAC1321 (IL10), #BMS231INST (IL13), #BMS2106 (IL15), and #BMS2017 (IL17).
Pancreatic biopsy collection
Pancreatic biopsies were collected from the same patients as described in blood sample collection using the endoscopic ultrasound-guided fine-needle aspiration (EUS-FNA) method [39]. The pancreatic cancer patients had no obvious inflammation in their pancreases according to endoscopic ultrasound results. All participants were aware of the purpose of this study and signed a consent form reviewed and approved by the ethical board of Jiangxi Provincial People's Hospital in China.
Cell culture and transfection
The human pancreatic epithelial cell line MIA PaCa-2 was obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA, #CRL-1420) and grown in DMEM (Dulbecco's modified Eagle's medium) (Sigma-Aldrich, #D6046) supplemented with 10% FBS (Fetal Bovine Serum) (Thermo Fisher Scientific, #16000044) and 1% PS (Penicillin-Streptomycin) (Thermo Fisher Scientific, #15140163). Cells under 80% confluence were used for transfections with plasmids or siRNAs, respectively, following a previous protocol [35]. The siRNAs used in this study were all purchased from the Thermo Fisher Scientific company and they were included siCASC2-1 (#HSS153939), siCASC2-2 (HSS153940), siCtBP1-1 (#114133), siCtBP1-2 (#2802), siCtBP2-1 (#30207), siCtBP2-2 (#289298), siDNMT1-1 (#S4215), siDNMT1-2 (#S4216), siDNMT3a-1 (#HSS176225), siDNMT3a-2 (#HSS176226), siDNMT3b-1 (#111744), siDNMT3b-2 (#111745), siSP1-1 (#116546), siSP1-2 (#116547), sip50-1 (#107296), sip50-2 (#107296), sip65-1 (#109424), sip65-2 (#116318), sic-JUN-1 (#106741) and sic-JUN-1 (#115273). After transfecting for 48 h, cells were collected and used in the required experiments.
Protein extraction and western blotting
Three-paired pancreatic biopsies from pancreatic cancer patients (stage 0) and AP patients and cultured cells were used for protein extraction with 1 × RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA, #R0278). Total protein concentrations were determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific, #ND-2000) at 280 nm. Equal amounts of total proteins were loaded onto 12% SDS-PAGE gels for electrophoretic separation. Proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Chicago, IL, USA, #10600023), followed by blocking with 5% milk for 1 h and then probing with primary antibodies including anti-c-MYC (Sigma-Aldrich, #06-340), anti-PCAF (Sigma-Aldrich, #SAB2101734), anti-CtBP1 (BD Biosciences, San Jose, CA, USA, #612042), anti-CtBP2 (BD Biosciences, #612044), anti-Myc (Abcam, Shanghai, China, #ab206486), anti-Flag (Sigma-Aldrich, #SAB4200071), anti-DNMT1 (Sigma-Aldrich, #D4692), anti-DNMT3a (Sigma-Aldrich, #SAB1410305), anti-DNMT3b (Sigma-Aldrich, #SAB2700189), and anti-GAPDH (Abcam, #ab8254). After washing with TBST buffer 5 times, the membrane was further probed with secondary antibodies. Protein signals were recorded with a ChemiDoc Imaging System from Bio-Rad (Hercules, CA, USA).
Total RNA isolation, microarray and qRT-PCR analyses
The pancreatic biopsies and freshly harvested cells were used for total RNA isolation using the TRIzolTM reagent (Thermo Fisher Scientific, #15596026) according to the manufacturer's instructions. Total RNA concentrations were determined using a Nanodrop spectrophotometer, followed by microarray and qRT-PCR analyses. For microarray analyses, 500 ng RNA of each sample was applied to experiments using an Arraystar Human LncRNA Array V4.0 Chip (Arraystar Inc., #AS-LNC-H-V4.0) and a GeneChip Human Genome U133 Plus 2.0 Array (ThermoFisher Scientific, #900467) following a protocol provided by the manufacturers, respectively. For qRT-PCR analyses, 1.0 μg RNA of each sample was used to synthesize cDNA using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, #4368814). After diluting 20-fold, cDNAs were subjected to qRT-PCR analyses to detect individual gene expression with primers listed in Supplementary Table-2. The PCR procedures included: 95°C for 3 min, 40 cycles of 95°C for 30 seconds and 68°C for 30 seconds, and 4°C for 10 min. The relative expression of individual genes was normalized to β-Actin using the 2-ΔΔCt method as described previously [35].
Vector constructions
The coding regions of CtBP1, CtBP2, PCAF, c-MYC, DNMT1, DNMT3a, and DNMT3b were amplified by PCR and then cloned into the BamHI and EcoRI sites of pCDNA3-2×Flag and pCDNA3-6×Myc empty vectors, respectively. The coding regions of SP1, p50, p65, and c-Jun were cloned into the BamHI and EcoRI sites of pCDNA3-2×Flag empty vectors. The full length of CASC2 transcript was cloned into the BamHI and EcoRI sites of pCDNA3 empty vector. The resulting vectors were sequenced to verify the correct constructions, followed by purification for transfection. The primers for these constructions were listed in Supplementary Table-3.
Quantitative methylation-specific PCR (qMSP) analysis
The qMSP analysis was performed following a previous protocol [40]. Briefly, the DNA methylation inhibitor AZA (5-Aza-2'-deoxycytidine)-treated cells were subjected to DNA isolation using a PureLinkTM Genomic DNA Mini Kit (Thermo Fisher Scientific, #K182001) according to the manufacturer's protocol. After quantification with a Nanodrop spectrophotometer, 1 µg of genomic DNA from each sample was treated with sodium bisulfite using the EZ DNA Methylation-GOLD Kit (Zymo Research, Tustin, CA, USA, #D5006) following a protocol provided by the manufacturer. The resulting genomic DNA was used for qMSP analysis using the TaqMan® Universal Master Mix II non-UNG Kit (Thermo Fisher Scientific, #4440038) with primers listed in Supplementary Table-4.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed following a previous protocol [35]. Briefly, cells under 80% confluence were crosslinked with 1% formaldehyde for 10 min at room temperature, followed by quenching with glycine at a final concentration of 0.125 M. After fixation, cells were used for immunoprecipitation with a kit (Abcam, #ab185913) according to the method provided by the manufacturer. The antibodies used for immunoprecipitation included anti-CtBP1, anti-CtBP2, anti-PCAF and anti-c-MYC, whose information was the same as descried in the western blotting. The purified input and output DNA were used for qRT-PCR analyses with the same PCR procedure as described in mRNA level detection. The primers were located in the promoter of CASC2 and their sequences were: forward, 5'-CCGACTTCCCTATGGCTGATGTC-3' and reverse, 5'-GTCCGCGCAGAGGCCTGCACGG-3'.
Immunohistochemistry (IHC) staining and quantification
Three-paired pancreatic tissues from pancreatic cancer (stage 0) and AP patients were subjected to IHC staining following a previous protocol [41]. In brief, the formalin-fixed tissues were sectioned at a thickness of 5 μm, followed by mounting on glass slides. After de-paraffinizing, rehydrating, quenching to remove endogenous peroxidase and antigen retrieval, the slides were incubated with anti-CtBP1 (1:100), anti-CtBP2 (1:100), anti-PCAF (1:200), anti-c-MYC (1:100), anti-IL6 (1:300), anti-IL17 (1:200), anti-DNMT1 (1:100) or anti-DNMT3a (1:100). These antibodies were the same as those used in western blotting assay. After incubating with secondary biotinylated antibodies, the signals of these proteins in tissue sections were visualized using a mouse and rabbit specific HRP/DAB detection kit (Abcam, #ab64264). The quantification of immunohistochemical scores was carried out using the histoscore (H-score) based on the definition and standards described previously.
Statistical analysis
All experiments were independently performed in triplicate. Statistical analyses of the experimental data were performed using a two-sided Student's t test. The significance levels were set at P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***). The Pearson correlation coefficient (r) and associated probability (P) were performed to determine the correlation between CASC2 expression and cytokines, DNMTs and CtBP-associated transcriptional complex members.
Results
The concentrations of circulating proinflammatory cytokines were significantly increased in AP patients
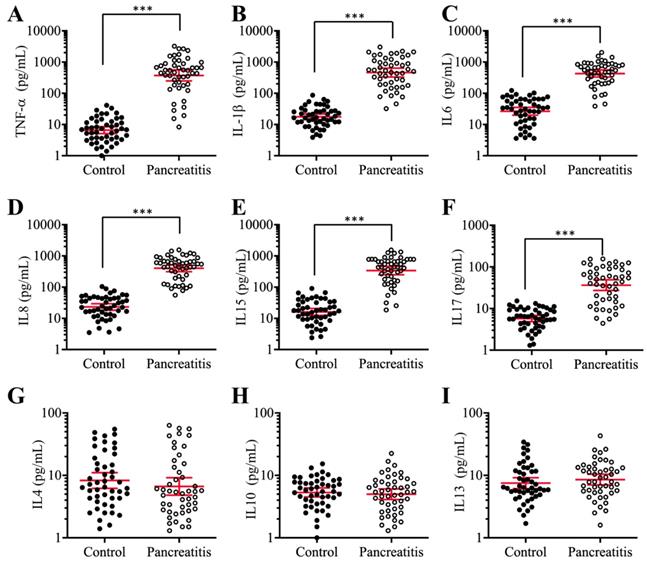
Previous publications have shown increased levels of some proinflammatory cytokines, such as TNF-α, IL-1β and IL8, in pancreatic patients and animal models [42, 43]. To verify that the elevated levels of proinflammatory cytokines are a common phenomenon in AP patients, we collected 48-paired blood samples from pancreatic cancer patients (stage 0, control) and AP patients and measured the serum concentrations of six proinflammatory cytokines (TNF-α, IL-1β, IL6, IL8, IL15 and IL17) and three anti-inflammatory cytokines (IL4, IL10 and IL13). The ELISA results indicated that the average levels of all six proinflammatory cytokines in the serum of AP patients were much higher than those in controls (Figures 1A-1F), while the average concentrations of the three anti-inflammatory cytokines were not significantly different between the two groups (Figures 1G-1I). In detail, the concentrations of these cytokines in controls compared to AP patients were as follows: TNF-α (9.23±1.05 pg/mL vs 537.65±78.22 pg/mL, P < 0.001) (Figure 1A), IL-1β (19.15±1.69 pg/mL vs 643.69±68.75 pg/mL, P < 0.001) (Figure 1B), IL6 (27.55±2.67 pg/mL vs 478.34±37.88 pg/mL, P < 0.001) (Figure 1C), IL8 (23.91±2.44 pg/mL vs 311.46±26.57 pg/mL, P < 0.001) (Figure 1D), IL15 (17.72±1.64 pg/mL vs 333.88±42.54 pg/mL, P < 0.001) (Figure 1E), IL17 (7.22±0.82pg/mL vs 51.43.16±4.25 pg/mL, P < 0.001) (Figure 1F), IL4 (8.32±1.13 pg/mL vs 6.87±1.55 pg/mL, P = 0.73) (Figure 1G), IL10 (5.09±0.87 pg/mL vs 4.64±0.79 pg/mL, P = 0.68) (Figure 1H), and IL13 (7.57±1.22 pg/mL vs 8.46±1.13 pg/mL, P = 0.46) (Figure 1I). These results suggested that the elevated levels of proinflammatory cytokines were associated with the occurrence of AP.
The serum concentrations of proinflammatory cytokines were significantly increased in AP patients. Circulating levels of TNF-α (A), IL-1β (B), IL6 (C), IL8 (D), IL15 (E), IL17 (F), IL4 (G), IL10 (H), and IL13 (I) were measured in blood samples collected from stage 0 pancreatic patients (control, n = 48) and AP patients (Pancreatitis, n=48) using ELISA assays. *** P < 0.001.
The lncRNA CASC2 was upregulated in AP patients
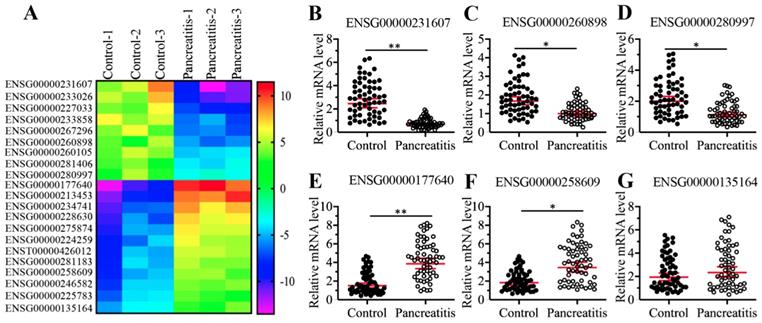
To identify differentially expressed lncRNAs in the pancreatic tissues, we conducted a microarray analysis using three-paired pancreatic tissues from AP patients and pancreatic cancer patients (stage 0, control). In total, we identified 21 lncRNAs that were consistently downregulated or upregulated in AP patients compared to controls (Supplementary Table-5). Of these differentially expressed lncRNAs, 9 were downregulated, and the remaining 12 were upregulated in AP samples (Figure 2A and Supplementary Table-5). To further evaluate whether the expression of these differentially expressed lncRNAs was associated with AP, we randomly selected three downregulated lncRNAs (ENSG00000251562, ENSG00000260898, and ENSG00000280997) and three upregulated lncRNAs (ENSG00000177640, ENSG00000258609, and ENSG00000135164) and measured their levels in 48-paired pancreatic tissues from the same controls and AP patients as described in blood collection. Consistent with the microarray results, we observed the downregulation of ENSG00000251562, ENSG00000260898 and ENSG00000280997 and the upregulation of ENSG00000177640 and ENSG00000258609 in the 48 AP patients compared to the controls (Figures 2B-2F). However, the expression of ENSG00000135164 was not significantly changed in the same RNA samples (Figure 2G), which suggested that ENSG00000135164 might be a false positive lncRNA in the microarray result. After checking the chromosome loci of these lncRNAs, we found that they were assigned different names and most of them were previously reported to function in the pathogenesis of tumorigenesis (Supplementary Table-5). The most upregulated lncRNA ENSG00000177640 was termed as CASC2, which has been characterized as a tumor suppressor in many cancer types [24-29, 44]. The significantly aberrant expression pattern of CASC2 in AP patients inspired us to investigate its role in this process in the following study.
Identification and verification of differentially expressed lncRNAs in AP patients. (A) The heatmap of aberrantly expressed lncRNAs. Total RNA from three-paired pancreatic tissues of pancreatic cancer patients (stage 0) (Control-1, -2 and -3) and AP patients (Pancreatitis-1, -2 and -3) were subjected to microarray analysis. The differentially expressed lncRNAs were shown. (B-G) Verification of lncRNA expression levels. Total RNA from 48-paired pancreatic tissues of pancreatic cancer patients (Control, stage 0) (Control) and AP patients (Pancreatitis) were subjected to qRT-PCR analyses to measure the expression levels of three downregulated lncRNAs including ENSG00000251562 (B), ENSG00000260898 (C), and ENSG00000280997 (D), and three upregulated lncRNAs including ENSG00000177640 (E), ENSG00000258609 (F), ENSG00000135164 (G). * P < 0.05 and ** P < 0.01.
CASC2 regulated the expression of IL6 and IL17
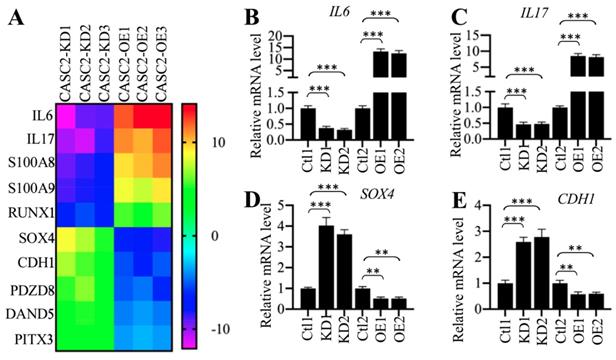
To identify the downstream target genes dependent on CASC2, we generated three CASC2 knockdown (KD) and overexpression (OE) cell lines, respectively, followed by performing a gene-based microarray analysis using these cell lines. Totally, we identified 25 genes that were conversely expressed in three CASC2-KD and CASC2-OE cell lines (Supplementary Table-6). Of these differentially expressed genes, 8 were downregulated in CASC2-KD cells but were upregulated in CASC2-OE cells, while the other 17 were upregulated in CASC2-KD cells but were downregulated in CASC2-OE cells (Supplementary Table-6). Interestingly, we only observed that the changes in CASC2 expression could affect the expression of IL6 and IL17 but not the other proinflammatory cytokine genes (Supplementary Table-6). As shown in Figure 3A, we selected 10 genes that were mostly changed when CASC2 was knocked down or overexpressed. To further verify our microarray results, we randomly selected four genes including IL6, IL17, SOX4 and CDH1 and determined their dependency on CASC2. Consistent with the microarray results, we observed the overexpression of CASC2 could induce the expression of IL6 and IL17 but repress the expression of SOX4 and CDH1 (Figures 3B-3F). In contrast, knockdown of CASC2 resulted in the reverse effects on the expression of these four genes (Figures 3B-3F). Besides, we also measured the concentrations of IL6 and IL17 in the supernatant of cell culture. The ELISA results indicated that both IL6 and IL17 concentrations were decreased in the supernatant of CASC2-KD cells but increased in the supernatant of CASC2-OE cells (Supplementary Figure 1). The dependency of IL6 and IL17 on CASC2 expression in vitro together with the results shown in Figures 1C, 1F and 2E suggested that CASC2 was a key regulator of IL6 and IL17 in the pathogenesis of AP. To investigate if the expression of CASC2 were correlated with circulating IL6 and IL17 concentrations in AP patients, we conducted a Pearson correlation assay and found that both of them were positively correlated with the CASC2 expression (Supplementary Figures 2A and 2B). In contrast, we did not identify a correlation between IL13 and CASC2 expression (Supplementary Figure 2C).
The expression of IL6 and IL17 was dependent on CASC2. (A) Identification of genes dependent on CASC2. Total RNA from three independent CASC2-KD (1, 2 and 3) and CASC2-OE (1, 2 and 3) cell lines were subjected to microarray analysis. The top 10 differentially expressed genes were shown. (B-E) Verification of gene expression levels. Total RNA from one Control-KD (Ctl1), two independent CASC2-KD cell lines (KD1 and KD2), one Control-OE (Ctl2), and two independent CASC2-OE (OE1 and OE2) were subjected to qRT-PCR analyses to measure the expression levels of IL6 (B), IL17 (C), SOX4 (D), and CDH1 (E). ** P < 0.01 and *** P < 0.001.
Transcription factor c-MYC specifically regulated the expression of CASC2
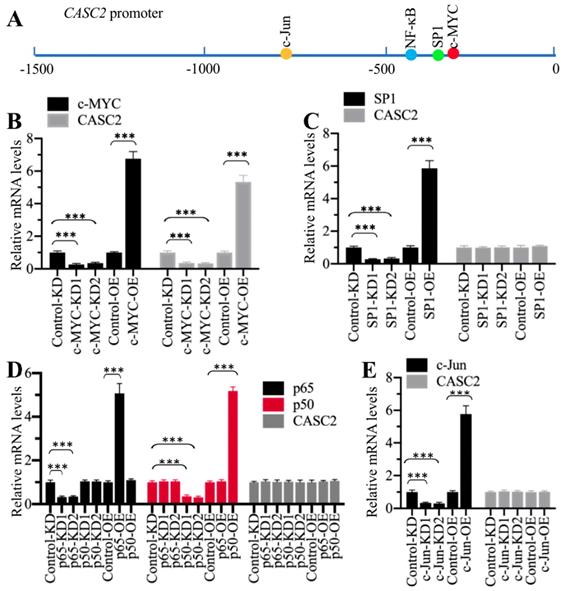
To reveal the mechanism underlying CASC2 overexpression in AP patients, we initially analyzed its promoter to determine if transcription factors could control the expression of CASC2. We selected a length of 1500-bp promoter region of CASC2 and scanned the potential transcription factor binding sites. As shown in Figure 4A, we found four conserved transcription factor binding sites including c-MYC [-356-(-361), CACGTG], SP1 [-390-(-399), CCCCCGCCCC], NF-κB [-414-(-423), GGGGGTCCCC], and c-JUN [-762-(-768), TGTGTCA] using their consensus sequences. We individually knocked down and overexpressed these transcription factors. After verifying their successful downregulation and upregulation with immunoblots (Supplementary Figure 3), we evaluated their effects on CASC2 expression. As shown in Figure 4B, knockdown or overexpression of c-MYC decreased or increased CASC2 level, respectively. However, knockdown or overexpression of the other three transcription factors could not change the expression of CASC2 (Figures 4C-4E). These results suggested that c-MYC could specifically regulate the expression of CASC2 at the transcriptional level.
c-MYC specifically regulated the expression of CASC2. (A) The potential transcription factor binding sites on the promoter of CASC2. A 1500-bp length of the CASC2 promoter was predicted the transcription factor binding sites. One c-MYC, one SP1, one NF-κB and one c-JUN binding sites were found, and their positions were shown. (B) Knockdown or overexpression of c-MYC changed the expression of CASC2. Total RNA from Control-KD, c-MYC-KD1, c-MYC-KD2, Control-OE, and c-MYC-OE cells were applied to qRT-PCR analyses to measure the mRNA levels of c-MYC and CASC2. ***P<0.001. (C) Knockdown or overexpression of SP1 could not change the expression of CASC2. Total RNA from Control-KD, SP1-KD1, SP1-KD2, Control-OE, and SP1-OE cells were applied to qRT-PCR analyses to measure the mRNA levels of SP1 and CASC2. ***P<0.001. (D) Knockdown or overexpression of NF-κB subunits could not change the expression of CASC2. Total RNA from Control-KD, p50-KD1, p50-KD2, p65-KD1, p65-KD2, Control-OE, p50-OE and p65-OE cells were applied to qRT-PCR analyses to measure the mRNA levels of p50, p65 and CASC2. ***P<0.001. (E) Knockdown or overexpression of c-JUN could not change the expression of CASC2. Total RNA from Control-KD, c-JUN-KD1, c-JUN-KD2, Control-OE, and c-JUN-OE cells were applied to qRT-PCR analyses to measure the mRNA levels of c-JUN and CASC2. ***P<0.001.
c-MYC recruited PCAF and CtBPs to assemble a transcriptional complex
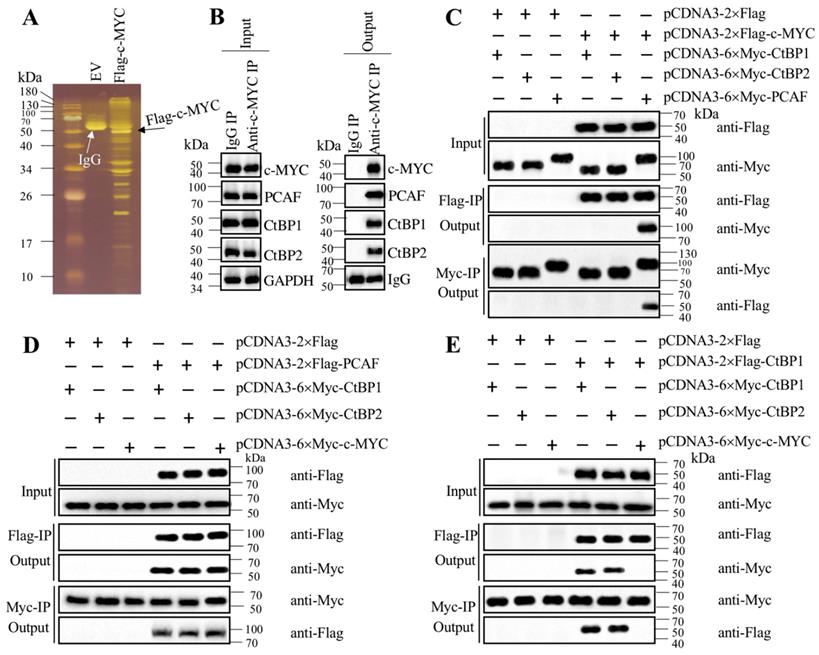
Transcription factors often recruit coactivators and corepressors to assemble transcriptional complexes [35-37]. To investigate how c-MYC activated CASC2 in the pathogenesis of AP, we performed immunoprecipitation in cells expressing pCDNA3-2×Flag (empty vector, EV) and pCDNA3-2×Flag-c-MYC (Figure 5A). The purified Flag-c-MYC complex was applied to mass spectrometry analysis to identify c-MYC-associated proteins. Totally, we obtained 35 proteins in this complex (Supplementary Table-7). After analyzing the candidate protein list, we found a histone acetyltransferase PCAF and two corepressors CtBP1 and CtBP2, which are well-known components of a transcriptional machinery. To determine if c-MYC could associate these three proteins to assemble a complex in vivo, we performed an immunoprecipitation assay in AP tissues using anti-c-MYC antibody. The purified protein complex was subjected to immunoblots to examine the existence of PCAF and CtBP1/2. The western blotting results indicated that c-MYC could pull down PCAF and CtBP1/2 together (Figure 5B), which suggested that c-MYC could assemble a complex with these three proteins in vivo. To further determine how this complex was assembled, we performed coimmunoprecipitation (Co-IP) assay to examine the direct interactions between c-MYC and PCAF, c-MYC and CtBPs, PCAF and CtBPs, as well as CtBP1 and CtBP2. The Co-IP results indicated that c-MYC could interact with PCAF directly but not with CtBPs (Figure 5C), and PCAF could directly interact with both c-MYC and CtBPs (Figures 5C and 5D). In addition, CtBP1 and CtBP2 could interact with themselves and each other (Figure 5E), suggesting that they could form a heterotetramer. These results clearly suggested that c-MYC directly recruited PCAF, which acted as a linker to bind to the CtBP heterotetramer to assemble the CtBPs-PCAF-c-MYC (CPM) complex. To determine if this complex specifically bond to the promoter of CASC2, we conducted ChIP assays in c-MYC-KD and c-MYC-OE cells to evaluate the occupancies of CPM components. The ChIP results showed that knockdown of c-MYC significantly decreased the enrichment of CPM components in the promoter of CASC2 (Supplementary Figure 4A), while overexpression of c-MYC increased their occupancies (Supplementary Figure 4B). These results suggested that the CPM complex was required for the regulation of CASC2 overexpression in the pathogenesis of AP.
c-MYC associated with PCAF and CtBPs to assemble a complex. (A) The Flag-c-MYC-associated complex. The pCDNA3-2×Flag (empty vector, EV) and pCDNA3-2×Flag-c-MYC plasmids were transfected into MIA PaCa-2 cells, respectively. The resulting cells were subjected to immunoprecipitation with the anti-Flag resin. The purified complexes were separated in an SDS-PAGE gel and incubated with a silver staining kit. The IgG and Flag-c-MYC were indicated by arrows. (B) c-MYC could pull down PCAF and CtBPs in vivo. Equal weight of pancreatic tissues from three AP patients was mixed and lysed, and 1/11 cell extracts were used as an input, and the other 10/11 cell extracts were equally divided into two parts, followed by immunoprecipitation with an IgG and anti-c-MYC antibody-associated protein A beads, respectively. The input and output proteins were used to determine protein levels of c-MYC, PCAF, CtBP1 and CtBP2, respectively. (C) c-MYC directly interacted with PCAF but not CtBPs in vitro. The MIA PaCa-2 cells were transfected with different plasmids including pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP2, pCDNA3-2×Flag + pCDNA3-6×Myc-PCAF, pCDNA3-2×Flag-c-MYC + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag-c-MYC + pCDNA3-6×Myc-CtBP2, and pCDNA3-2×Flag-c-MYC + pCDNA3-6×Myc-PCAF. The resulting cells were lysed and immunoprecipitated with an anti-Flag and anti-Myc resins, respectively, followed by immunoblots to examine the input and output proteins levels using anti-Flag and anti-Myc antibodies. (D) PCAF directly interacted with both c-MYC and CtBPs in vitro. The MIA PaCa-2 cells were transfected with different plasmids including pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP2, pCDNA3-2×Flag + pCDNA3-6×Myc-c-MYC, pCDNA3-2×Flag-PCAF + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag-PCAF + pCDNA3-6×Myc-CtBP2, and pCDNA3-2×Flag-PCAF + pCDNA3-6×Myc-c-MYC. The resulting cells were lysed and immunoprecipitated with an anti-Flag and anti-Myc resins, respectively, followed by immunoblots to examine the input and output protein levels using anti-Flag and anti-Myc antibodies. (E) CtBPs assembled a heterotetramer in vitro. The MIA PaCa-2 cells were transfected with different plasmids including pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag + pCDNA3-6×Myc-CtBP2, pCDNA3-2×Flag + pCDNA3-6×Myc-c-MYC, pCDNA3-2×Flag-CtBP1 + pCDNA3-6×Myc-CtBP1, pCDNA3-2×Flag-CtBP1 + pCDNA3-6×Myc-CtBP2, and pCDNA3-2×Flag-CtBP1 + pCDNA3-6×Myc-c-MYC. The resulting cells were lysed and immunoprecipitated with an anti-Flag and anti-Myc resins, respectively, followed by immunoblots to examine the input and output proteins levels using anti-Flag and anti-Myc antibodies.
CtBPs functioned as coactivators to regulate the expression of CSAC2
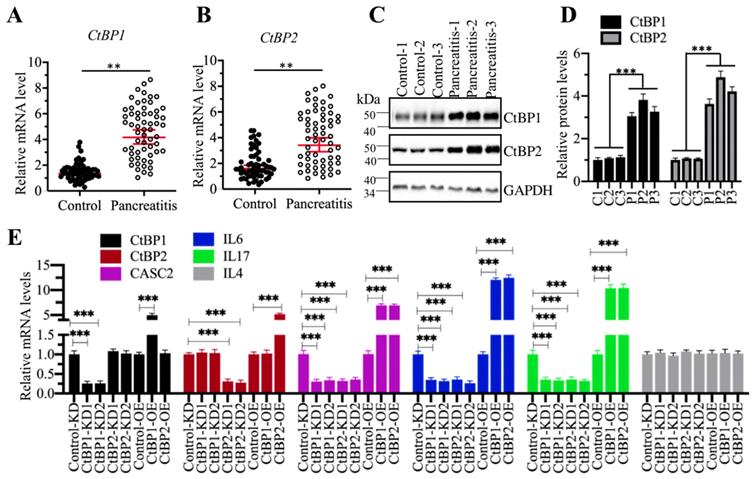
CtBPs can function as either corepressors or coactivators in different biological processes [35-37]. To determine how CtBPs functioned in the regulation of CASC2 expression, we primarily detected the mRNA and protein levels of CtBPs in the pancreatic samples from AP and control patients. The qRT-PCR results showed that both CtBP1 and CtBP2 mRNA levels were significantly upregulated in comparison to controls (Figures 6A and 6B). We then randomly selected three-paired tissues as represent to detect the protein levels CtBP1 and CtBP2. Similarly, we also observed the accumulation of CtBP protein levels in AP samples compared to controls (Figures 6C and 6D). The similar expression patterns of CtBPs with CASC2 implied that they might transactivate CASC2 expression. To verify this hypothesis, we created CtBP-KD and CtBP-OE cell lines and then examined the expression levels of CASC2 and its downstream targets IL6 and IL17 in these cells. As expected, we observed that knockdown of CtBPs repressed the expression of CASC2, IL6 and IL17 while overexpression of CtBPs caused the reverse effects on the expression of CASC2 and its targets (Figure 6E). In addition, we also conducted a Pearson correlation assay to determine the correlation between CtBPs and CASC2 in pancreatic samples from AP patients. As shown in Supplementary Figure 5, the expression of both CtBP1 and CtBP2 was positively correlated with CASC2. These results together supported the conclusion that CtBPs acted as coactivators instead of corepressor in the regulation of CASC2 expression.
CtBPs were overexpressed in AP patients. (A and B) CtBPs were overexpressed in AP patients. Total RNA from 48-paired pancreatic tissues of pancreatic cancer patients (Control, stage 0) and AP patients (Pancreatitis) were subjected to qRT-PCR analyses to measure the mRNA levels of CtBP1 (A) and CtBP2 (B). **P < 0.01. (C) CtBP protein levels were elevated in AP patients. Three-paired pancreatic tissues from controls (1, 2 and 3) and AP patients (1, 2 and 3) were used to determine protein levels of CtBP1 and CtBP2. GAPDH was probed as a loading control. (D) The relative protein levels of CtBPs. The protein bands in (C) were quantified and normalized to GAPDH. ***P < 0.001. (E) The effects of knockdown or overexpression of CtBPs on the expression of CASC2, IL6, IL17 and IL4. Total RNA from Control-KD, CtBP1-KD1, CtBP1-KD2, CtBP2-KD1, CtBP2-KD2, Control-OE, CtBP1-OE and CtBP2-OE were used to examine mRNA levels of CtBP1, CtBP2, CASC2, IL6, IL17 and IL4 by qRT-PCR analyses. ***P < 0.001.
Decreased DNA methylation levels in the promoters of CtBPs contributed to their overexpression in AP pancreatic tissues
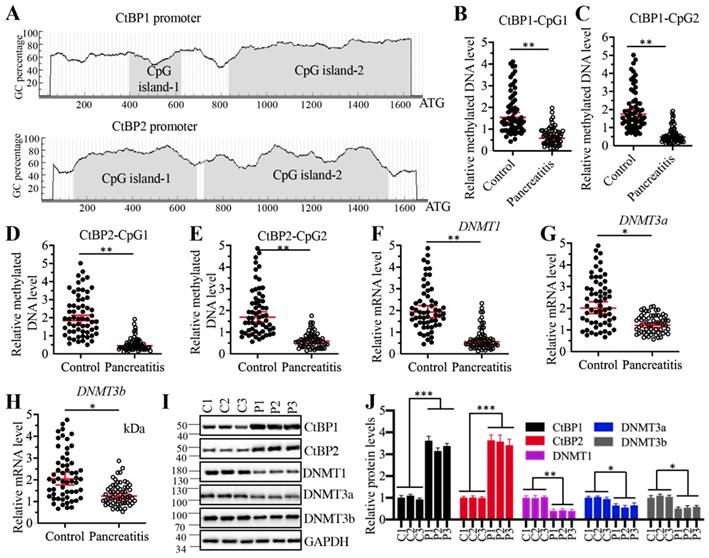
Given that CtBPs were upregulated at the transcriptional level, we next aimed to investigate the underlying mechanisms of their overexpression. DNA methylation is a fundamental mechanism that regulates gene expression. To investigate if DNA methylation was involved in the regulation of CtBP overexpression, we selected a 1700-bp length of CtBP promoters and predicted if they were abundant in GC nucleotides in a website (http://www.urogene.org). As shown in Figure 7A, both CtBP1 and CtBP2 promoters were abundant in GC nucleotides and they had two CpG islands. We then examined the methylated levels in the CpG islands. Surprisingly, we observed that the methylated levels of CpG islands in both CtBP1 and CtBP2 promoters were significantly decreased in AP pancreatic samples compared to controls (Figures 7B-7E). These results implied that the expression of DNMTs might be changed in AP tissues. To validate this hypothesis, we examined the mRNA and protein levels of three DNMTs including DNMT1, DNMT3a and DNMT3b in three-paired pancreatic tissues from AP patients and controls. The results indicated that all three DNMTs were downregulated in their mRNA and protein levels in AP patients (Figure 7F-7J). To determine if changes of DNMT levels affected the expression of CtBPs, we generated two independent knockdown and one overexpression cell lines of each DNMT genes. After confirming their successful knockdown and overexpression (Supplementary Figures 6A and 6B), we measured CtBP levels in these cells. The qRT-PCR results indicated that knockdown of DNMTs caused the upregulation of CtBPs while overexpression of DNMTs resulted in the downregulation of CtBPs (Supplementary Figures 6A and 6B). We also observed similar patterns of CtBP protein levels in DNMT-KD and DNMT-OE cells (Supplementary Figures 6C and 6D). These results suggested that decreased DNA methylation levels in the CtBP promoters were responsible for their overexpression. In addition, we also measured the mRNA levels of CASC2, IL6 and IL17 in these DNMT-KD and DNMT-OE cells. Consistent with the expression of CtBPs in these cells, we also observed the increase or decrease of CASC2, IL6 and IL17 levels in DNMT-KD or DNMT-OE cells, respectively (Supplementary 7). To determine if the expression of DNMTs was correlated with CASC2, we also conducted a Pearson correlation assay and found DNMT mRNA levels were negatively correlated with CASC2 expression (Supplementary Figure 8). Thus, we revealed a mechanism in which the downregulation of DNMTs caused the induction of CtBPs, which further assembled a CPM complex to activate CASC2 expression. To further support this conclusion, we also carried out IHC staining assay to determine the protein levels of DNMTs, CtBPs, PCAF, c-MYC and two proinflammatory cytokines IL6 and IL17 in the pancreatic tissues from AP patients and controls. The IHC staining results of these proteins and their corresponding quantification results also showed CtBPs, PCAF, c-MYC, IL6 and IL17 protein levels were much higher while DNMT protein levels were much lower in AP patients compared to controls (Supplementary Figure 9).
Decreased DNA methylation levels in the promoters of CtBPs were responsible for their overexpression. (A) Both CtBP1 and CtBP2 promoters had two CpG islands. A 1700-bp length of CtBP promoters was selected to predict the CpG island and two CpG islands were identified in each gene. (B-E) The relative DNA methylation levels of CpG islands in AP patients. Pancreatic tissues from 48-paired controls and AP patients were subjected to isolate genomic DNA, followed by treating with sodium bisulfite. The qMSP analyses were performed to examine the methylated DNA levels in the CpG islands of CtBP1 (B and C) and CtBP2 (D and E) promoters. ** P < 0.01. (F-H) DNMTs were downregulated in AP patients. Total RNA from 48-paired pancreatic tissues of controls and AP patients were subjected to qRT-PCR analyses to measure the mRNA levels of DNMT1 (F), DNMT3a (G) and DNMT3b (H). *P < 0.05 and **P < 0.01. (I) DNMT protein levels were decreased in AP patients. Three-paired pancreatic tissues from controls (1, 2 and 3) and AP patients (1, 2 and 3) were used to determine protein levels of CtBP1, CtBP2, DNMT1, DNMT3a and DNMT3b, respectively. GAPDH was probed as a loading control. (J) The relative protein levels of DNMTs. The protein bands in (I) were quantified and normalized to GAPDH. *P < 0.05, **P < 0.01 and ***P < 0.001.
Inflammation stimuli induced the expression of CtBPs
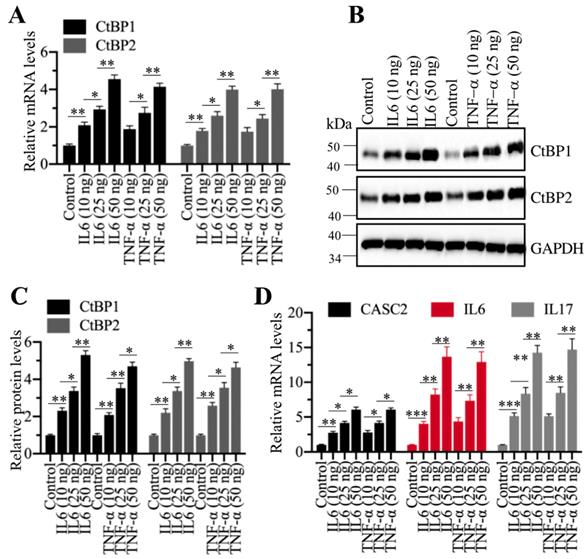
Given that CtBPs were significantly amplified in AP tissues, we were curious if inflammation stimuli could induce their expression. For this purpose, we treated cells with different concentrations (0, 10, 25 and 50 ng/mL) of recombinant IL6 or TNF-α, followed by examining CtBP mRNA and protein levels. Interestingly, the qRT-PCR results showed that both IL6 and TNF-α treatments could induce the expression of CtBPs in a dose-dependent manner (Figure 8A). Similarly, we also observed a dose-dependent induction of CtBP protein levels in IL6 or TNF-α-treated cells (Figures 8B and 8C). Moreover, we also measured the mRNA levels of CASC2, IL6 and IL17 in IL6- or TNF-α-treated cells. Consistent with CtBP expression patterns, we also found a dose-dependent induction of CASC2, IL6 and IL17 (Figure 8D). These results suggested that inflammation microenvironment in AP patients might also contribute to the overexpression of CtBPs.
Recombinant IL6 and TNF-α induced the expression of CtBPs at both transcriptional and protein levels. (A) IL6 and TNF-α induced the mRNA levels of CtBPs. The MIA PaCa-2 cells were treated with IL6 and TNF-α at the concentrations of 0, 10, 25 and 50 ng/mL for 6 h. The resulting cells were used for RNA isolation, followed by qRT-PCR analyses to determine the mRNA levels of CtBP1 and CtBP2. **P<0.01 and ***P<0.001. (B) IL6- and TNF-α induced the protein levels of CtBPs. Cells used in (A) were subjected to immunoblots to examine protein levels of CtBP1 and CtBP2. GAPDH was probed as a loading control. (C) The relative protein levels of CtBPs. The protein bands in (B) were quantified and normalized to GAPDH. **P < 0.01 and ***P < 0.001. (D) IL6 and TNF-α induced the mRNA levels of CASC2, IL6 and IL17. RNA samples used in (A) were subjected to examine mRNA levels of CASC2, IL6 and IL17 by qRT-PCR analyses. **P < 0.01 and ***P < 0.001.
DNA methylation and inflammation stimuli coordinately regulated the expression of CtBPs
Our above results indicated that both DNA methylation and inflammation stimuli could regulate the expression of CtBPs. To determine if DNA methylation and inflammation stimuli could function coordinately, we treated Control-KD and DNMT1-KD cells with 50 ng/mL recombinant IL6 and TNF-α, respectively, followed by measuring mRNA levels of CtBPs. The qRT-PCR results indicated that CtBP mRNA levels were significantly upregulated in IL6- or TNF-α-treated DNMT1-KD cells in comparison to untreated DNMT1-KD cells or in IL6- or TNF-α-treated Control-KD cells (Supplementary Figure 10A). We also observed similar patterns of CtBP protein levels (Supplementary Figures 10B and 10C). In addition, we also found that the expression of CASC2, IL6 and IL17 was significantly upregulated in IL6- or TNF-α-treated DNMT1-KD cells compared to DNMT1-KD cells or IL6- or TNF-α-treated Control-KD cells (Supplementary Figure 10D). These results demonstrated that the decreased DNA methylation levels and inflammation stimuli could coordinately regulate the expression of CtBPs and CASC2. To determine if knockdown of DNMT1 and inflammation stimuli affected the occupancies of CPM members on the promoter of CASC2, we carried out ChIP assays using antibodies against CPM members. The qRT-PCR results revealed that either knockdown of DNMT1 or IL6/TNF-α treatment could significantly increase the occupancies of CPM members on the promoter of CASC2, and this increase could be further enhanced in DNMT1-KD cells treated with The IL6 or TNF-α (Supplementary Figure 11).
Discussion
Although lncRNAs are involved in the pathogenesis of many diseases, it is still unknown their roles in AP. In the present study, we identified a lncRNA CASC2 was significantly upregulated in the pancreatic tissues of AP patients. Screening genes dependent on CASC2, we found that two proinflammatory cytokine genes IL6 and IL17 were positively regulated by CASC2. We then revealed a transactivation mechanism in which the decreased DNA methylation levels and inflammation stimuli coactivated the expression of CtBPs, and the increased CtBPs associated with PCAF and c-MYC to assemble the CPM complex, binding to the promoter of CASC2 and activating its expression (Figure 9). The clear description of this new signaling pathway will provide more potential targets for developing therapeutic strategies to prevent AP progression.
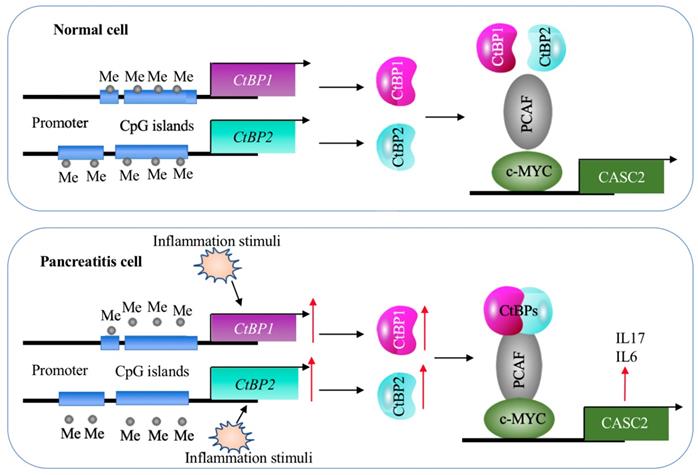
A schematic model of CASC2 in normal cells and pancreatitis cells. In normal cells, the DNA methylation levels in the promoters of CtBPs are very low and there are no intracellular and environmental inflammation stimuli. Thus, the expression of CtBPs is maintained at a basal level. As a result, CtBPs proteins cannot efficiently associate with PCAF and c-MYC to assemble a complex to activate the expression of CASC2 and its targets IL6 and IL17. In pancreatitis cells, the decreased DNA methylation levels in the promoters of CtBPs and environmental inflammation stimuli activate the expression of CtBPs. The amplified CtBPs form a heterotetramer, which is then recruited by the c-MYC-PCAF complex to assemble the CPM transcriptional machinery. The activation of CPM complex specifically binds to the promoter CASC2 to induce its expression, further leading to the induction of IL6 and IL17.
CtBP1 and CtBP2 are two conserved proteins with more than 80% amino acid sequence identify [35-37]. However, these two proteins do not show obvious redundancy in their functions [35-37]. Both of them are separately amplified in different cancers. In the tumorigenesis, CtBPs function as corepressors and they can cooperate with transcription factors to repress the expression of tumor suppressors such as Bax (BCL2 Associated X protein), Bim (also known as BCL2L11, BCL2 Like 11), BRCA1 (Breast Cancer Type 1), and CDH1 (Cadherin 1) [37]. Moreover, CtBPs also have transactivation roles. For example, CtBP2 can activate Tiam1 (T cell lymphoma Invasion and Metastasis 1) in an NADH-dependent manner, thereby promoting cancer cell migration [45]. CtBP2 physically interacts with TCF4 (Transcription factor 4) and activates TCF4-mediated signaling to regulate cancer stem cell growth and self-renewal [46]. CtBP1 can transcriptionally activate the expression of MDR (Multidrug resistance) gene, thereby contributing to chemotherapeutic drug resistance [47]. CtBP2 assembles a complex with p300 and AP1 to activate the expression of TGFB, causing chronic renal failure [48]. Except for regulating gene expression, CtBP-coupled transcriptional complexes have also been found to regulate the expression of miRNAs and lncRNAs. For instance, CtBP2 associates with HDAC1 and FOXP3 (Forkhead Box P3) to assemble a complex to repress miR-199a-3p, thereby attenuating its inhibition of NLRP1 (NLR Family Pyrin Domain Containing 1) and causing acute lung injury [35]. CtBP1 couples with HDAC1/2 and IRF1 to repress the expression of GAS5 in human osteosarcoma [38]. Interestingly, our current study revealed that both decreased DNA methylation levels in the promoters of CtBPs and inflammation stimuli contribute to the overexpression of CtBPs. To our knowledge, our results for the first time found this interesting regulatory effect. These results demonstrate that the diverse roles of CtBPs in different biological processes. Several CtBP inhibitors such as MTOB (2-Keto-4-butyric acid), HIPP (2-Hydroxyimino-3-phenyl-propionic acid), CP61 and NSC95397 have been reported to disrupt CtBP-mediated signaling [49]. Thus, it is worthwhile to evaluate the effects of these compounds to prevent the occurrence and progression of AP in a mouse model.
CASC2 is well studied in different cancers in which it acts as a tumor-suppressive lncRNA through regulating the expression of oncogenic microRNAs such as miR‐18a, miR‐21 and miR‐181a and affecting oncogenic pathways such as Wnt/β‐catenin, Ras-Raf-MEK-ERK and JNK signaling pathways [24-29, 43]. Recently, CASC2 is reported to be upregulated in osteoarthritis patients and its overexpression is positively correlated with IL17 level [23]. Overexpression of CASC2 in vitro resulted in the upregulation of IL17 mRNA level [23]. Consistent with this result, we also observe that overexpression or knockdown of CASC2 can change the IL17 mRNA level. Except for IL17, we also found that the expression of another important proinflammatory gene IL6 is dependent on CASC2. Our results for the first time reveal the upstream regulatory mechanism of CASC2 dysregulation, which will provide a reference for elucidating the mechanism of aberrant expression of CASC2 in other biological processes. Meanwhile, our results will also benefit to deepen the understanding of regulatory mechanisms of other lncRNAs (e.g., MALAT1, Mirt2 and ROCK1) involved in inflammation response. Except for CASC2, we will explore the functions of other dysregulated lncRNAs in the pathogenesis of AP in the future.
In summary, our results reveal that DNA methylation and inflammation stimuli coactivate the expression of CtBPs. Their encoding proteins associates with PCAF and c-MYC to assemble the CPM complex, which subsequently activates the expression of CASC2, IL6 and IL17, aggravating inflammation response and eventually leading to the occurrence of AP. Our results will benefit the research in this field and provide new avenues for the diagnosis and therapy of AP.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by a grant from Medical Scientific Research of Jiangxi Province, China (No: 2018A136).
Availability of the data and materials
All data analyzed during this study are included in this published article and its supplementary information files.
Author Contributions
Dr. Zhi Chen designed the experiments. Jun Zeng carried out the majority of the experiments. Jian-Yong Chen and Jun Meng performed some of the experiments. Zhi Chen and Jun Zeng analyzed the data, organized the figures and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Manohar M, Verma AK, Venkateshaiah SU, Sanders NL, Mishra A. Pathogenic mechanisms of pancreatitis. World J Gastrointest Pharmacol Ther. 2017;8:10-25
2. Tonsi AF, Bacchion M, Crippa S, Malleo G, Bassi C. Acute pancreatitis at the beginning of the 21st century: the state of the art. World J Gastroenterol. 2009;15:2945-59
3. Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol. 2015;31:395-9
4. Petrov M. Nutrition, inflammation, and acute pancreatitis. ISRN Inflamm. 2013;2013:341410
5. Liou GY, Storz P. Inflammatory macrophages in pancreatic acinar cell metaplasia and initiation of pancreatic cancer. Oncoscience. 2015;2:247-51
6. Shrivastava P, Bhatia M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol. 2010;16:3995-4002
7. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ. et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015;6:7158
8. Li G, Wu X, Yang L, He Y, Liu Y, Jin X. et al. TLR4-mediated NF-kappaB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int J Mol Med. 2016;37:99-107
9. Riesle E, Friess H, Zhao L, Wagner M, Uhl W, Baczako K. et al. Increased expression of transforming growth factor beta s after acute oedematous pancreatitis in rats suggests a role in pancreatic repair. Gut. 1997;40:73-9
10. Irrera N, Bitto A, Interdonato M, Squadrito F, Altavilla D. Evidence for a role of mitogen-activated protein kinases in the treatment of experimental acute pancreatitis. World J Gastroenterol. 2014;20:16535-43
11. Komar HM, Serpa G, Kerscher C, Schwoegl E, Mace TA, Jin M. et al. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci Rep. 2017;7:1787
12. Xiang H, Tao X, Xia S, Qu J, Song H, Liu J. et al. Targeting MicroRNA Function in Acute Pancreatitis. Front Physiol. 2017;8:726
13. Liu P, Xia L, Zhang WL, Ke HJ, Su T, Deng LB. et al. Identification of serum microRNAs as diagnostic and prognostic biomarkers for acute pancreatitis. Pancreatology. 2014;14:159-66
14. Kusnierz-Cabala B, Nowak E, Sporek M, Kowalik A, Kuzniewski M, Enguita FJ. et al. Serum levels of unique miR-551-5p and endothelial-specific miR-126a-5p allow discrimination of patients in the early phase of acute pancreatitis. Pancreatology. 2015;15:344-51
15. An F, Zhan Q, Xia M, Jiang L, Lu G, Huang M. et al. From moderately severe to severe hypertriglyceridemia induced acute pancreatitis: circulating miRNAs play role as potential biomarkers. PLoS One. 2014;9:e111058
16. Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017;18:558-76
17. Delas MJ, Hannon GJ. lncRNAs in development and disease: from functions to mechanisms. Open Biol. 2017 7
18. DiStefano JK. The Emerging Role of Long Noncoding RNAs in Human Disease. Methods Mol Biol. 2018;1706:91-110
19. Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017;292:12375-82
20. Zhao G, Su Z, Song D, Mao Y, Mao X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016;590:2884-95
21. Du M, Yuan L, Tan X, Huang D, Wang X, Zheng Z. et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat Commun. 2017;8:2049
22. Zhang Q, Chao TC, Patil VS, Qin Y, Tiwari SK, Chiou J. et al. The long noncoding RNA ROCKI regulates inflammatory gene expression. EMBO J. 2019 38
23. Huang T, Wang J, Zhou Y, Zhao Y, Hang D, Cao Y. LncRNA CASC2 is up-regulated in osteoarthritis and participates in the regulation of IL-17 expression and chondrocyte proliferation and apoptosis. Biosci Rep. 2019 39
24. He X, Liu Z, Su J, Yang J, Yin D, Han L. et al. Low expression of long noncoding RNA CASC2 indicates a poor prognosis and regulates cell proliferation in non-small cell lung cancer. Tumour Biol. 2016;37:9503-10
25. Zhou J, Huang H, Tong S, Huo R. Overexpression of long non-coding RNA cancer susceptibility 2 inhibits cell invasion and angiogenesis in gastric cancer. Mol Med Rep. 2017;16:5235-40
26. Huang G, Wu X, Li S, Xu X, Zhu H, Chen X. The long noncoding RNA CASC2 functions as a competing endogenous RNA by sponging miR-18a in colorectal cancer. Sci Rep. 2016;6:26524
27. Pei Z, Du X, Song Y, Fan L, Li F, Gao Y. et al. Down-regulation of lncRNA CASC2 promotes cell proliferation and metastasis of bladder cancer by activation of the Wnt/beta-catenin signaling pathway. Oncotarget. 2017;8:18145-53
28. Zhang Y, Qian W, Feng F, Cao Q, Li Y, Hou Y. et al. Upregulated lncRNA CASC2 May Inhibit Malignant Melanoma Development Through Regulating miR-18a-5p/RUNX1. Oncol Res. 2019;27:371-7
29. Liao Y, Shen L, Zhao H, Liu Q, Fu J, Guo Y. et al. LncRNA CASC2 Interacts With miR-181a to Modulate Glioma Growth and Resistance to TMZ Through PTEN Pathway. J Cell Biochem. 2017;118:1889-99
30. Heilmann K, Toth R, Bossmann C, Klimo K, Plass C, Gerhauser C. Genome-wide screen for differentially methylated long noncoding RNAs identifies Esrp2 and lncRNA Esrp2-as regulated by enhancer DNA methylation with prognostic relevance for human breast cancer. Oncogene. 2017;36:6446-61
31. Lai F, Shiekhattar R. Where long noncoding RNAs meet DNA methylation. Cell Res. 2014;24:263-4
32. Li J, Han W, Shen X, Han S, Ye H, Huang G. DNA methylation signature of long noncoding RNA genes during human pre-implantation embryonic development. Oncotarget. 2017;8:56829-38
33. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23-38
34. Jin B, Ernst J, Tiedemann RL, Xu H, Sureshchandra S, Kellis M. et al. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012;2:1411-24
35. Chen Z, Dong WH, Chen Q, Li QG, Qiu ZM. Downregulation of miR-199a-3p mediated by the CtBP2-HDAC1-FOXP3 transcriptional complex contributes to acute lung injury by targeting NLRP1. Int J Biol Sci. 2019;15:2627-40
36. Zhang W, Duan N, Zhang Q, Song T, Li Z, Chen X. et al. The intracellular NADH level regulates atrophic nonunion pathogenesis through the CtBP2-p300-Runx2 transcriptional complex. Int J Biol Sci. 2018;14:2023-36
37. Blevins MA, Huang M, Zhao R. The Role of CtBP1 in Oncogenic Processes and Its Potential as a Therapeutic Target. Mol Cancer Ther. 2017;16:981-90
38. Zhang X, Du K, Lou Z, Ding K, Zhang F, Zhu J. et al. The CtBP1-HDAC1/2-IRF1 transcriptional complex represses the expression of the long noncoding RNA GAS5 in human osteosarcoma cells. Int J Biol Sci. 2019;15:1460-71
39. Vilmann P, Saftoiu A. Endoscopic ultrasound-guided fine needle aspiration biopsy: equipment and technique. J Gastroenterol Hepatol. 2006;21:1646-55
40. Kahn SL, Ronnett BM, Gravitt PE, Gustafson KS. Quantitative methylation-specific PCR for the detection of aberrant DNA methylation in liquid-based Pap tests. Cancer. 2008;114:57-64
41. Numata M, Morinaga S, Watanabe T, Tamagawa H, Yamamoto N, Shiozawa M. et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int J Oncol. 2013;42:403-10
42. Granger J, Remick D. Acute pancreatitis: models, markers, and mediators. Shock. 2005;24(Suppl 1):45-51
43. Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47:546-52
44. Yu X, Zheng H, Tse G, Zhang L, Wu WKK. CASC2: An emerging tumour-suppressing long noncoding RNA in human cancers and melanoma. Cell Prolif. 2018;51:e12506
45. Paliwal S, Ho N, Parker D, Grossman SR. CtBP2 Promotes Human Cancer Cell Migration by Transcriptional Activation of Tiam1. Genes Cancer. 2012;3:481-90
46. Patel J, Baranwal S, Love IM, Patel NJ, Grossman SR, Patel BB. Inhibition of C-terminal binding protein attenuates transcription factor 4 signaling to selectively target colon cancer stem cells. Cell Cycle. 2014;13:3506-18
47. Jin W, Scotto KW, Hait WN, Yang JM. Involvement of CtBP1 in the transcriptional activation of the MDR1 gene in human multidrug resistant cancer cells. Biochem Pharmacol. 2007;74:851-9
48. Zhou P, Wan X, Zou Y, Chen Z, Zhong A. Transforming growth factor beta (TGF-β) is activated by the CtBP2-p300-AP1 transcriptional complex in chronic renal failure. Int J Biol Sci. 2020;16:204-15
49. Dcona MM, Morris BL, Ellis KC, Grossman SR. CtBP- an emerging oncogene and novel small molecule drug target: Advances in the understanding of its oncogenic action and identification of therapeutic inhibitors. Cancer Biol Ther. 2017;18:379-91
Author contact
![]() Corresponding author: zhichenedu.cn
Corresponding author: zhichenedu.cn