Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Origin and classification of...
Topology and polarization of...
Roles of activated Kupffer cells...
Intervention and treatment
Conclusions and Prospects
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(13):2367-2378. doi:10.7150/ijbs.47143 This issue Cite
Review
Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe?
Jiajia Chen1,2, Xiaoyi Deng1,2, Yongjian Liu1,2, Qiuhua Tan1,2, Guidong Huang3, Qishi Che4, Jiao Guo2 ![]() , Zhengquan Su1
, Zhengquan Su1 ![]()
1. Guangdong Engineering Research Center of Natural Products and New Drugs, Guangdong Provincial University Engineering Technology Research Center of Natural Products and Drugs, Guangdong Pharmaceutical University, Guangzhou 510006, China.
2. Guangdong Metabolic Diseases Research Centre of Integrated Chinese and Western Medicine, Guangdong TCM Key Laboratory for Metabolic Diseases, Key Laboratory of Modulating Liver to Treat Hyperlipemia SATCM, Level 3 Laboratory of Lipid Metabolism SATCM, Institute of Chinese Medicinal Sciences, Guangdong Pharmaceutical University, Guangzhou 510006, China.
3. Department of Pharmacy, Affiliated Hospital of Guilin Medical University; 15# Lequn Road, Guilin, Guangxi Zhuang Autonomous Region 54101, China
4. Guangzhou Rainhome Pharm & Tech CO., LTD 5F, No.10 Yongsheng Road, Yonghe Econoic region, Science City, Guangzhou 510663, China.
Received 2020-4-18; Accepted 2020-6-12; Published 2020-6-23
Abstract

The prevalence of non-alcoholic fatty liver disease (NAFLD) is increasing all around the world and it may become the primary cause of terminal liver disease in adults and children in the next few decades. However, the pathogenesis of NAFLD is complex, and the Food and Drug Administration (FDA) has not approved any drugs for its treatment. Kupffer cells are the key cells regulating immunity in the liver, and the effect of their unique polarization on NAFLD has received increasing attention. Kupffer cells mainly reside in the lumen of hepatic sinusoids and account for 80% to 90% of colonized macrophages in the human body. They are phagocytic cells with the capacity for self-renewal that rarely migrate from their niche in the liver, and play a crucial role in regulating and maintaining homeostasis. Upon liver damage, Kupffer cells will be activated, releasing a good deal of inflammatory cytokines and chemokines. This review summarizes the multiple roles of Kupffer cells in the pathogenesis of NAFLD, the role of infiltrating macrophages in the pathogenesis of NAFLD is also briefly discussed, and aims to provide a theoretical basis for designing an NAFLD treatment strategy with Kupffer cells as the therapeutic target.
Keywords: non-alcoholic fatty liver, Kupffer cells, inflammation, lifestyle interventions
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a public health problem worldwide, and 25% to 30% of the global population suffers from different degrees of it [1]. Due to the increasing prevalence of NAFLD, the clinical and economic burden of this disease is also increasing [2]. Furthermore, a growing number of patients with cirrhosis and end-stage liver disease require liver transplantation, and the incidence of hepatocellular carcinoma (HCC) is expected to increase dramatically [3, 4]. By 2030, the burden of late-stage liver disease in both Western countries and several Asian countries is estimated that increase 2-3-fold [5, 6].
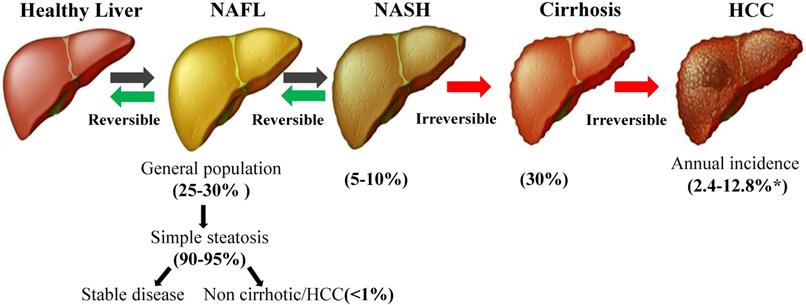
As the name implies, NAFLD occurs without excessive alcohol consumption, which is defined using the widely used high alcohol intake thresholds of 20 g/day for women and 30 g/day for men [7]. The hallmark pathological feature of NAFLD is the accumulation of triglycerides in the cytoplasm [8], and the gold standard for diagnosing the presence and severity of NAFLD is a liver biopsy [9]. The spectrum of histological findings in the liver attributed to NAFLD includes simple steatosis without obvious signs of inflammation or fibrosis [non-alcoholic fatty liver (NAFL)], hepatic triglyceride accumulation with inflammation and hepatocyte damage (ballooning) [non-alcoholic steatohepatitis (NASH)], liver fibrosis and cirrhosis and/or HCC [10-12] (Figure 1). Moreover, NAFLD is also a systemic disease related to obesity, type 2 diabetes mellitus (T2DM), insulin resistance (IR), cardiovascular disease (CVD), and metabolic syndrome (MetS) [13, 14].The pathogenesis of NAFLD is a multistep process that begins with triglyceride accumulation and leads to hepatic steatosis, which was originally proposed in the “two-hit hypothesis” [15]. According to this hypothesis, the second hit is the induction of oxidative stress and inflammation, which promote pathological progression [16]. In fact, the pathogenesis of NAFLD, particularly in the smaller subgroup of patients with hepatocyte injury, inflammation and/or fibrosis, is not completely understood [17].
The progression of NAFLD. NAFLD covers a wide range of conditions, from simple fat accumulation (fatty liver or steatosis) to NASH, cirrhosis and HCC. Although the prevalence of NAFLD is very high in the general population, approximately 25-30%, the vast majority of patients present with simple steatosis, which not associated with survival or liver damage. Only 5-10% of patients with NAFLD develop NASH, and 30% of patients with NAFLD develop cirrhosis. *Depending on the disease state (with or without cirrhosis), the incidence of HCC may range from 2.4% to 12.8% [118]. NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; HCC: hepatocellular carcinoma.
In the past 20 years, numerous studies of basic pathology and clinical trials have been conducted worldwide to better understand and prevent the development of NAFLD, but the Food and Drug Administration (FDA) has been approved no drugs. Currently, the general therapeutic strategies for NAFLD are as follows: (1) implementation of lifestyle interventions, including exercise and a proper diet; (2) active treatment of MetS to improve IR, weight loss, blood pressure and dyslipidemia; (3) treatment of liver injury to inhibit oxidative stress and improve liver fibrosis; and (4) perform liver transplantation in patients with advanced end-stage liver disease. Considering the increasing clinical burden and poor quality of life of patients with NAFLD, studies aiming to identify effective strategies that slow the increase in the number of NAFLD cases and improve the therapeutic options are urgently needed.
Origin and classification of hepatic macrophages
Hepatic macrophages mainly consist of two types: Kupffer cells, which originate from yolk sac-derived red bone marrow progenitor cells and reside in the liver, and monocyte-derived macrophages, which are derived from bone marrow hematopoietic stem cells and transplanted through the blood circulation to the liver [18]. Essentially, monocyte-derived macrophages are immunogenic macrophages that can differentiate under the influence of microenvironment; Kupffer cells are a self-sustaining, locally proliferating, and usually susceptible population of phagocytic cells [19]. More importantly, a recent study reported differences in the transcriptome between infiltrating macrophages and Kupffer cells in NASH mouse models, and the expression of certain genes was significantly different; for example, 1500 genes were enriched in infiltrating macrophages, and 1690 genes were enriched in Kupffer cells [20]. In general, Kupffer cells are able to be distinguished from monocyte-derived macrophages. CD68, a scavenger receptor (SR) for lipoproteins on macrophages is used to indicate the presence of macrophages in the liver [21]. Moreover, Kim et al. performed dual staining of mouse liver section using ionized calcium binding adapter molecule 1 (IBA-1) and C‐type lectin domain family 4 member F (CLEC‐4F), which adequately differentiated between infiltrating macrophages and Kupffer cells [22].
In addition to the above-mentioned hepatic macrophages, other macrophages also play more and more roles in the pathogenesis of NAFLD. For example, adipose tissue macrophages (ATM) promote the recruitment of blood mononuclear macrophages to the liver during the occurrence of NAFLD, and the activation of adipocyte death-related macrophage may promote liver injury by stimulating neutrophil recruitment, thereby exacerbating lipolysis-mediated lipotoxicity and liver injury [22, 23]. When Nod-like receptor protein 3 (NLRP3) inflammatory bodies are activated, they promote macrophage activation in subjects with NAFLD and accelerate liver inflammation [24]. In high fat diet (HFD) fed mice, the neutrophil‐derived reactive oxygen species and activation of stress kinases lead to the hepatic overexpression of C-X-C motif chemokine ligand 1 (CXCL1) and drive the progression of steatosis-to-NASH [25]. In summary, macrophages play an essential role in maintaining homeostasis and defense functions.
Topology and polarization of Kupffer cells
Kupffer cells are the resident liver macrophages and account for 80-90% of all macrophages in the body. They are the largest component of the reticular endothelial system [26] and represent a resident self-renewing phagocytic population in the liver. Kupffer cells migrate between liver sinusoids and the space of Disse, directing and regulating interactions between various resident and recruited cells in the liver [27]. It was found that Kupffer cells lost in the early stages of the mouse NASH model fed by a methionine-choline-deficient (MCD) diet, and then the Ly6C+ monocyte-derived macrophages replacing the resident Kupffer cells as the dominant macrophage population [28]. Kupffer cells express Toll-like receptor (TLR) 4 and bind to lipopolysaccharide (LPS), resulting in the activation of nuclear factor-kappa B (NF-κB), mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase 1 (ERK1), p38, Jun N-terminal kinase (JNK) and interferon regulatory factor 3 (IRF3); subsequently, the production of large amounts of pro-inflammatory cytokines leads to liver damage, leukocyte infiltration and the activation of hepatic stellate cells [29].
Historically, macrophages have been considered to undergo phenotypic differentiation in response to different microenvironmental signals, and the phenotypes have been traditionally oversimplified as classically activated macrophages (M1) and alternatively activated macrophages (M2) [30, 31]. M1 macrophages are induced by pro-inflammatory mediators, have a high antigen presentation capability, and induce the release of large amounts of cytokines, such as tumor necrosis factor (TNF), inducible nitric oxide synthase (iNOS), interleukin (IL)-1β, and reactive oxygen species (ROS) while simultaneously activating the Th1 response. In addition, M1 macrophages exert anti-proliferative and cytotoxic effects, and promote macrophage-mediated tissue damage. In contrast, M2 macrophages show anti-inflammatory and pro-degradation activity, such as arginase 1 expression, IL-4 and IL-10 secretion and high levels of phagocytosis [32, 33]. M2 macrophages are further classified as M2a, M2b, and M2c and each subtype of cells secretes different cytokines and expresses different epitopes. M2 macrophages, which balance the activity of M1 macrophages, are involved in downregulating the inflammatory response and initiating tissue repair, and these cells synthesize important mediators that promote tissue remodeling and vasculogenesis to control the inflammatory response; these cells also promote the Th2 immune response and play a key role in regulating T cell function [34, 35].
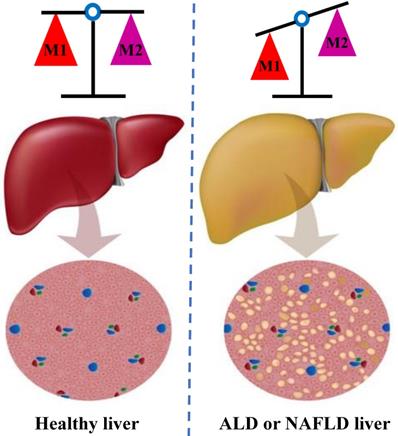
In individuals with alcoholic liver disease (ALD) and NAFLD, the balance between classic pro-inflammatory M1 Kupffer cells and selectively activated anti-inflammatory M2 Kupffer cells is crucial to strictly regulate the occurrence and development of liver injury [36] (Figure 2). Through in vivo and in vitro experiments, J. Wan and colleagues showed that the selective induction of M1 Kupffer cells apoptosis enabled M2-polarized Kupffer cells to protect against alcoholic liver injury by neutralizing the remaining M1 Kupffer cells; these researchers concluded that limiting the number of M1 Kupffer cells while promoting M2 Kupffer cells polarization may be a valuable therapeutic strategy for ALD [37]. Moreover, strategies that promote the polarization of anti-inflammatory M2 Kupffer cells prevented alcohol-induced steatosis and hepatocyte apoptosis, and IL-6 is a mediator of hepatocyte senescence induced by M2 Kupffer cells [38]. Interestingly, the occurrence of NAFLD/NASH is often accompanied by an increase in the proportion of M1/M2 macrophages in the liver tissue [39]. Experiments conducted in a mouse model of NAFLD further showed that M2 macrophages induce the apoptosis of M1 macrophages by secreting arginase and the anti-inflammatory factor IL-10, which act in a paracrine manner; in addition, these M2 macrophages reduced the proinflammatory effects of M1 macrophages and then mitigated the inflammatory response and cell damage [40]. In summary, Kupffer cells exert various regulatory effects on different stages of liver injury and repair. The existing evidence implies that therapeutic interventions targeting M2 Kupffer cells polarization may be an appealing strategy for limiting inflammation and hepatocyte injury in the initial stages of ALD and NAFLD.
The balance of M1 and M2 Kupffer cells. Kupffer cells maintain a balance between the M1 and M2 phenotypes to control inflammation in individuals ALD and NAFLD. M1 Kupffer cells secrete a large amount of pro-inflammatory factors, such as IL-1β, TNF-α and IL-6; in contrast, the M2 Kupffer cells secrete a numerous anti-inflammatory factor, such as IL-4and IL-10. M2 Kupffer cells balance the activity of M1 Kupffer cells, initiate tissue repair, synthesize important mediators to promote tissue remodeling and angiogenesis, and control the inflammatory response. ALD: alcoholic liver disease; NAFLD: non-alcoholic fatty liver disease. IL: interleukin; TNF: tumor necrosis factor.
Roles of activated Kupffer cells in NAFLD
Kupffer cells, with their high heterogeneity and plasticity, are excellent at maintaining homeostasis and defense functions. In the body, factors such as lipids, lipids metabolites, and LPS, activate Kupffer cells. Once activated, Kupffer cells increase the expression of inflammatory cytokines, exacerbate the degree of necrotic inflammation among liver cells, and alter the expression of genes associated with fibrosis and oxidative damage, leading to the development of NAFLD. The role of Kupffer cells in NAFLD, as elucidated in recent years, is summarized in four pathological processes, including inflammation, steatosis, oxidative stress, and fibrosis, which are discussed to provide a theoretical basis for designing treatment strategies for NAFLD with Kupffer cells as the therapeutic target.
Inflammation
Inflammation and the gut-liver axis
The intestinal barrier acts as the body's first line of defense to protect against damage caused by bacteria, food antigens, toxins and other factors in the intestinal cavity [41]. The liver, as the body's second line of defense, further regulates the immune function of the intestinal barrier. Together, the liver and intestinal barrier forms the body's defense system as the gut-liver axis [42]. The pathogenesis of NAFLD, a multisystem disease, is related to the permeability of the intestinal barrier. The intestinal flora and related metabolites directly penetrate the intestinal barrier, enter the blood circulation and reach the liver, where they elicit an inflammatory response, and promote and exacerbate the development of NAFLD. For example, intestinal endotoxins (such as LPS) stemming from flora disorders, bacterial transmigration or changes in the intestinal mucosal barrier play a large role in the pathogenesis of NAFLD [43, 44]. LPS is one of the most potent activators of the mammalian immune system and is released from the surface of bacterial cells during proliferation, lysis and death [45].Obese individuals or patients with NAFLD present with intestinal bacteria overgrowth compared with normal-weight or unaffected people, which results in an increase in the number of bacteria producing LPS [46, 47]. Upon accumulation, LPS binds to Kupffer cell surface receptor complexes, leading to the production of a good deal of factors and the induction of immune responses and inflammatory reactions, thereby facilitating liver inflammation and pathological damage. The detailed mechanism is described below.
LPS binds to CD14 on the surface of Kupffer cells through the TLR4 signaling pathway, which subsequently recruits T lymphocytes, B lymphocytes and other leukocytes, resulting in the release of inflammatory cytokines and chemokines, such as TNF-α, IL-1β, IL-6, IL-12 and interferon-γ (IFN-γ) [48, 49]. Next, a large multiprotein complex containing myeloid differentiation factor 88 (MyD88), TNFR-associated factors (TRAFs), IL-1 receptor-associated kinases (IRAKs) and transforming growth factor (TGF)-beta-activated kinase 1 (TAK1) forms [50, 51]. Activated TLR4 is also translocated to an endosomal compartment, which results in the recruitment of TIR domain-containing adaptor inducing IFN-β (TRIF), RNA modifying proteins containing the conserved TRAM domain, TNF receptor-associated factor 3 (TRAF3) and other proteins [52]. These events induce the phosphorylation of IRF3 and the IFN response [53]. Activated Kupffer cells produce TNF-α, IL-6 and other inflammatory factors, recruit infiltrating inflammatory cells, and play multiple roles in NAFLD [54].
Inflammation and pyroptosis
Based on accumulating evidence, the liver is exposed to various microbial molecules produced in the digestive tract through the local circulation and can be infected by various hepadnaviruses, leading to the activation of inflammatory bodies and pyroptosis, which mediates the inflammatory defense mechanism against pathogens. During the pathogenesis of NASH, the levels of NLRP3 inflammatory bodies in the intestine are reduced, which weakens the intestinal barrier function against bacterial translocation, allowing a large amount of LPS to enter the liver and exacerbating the development of NASH [55].
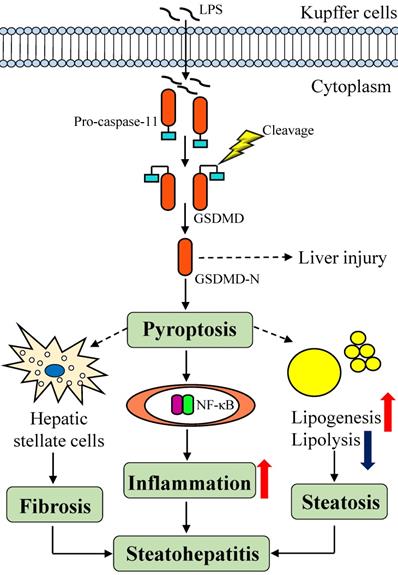
One study revealed a new mechanism by which macrophages are activated by cytoplasmic LPS via a TLR4-independent pathway, thereby activating the inflammatory enzyme caspase-11 [56] (Figure 3). As shown in the study by Shi and colleagues, caspase-11 exposes the N-terminal fragment of gasdermin D (GSDMD), which triggers an inflammatory cell death response called pyroptosis in response to cytoplasmic LPS that is essential for endotoxic shock in mice [57, 58]. As the executor of pyroptosis, caspase-cleaved GSDMD directly increases the production of pro-inflammatory cytokines, indirectly activates the NF-κB signaling pathway and recruits macrophages [59]. In this context, the discovery that GSDMD both indirectly and directly promotes lipid production through signaling pathways provides a new insight into the associated mechanism. Pyroptosis is correlated with NASH-associated hepatic fibrosis, which activates hepatic stellate cells to induce liver fibrosis and even hepatocyte death [60, 61]. Bing Xu and colleagues analyzed human NAFLD/NASH liver tissues and showed that the levels of GSDMD and the pyrophosphorylation-induced fragment GSDMD-N were both increased, and more importantly, hepatic levels of the GSDMD-N protein were significantly increased patients with NASH and were related to NAFLD activity scores and fibrosis [62]. Although the role of GSDMD in the pathogenesis of NAFLD and the potential underlying mechanisms remain unclear, GSDMD represents a potential therapeutic target or biomarker of disease progression for human NAFLD that deserves further attention in the future [63]. The development of compounds or biopharmaceuticals that target cell death and inflammatory bodies represents an important approach for treating NAFLD /NASH.
The LPS-induced signaling pathways in Kupffer cells and mechanisms of GSDMD-induced pyroptosis. LPS is sensed in a TLR4-independent manner in the cytoplasm through a directly interaction with caspase-11. Then, caspase-11 cleaves and activates GSDMD, which promotes pyroptosis. GSDMD acts as a direct executor of pyroptosis by causing increased production of in pro-inflammatory cytokines, and indirectly activates the NF-κB signaling pathway and subsequent macrophage recruitment. GSDMD also contributes to steatohepatitis by increasing lipogenesis and decreasing lipolysis. At the same time, pyroptosis activates hepatic stellate cells to induce liver fibrosis and even the death of hepatocytes in individuals with NASH. LPS: lipopolysaccharides; TLR4: toll-like receptor 4; GSDMD: gasdermin D.
Steatosis
The liver has a high capacity for fatty acid uptake and plays a significant role in the metabolism of lipids and lipoproteins [64]. The biological activity of Kupffer cells in individuals with NAFLD may be regulated by different mechanisms, such as the lipid abundance and composition and changes in the liver tissue [65]. In a marginal-copper high-fructose diet (CuMF) rats, Kupffer cell ablation decreases hepatic triglyceride accumulation and plasma macrophage chemotactic protein 1 (MCP-1) level, suggesting that Kupffer cells protect liver cells lipid metabolism disorders and steatosis [66].
As an early stage of NAFLD, hepatic steatosis occurs when an imbalance exists between the inward and outward fluxes of free fatty acids (FFAs) in the liver and is defined as triglyceride accumulation in more than 5% of hepatocytes [67]. Asanuma et al., performed superparamagnetic iron oxide (SPIO) magnetic resonance imaging and reported a significantly impairment in the phagocytosis activity of Kupffer cells in patients with NAFLD and NASH animal models without a reduction in cell number, and the degree of functional impairment was positively correlated with hepatic steatosis [68]. Thus, the TLR-mediated recognition of fatty acid groups is a primary mechanism of lipid regulation in inflammation and innate immunity [69]. In the Kupffer cell extracellular matrix, the binding of FFAs to TLRs activates the C-JNK and NF-κB pathways [70]. Activated NF-κB upregulates the levels of adhesion molecules and MCP-1, thus recruiting CD11b+ macrophages and promoting lipid synthesis, which increases the transcription of activating protein 1 (AP-1) and pro-inflammatory cytokines [71]. Excess FFAs that accumulate in Kupffer cells impair not only β-oxidation but also other mitochondrial functions [72].
Interestingly, increasing experimental and clinical evidence suggests an association between the pathogenesis of NASH and changes in intrahepatic cholesterol homeostasis and the accumulation of free cholesterol [73, 74]. Rats fed choline-deficient (but methionine-sufficient) and high-cholesterol (2%) diets suffer from impaired mitochondrial function, which characterizes were the accumulation of free cholesterol and the depletion of glutathione, and increased susceptibility to TNF-α and fatty acid synthase (Fas)-mediated liver damage [75]. Cholesterol synthesis pathways and hepatocyte uptake must be perfectly balanced with cholesterol clearance by hepatocytes to maintain liver cholesterol homeostasis [76]. However, when Kupffer cells lack the cholesterol biosynthesis capacity, they must obtain cholesterol from circulating cholesterol-rich lipoproteins. The main cholesterol uptake pathway in Kupffer cells involves oxidized low-density lipoprotein (oxLDL) internalization by the SR clustering protein CD36 and SR-A. In patients with MetS or NASH, plasma oxLDL levels are significantly increased, particularly after a meal, and the SR-mediated uptake of these lipoproteins may be a major cause of Kupffer cells activation and inflammatory factor infiltration in the liver [77, 78]. Consistent with these findings, strategies targeting defective oxLDL uptake secondary to the inactivation of Kupffer cells by reversing the genetic deletion of lipoprotein receptors or administering anti-oxLDL antibodies restore the resting Kupffer cells phenotype, reverse liver inflammation and reduce the incidence of NAFLD [79, 80].
Oxidative stress
A common feature of chronic liver disease is oxidative stress, which affects the development of NASH [81]. By observing changes in various oxidative stress markers in liver biopsies or blood samples, researchers observed elevated levels of oxidative stress in patients with NAFLD, and this level was related to the severity of NAFLD [82]. Absent or dysfunctional FFA β-oxidation leads to the generation of ROS, which induce mitochondrial lipotoxicity and activate the NF-κB/JNK pathway, high mobility group box 1(HMGB-1)/TLRs, cytokines and chemokines [83, 84]. In the liver, the main source of ROS is Kupffer cells, in which ROS are usually produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [85]. Since the discovery of uncoupling protein 2(UCP2), this protein has been considered one component of the pathogenic mechanisms of NAFLD [86]. According to previous reports, genetically obese (ob/ob) mice fed a high-fat diet show a significant enrichment of UCP2 in hepatocytes and a reduction in the numbers of Kupffer cells and other macrophages [87, 88]. As described below, the overexpression of UCP2 inhibits ROS production and the activation of Kupffer cells and other macrophages; however, the inhibition or ablation of UCP2 leads to increased ROS levels and pro-inflammatory cytokine release and the continued activation of NF-κB [89, 90].
Oxidases in different cellular compartments, such as mitochondrial, NADPH oxidase and cytoplasmic xanthine oxidase, inhibit ROS production and damage the antigenicity of Kupffer cells to a similar extent [91]. Moreover, because of the inability of UCP2-/- macrophages to control the mitochondrial ROS production, these cells show markedly increased antimicrobial and pro-inflammatory activities [92]. Interestingly, any cell-derived ROS may affect the antioxidant defense and redox-sensitive signaling pathways in Kupffer cells. Fortunately, hepatocytes have a higher antioxidant capacity than Kupffer cells, and thus ROS-mediated regulatory mechanisms are thought to exert a greater effect [93]. In addition, ROS and other oxidative stress mediators, such as the reactive aldehyde end products of lipid peroxidation, are postulated to maintain fibrosis progression in individuals with chronic liver diseases of different etiologies by inducing cirrhosis, which triggers cell damage and death, as well as fibrosis and inflammatory reactions [94]. This approach may be a valuable avenue for future research on Kupffer cells.
Fibrosis
During steatohepatitis, activated Kupffer cells secrete a large number of pro-inflammatory cytokines, such as TNF-α and IL-6, to recruit T lymphocytes, natural killer T (NKT) cells and other inflammatory cells, indirectly participating in inflammation and the immune response [95]. NKT cells release perforin and granulase from fine particles that damage liver cells. The recruitment of NKT cells also promotes the activation of hepatic stellate cells and myofibroblasts, leading to the progression of NASH-associated fibrosis [95]. In addition, lipid peroxides hydrolyze apolipoprotein B100, aggravate inflammatory necrosis, activate Kupffer cells, and induce the secretion of TGF-β1, an important anti-proliferative and profibrotic cytokine. Then, the TGF-β signal is transmitted through the TGF-β1 receptor (TβR) at the cell surface to promote the phosphorylation of the Smad protein in the nucleus, which leads to the transformation of hepatic stellate cells into muscle fiber cells and aggravates liver fibrosis [96, 97].
Summary
The above description clearly highlights the multiple functions and various regulatory activities of Kupffer cells in the proregression of NAFLD and their important roles in pathological processes (Table 1). During the inflammatory response, LPS binds to CD14 on the membrane and signals through the TLR4 pathway to activate Kupffer cells and induce the production of various inflammatory factors; LPS also directly binds to Kupffer cells and activates the inflammatory enzyme caspase-11, causing pyroptosis and the release of inflammatory factors, highlighting the multiple roles of this molecule. During steatosis, FFAs mediate Kupffer cells activation and increased MCP-1 expression to promote lipid accumulation. During oxidative stress, Kupffer cell activation and ROS production form a positive feedback loop. In the process of fibrosis, Kupffer cells produce cytokines such as TGF-β1 and IL-6 to promote fibrosis.
The role of Kupffer cells in the various pathological processes of NAFLD
| Pathological process | Activated pathway | Resulting effects |
|---|---|---|
| Inflammation | LPS binds CD14 via TLR4 pathway to activate IKK | Secretes inflammatory factors: TNF-α: involved in IR, promotes lipid synthesis, induces hepatocyte apoptosis IL-6: improves hepatic microcirculation and enhances immunity TL-1β: promotes lipid synthesis and accumulation IL-12: promotes the differentiation of Th1 cells and participates in NKT cell depletion |
| LPS directly activates the inflammatory enzyme caspase-11 and indirectly activates NF-κB | Causes pyroptosis, recruits macrophages, activates hepatic stellate cells, and causes liver fibrosis and even liver death | |
| Steatosis | Accumulation of FFAs that binds to TLRs and activate the C-JNK and NF-κB pathways | Destroys the liver cell membrane, up-regulates MCP-1 expression, recruits macrophages and promotes lipid synthesis |
| Oxidative stress | Excessive ROS activates Kupffer cells | Cells secrete cytokines and participate in the inflammatory response; UCP-2 expression is upregulated |
| Fibrosis | Increased lipid peroxides and ApoB100 hydrolysis | Cells secrete TGF-β1 to activate hepatic stellate cells and aggravate fibrosis |
Intervention and treatment
Lifestyle interventions
Currently, the FDA has been approved no drugs for the treatment of NAFLD/NASH. Lifestyle improvements, weight loss and increased exercise are the currently available and effective non-pharmacological treatments for NAFLD/NASH [98, 99]. Comprehensive lifestyle changes, including a reduction in energy intake and an increase in physical labor, improve liver enzyme levels [alanine aminotransferase (AST), aspartate aminotransferase (ALT) and gamma-glutamyl transpeptidase (GGT)], metabolic parameters (fasting plasma glucose level and insulin sensitivity), and the triglyceride concentration in the liver [via proton nuclear magnetic resonance (NMR) spectroscopy] in 6 to 12 months [100, 101]. These interventions have also been shown to improve steatosis and necrotic inflammation, including swelling and fibrosis, in paired liver biopsies [102, 103]. Dietary interventions improve glucose and lipid metabolism in patients, reduce the body weight of obese patients, reduce IR, improve insulin sensitivity in peripheral tissues, and promote glucose uptake and utilization by muscle cells. Exercise increases energy consumption, reduces the conversion of energy into fat, reduces body mass, excite sympathetic nerves, increases insulin secretion, promotes fat decomposition, and reduces the occurrence of visceral obesity. Exercise also increases high-density lipoprotein levels and insulin sensitivity, thereby reducing the degree of fatty liver. Therefore, the formulation of scientific evidence-based exercise programs and the mastery of reasonable dietary principles are very important for reducing the morbidity of fatty liver disease, improving the reversal rate and quality of life of people with fatty liver disease, and reducing the burden of fatty liver disease on human health.
Strategies targeting Kupffer cells
Kupffer cells are phagocytes with the capacity for self-renewal that rarely migrate from their niche and targeting Kupffer cells for the treatment of NAFLD has great significance. In an experimental NASH model established in mice, Rivera et al., found that disrupting Kupffer cells reduces histological manifestations in the liver, such as steatosis, inflammation, and necrosis [104]. Similarly, Zeng et al., reduced the number of Kupffer cells with Gdcl3 in a high-fat diet mouse model and observed significant reductions in steatosis and IR [105]. Therefore, strategies targeting Kupffer cells are an important aspect of NAFLD treatment (Table 2).
Therapeutic approaches targeting Kupffer cells in individuals with liver diseases
| Categorization | Subsets | Main findings | Ref |
|---|---|---|---|
| Activation of Kupffer cells | Benzyl isothiocyanate | Inhibits cholesterol crystal-activated NLRP3 inflammasomes in Kupffer cells to improve high-fat/high-cholesterol induced changes in the liver | Chen et al., (2020) |
| Alagebrium | Increases AGEs clearance and indirectly inhibits hepatic stellate cell activation | Fernando et al., (2019) | |
| Resveratrol | Improvs the inflammation and fibrosis by inhibiting LPS reactivity controlled by CD14 expression in Kupffer cells | Takaomi et al., (2016) | |
| M1 and M2 Kupffer cells polarization | β-Cryptoxanthin | Prevents and reverses insulin resistance and steatohepatitis | Ni et al., (2015) |
| Astaxanthin | Regulates macrophage/Kupffer cells polarization and hepatic homeostasis | Ni et al., (2016) | |
| RORα | Enhances M2 polarization in Kupffer cells | Han et al., (2017) | |
| Liraglutide | Regulates the M2 polarization of Kupffer cells to reduce liver steatosis | Li et al., (2019) | |
| Saxagliptin | Regulates the polarization of M1 / M2 macrophages / Kupffer cells | Yang et al., (2018) |
Kupffer cells activation: As mentioned above, activated Kupffer cells show increased expression of inflammatory cytokines and thereby exacerbate the degree of necrotic inflammation of liver cells, leading to the occurrence of NAFLD. The inhibition of Kupffer cell overactivation may reverse the course of this disease. Benzyl isothiocyanate (BITC) is an organic sulfur compound that is abundant in cruciferous vegetables. Recently, Chen et al. showed that BITC ameliorates the effects of a high-fat/high-cholesterol diet by inhibiting cholesterol crystal-activated NLRP3 inflammasomes in Kupffer cells, thereby preventing the development of diet-induced NASH [106]. Alagebrium was shown to impair the progression of NAFLD in mice fed a diet containing advanced glycation end products (AGEs) by directly inhibiting Kupffer cells and indirectly inhibiting hepatic stellate cell activation; Alagebrium increases AGEs clearance and may inhibit the progression of NAFLD to liver fibrosis [107]. In addition, the natural polyphenolic compound resveratrol (RSV) is considered to possess a wide range of bioactivities that are beneficial to human health. According to Takaomi et al., RSV improvs the inflammation and fibrosis in mice with HFD-induced NAFL by inhibiting LPS reactivity controlled by CD14 expression in Kupffer cells [108].
M1 and M2 Kupffer cells polarization: Kupffer cells are the key cells regulating hepatic immune responses, and their specific polarization has received increasing attention in research on NAFLD. In one experiment of mice fed the antioxidant β-cryptoxanthin, the total population of hepatic macrophages and T cells was reduced, but the population of M2 macrophages was increased. β-Cryptoxanthin may at least partially prevent and reverse IR and steatohepatitis by activating M2 macrophages/Kupffer cells [109]. Similarly, Ni et al. proposed that dietary carotenoids, such as β-cryptoxanthin and astaxanthin, prevent or improve NAFLD by regulating macrophage/Kupffer cells polarization and hepatic homeostasis [110]. As a key regulator, retinoic acid-associated orphan receptor α (RORα) can regulate M1/M2 polarization in Kupffer cells; RORα enhances the M2 polarization of Kupffer cells by inducing Kruppel-like factor 4 and may be a vulnerable therapeutic target in liver macrophages for the treatment of NASH [111]. In recent years, the repurposing of old medicines has received considerable attention. For example, Li et al., found that liraglutide, the first-line drug for T2DM, regulates the M2 polarization of Kupffer cells through the cAMP-PKA-STAT3 signaling pathway, thereby reducing liver steatosis caused by a HFD [112]. Analogously, Yang et al., found that Saxagliptin, a hypoglycemic drug, regulates the polarization of M1/M2 macrophages/Kupffer cells through the CaMKKβ/AMPK pathway, thereby reducing NAFLD [113].
Conclusions and Prospects
In the past few years, our basic understanding of Kupffer cells has substantially improved Kupffer cells are a “double-edged sword”; they promote inflammation that leads to liver cell damage and activate hepatic stellate cells, causing fibrosis, angiogenesis and tumor development, but they also stimulate fibrosis regression and limit disease progression. Important issues to consider in future research include controlling the ratio of M1 to M2 Kupffer cells and therapeutically interfering with the M2 polarization of Kupffer cells. Although pyroptosis has been shown to be closely related to the progression of liver disease, many details remain unknown, and further research is required. For example, researchers have not clearly determined how pyroptosis causes liver damage and affects disease progression. Currently, more systematic research is needed to clarify the function of Kupffer cells in NAFLD and resolve the controversy. These studies will not only facilitate the further elaboration of NAFLD pathogenesis but also provide new research ideas and targets for NAFLD in the near future.
In recent years, research on the use of natural drugs in the treatment of NAFLD has expanded. Some herbs also have potential value as treatment for NAFLD. According to modern pharmacological studies, Salvia miltiorrhiza promotes blood circulation, improves the microcirculation, inhibits collagen fiber hyperplasia, and exerts anti-fibrotic, anti-free radical and anti-peroxidation activities [114, 115]. The stilbene component of Polygonum multiflorum improves the function of damaged fatty livers in rats and reduces lipid peroxide contents in the liver. Moreover, P. multiflorum is rich in phospholipids that promote cholesterol and lipids metabolism in the liver, thereby reducing intrahepatic fat accumulation [116, 117]. Perhaps further in-depth research will determine whether the aforementioned herbs slow the inflammatory response by regulating Kupffer cells and other macrophages to achieve anti-oxidant and anti-liver fibrosis effects.
In general, the therapeutic effects of certain drugs mentioned above on patients with NAFLD are controversial or unclear. The prevention and treatment of NAFLD, a metabolic disorder caused by an unhealthy lifestyle, must not rely solely on medication. A reasonable recommendation is to improve detrimental lifestyles and poor eating habits, increase aerobic exercise, and control body weight. The combination of lifestyle interventions and drug therapy is the basis for the treatment of NAFLD, and the efficacy of therapeutic drugs will depend on the success of lifestyle interventions.
Abbreviations
NAFLD: non-alcoholic fatty liver disease; FDA: Food and Drug Administration; HCC: hepatocellular carcinoma; NAFL: non-alcoholic fatty liver; NASH: non-alcoholic steatohepatitis; T2DM: type 2 diabetes mellitus; IR: insulin resistance; CVD: cardiovascular disease; MetS: metabolic syndrome; SR: scavenger receptor; IBA-1: ionized calcium binding adapter molecule 1; CLEC‐4F: C‐type lectin domain family 4 member F; ATM: adipose tissue macrophages; NLRP3: Nod-like receptor protein 3; HFD: high fat diet; CXCL1: C‐X‐C motif chemokine ligand 1; MCD: methionine-choline deficient; TLR: Toll-like receptor; LPS: lipopolysaccharide; NF-κB: nuclear factor-kappa B; MAPK: mitogen-activated protein kinase; ERK1: extracellular signal-regulated kinase 1; JNK: Jun N-terminal kinase; IRF3: interferon regulatory factor 3; iNOS: inducible nitric oxide synthase; TNF: tumor necrosis factor; IL: interleukin; ROS: reactive oxygen species; ALD: alcoholic liver disease; IFN-γ: interferon-γ; MyD88: myeloid differentiation molecular 88; TRAFs: TNFR-associated factors; IRAKs: IL-1 receptor-associated kinases; TGF: transforming growth factor; TAK1: TGF-beta-activated kinase 1; TRAF3: TNF receptor associated factor 3; GSDMD: gasdermin D; CuMF: marginal-copper high-fructose diet; FFAs: free fatty acids; SPIO: superparamagnetic iron oxide; MCP-1: macrophage chemotactic protein 1; AP-1: activating protein 1; Fas: fatty acid synthase; oxLDL: oxidized low-density lipoprotein; HMGB-1: high mobility group box 1; NADPH: nicotinamide adenine dinucleotide phosphate; UCP2: uncoupling protein 2; NKT: natural killer T; TβR: TGF-β1 receptor; AST: alanine aminotransferase; ALT: aspartate aminotransferase; GGT: gamma-glutamyl transpeptidase; NMR: nuclear magnetic resonance; BITC: Benzyl isothiocyanate; AGEs: advanced glycation end products; RSV: resveratrol; RORα: retinoic acid-associated orphan receptor α.
Acknowledgements
This work was supported by the Innovation Strong School Project of Department of Education of Guangdong Province and Guangdong Pharmaceutical University, China; the Science and Technology Planning Project of Guangdong, China (No.201806040009); the Science and Technology Planning Project of Yunfu, Guangdong, China (No. 201702-9) and the Health Committee of Guangxi Zhuang Autonomous Region (no. Z20190744).
Competing Interests
No conflict of interest exits in the submission of this manuscript, and the manuscript has been approved by all authors for publication. I would like to declare on behalf of my co-authors that the work described is an original review that has not been published previously and is not under consideration for publication elsewhere, in whole or in part. All listed authors have approved the enclosed manuscript.
References
1. CD B, G T. NAFLD as a driver of chronic kidney disease. Journal of hepatology. 2020;72:785-801
2. Muthiah MD, Sanyal AJ. Burden of Disease due to Nonalcoholic Fatty Liver Disease. Gastroenterology Clinics of North America. 2020;49:1-23
3. Mantovani A, Zusi C. The dawn of a new era for nonalcoholic fatty liver disease? Hepatobiliary Surgery and Nutrition. 2019;8:629-31
4. Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC. et al. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology. 2017;152:1090-9 e1
5. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123-33
6. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018;69:896-904
7. Stein E, Cruz-Lemini M, Altamirano J, Ndugga N, Couper D, Abraldes JG. et al. Heavy daily alcohol intake at the population level predicts the weight of alcohol in cirrhosis burden worldwide. J Hepatol. 2016;65:998-1005
8. Gehrke N, Schattenberg JM. Metabolic Inflammation-A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease? Gastroenterology. 2020;158:1929-47.e6
9. Yoneda M, Imajo K, Nakajima A. Will the magnetic resonance imaging proton density fat fraction replace liver biopsy as the gold standard for detecting steatosis? Hepatobiliary Surgery and Nutrition. 2018;7:292-3
10. Yoneda M, Ogawa Y, Imajo K, Nakajima A. Nonalcoholic fatty liver disease with advanced fibrosis as a multi-systemic disease: proceed with caution. Hepatobiliary Surgery and Nutrition. 2018;8:170-2
11. Romero-Gomez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67:829-46
12. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P. et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149:389-97 e10
13. Petrucciani N, Gugenheim J. Molecular pathways between obesity, non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC). Hepatobiliary Surgery and Nutrition. 2019;8:395-7
14. Schulfer A, Santiago-Rodriguez TM, Ly M, Borin JM, Chopyk J, Blaser MJ. et al. Fecal Viral Community Responses to High-Fat Diet in Mice. mSphere. 2020;5:e00833-19
15. Day C P JOF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842-5
16. Baiceanu Andrei MP, Lagouge Marie. et al. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol. 2016;12:710-722
17. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68:280-95
18. Gomez PE, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547
19. CL S, F Z, P DB, L M, Y S, S DP. et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nature communications. 2016;7:10321
20. B M, R M, D O, M B, T D, P F. et al. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology (Baltimore, Md). 2019;70:67-83
21. Antoniades CG, Quaglia A, Taams LS, Mitry RR, Hussain M, Abeles R. et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. 2012;56:735-46
22. Kim S-J, Feng D, Guillot A, Dai S, Liu F, Hwang S. et al. Adipocyte Death Preferentially Induces Liver Injury and Inflammation Through the Activation of Chemokine (C-C Motif) Receptor 2-Positive Macrophages and Lipolysis. Hepatology. 2019;69:1965-82
23. Bijnen M, Josefs T, Cuijpers I, Maalsen CJ, José VDG, Vroomen M. et al. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut. 2017: gutjnl-2016-313654.
24. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM. et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. Journal of Hepatology. 2017;66:1037-46
25. S H, Y H, X X, W S, SJ K, J M. et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology (Baltimore, Md). 2019
26. Bouwens L BM, Zanger R D. et al. Quantitation, tissue distribution and proliferation kinetics of kupffer cells in normal rat liver. Hepatology. 1986 6(4)
27. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver International. 2006 26
28. DT R, JL R, BA M, T V, RA R, B E. Kupffer Cells Undergo Fundamental Changes during the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PloS one. 2016;11:e0159524
29. V B, C T. The innate immune response during liver inflammation and metabolic disease. Trends in immunology. 2013;34:446-52
30. Gordon S. Alternative activation of macrophages. Nature reviews Immunology. 2003;3:23-35
31. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage Polarization in Inflammatory Diseases. International Journal of Biological Sciences. 2014;10:520-9
32. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nature Reviews Immunology. 2017;17:306-21
33. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N. et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060-72
34. Smigiel KS, Parks WC. Macrophages, Wound Healing, and Fibrosis: Recent Insights. Current rheumatology reports. 2018;20:17
35. Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annual review of pharmacology and toxicology. 2011;51:267-88
36. Smith K. Liver disease: Kupffer cells regulate the progression of ALD and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:503
37. Wan J, Benkdane M, Teixeira-Clerc F, Deveaux V, Louvet A, Lafdil F. et al. 93 Interleukin-10 Released by M2 Kupffer Cells Promotes M1 Kupffer Cell Apoptosis: A Novel Protective Mechanism against Alcoholic Liver Disease. Journal of Hepatology. 2012;56:S40-S1
38. Wan J, Benkdane M, Alons E, Lotersztajn S, Pavoine C. M2 kupffer cells promote hepatocyte senescence: an IL-6-dependent protective mechanism against alcoholic liver disease. The American journal of pathology. 2014;184:1763-72
39. Maina V, Sutti S, Locatelli I, Vidali M, Mombello C, Bozzola C. et al. Bias in macrophage activation pattern influences non-alcoholic steatohepatitis (NASH) in mice. 2012; 122: 545.
40. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F. et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130-42
41. Chen P. Enteric dysbiosis, gut barrier and liver disease. Hepatobiliary Surgery and Nutrition. 2018;7:38-40
42. Bieghs V, Trautwein C. Innate immune signaling and gut-liver interactions in non-alcoholic fatty liver disease. Hepatobiliary Surgery and Nutrition. 2014;3:377-85
43. M U, M M, H Y. Gut microbiota and host metabolism in liver cirrhosis. World journal of gastroenterology. 2015;21:11597-608
44. Harte AL, Silva NFD, Creely SJ, Mcgee KC, Billyard T, Youssef-Elabd EM. et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. Journal of Inflammation. 2010;7(1):15
45. Whitfield C, Clarke BR. Lipopolysaccharides (Endotoxins). 2019.
46. Crane RJ, Jones KDJ, Berkley JA. Environmental Enteric Dysfunction: An Overview. Food & Nutrition Bulletin. 2015;36:S76-87
47. Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206-11
48. Wang HD, Lu DX, Qi RB. Therapeutic strategies targeting the LPS signaling and cytokines. Pathophysiology: the official journal of the International Society for Pathophysiology. 2009;16:291-6
49. Xu FL, You HB, Li XH, Chen XF, Liu ZJ, Gong JP. Glycine attenuates endotoxin-induced liver injury by downregulating TLR4 signaling in Kupffer cells. American journal of surgery. 2008;196:139-48
50. Lu Y C YWC, Ohashi P S LPS/TLR4 signal transduction pathway. Cytokine. 2008, 42(2):0-151.
51. Yao H, Hu C, Yin L, Tao X, Xu L, Qi Y. et al. Dioscin reduces lipopolysaccharide-induced inflammatory liver injury via regulating TLR4/MyD88 signal pathway. International immunopharmacology. 2016;36:132-41
52. Li P, He K, Li J, Liu Z, Gong J. The role of Kupffer cells in hepatic diseases. Mol Immunol. 2017;85:222-9
53. Wang YQ, Mao JB, Zhou MQ, Jin YW, Lou CH, Dong Y. et al. Polysaccharide from Phellinus Igniarius activates TLR4-mediated signaling pathways in macrophages and shows immune adjuvant activity in mice. International journal of biological macromolecules. 2019;123:157-66
54. Leroux A, Godié V, Cailleux F, Naveau S, Prévot S, Makhzami S. et al. 1245 TOXIC LIPIDS STORED BY KUPFFER CELLS CORRELATES WITH THEIR PRO-INFLAMMATORY PHENOTYPE AT AN EARLY STAGE OF STEATOHEPATITIS. Journal of Hepatology. 2012;56:S493
55. De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L. et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738-49
56. Nobuhiko K, Wong MT, Stowe IB, Sree Ranjani R, Gonzalez LC, Sachiko AT. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246-9
57. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187-92
58. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666-71
59. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5
60. JP A, D C, TS S, N J-S, VK V, D S. et al. Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. Journal of hepatology. 2020
61. Guo H, Xie M, Zhou C, Zheng M. The relevance of pyroptosis in the pathogenesis of liver diseases. Life sciences. 2019;223:69-73
62. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X. et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2018;68:773-82
63. JI B, JM B. Pyroptosis: An inflammatory link between NAFLD and NASH with potential therapeutic implications. Journal of hepatology. 2018;68:643-5
64. Ramadori P, Kroy D, Streetz KL. Immunoregulation by lipids during the development of non-alcoholic steatohepatitis. Hepatobiliary Surgery and Nutrition. 2015;4:11-23
65. Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. 2009;51:212-23
66. Song M, Schuschke DA, Zhou Z, Zhong W, Zhang J, Zhang X. et al. Kupffer cell depletion protects against the steatosis, but not the liver damage, induced by marginal-copper, high-fructose diet in male rats. Am J Physiol Gastrointest Liver Physiol. 2015;308:G934-G45
67. Hu M, Phan F, Bourron O, Ferre P, Foufelle F. Steatosis and NASH in type 2 diabetes. Biochimie. 2017;143:37-41
68. Asanuma T, Ono MK, Hirose A, Hayashi Y, Saibara T, Inanami O. et al. Super paramagnetic iron oxide MRI shows defective Kupffer cell uptake function in non-alcoholic fatty liver disease. Gut. 2010;59:258-66
69. Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through Toll-like receptors. Molecules & Cells. 2006;21:174-85
70. Han R, Zhang F, Wan C, Liu L, Zhong Q, Ding W. Effect of perfluorooctane sulphonate-induced Kupffer cell activation on hepatocyte proliferation through the NF-kappaB/TNF-alpha/IL-6-dependent pathway. Chemosphere. 2018;200:283-94
71. Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360-9
72. Nolan CJ, Larter CZ. Lipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it? Journal of gastroenterology and hepatology. 2009;24:703-6
73. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Progress in lipid research. 2013;52:175-91
74. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. Journal of lipid research. 2013;54:1326-34
75. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A. et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185-98
76. Ioannou GN. The Role of Cholesterol in the Pathogenesis of NASH. Trends in endocrinology and metabolism: TEM. 2016;27:84-95
77. Musso G, Gambino R, Pacini G, De Michieli F, Cassader M. Prolonged saturated fat-induced, glucose-dependent insulinotropic polypeptide elevation is associated with adipokine imbalance and liver injury in nonalcoholic steatohepatitis: dysregulated enteroadipocyte axis as a novel feature of fatty liver. The American journal of clinical nutrition. 2009;89:558-67
78. Bieghs V, Wouters K, van Gorp PJ, Gijbels MJ, de Winther MP, Binder CJ. et al. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. 2010;138:2477-86 86 e1-3
79. Bieghs V, van Gorp PJ, Walenbergh SM, Gijbels MJ, Verheyen F, Buurman WA. et al. Specific immunization strategies against oxidized low-density lipoprotein: a novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. 2012;56:894-903
80. Bieghs V, Verheyen F, Gorp PJV, Hendrikx T, Wouters K, Lütjohann D. et al. Internalization of Modified Lipids by CD36 and SR-A Leads to Hepatic Inflammation and Lysosomal Cholesterol Storage in Kupffer Cells. PloS one. 2012 7
81. Magee N, Zou A, Zhang Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed Res Int. 2016;2016:5170402
82. W L, SS B, RD B, L Z. Antioxidant Mechanisms in Nonalcoholic Fatty Liver Disease. Current drug targets. 2015;16:1301-14
83. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F. et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57:141-9
84. Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clinica Chimica Acta. 2011;412:1297-305
85. Clària J. New insights into the role of macrophages in adipose tissue inflammation and fatty liver disease: modulation by endogenous omega-3 fatty acid-derived lipid mediators. Frontiers in Immunology. 2011 2
86. Baffy G. Uncoupling protein-2 and non-alcoholic fatty liver disease. Frontiers in Bioscience A Journal & Virtual Library. 2005;10:2082
87. Péter FP, Zoltán D, Anthony S, Edmond S, Berthiaume EP, Resnick MB. et al. Lack of UCP2 reduces Fas-mediated liver injury in ob/ob mice and reveals importance of cell-specific UCP2 expression. Hepatology. 2010;44:592-601
88. Evans ZP, Ellett JD, Schmidt MG, Schnellmann RG, Chavin KD. Mitochondrial uncoupling protein-2 mediates steatotic liver injury following ischemia/reperfusion. Journal of Biological Chemistry. 2008;283:8573
89. Ryu JW, Hong KH, Maeng JH, Kim JB, Ko J, Park JY. et al. Overexpression of uncoupling protein 2 in THP1 monocytes inhibits beta2 integrin-mediated firm adhesion and transendothelial migration. Arteriosclerosis Thrombosis & Vascular Biology. 2004;24:864
90. Yushi B, Hiroki O, Xu B, Medvedev AV, Mary M, J Brice W. et al. Persistent nuclear factor-kappa B activation in Ucp2-/- mice leads to enhanced nitric oxide and inflammatory cytokine production. Journal of Biological Chemistry. 2005;280:19062-9
91. Kosei M, Qizhi Z, Tatehiko W, Michitaka O, Sonshin T, Takashi A. et al. Reactive oxygen species are essential mediators in antigen presentation by Kupffer cells. Immunology & Cell Biology. 2005;83:336
92. Turrens JF. Mitochondrial formation of reactive oxygen species. Journal of Physiology. 2004;552:335-44
93. Luckey SW, Petersen DR. Metabolism of 4-hydroxynonenal by rat Kupffer cells. Archives of Biochemistry & Biophysics. 2001;389:77-83
94. Parola M RG. Oxidative stress-related molecules and liver fibrosis. Journal of Hepatology. 2001;35(2):297-306
95. Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Seminars in Liver Disease. 2008;28:370-9
96. Kiagiadaki F, Kampa M, Voumvouraki A, Castanas E, Kouroumalis E, Notas G. Activin-A causes Hepatic stellate cell activation via the induction of TNFalpha and TGFbeta in Kupffer cells. Biochimica et biophysica acta Molecular basis of disease. 2018;1864:891-9
97. Schon H-T, Weiskirchen R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surgery and Nutrition. 2014;3:386-406
98. Loomba R, Cortez-Pinto H. Exercise and improvement of NAFLD: Practical recommendations. J Hepatol. 2015;63:10-2
99. Trovato FM, Castrogiovanni P, Malatino L, Musumeci G. Nonalcoholic fatty liver disease (NAFLD) prevention: role of Mediterranean diet and physical activity. Hepatobiliary Surgery and Nutrition. 2018;8:167-9
100. Patel NS, Doycheva I, Peterson MR, Hooker J, Kisselva T, Schnabl B. et al. Effect of weight loss on magnetic resonance imaging estimation of liver fat and volume in patients with nonalcoholic steatohepatitis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2015;13:561-8 e1
101. Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL. et al. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes care. 2010;33:2156-63
102. Eckard C CR, Lockwood J. et al. Prospective histopathologic evaluation of lifestyle modification in nonalcoholic fatty liver disease: a randomized trial. Therapeutic Advances in Gastroenterology. 2013;6(4):249-259
103. Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology. 2009;49:80-6
104. CA R, P A, N vR, A T, M A, M W. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. Journal of hepatology. 2007;47:571-9
105. Zeng TS, Liu FM, Jing Z, Pan SX, Chen LL. Depletion of Kupffer cells attenuates systemic insulin resistance, inflammation and improves liver autophagy in high-fat diet fed mice. Endocrine Journal. 2015;62:615
106. Chen H-W, Yen C-C, Kuo L-L, Lo C-W, Huang C-S, Chen C-C. et al. Benzyl isothiocyanate ameliorates high-fat/cholesterol/cholic acid diet-induced nonalcoholic steatohepatitis through inhibiting cholesterol crystal-activated NLRP3 inflammasome in Kupffer cells. Toxicology and Applied Pharmacology. 2020: 114941.
107. Fernando HKDH, Leung C, Angus P, Herath C. SAT-343-Alagebrium ameliorates NAFLD progression in mice fed a high advanced glycation end products diet via direct inhibition of Kupffer cells and indirect inhibition of hepatic stellate cell activation. Journal of Hepatology. 2019;70:e789-e90
108. Kessoku T, Imajo K, Honda Y, Kato T, Nakajima A. Resveratrol ameliorates fibrosis and inflammation in a mouse model of nonalcoholic steatohepatitis. Scientific reports. 2016;6:22251
109. Ni Y, Nagashimada M, Zhan L, Nagata N, Kobori M, Sugiura M. et al. Prevention and reversal of lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice by an antioxidant carotenoid, beta-cryptoxanthin. Endocrinology. 2015;156:987-99
110. Ni Y, Zhuge F, Nagashimada M, Ota T. Novel Action of Carotenoids on Non-Alcoholic Fatty Liver Disease: Macrophage Polarization and Liver Homeostasis. Nutrients. 2016 8
111. Han Y-H, Kim H-J, Na H, Nam M-W, Kim J-Y, Kim J-S. et al. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Reports. 2017;20:124-35
112. Li Z, Feng P-P, Zhao Z-B, Zhu W, Gong J-P, Du H-M. Liraglutide protects against inflammatory stress in non-alcoholic fatty liver by modulating Kupffer cells M2 polarization via cAMP-PKA-STAT3 signaling pathway. Biochemical and Biophysical Research Communications. 2019;510:20-6
113. Yang Y, Lu Y, Han F, Chang Y, Li X, Han Z. et al. Saxagliptin regulates M1/M2 macrophage polarization via CaMKKbeta/AMPK pathway to attenuate NAFLD. Biochem Biophys Res Commun. 2018;503:1618-24
114. Ji XY, Tan BK, Zhu YZ. Salvia miltiorrhiza and ischemic diseases. Acta pharmacologica Sinica. 2000;21:1089-94
115. Su CY, Ming QL, Rahman K, Han T, Qin LP. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chinese journal of natural medicines. 2015;13:163-82
116. Nonaka GI, Miwa N, Nishioka I. Stilbene glycoside gallates and proanthocyanidins from Polygonum multiflorum. Phytochemistry. 21: 429-32.
117. Liu Y, Wang W, Sun M, Ma B, Pang L, Du Y. et al. Polygonum multiflorum-Induced Liver Injury: Clinical Characteristics, Risk Factors, Material Basis, Action Mechanism and Current Challenges. Frontiers in pharmacology. 2019;10:1467
118. DL W, F K, HB E-S. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2012;10:1342-59.e2
Author contact
![]() Corresponding author: Professor Zhengquan Su. Guangdong Engineering Research Center of Natural Products and New Drugs, Guangdong Provincial University Engineering Technology Research Center of Natural Products and Drugs, Guangdong Pharmaceutical University, Guangzhou 510006, China; Jiao Guo. Guangdong Metabolic Diseases Research Centre of Integrated Chinese and Western Medicine, Guangdong TCM Key Laboratory for Metabolic Diseases, Key Laboratory of Modulating Liver to Treat Hyperlipemia SATCM, Level 3 Laboratory of Lipid Metabolism SATCM, Institute of Chinese Medicinal Sciences, Guangdong Pharmaceutical University, Guangzhou 510006, China. E-mails: suzhqedu.cn (Z.-Q.S.); gyguoyzcom (J.G.); Tel.: +86-20-3935-2067; Fax: +86-20-3935-2067.
Corresponding author: Professor Zhengquan Su. Guangdong Engineering Research Center of Natural Products and New Drugs, Guangdong Provincial University Engineering Technology Research Center of Natural Products and Drugs, Guangdong Pharmaceutical University, Guangzhou 510006, China; Jiao Guo. Guangdong Metabolic Diseases Research Centre of Integrated Chinese and Western Medicine, Guangdong TCM Key Laboratory for Metabolic Diseases, Key Laboratory of Modulating Liver to Treat Hyperlipemia SATCM, Level 3 Laboratory of Lipid Metabolism SATCM, Institute of Chinese Medicinal Sciences, Guangdong Pharmaceutical University, Guangzhou 510006, China. E-mails: suzhqedu.cn (Z.-Q.S.); gyguoyzcom (J.G.); Tel.: +86-20-3935-2067; Fax: +86-20-3935-2067.