Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structural Architecture at...
Tissue-specific role of FXR and...
Specific bile acid as endogenous...
Bile acid a link between FXR and...
Curation of leads with their...
Conclusion
Future Perspective
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(13):2308-2322. doi:10.7150/ijbs.44774 This issue Cite
Review
Bile acids mediated potential functional interaction between FXR and FATP5 in the regulation of Lipid Metabolism
Anita Kumari1,3, Dharam Pal Pathak2,3, Shailendra Asthana1 ![]()
1. Translational Health Science and Technology Institute (THSTI), Faridabad, Haryana, India.
2. Delhi Institute of Pharmaceutical Sciences and Research (DIPSAR), New Delhi, India.
3. Delhi Pharmaceutical Sciences and Research University (DPSRU), New Delhi, India.
Received 2020-2-12; Accepted 2020-5-17; Published 2020-6-14
Abstract

Perturbation in lipid homeostasis is one of the major bottlenecks in metabolic diseases, especially Non-alcoholic Fatty Liver Disease (NAFLD), which has emerged as a leading global cause of chronic liver disease. The bile acids (BAs) and their derivatives exert a variety of metabolic effects through complex and intertwined pathways, thus becoming the attractive target for metabolic syndrome treatment. To modulate the lipid homeostasis, the role of BAs, turn out to be paramount as it is essential for the absorption, transport of dietary lipids, regulation of metabolic enzymes and transporters that are essential for lipid modulation, flux, and excretion. The synthesis and transport of BAs (conjugated and unconjugated) is chiefly controlled by nuclear receptors and the uptake of long-chain fatty acids (LCFA) and BA conjugation via transporters. Among them, from in-vivo studies, farnesoid X receptor (FXR) and liver-specific fatty acid transport protein 5 (FATP5) have shown convincing evidence for their key roles in lipid homeostasis and reversal of fatty liver disease substantially. BAs have a wider range of biological effects as they are identified as modulators for FXR and FATP5 both and therefore hold a significant promise for altering the lipid content in the treatment of a metabolic disorder. BAs also have received noteworthy interest in drug delivery research due to its peculiar physicochemical properties and biocompatibility. Here, we are highlighting the connecting possibility of BAs as an agonist for FXR and antagonist for FATP5, paving an avenue to target them for designing synthetic small molecules for lipid homeostasis.
Keywords: Fatty liver diseases, FXR, FATP5, Bile acids, cyp7a1, Triglycerides
Introduction
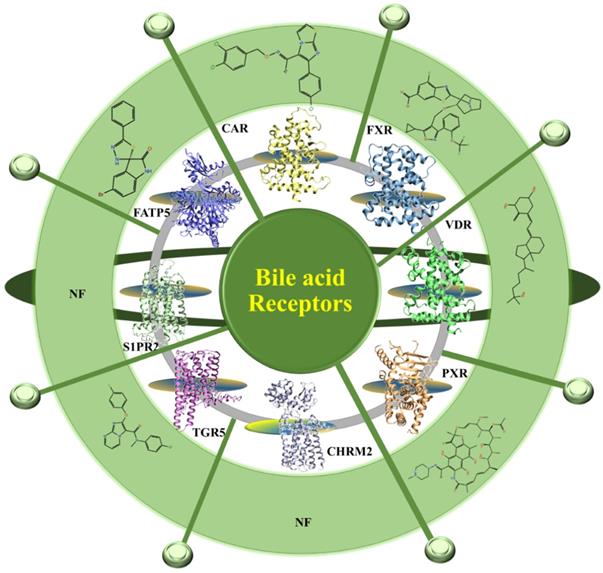
The prevalence of the fatty liver disease in association with the epidemic of obesity and type 2 diabetes has increased worldwide and affects 15-40% of the general population [1,2]. The fatty liver disease clusters with metabolic abnormalities, including type 2 diabetes, obesity, hypertension, hyperlipidemia which affects ~25% of the adult population worldwide [3,4].These metabolic abnormalities are linked to the alternation in the bile acid metabolism. BAs are endocrine molecules that contribute to several essential functions including cholesterol catabolism and intestinal lipid emulsification and, known for regulating the BA pool and lipid metabolism [3]. The liver is the site for the BAs synthesis and converts cholesterol to BAs through the classic and alternative pathways [5,6]. Here, the hepatic enzymes generate free primary BAs such as chenodeoxycholic acid (CDCA) and cholic acid (CA). In gut, the action of bacterial enzymes converts primary BAs into secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA). These primary as well as secondary BAs activate the nuclear receptors such as FXR, PXR (pregnane X receptor), VDR (vitamin D receptor), CAR (constitutive androstane receptor) and membrane-bound receptors including G protein-coupled receptor (TGR5), sphingosine-1-phosphate receptor 2 (S1PR2), cholinergic receptor muscarinic 2 (CHRM2) (7,8) (Figure 1) and various cell signaling pathways (c-jun N-terminal kinase 1/2, AKT, and ERK 1/2 (extracellular signal-regulated kinases)). The activation of these receptors alters the expression of various genes involved in the regulation of BAs, glucose, fatty acid, lipoprotein synthesis, metabolism, transport, and energy metabolism. In the liver, the BAs regulate the expression of a number of transport proteins and biosynthetic enzymes which are crucial for the maintenance of BA and lipid homeostasis [9]. These include NTCP (Na+ dependent taurocholate cotransport peptide) [10], BSEP (bile salt export pump) [11] and cyp7a (Cholesterol 7 alpha-hydroxylase) which is the rate-limiting enzyme for the production of BAs from cholesterol via the neutral biosynthetic pathway in the liver [12]. The function of these dedicated bile acid receptors has been well documented and studied before [7,13]. In Figure 1 we have given the available 3D structure for the receptors and their available synthetic molecules against them. Besides all this the liver also plays as a central organ for coordinating metabolism and has a large capacity for fatty acid (FA) uptake [14,15]. Among these targets where expression and/or activity modulate lipid homeostasis, there is evidence of the BA-mediated link between FXR and FATP5 in the liver and lipid specific manner, which provide an idea about their involvement in NAFLD [16]. It embraces two disease states i.e. Non-alcoholic Fatty Liver (NAFL) which is a broad, mostly benign liver disease and Non-alcoholic steatohepatitis (NASH) an inflammatory and progressive condition. For the first time, Stahl et.al. reported that the BAs modulates the FXR and FATP5, respectively, which is considered to have a large impact on metabolic disorders, especially those related to the metabolic syndrome [17]. These two targets majorly modulate the lipid homeostasis in the liver [15,17-19]. Therefore, it is demanding to explore the functional and biological link between BA-FXR-FATP5 to rationally mimic the endogenous BA for the discovery of small synthetic compounds that can narrow down the lipid uptake as an antagonist of FATP5 and enhance liver health and resensitize the BA pool substantially as FXR agonist in pathological condition.
Schematic overview of BA receptors. The available crystal structures of BA receptors are FXR, PXR, VDR, CAR, CHMR2 are shown in 3D and in cartoon representation. The homology models of TGR5 and FATP5 were developed in the absence of their crystal structure. All the BA receptors are summarized in the Figure. The 2D structures of the available modulators of BAs receptors are shown (the small molecule which is reported against metabolic disorder are only picked). NF is not found.
Structural Architecture at Atomic Level and elucidation of possible Molecular Mechanism can pave a path for Lead Discovery
In the current scenario the cornerstone of modern medicinal chemistry is ligand- and Structure-based drug discovery (LB/SBDD) which has inspired numerous small molecule lead optimization efforts and play a role in the discovery of drugs and drug like molecules immensely [20,21]. The protein structures have increased exponentially and now confine highly dynamic targets also which was previously impervious to crystallography. X-ray and NMR structures of proteins (APOs) and co-crystals with ligand complexes (HOLOs) are generally available and provide a platform for structure-guided drug discovery as the structures are typical of sufficient resolution at the atomic-level to stimulate effective hit identification and design and their conversion from identified hit-to-lead molecules i.e. lead optimization [22]. However, the interaction maps of frozen state of protein-ligand observed in X-ray structures are in general not good enough to explain the structure-activity relationships (SAR) well and this can lead to inaccurate and futile synthesis cycles. Nevertheless, molecular dynamics simulation appears a strong tool to understand the dynamical changes of different states of proteins such as transition state, open-to-close state, conformational change states like recruitment of co-factors, prosthetic group, post-translational modifications, role of mutations, changes in the binding site architecture and allostery. Therefore, a curation of possible structural and ligands information seems to be a priority to explore the key targets for drug discovery.
About FXR
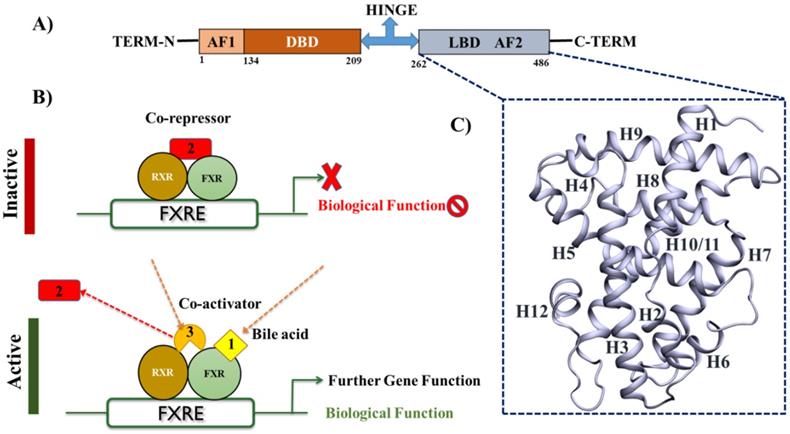
FXR is a ligand-activated transcription factor, belongs to the NR (nuclear receptor) superfamily and is essential in regulating a network of genes involved in maintaining BA and lipid homeostasis. The molecular mechanism of FXR is extensively studied; however, limited information is available about FATP5 and its modulation. FXR exerts its function by eliciting transcriptional alterations [23]. It binds to DNA as a heterodimer with RXR (retinoid X receptor) or as a monomer to regulate the expression of various genes. In common with other NRs, the FXR protein exhibits a modular structure, and contain several autonomous functional domains: N-terminal region with a ligand-independent activation function (AF1), a highly conserved zinc-finger DNA-binding domain (DBD) that is connected to the ligand-binding domain (LBD) by a flexible hinge region (Figure 2A) [24]. Upon agonist binding to FXR creates the huge conformational rearrangement due to which the dissociation of co-repressors and the recruitment of co-activators occur to promote the transcriptional initiation (Figure 2B). The LBD contains two well-conserved regions: a signature motif and the AF2 motif located at the carboxy-terminal end of the domain, which is responsible for the ligand-dependent transactivation function (Figure 2C).
Structural-functional organization of FXR A) The schematic representation of the FXR domains structure B) FXR forms a heterodimer with retinoid X receptor (RXR) that binds to the FXR-response element (FXRE), a DNA sequence in the promoter of its target genes. In the absence of ligands, the FXR heterodimer is associated in complex with corepressor, leading to transcriptional repression. Following BAs binding to the LBD of FXR, the heterodimer undergoes conformational changes, leading to the recruitment of co-activators to replace corepressors, which results in the transactivation of target genes expression. C) The 3D structures of LBD of FXR by using the PDB-ID: 5Q0K.
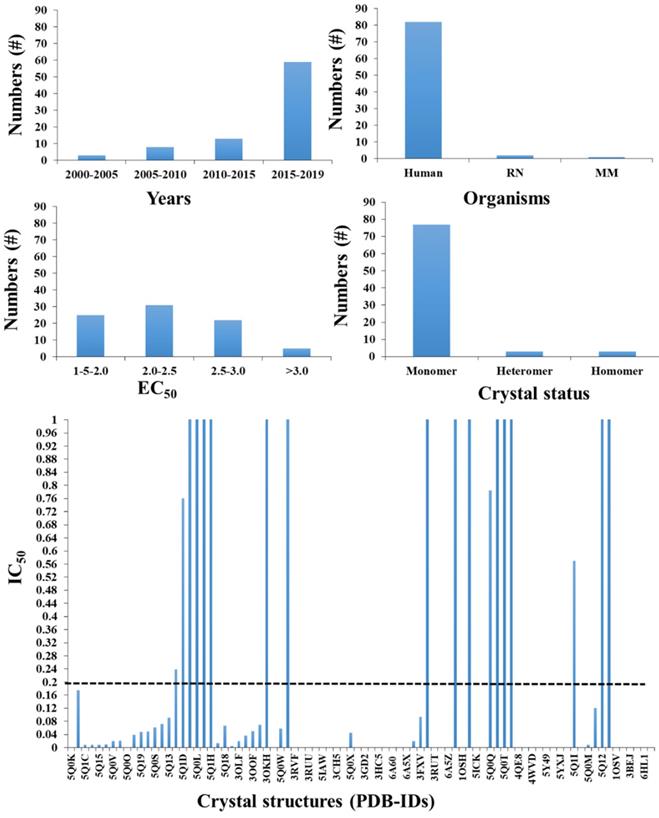
To date, 84 FXR structures have been reported to the Protein Data Bank (PDB), which was up to ~25 in number till 2015, indicating the importance of FXR study in the current scenario (Figure 3). Considerable interest in drug discovery and pharmacology in the recent past is due to its important role in metabolism and its value as a drug target to treat liver disorders and metabolic diseases. The details of the available crystal structures of FXR with their available biological activity are reported in Figure 3. Despite all these available crystal structures, there are several areas which need structural explanations such as flexibility of FXR, which reveals clearly from different crystals/co-crystals about its structural conformations. The activity data shows that there are molecules which are having potent IC50 and EC50 values (Figure 3).
The available crystal structure for FXR (different species) with their biological activity and their existing form with RXR: A) the total number of FXR crystals published year-wise, B) number of organisms, C) The co-crystal structures with reported agonist and antagonist based on their EC50, D) the type of FXR crystal reported and E) the best compounds (below 1uM) is only shown here. The dotted line is used as a cutoff for low micromolar compounds for a ligand-based approach.
This information possibly provides a rational basis for FXR structure- and ligand-based drug design which has enormous potential to yield a novel molecule with optimal selectivity, potency, and efficacy profiles. It is also addressable whether FXR agonism is required and/or partial-agonism is good enough for lipid modulation in a diseased condition. Recently, it is reported that the pharmacological administration of full FXR-agonist has been plagued by mechanism-based side effects [25]. Since NR is known to participate in various endocrine functions, FXR modulation is prone to cause mechanism-based side-effects. Clinical trials of OCA have already shown that extensive FXR activation disrupts cholesterol homeostasis with FXR activation blocking the metabolic conversion of cholesterol to BAs via SHP/FGF19 up- and cyp7a1 down-regulation. Since this pathway constitutes the main route of metabolic cholesterol elimination, its long-term pharmacological blockade may have serious consequences. Partial activation appears to be an avenue to safely exploit FXR as a drug target as it could reduce side effects such as loss of metabolic cholesterol degradation. Similar strategies have been proposed for other BAs sensors that are known for causing unwanted effects. However, to explore this strategy, a better understanding of partial FXR activation on a molecular level is necessary. The comparison of FXR-LBD co-crystal structures in complex with agonists, antagonists, and partial agonists revealed several significant differences and these differences seem to affect the conformation of the FXR-LBD explaining different pharmacological effects.
From the structure point of view, it is essential to know about key residue and structural determinants for partial agonism, the positions of H12 and the co-activator/co-repressor bound to the FXR-LBD and loss/gain of interaction networks (like hydrogen bonds, VdW interaction, and non-bonded contacts, etc.), as it has been seen that recruitment of co-activator/co-repressor itself inducing the significant conformational changes. Since a co-crystal structure provides only a single snapshot of dynamic binding equilibrium and here, it is not sufficient to gain mechanistic understanding.
From structures, it is well evidential that loops between (H1-H2, H5-H6, and H6- H7) and helices H11 and H12 are the key determinants for the recruitment of agonists, partial-agonist and antagonist, and their biological function. NMR and x-ray crystallography provides enough clues about structural and conformational variations of FXR with different interacting partners, co-activators and co-repressors. Since NMR has its own limitations and crystal structures are static in nature, a multidisciplinary approach is required to unravel the molecular changes in the FXR structure that may lead to the discovery of novel selective FXR modulators. A better understanding of FXR's molecular mechanism may significantly support future drug discovery of safer, optimal selectivity, potency and effective FXR modulating agents [25].
About FATP5
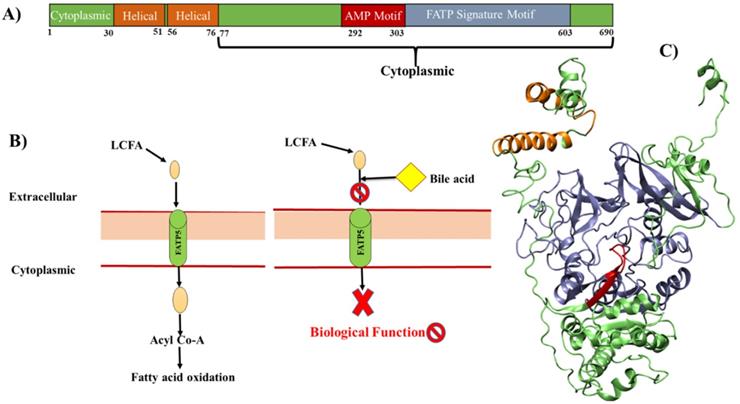
The FATPs comprise a family of 6 members, which encode FATP1-6 [26]. Among all the family members, mainly FATP5 has been known to exert tissue-specific effects in regulating BA synthesis and LCFAs transport and can reduce the lipid accumulation; therefore, it appears as a potential target for rational drug design [17]. FATPs are integral membrane proteins, ranging from 63-80 kilo Daltons (kDa) in size with the transmembrane domain. The N-terminus and C-terminus are located on the extracellular/luminal side and on the cytosolic side respectively (Figure 4A) [27,28]. All FATPs members are characterized by the presence of a highly conserved, 311-amino acid signature sequence known as the FATP sequence, as well as an AMP binding domain (292-303), located on the C-terminus (Figure 4A). This region is responsible for the binding and uptake of LCFA and is commonly found in members of the ACSL family [29-31]. The cellular capacity of LCFA uptake depends on transporter proteins and BA acts on the extracellular domain of FATP5 as competitive inhibitors (Figure 4B), further studies are needed to explore the mechanism of BAs on FATP5 [17].
A model for cellular fatty acid uptake: A) The domain structures of FATP5 receptor, B) extracellular LCFAs bind directly to FATP5 and be transported into cells. Intracellular LCFAs would then be coupled to CoA and further got oxidized. The BAs bind the extracellular domain of FATP5 and inhibit the uptake of fatty acids. C) 3D model is generated based on homology and fragment-based modeling approach. The FATP5 is rendered in cartoon and the color code is the same as shown in panel (A).
Fatty acid uptake factors such as CD36, FATP2, and FATP5 actively function when excessive fatty acids are present owing to high fat intake or obesity, but not in normal conditions. It is also studied that there is a decrease in hepatic FATP5 expression in NAFLD that is associated with the hepatic fat loss during NASH progression to cirrhosis [32]. The role of FATP5 as a tumor suppressor in HCC (Hepatocellular carcinoma), both in vivo and in vitro, has been studied and provides a mechanistic link between disrupted lipid metabolism and redox homeostasis [33]. From the structural point of view, there is no crystal structure available till now. But the helical and AMP binding sites possibly appear as the modulating sites in FATP5 either for BA's mimics and/or for small molecules (Figure 4). There are few molecules known for FATP5 as compared to FXR, but there are secondary bile acids (DCA and UDCA) which act on FATP5 and ability to inhibit the TG accumulation [17]. There are also some preliminary findings which showed that the bile acid (chenodiol and ursodiol) selectively target the FATP5 by employing the HTS (high throughput screening) assay [34]. The structure and their details we have discussed further in the paper. This structural based knowledge of FATP5 protein would help the discovery of new drug leads against metabolic disorder.
Tissue-specific role of FXR and FATP5 in lipid metabolism
In Liver
Selective targeting of FXR and FATP5 in specific tissues seems a promising strategy to increase the therapeutic index of their modulators and reduce the side effects compared to whole-body targeting. The role of nuclear receptors in BA metabolism is well explained by Li and Chiang et.al. [5], where in the liver, FXR is a key sensor of BA and has a central role in maintaining BA homeostasis and to protect liver cells from potential deleterious consequences of cellular BA overload and therefore it becomes an attractive target for the metabolic disorder [19]. FXR is dominantly expressed in enterohepatic system; however, it is also expressed in other tissues including adrenal glands, kidney, stomach, heart, and macrophages [35] as well as in white and brown adipose depots [36,37], that's why by restricting the tissue-specific activity. In enterohepatic circulation of BAs, FXR plays the key mediator role in the BA feedback repression mechanism by primarily inducing the expression of small heterodimer partner (SHP), which further inhibit transcription of the cyp7a1 gene that allows the liver to downregulate the BA synthesis in response to an increase in BA levels and thus maintains a constant BA pool [38,39] (Figure 5). It also works as an important component of lipid homeostasis, most likely in the regulation of enzymes and transporters that are critical for lipid catabolism and excretion [9]. Upon BA activation of FXR induces BSEP (bile salt export protein) [40] and MRP2 (multidrug resistance-associated protein 2) in hepatocytes [41] (Figure 5). On the other hand, FXR also repressed the NTCP as a feedback inhibition of BA uptake to prevent liver injury [42]. The studies of FXR null mice have revealed that the reduced capacity to excrete BAs and the level of hepatic TGs also significantly greater than that for wild-type mice, which provides convincing evidence for a central role of FXR in BAs homeostasis [9]. Activation of FXR inhibits cyp7a1 and reduces BA synthesis, and inhibits NTCP and OATPs to reduce sinusoidal uptake of BAs to maintain the BAs homeostasis [9]. There are a number of in-vitro and in-vivo studies using mouse models for FXR that have elucidated the positive relationship with fatty liver disease and its effect on the regulation of lipid metabolism [9,43-45]. The study of FXR -/- mice confirmed that it was critically involved in lipid homeostasis through regulating the cholesterol catabolism, transport, and lipoprotein metabolism [9]. FXR suppresses sterol regulatory element-binding protein-1c (SREBP-1c), reduces the hepatic triglyceride (TG) level (Figure 5). The treatment of mice with FXR agonists results in the repression of SREBP-1c. So SREBP-1c functions as a critical transcription factor that regulates many genes involved in both fatty acid and TG synthesis [46]. The activation of FXR also represses hepatic de novo lipogenesis and stimulates fatty acid β-oxidation by inducing expression of Peroxisome proliferator-activated receptor -alpha (PPARα), limiting hepatic lipid accumulation [47-49] can also induce the expression of ApoCII (apolipoprotein CII), an activator of lipoprotein lipase, so that promote plasma very-low-density lipoprotein TG clearance and suppressing the expression of ApoCIII (apolipoprotein CIII), an inhibitor of lipoprotein lipase activity [50,51]. Altogether, these data suggest that FXR activation lowers plasma TG levels via both repressing hepatic lipogenesis and TG secretion, and increasing the clearance of TG-rich lipoproteins from the blood (Figure 6). These studies suggesting the expression level of FXR also acts as a determinant of lipid metabolism and has been suggested as a promising therapeutic target for hepatic metabolic disorders [52].
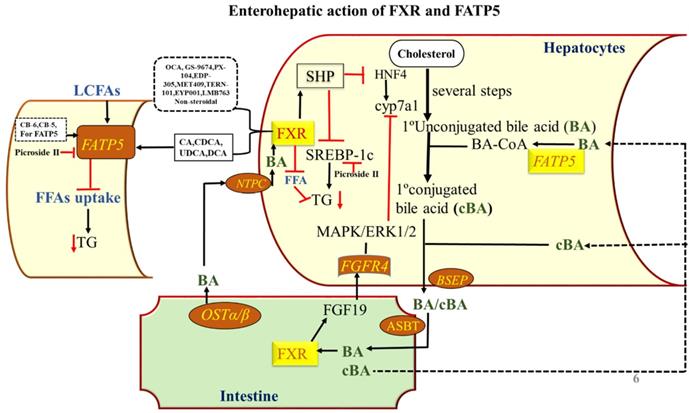
Enterohepatic action of FXR and FATP5. Role of FXR and FATP5 in synthesis, transport, and enterohepatic circulation of BAs and the effects of steroidal and non-steroidal agonists on the activity of the same. In the liver, activation of FXR by agonists induces SHP to inhibit cyp7a1 gene transcription that further allows the liver to downregulate the BA synthesis in response to maintaining a constant BA pool. The TG level is reduced by SHP acting on SREBP-1c. BAs are secreted via BSEP into the gallbladder and reabsorbed via ASBT in terminal ileum enterocytes. Here, they bind and activate FXR, which stimulates production and secretion of FGF15/19 into the portal circulation. BAs activate FXR in the intestine to induce FGF15/19 which is transported to hepatocytes to activate FGFR4, which further activates a signaling pathway involving MAP kinases and causes repression of cyp7a1 transcription, thus downregulating BA synthesis. After this the OSTα/β-mediated secretion into the portal circulation, BAs are taken up by the liver via NTCP, completing the enterohepatic cycle. The FATP5 involved in the uptake of fatty acids and the conjugation of BAs in the liver. The FATP5 conjugates the BA by BA-CoA enzymatic activity. The BAs inhibit the uptake of long-chain fatty acid in FATP5 dependent manner and inhibits the TG level in the liver and maintain the lipid homeostasis in the liver. The endogenous ligand and the synthetic ligand for both the receptors are given in the figure.
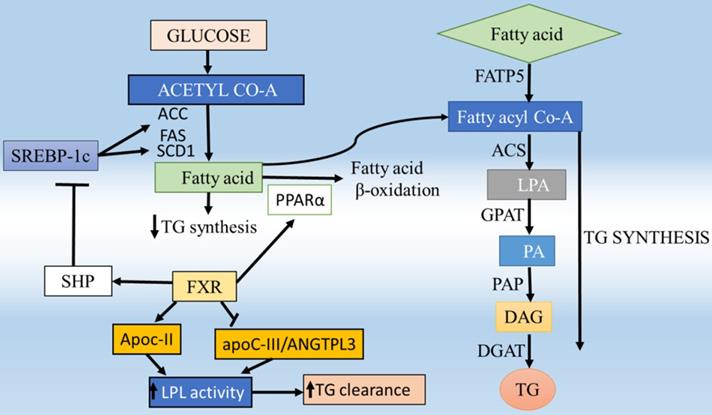
Regulation of lipid homeostasis by hepatic FXR and FATP5 and their involvement in hepatic steatosis. FFA can be synthesized de novo from glucose, mainly regulated by SREBP-1c. Activation of hepatic FXR lowers plasma FFA and TG, resulting from (i) repression of hepatic TG and fatty acid (FA) synthesis as a result of SHP-dependent inhibition of SREBP-1c; (ii) induction of ApoC-II and repression of ApoC-III and ANGTPL3 in the liver, resulting in enhanced lipoprotein lipase activity; thus promoting clearance of TG-rich lipoproteins and (iv) induction of human PPARα and FA β-oxidation. The FATP5 plays an important role in fatty acid uptake to the liver upon a high-fat diet which contributes to the development and/or aggravation of fatty liver. Consequently, the incorporation of fatty acyl CoA into TG is greatly enhanced and lipid accumulation occurs in the liver.
The study also shows that the lipid homeostasis, in general, is largely a protein-mediated process requiring FATP5 and helps in the understanding of the liver function and disease [53]. Disturbed fatty acid metabolism is one of the causes involved in the pathogenesis of NASH [54]. The uptake of circulating fatty acids to the liver is largely dependent on fatty acid transporter protein (FATP), CD36/fatty acid translocase (CD36/FAT) and fatty acid-binding protein-plasma membrane-bound (FABPpm) which play an important role in the trafficking of the FFA [55,56]. Among these, the FATP5 is widely expressed in liver and localized to the basal plasma membrane of the hepatocytes [53] and the deletion of FATP5 influence the development of hepatic steatosis [53] but in CD36 it does not show the same effect [57] which clearly indicates the important role of FATP5 over other transporters. Consequently, the incorporation of fatty acyl CoA into TG is greatly enhanced and the machinery of lipid accumulation occurs in the liver (Figure 5). In humans, the FATPs comprise a family of 6 members that contain a common motif for fatty acid uptake and fatty Acyl-CoA synthetase function [58]. All these transporters are helping in the uptake of FAs in the multiple body tissues like kidney, skeleton muscles, heart, and adipose tissues furthermore the oxidation of FA occurs. It mainly affects the liver where the FATP5 protein plays an important role in the uptake of fatty acid. The detailed analysis of the hepatic FATP5 knockout in livers shows that the alternation in the lipid homeostasis which supports the hypothesis that efficient hepatocellular uptake of LCFAs, and thus liver lipid homeostasis in general, is largely a protein-mediated process requiring FATP5. These new insights into the physiological role of FATP5 should lead to an improved understanding of liver function and disease [53]. The altered fatty acid metabolism is a hallmark of numerous metabolic diseases and pathological conditions such as NAFLD, which is linked to obesity and types 2 diabetes and insulin resistance [59]. So the fatty acid transporters can be used as a target to rectify lipid fluxes in the human body, specifically in the liver and regain metabolic homeostasis. The knockdown studies of FATP5 is able to reverse NAFLD, results in considerably improved glucose homeostasis in an animal model (fed HFD), therefore, proposed critical for the sustained caloric uptake and fatty acid flux into the liver [59]. Inhibition of uptake of FATP5 mediated fatty acids in the liver seems to be a potential avenue for the treatment of NAFLD. FATP5 presence is liver-specific and its knockout study reported that there is a significant drop (~50%) of lipid uptake which suggests that FATP5 contributes significantly to fatty acid uptake in primary hepatocytes [53]. Moreover, it also has shown that it perturbed sugar homeostasis [59]. The synthetic molecules which can block the cellular uptake of LCFA could be a key to identifying potential therapies for metabolic diseases. Therefore, targeting the FATP5 can rectify the lipid fluxes in the human body, specifically in the liver and regain metabolic homeostasis. Therefore, selective targeting of receptors in specific tissues also seems a promising strategy to increase the therapeutic index of modulators. The previous studies also report that the FATP5 also exhibits the bile acid CoA synthetase (BACS) activity, hence, essential for proper BA conjugation and involved in lipid metabolism [60]. The gene expression analyses of the BA-CoA ligase FATP5 revealed that there is impaired BA amidation due to the decreased expression of this [61]. The knockout and knockdown mice models for FATP5 also confirm that it is responsible for the majority of BACS in the liver and show a dramatic increase in unconjugated BAs which is suggesting that it plays a major role during reconjugation of BAs in the enterohepatic recirculation [18,60] (Figure 5). It was also investigated the severe conjugation defect in FATP5 knockout mice in contrast to wild-type mice, where 95% of BAs are conjugated, whereas only 17% of BAs are conjugated in FATP5 deletion mice [60]. Moreover, in FATP5 knockout mice, there is an up-regulation of the SHP (Table 1) which mediates the inhibition of cyp7a1 expression in response to high BA concentration although this regulation did not reach statistical significance. The FATP5 knockdown also resulted in a significant increase in genes involved in hepatic cholesterol biosynthesis (SREBP2 pathway) and fatty acid synthesis (SREBP-1 pathway) [18]. The deletion of FATP5 also causes protection from obesity and hepatic TG accumulation and improved insulin sensitivity [3] and a significant reduction in lipid uptake (Figure 6). It was also examined in KO mice the total diglyceride and TG content was reduced by 59% and the LCFA uptake by 50% by using the gas chromatography/mass spectrometry-based analyses and FACS-based measurements, respectively [53]. The loss of FATP5 causes the redistribution of dietary lipids away from the liver to other FFA metabolizing tissues. The in-vivo study for these receptors has shown the up and down-regulation of the target genes which involves the BA synthesis and uptake of fatty acids (Table 1) (Figure 5). The data suggest that the alterations in gene expression have a role in BA and lipid metabolism. Overall, FATP5 is a protein with multiple activities and known to play an essential role in the BA reactivation, hepatic FA uptake, and lipid accumulation. Henceforth, from all these observations, we found that gene expression level gets affected differently in FXR and FATP5.
The regulation of key targets in in-vivo studies. The effective level of target genes involved in BA synthesis and fatty acid uptake. The expression level of these genes is studied for FATP5 and the same has been evaluated against FXR, too.
| Changes in Gene Expression | FXR (KO*) | FXR (CA) | FATP5 (KO) | References |
|---|---|---|---|---|
| cyp7a1 | Up | Down | Up | (9,48,60,74) |
| SHP | Down | Up | Down | |
| NTCP | Up | Down | Up | |
| BSEP | Down | Up | Up | |
| OATP | Up | No change | No change |
In Intestine
In the intestine the BAs are actively reabsorbed by the Apical Sodium-dependent Bile Acid Transporter (ASBT). FXR modulation provides significantly increased therapeutic benefits. FXR signaling provides cross-talk between the intestine and the liver via activating the expression of the enterokinase FGF15/19 (Figure 5). Activation FXR in intestine increase FGF15/19 expression, which further activates FGFR4, causes repression of cyp7a1 transcription by signaling pathway involving the MAP kinase (A mitogen-activated protein kinase), thereby reducing hepatic BAs overload [62] (Figure 5). Constitutively active intestinal FXR improved BA homeostasis and reduced cellular proliferation, hepatic inflammation, and fibrosis in young FXR null mice [62,63] The therapeutic indications for the use of intestinal-specific FXR modulators may also be extended to liver disorders. In the intestine the BSEP and NTCP are up-regulated in FATP5 deleted mice (Table 1) which possibly allows increasing the transport of the remaining conjugated BAs [60]. These tissue-specific roles of FATP5 and FXR suggest that both play a role in BAs metabolism and lipid homeostasis. These new insights into the physiological role of FATP5 and FXR might lead to an improved understanding of liver function and disease. With an in-depth understanding of FXR and FATP5 function and regulation at the cell-, gene- and tissue-specific levels, we will have more tools and be more confident in designing FXR modulators in the future to prevent and/or treat BA and lipid-related abnormalities in humans. Henceforth, the reduction of hepatic FATP5 through different routes such as via gene therapy or small molecular inhibitors is a novel tool to dynamically redirect lipid fluxes and may provide novel approaches for lipid homeostasis. These provide new insights into the physiological role of FATP5 which is poorly characterized as a drug target.
Specific bile acid as endogenous modulators of FXR and FATP5
There are numerous pharmacological FXR modulators including agonists and antagonists, which have demonstrated that FXR plays a central role in controlling lipid homeostasis by reducing plasma TG and HDL (high-density lipoproteins) levels. The obeticholic acid (OCA) is ~100-fold more agonistic activity than the endogenous ligands chenodeoxycholic acid (CDCA) [64] and many agonists have been tested in clinical trials for treating the metabolic disorder [65] (Table 2). The steroidal (BA and their derivative) and non-steroidal (synthetic) agonist and antagonist are well known for the BA receptors as discussed in Table 2 and have the modulation action on FXR and FATP5. The activation of FXR with BAs or synthetic activators has shown the reduced secretion of TG level in the liver and maintaining lipid homeostasis. The order of potency of bile BAs activating FXR is chenodeoxycholic acid (CDCA) > lithocholic acid (LCA) = deoxycholic acid (DCA) > cholic acid (CA), whereas one of the secondary BAs, LCA (lithocholic acid), is the most potent activator of TGR5, PXR and VDR [5]. Among BAs, UDCA cannot activate FXR [66] and has no suppressive effect on cyp7a1 in human hepatocytes up to ≤100 μmol/L [67]. It is well documented that the BA inhibits FATP5 and reduces the TG level in in-vivo and in-vitro studies [17]. The order of potency of BAs inhibiting FATP5 is: deoxycholic acid (DCA) > ursodeoxycholic acid (UDCA) > chenodeoxycholic acid (CDCA) > cholic acid (CA) [17]. It is also noticeable that the BA like CA, CDCA, and DCA activate FXR but the UDCA was found to act as an FXR antagonist rather than agonist, having effects on BA and lipid metabolism in morbid obesity [68]. The patients with NAFLD have shown the expression of FXR is less, which is associated with hepatic TG accumulation and hepatic steatosis [43]. The available agonist and antagonist can alter the bile- and lipid- acid metabolism by acting on FXR at the same time BA can inhibit the uptake of fatty acid by acting on FATP5. The available data also suggests that the association of FATP5 in the pathogenesis of metabolic syndrome and steatosis [69]. Interestingly, other than the BAs, Picroside II attenuated FFA accumulation in HepG2 (human liver cancer cell line) cells via downregulation of FATP5, SREBP-1c decreasing FFAs uptake and lipid synthesis [70] (Figure 5). Henceforth, to mimic the BAs which can work as an agonist for FXR and antagonist for FATP5.
The steroidal and nonsteroidal agonist and antagonist for the bile acid receptors
| Receptor | Target Tissue | Crystal information* | Steroidal | Activity | Non-Steroidal | Activity | References |
|---|---|---|---|---|---|---|---|
| FXR | Liver, intestine, kidney | Yes, 84* | CDCA > DCA > LCA > CA > UDCA, 5β-cholanoic acid, 5β-norcholanoic acid, 5α-cholanoic acid 6α-ethyl-CDCA, 6α-ethyl-3α,7α, 23-trihydroxy-24-nor-5β-cholan-23-sulphate (INT-767) | Agonist UDCA (antagonist) LCA (antagonist) | GW4064, Fexaramine, Way362450, PX-102,Cilofexor,Px-104,TERN-101, EYP001, Nidufexor, Tropifexor | Agonist | (7),(24),(13,68) |
| VDR | Intestine, kidney, bone | Yes, 49* | LCA, 3-keto-LCA | Agonist | LY2108491, LY2109886,vitamin D3 (Calcitriol) and Synthetic analogues (e.g. 2MbisP, BXL-01-0772) | Agonist | (7) ,(13),(75-77) |
| PXR | Liver, intestine | Yes, 23* | LCA, 3-keto-LCA >> CDCA, DCA, CA7α-hydroxy-4-cholesten-3-one | Agonist | Herbal medicine (e.g. Hyperforin, Guggulsterone), drugs (e.g. Rifampicin, Meclizine, Rifaximin Paclitaxel, Lovastatin | Agonist | (7),(13),(78-81) |
| CAR | Liver | Yes, 4* | CA, 6-keto-LCA, 12-keto-LCA | Agonist | Xenobiotic ligands: CITCO, TCPOBOP, Herbal medicine (e.g.6,7-dimethylesculetin), drugs (e.g. Phenobarbital. | Agonist | (7),(13),(80),(82) |
| TGR5 | Liver, intestine, gallbladder, muscle, brain | No | LCA > DCA > CDCA > CA > UDCA TLCA BA analogs: INT-767, 6α-ethyl-23(S)-methyl-3α,7α,12α-trihydroxy-5β-cholan-24-oic acid (INT-777) | Agonist | Xenobiotic ligands: Oleanolic, Synthetic agonists: WB403, TRC210258,RDX8940 | Agonist | (7),(13),(83-87) |
| S1PR2 | Liver, intestine, Heart, brain | Yes, 3* | TCA, GCA, TCA, GDCA, TDCA, TUDCA | Agonist | NF | NF | (7),(88-90) |
| CHMR2 | Heart | Yes, 9* | TCA | Partial agonist | NF | NF | (7),(91,92) |
| FATP5 | Liver | No | DCA >UDCA>CDCA>CA | Antagonist | CB-2, CB-5, CB-6 and J3 | Antagonist | (17), (34,93) |
NF; not found; *Total number of crystal structures available in the Protein Data Bank for each bile acid receptors. >, higher affinity than; >>, much higher affinity than; TCA, taurocholic acid; GCA, glycocholic acid; GDCA, glycodeoxycholic acid; TUDCA, Tauroursodeoxycholic acid; CB-2((5E)-5-[(3-bromo-4-hydroxy-5-methoxyphenyl) methylene]-3-(3-chlorophenyl)-2thioxothiazolidin-4-one); CB-5, (2-benzyl-3-(4-chlorophenyl)-5-(4-nitrophenyl)-1H-pyrazolo[5,1-b] pyrimidin-7-one); CB-6(2-[7-(trifluoromethyl)-2,3-dihydro-1H-1,4-diazepin-5-yl] naphthalen-1-ol; J3,4-aryl-dihydropyrimidinones.
Bile acid a link between FXR and FATP5 for small molecule modulators
BAs are not the only function as physiological detergents to aid digestion of lipid nutrients but also function as signaling molecules that profoundly impact metabolism by activating nuclear and membrane BA receptors [13] and also by mediating induction of liver-specific FATP5 [17]. In excess amounts, BAs are toxic to the cell; therefore, their levels must be tightly controlled for the homeostasis of the cell. The nuclear receptor FXR and transporter protein FATP5 plays a key role in this regulation. Despite recent advances, the mechanisms underlying the interplay between the liver and intestine mediated by BAs-FXR and FATP5 to regulate BA levels, in particular, hepatic expression of cyp7a1, the rate-limiting BA synthetic enzyme, are not well understood. Studies have shown that BA can activate or suppress the FXR and well known for the treatment of metabolic disorder [71] Overexpression studies with hepatic FATP5 have shown that they not only enhance the LCFA uptake [53] but also activates the primary bile precursors [72]. Here, we are exploring the BA-mediated link of FXR with FATP5 and a summary of the regulatory role of FXR and FATP5 in BAs transport, biosynthesis, and fatty acid uptake is presented in Figure 5. It is well documented that endogenous BAs like UDCA and DCA interferes with protein-mediated hepatic LCFA uptake and reduces the TG level up to 50% [17], and it is also well mounted that FXR and FATP5 are modulated significantly with BA [17,73]. Thus these findings put forth a rationale to establish a link between hepatic FXR and hepatic FATP5 proteins. The studies suggest that FATP5 plays a role in hepatic TG metabolism via FXR [18]. In the current scenario, there are few leads working as agonists against FXR [65], antagonists against FATP5 [17], and few endogenous BAs are reported to work as a dual character such as agonist and antagonist both for FXR and FATP5, respectively [17,73]. Therefore, there is a curiosity to unveil the functional and mechanistic biology to explore the molecular mechanism between FXR and FATP5.
Curation of leads with their chemo types for ligand-based approach
From the known ligands chemical moiety, it would be possible for medicinal chemists to design and modify the possible chemical changes for better efficacy. The discovery of OCA is the best example of mimicking natural BAs. At present, several molecules for FXR evaluated in pre-clinical or clinical trials as agonist, antagonist and partial agonist as discussed in Table 2. Indeed, there is no specific molecule for FATP5 that has been reported to date and few kinds of literature claim the BAs and its mimics possibly modulate the FATP5. The steroidal and non-steroidal ligands for FXR and FATP5 are shown in Figure 7. The availability of these compounds or the structurally related derivatives has contributed to characterize the receptor from a structural, pathophysiological and therapeutic point of view. This structural information may provide a room to optimize the compounds for their affinity, selectivity of receptor recognition and target gene modulation. These are the prerequisite for many of the future developments as FXR and FATP5 modulators.
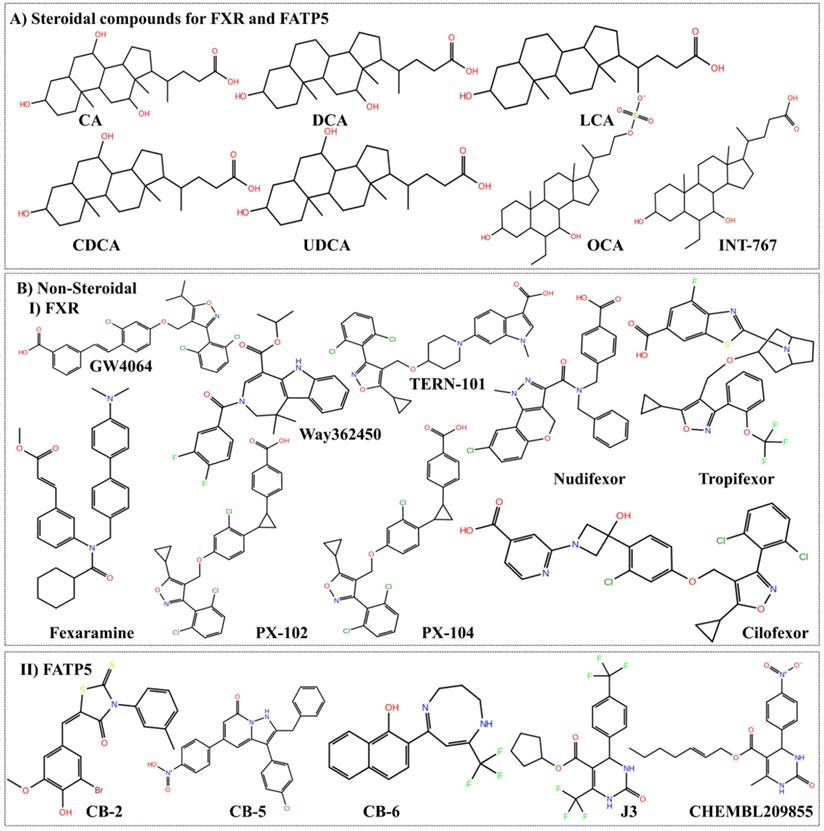
The chemical structure of lead compounds. A) The structure of BAs and their derivatives, which acts upon FXR and FATP5 both as highlighted in Table 2. B-I) and B-II) The non-steroidal compounds for FXR and FATP5, respectively. We here showed the molecules in advance phases for FXR. For FATP5 only those molecules are picked which have substantially potent and can be used for lead discovery.
Conclusion
Extensive research over the last several decades has unveiled the several unrecognized functions of BAs which are mediated by activation of a group of its receptors and pathways. Here, we are trying to explore the possible insights for a targets like FATP5 which is less explored, indeed, it is a challenging target for drug ability but efforts required to crack it for the discovery of novel leads (especially non-steroidal and synthetic new chemical moieties) that selectively can target the metabolic pathways involved diseases and set the stage for the development of a novel generation of FXR and FATP5 targeting drugs with improved pharmacological actions. Therefore, it is required to unveil some specific properties of BAs relevant to their intrinsic potency and selectivity for particular receptors and to design novel modulators of these receptors with improved pharmacokinetic and pharmacodynamic profiles. In this regard, the structural, dynamical and functional understanding of established BAs which are known to be potent against FXR, must be tested against FATP5 also, in the pursuit of designing selective ligands that can regulate the lipid content by modulating these two targets.
Future Perspective
BAs function in digestion and solubilization of lipophilic nutrients and as drugs in the small intestine, which are well documented. From the past two decades, there is emerging evidence which identified BAs as signaling molecules exerting the multiple physiological functions through complex and intertwined pathways that are largely mediated by modulation of different receptors as discussed above. There is extensive literature supporting the role of BAs in regulating metabolic processes. The role of tissue-specific FATP5 functions in the regulation of BA and NAFLD is emerging. BAs lower TGs level via a pathway involving FXR, SHP, and SREBP-1c. Basic research in BA metabolism and signaling suggest a scientific rationale for targeting the fatty liver diseases by inhibiting the FATP5 and activating the FXR. In this regard, we proposed that the BAs can modulate the FXR activity and inhibit the uptake of fatty acids through a protein-mediated process requiring FATP5, which appears promising for fatty acid modulation. These new insights into the physiological role of FATP5 should lead to an improved understanding of liver function and disease by modulating the close relationships of the BA and with the FXR. Although the signaling function of BAs and insights into the 3D structures (crystal structures) of BA receptors and their known ligands have accelerated the pace of discovery of new drugs for metabolic and liver disorders, limited has been explored for FATP5. The discovery and development of a therapeutic drug requires a more in-depth insight into the metabolic effects of each of the endogenous and synthetic BA, also it requires the pharmacological mechanism particularly at the molecular level on both FXR and FATP5. The clinical trial shows that the OCA disrupts cholesterol homeostasis by doing extensive FXR activation and blocking the metabolic conversion of cholesterol to BAs via SHP/FGF15/19 up- and cyp7a1 down-regulation as discussed above. Also, there are some potent FATP5 inhibitors that are also reported, moreover, their effect has not been exploited for FXR and vice versa. In the current scenario, the industry and academia are more focused to design agonists to upregulate the target proteins to handle the diseased state similar to BA which is known for lipid homeostasis and its regulation. Since OCA found with several drawbacks like itching and elevated LDL levels and its marketed cost, also in pathological condition do we need agonist or partial agonist? or only the requirement of modulators that can resensitize the BA pool. Undoubtedly more pathways involved and more research needs to be done to understand the mechanism behind insulin resistance, an accumulation of hepatic lipids, initiation of hepatocyte apoptosis, an increase in inflammation, and an increase in extracellular matrix that eventually results in fibrosis, steatosis to steatohepatitis progression so that drugs that successfully treat NAFLD and NASH can be developed. Therefore, new targets involved in BA pathways must be explored by combining the arsenal of structure-based, ligand-based, medicinal chemistry and computational drug designing approaches to design the non-steroidal mimics of BA's as a polypharmacological agent.
Acknowledgements
AK and SA thanks to the Translational Health Science and Technology Institute (THSTI) for providing core grant funds to support this research.
Abbreviations
FXR: Farnesoid X receptor; NR: Nuclear receptor; FATP5: Fatty acid transport protein 5; TG: Triglyceride; ASBT: apical sodium-dependent bile salt transporter; BSEP: bile salt export pump; cyp7a1: cholesterol 7α-hydroxylase; ASBT: apical sodium-dependent bile salt transporter; BSEP: bile salt export pump; FGF19: fibroblast growth factor 19; FGFR4: fibroblast growth factor receptor 4; SREBP-1c: sterol regulatory element-binding protein 1c; NTCP: Na+-dependent taurocholate cotransport peptide; SHP: small heterodimer partner; BA-CoA: Bile acyl co-A; PXR: Pregnane X receptor; VDR: Vitamin D receptor; CAR: Constitutive androstane receptor; TGR5: Takeda G protein receptor 5; S1PR2: Sphingosine-1-phosphate receptor 2; CHRM2: Cholinergic receptor muscarinic 2; ACC: Acetyl CoA carboxylase; FAS: Fatty acid synthase; SCD1: stearyl CoA desaturase; ACS: Acyl Co-A synthetase; DAG: Diacylglycerol; GPAT: Glycerol-3-phosphate acyltransferases; PA: Lysophosphatidic acid.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kwok R, Tse Y-K, Wong GL-H, Ha Y, Lee AU, Ngu MC. et al. Systematic review with meta-analysis: non-invasive assessment of non-alcoholic fatty liver disease-the role of transient elastography and plasma cytokeratin-18 fragments. Aliment Pharmacol Ther. 2014Feb;39(3):254-69
2. Yu Y, Cai J, She Z, Li H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv Sci. 2019Feb20;6(4):1801585
3. Xu J-Y, Li Z-P, Zhang L, Ji G. Recent insights into farnesoid X receptor in non-alcoholic fatty liver disease. World J Gastroenterol. 2014Oct7;20(37):13493-500
4. Yu Y, Cai J, She Z, Li H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv Sci. 2019Feb20;6(4):1801585
5. Li T, Chiang JYL. Nuclear receptors in bile acid metabolism. Drug Metab Rev. 2013;45(1):145-55
6. Chiang JYL, Ferrell JM. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018May18;18(2):71-87
7. Wan Y-JY, Sheng L. Regulation of bile acid receptor activity [Internet]. Liver Research. 2018;2:180-5 Available from: http://dx.doi.org/10.1016/j.livres.2018.09.008
8. Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Bile acid receptors and gastrointestinal functions [Internet]. Liver Research. 2019;3:31-9 Available from: http://dx.doi.org/10.1016/j.livres.2019.01.001
9. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell. 2000;102(6):731-44
10. Cattori V, Eckhardt U, Hagenbuch B. Molecular cloning and functional characterization of two alternatively spliced Ntcp isoforms from mouse liver1. Biochim Biophys Acta. 1999Apr14;1445(1):154-9
11. Green RM, Hoda F, Ward KL. Molecular cloning and characterization of the murine bile salt export pump. Gene. 2000Jan4;241(1):117-23
12. Jelinek DF, Andersson S, Slaughter CA, Russell DW. Cloning and regulation of cholesterol 7 alpha-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J Biol Chem. 1990May15;265(14):8190-7
13. Schaap FG, Trauner M, Jansen PLM. Bile acid receptors as targets for drug development [Internet]. Nature Reviews Gastroenterology & Hepatology. 2014;11:55-67 Available from: http://dx.doi.org/10.1038/nrgastro.2013.151
14. Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50(8):1509-20
15. Molinaro A, Wahlström A, Marschall H-U. Role of Bile Acids in Metabolic Control. Trends Endocrinol Metab. 2018;29(1):31-41
16. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015Mar14;385(9972):956-65
17. Nie B, Park HM, Kazantzis M, Lin M, Henkin A, Ng S. et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology. 2012Oct;56(4):1300-10
18. Ason B, Castro-Perez J, Tep S, Stefanni A, Tadin-Strapps M, Roddy T. et al. ApoB siRNA-induced liver steatosis is resistant to clearance by the loss of fatty acid transport protein 5 (Fatp5). Lipids. 2011Nov;46(11):991-1003
19. Fiorucci S, Cipriani S, Baldelli F, Mencarelli A. Bile acid-activated receptors in the treatment of dyslipidemia and related disorders. Prog Lipid Res. 2010Apr;49(2):171-85
20. Asthana S, Shukla S, Ruggerone P, Vargiu AV. Molecular mechanism of viral resistance to a potent non-nucleoside inhibitor unveiled by molecular simulations. Biochemistry. 2014Nov11;53(44):6941-53
21. Mittal L, Srivastava M, Asthana S. Conformational Characterization of Linker Revealed the Mechanism of Cavity Formation by 227G in BVDV RDRP. J Phys Chem B. 2019Jul25;123(29):6150-60
22. Mittal L, Kumari A, Suri C, Bhattacharya S, Asthana S. Insights into structural dynamics of allosteric binding sites in HCV RNA-dependent RNA polymerase. J Biomol Struct Dyn. 2020Apr;38(6):1612-25
23. Shaik FB, Prasad DVR, Narala VR. Role of farnesoid X receptor in inflammation and resolution [Internet]. Inflammation Research. 2015;64:9-20 Available from: http://dx.doi.org/10.1007/s00011-014-0780-y
24. Massafra V, Pellicciari R, Gioiello A, van Mil SWC. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol Ther. 2018Nov;191:162-77
25. Merk D, Sreeramulu S, Kudlinzki D, Saxena K, Linhard V, Gande SL. et al. Molecular tuning of farnesoid X receptor partial agonism. Nat Commun. 2019Jul2;10(1):2915
26. Anderson CM, Stahl A. SLC27 fatty acid transport proteins [Internet]. Molecular Aspects of Medicine. 2013;34:516-28 Available from: http://dx.doi.org/10.1016/j.mam.2012.07.010
27. Lewis SE, Listenberger LL, Ory DS, Schaffer JE. Membrane Topology of the Murine Fatty Acid Transport Protein 1 [Internet]. Journal of Biological Chemistry. 2001;276:37042-50 Available from: http://dx.doi.org/10.1074/jbc.m105556200
28. Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994Nov4;79(3):427-36
29. Faergeman NJ, DiRusso CC, Elberger A, Knudsen J, Black PN. Disruption of the Saccharomyces cerevisiae homologue to the murine fatty acid transport protein impairs uptake and growth on long-chain fatty acids. J Biol Chem. 1997;272(13):8531-8
30. Hirsch D, Stahl A, Lodish HF. A family of fatty acid transporters conserved from mycobacterium to man [Internet]. Proceedings of the National Academy of Sciences. 1998;95:8625-9 Available from: http://dx.doi.org/10.1073/pnas.95.15.8625
31. Milger K, Herrmann T, Becker C, Gotthardt D, Zickwolf J, Ehehalt R. et al. Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4 [Internet]. Journal of Cell Science. 2006;119:4678-88 Available from: http://dx.doi.org/10.1242/jcs.03280
32. Enooku K, Tsutsumi T, Kondo M, Fujiwara N, Sasako T, Shibahara J. et al. Hepatic FATP5 expression is associated with histological progression and loss of hepatic fat in NAFLD patients. J Gastroenterol [Internet]. 2019 Available from: http://dx.doi.org/10.1007/s00535-019-01633-2
33. Gao Q, Zhang G, Zheng Y, Yang Y, Chen C, Xia J. et al. SLC27A5 deficiency activates NRF2/TXNRD1 pathway by increased lipid peroxidation in HCC [Internet]. Cell Death & Differentiation. 2019 Available from: http://dx.doi.org/10.1038/s41418-019-0399-1
34. Zhou W, Madrid P, Fluitt A, Stahl A, Xie XS. Development and validation of a high-throughput screening assay for human long-chain fatty acid transport proteins 4 and 5. J Biomol Screen. 2010Jun;15(5):488-97
35. Wang H, He Q, Wang G, Xu X, Hao H. FXR modulators for enterohepatic and metabolic diseases. Expert Opin Ther Pat. 2018Nov;28(11):765-82
36. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T. et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995Jun2;81(5):687-93
37. Cho SW, An JH, Park H, Yang J-Y, Choi HJ, Kim SW. et al. Positive regulation of osteogenesis by bile acid through FXR. J Bone Miner Res. 2013Oct;28(10):2109-21
38. Chiang JYL. Regulation of bile acid synthesis. Front Biosci. 1998;3(4):d176-93
39. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB. et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000Sep;6(3):517-26
40. Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001Aug3;276(31):28857-65
41. Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM. et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002Jan25;277(4):2908-15
42. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017Jun;1(1):3-9
43. Yang Z-X, Shen W, Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int. 2010Aug12;4(4):741-8
44. Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005Oct;25(10):2020-30
45. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J. et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000Sep;6(3):507-15
46. Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS Lett. 2008Jan9;582(1):10-8
47. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart J-C, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003Feb;17(2):259-72
48. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA. et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c [Internet]. Journal of Clinical Investigation. 2004;113:1408-18 Available from: http://dx.doi.org/10.1172/jci200421025
49. Savkur RS, Bramlett KS, Michael LF, Burris TP. Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochem Biophys Res Commun. 2005Apr1;329(1):391-6
50. Mazuy C, Helleboid A, Staels B, Lefebvre P. Nuclear bile acid signaling through the farnesoid X receptor [Internet]. Cellular and Molecular Life Sciences. 2015;72:1631-50 Available from: http://dx.doi.org/10.1007/s00018-014-1805-y
51. Fuchs C, Traussnigg S, Trauner M. Nuclear Receptor Modulation for the Treatment of Nonalcoholic Fatty Liver Disease [Internet]. Seminars in Liver Disease. 2016;36:069-86 Available from: http://dx.doi.org/10.1055/s-0036-1571296
52. Correia JC, Massart J, de Boer JF, Porsmyr-Palmertz M, Martínez-Redondo V, Agudelo LZ. et al. Bioenergetic cues shift FXR splicing towards FXRα2 to modulate hepatic lipolysis and fatty acid metabolism. Mol Metab. 2015Dec;4(12):891-902
53. Doege H, Baillie RA, Ortegon AM, Tsang B, Wu Q, Punreddy S. et al. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology. 2006Apr;130(4):1245-58
54. de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002Jun;21(3):219-23
55. Lee YJ, Yu JH, Kim W-H, Kim J-W. New mechanisms contributing to hepatic steatosis: glucose, insulin, and lipid signaling [Internet]. Animal Cells and Systems. 2014;18:77-82 Available from: http://dx.doi.org/10.1080/19768354.2014.906502
56. Black PN, Sandoval A, Arias-Barrau E, DiRusso CC. Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism. Immunol Endocr Metab Agents Med Chem. 2009Sep;9(1):11-7
57. Yu J, Peng J, Luan Z, Zheng F, Su W. MicroRNAs as a Novel Tool in the Diagnosis of Liver Lipid Dysregulation and Fatty Liver Disease. Molecules. 2019;24(2):230
58. DiRusso CC, Darwis D, Obermeyer T, Black PN. Functional domains of the fatty acid transport proteins: studies using protein chimeras. Biochim Biophys Acta. 2008Mar;1781(3):135-43
59. Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H. et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008Aug8;283(32):22186-92
60. Hubbard B, Doege H, Punreddy S, Wu H, Huang X, Kaushik VK. et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology. 2006Apr;130(4):1259-69
61. Penno CA, Morgan SA, Rose AJ, Herzig S, Lavery GG, Odermatt A. 11β-Hydroxysteroid dehydrogenase-1 is involved in bile acid homeostasis by modulating fatty acid transport protein-5 in the liver of mice. Mol Metab. 2014Aug;3(5):554-64
62. Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal. 2010Nov19;8:e005
63. Degirolamo C, Modica S, Vacca M, Di Tullio G, Morgano A, D'Orazio A. et al. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology. 2015Jan;61(1):161-70
64. Pellicciari R, Costantino G, Camaioni E, Sadeghpour BM, Entrena A, Willson TM. et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. 2004Aug26;47(18):4559-69
65. Ali AH, Carey EJ, Lindor KD. Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med. 2015Jan;3(1):5
66. Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. 2013;86(11):1517-24
67. Zhang Y, LaCerte C, Kansra S, Jackson JP, Brouwer KR, Edwards JE. Comparative potency of obeticholic acid and natural bile acids on FXR in hepatic and intestinal in vitro cell models. Pharmacol Res Perspect [Internet]. 2017 5(6). Available from: http://dx.doi.org/10.1002/prp2.368
68. Mueller M, Thorell A, Claudel T, Jha P, Koefeler H, Lackner C. et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. 2015Jun;62(6):1398-404
69. Auinger A, Valenti L, Pfeuffer M, Helwig U, Herrmann J, Fracanzani AL. et al. A Promoter Polymorphism in the Liver-specific Fatty Acid Transport Protein 5 is Associated with Features of the Metabolic Syndrome and Steatosis. Horm Metab Res. 2010;42(12):854-9
70. Dhami-Shah H, Vaidya R, Udipi S, Raghavan S, Abhijit S, Mohan V. et al. Picroside II attenuates fatty acid accumulation in HepG2 cells via modulation of fatty acid uptake and synthesis [Internet]. Clinical and Molecular Hepatology. 2018;24:77-87 Available from: http://dx.doi.org/10.3350/cmh.2017.0039
71. Han C. Update on FXR Biology: Promising Therapeutic Target? [Internet]. nternational Journal of Molecular Sciences. 2018;19:2069. Available from: http://dx.doi.org/10.3390/ijms19072069
72. Mihalik SJ, Steinberg SJ, Pei Z, Park J, Kim DG, Heinzer AK. et al. Participation of two members of the very long-chain acyl-CoA synthetase family in bile acid synthesis and recycling. J Biol Chem. 2002Jul5;277(27):24771-9
73. Tu H. FXR, a Bile Acid Receptor and Biological Sensor [Internet]. Trends in Cardiovascular Medicine. 2000;10:30-5 Available from: http://dx.doi.org/10.1016/s1050-1738(00)00043-8
74. Kim I, Ahn S-H, Inagaki T, Choi M, Ito S, Guo GL. et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007Dec;48(12):2664-72
75. Ma Y, Khalifa B, Yee YK, Lu J, Memezawa A, Savkur RS. et al. Identification and characterization of noncalcemic, tissue-selective, nonsecosteroidal vitamin D receptor modulators. J Clin Invest. 2006Apr;116(4):892-904
76. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM. et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002May17;296(5571):1313-6
77. Barzegari M, Sarbakhsh P, Mobasseri M, Noshad H, Esfandiari A, Khodadadi B. et al. The effects of vitamin D supplementation on lipid profiles and oxidative indices among diabetic nephropathy patients with marginal vitamin D status. Diabetes Metab Syndr. 2019Jan;13(1):542-7
78. Effects of Pregnane X Receptor (PXR) Activation on Human Glucose, Lipid and Hormone Homeostasis - Full Text View - ClinicalTrials.gov. [cited 2020 Jan 1]. Available from:https://clinicaltrials.gov/ct2/show/NCT01293422
79. Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A. et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity [Internet]. Proceedings of the National Academy of Sciences. 2001;98:3369-74 Available from: http://dx.doi.org/10.1073/pnas.051551698
80. Chang TKH. Activation of Pregnane X Receptor (PXR) and Constitutive Androstane Receptor (CAR) by Herbal Medicines [Internet]. The AAPS Journal. 2009;11:590 Available from: http://dx.doi.org/10.1208/s12248-009-9135-y
81. Goodwin B, Gauthier KC, Umetani M, Watson MA, Lochansky MI, Collins JL. et al. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A. 2003;100(1):223-8
82. Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA. et al. Pregnane X Receptor (PXR), Constitutive Androstane Receptor (CAR), and Benzoate X Receptor (BXR) Define Three Pharmacologically Distinct Classes of Nuclear Receptors [Internet]. Molecular Endocrinology. 2002;16:977-86 Available from: http://dx.doi.org/10.1210/mend.16.5.0828
83. Guo C, Chen W-D, Wang Y-D. TGR5, Not Only a Metabolic Regulator [Internet]. Frontiers in Physiology. 2016;7:646. Available from: http://dx.doi.org/10.3389/fphys.2016.00646
84. Finn PD, Rodriguez D, Kohler J, Jiang Z, Wan S, Blanco E. et al. Intestinal TGR5 agonism improves hepatic steatosis and insulin sensitivity in Western diet-fed mice [Internet]. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2019;316(G):412-24 Available from: http://dx.doi.org/10.1152/ajpgi.00300.2018
85. Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G. et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol. 2010Oct;78(4):617-30
86. Zheng C, Zhou W, Wang T, You P, Zhao Y, Yang Y. et al. A Novel TGR5 Activator WB403 Promotes GLP-1 Secretion and Preserves Pancreatic β-Cells in Type 2 Diabetic Mice. PLoS One. 2015Jul24;10(7):e0134051
87. Zambad SP, Tuli D, Mathur A, Ghalsasi SA, Chaudhary AR, Deshpande S. et al. TRC210258, a novel TGR5 agonist, reduces glycemic and dyslipidemic cardiovascular risk in animal models of diabesity. Diabetes Metab Syndr Obes. 2013Dec21;7:1-14
88. Efficacy and Safety of FTY720 for Acute Stroke - Full Text View - ClinicalTrials.gov. [cited 2019 May 3]. Available from:https://clinicaltrials.gov/ct2/show/NCT02002390
89. Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M. et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012Jan;55(1):267-76
90. Chiang JYL. Sphingosine-1-phosphate receptor 2: A novel bile acid receptor and regulator of hepatic lipid metabolism? Hepatology. 2015;61(4):1118-20
91. Muscarinic Receptor Antagonists as a Therapy for Diabetic Neuropathy - Full Text View - ClinicalTrials.gov. [cited 2019 May 9]. Available from:https://clinicaltrials.gov/ct2/show/NCT03050827
92. Sheikh Abdul Kadir SH, Miragoli M, Abu-Hayyeh S, Moshkov AV, Xie Q, Keitel V. et al. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS One. 2010Mar15;5(3 e):9689
93. Sandoval A, Chokshi A, Jesch ED, Black PN, Dirusso CC. Identification and characterization of small compound inhibitors of human FATP2. Biochem Pharmacol. 2010;79(7):990-9
Author contact
![]() Corresponding author: Dr. Shailendra Asthana, Scientist C, Translational Health Science and Technology Institute (THSTI); NCR Biotech Science Cluster, 3rd Milestone; Faridabad - Gurgaon Expressway, Haryana-121001, India; E-Mail: sasthanares.in.
Corresponding author: Dr. Shailendra Asthana, Scientist C, Translational Health Science and Technology Institute (THSTI); NCR Biotech Science Cluster, 3rd Milestone; Faridabad - Gurgaon Expressway, Haryana-121001, India; E-Mail: sasthanares.in.