Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Coronavirus heterogeneity
SARS-CoV-2 entry points
The immune response in COVID-19
Interferons and SARS-CoV-2
Antibody and plasma therapy
Coagulation abnormalities:...
Outlook
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(14):2479-2489. doi:10.7150/ijbs.48400 This issue Cite
Review
Immune Response and COVID-19: A mirror image of Sepsis
Eduardo López-Collazo1,2,3 ![]() , José Avendaño-Ortiz1,2, Alejandro Martín-Quirós4, Luis A. Aguirre1,2
, José Avendaño-Ortiz1,2, Alejandro Martín-Quirós4, Luis A. Aguirre1,2 ![]()
1. The Innate Immune Response Group, IdiPAZ, La Paz University Hospital, Paseo de la Castellana 261, Madrid 28046, Spain.
2. Tumor Immunology Laboratory, IdiPAZ, La Paz University Hospital, Paseo de la Castellana 261, Madrid 28046, Spain.
3. CIBER of Respiratory Diseases (CIBERES), Madrid, Spain.
4. Emergency Department and Emergent Pathology Research Group, IdiPAZ La Paz University Hospital, Paseo de la Castellana 261, Madrid 28046, Spain.
Received 2020-5-19; Accepted 2020-6-22; Published 2020-7-9
Abstract

The emergence of SARS-CoV-2 virus and its associated disease COVID-19 have triggered significant threats to public health, in addition to political and social changes. An important number of studies have reported the onset of symptoms compatible with pneumonia accompanied by coagulopathy and lymphocytopenia during COVID-19. Increased cytokine levels, the emergence of acute phase reactants, platelet activation and immune checkpoint expression are some of the biomarkers postulated in this context. As previously observed in prolonged sepsis, T-cell exhaustion due to SARS-CoV-2 and even their reduction in numbers due to apoptosis hinder the response to the infection. In this review, we synthesized the immune changes observed during COVID-19, the role of immune molecules as severity markers for patient stratification and their associated therapeutic options.
Keywords: COVID-19, Sepsis, Immune response, Immune-checkpoints, T-cell exhaustion
Introduction
The emergence of the new coronavirus, known as SARS-CoV-2, poses a substantial threat to public health and a major impact to worldwide economies and societies. The infection caused by this virus leads to a primary viral pneumonia identified as COVID-19, which resembles the Severe Acute Respiratory Syndrome (SARS) [1]. Despite similarities with the seasonal influenza, severe disease behaves quite differently. The data indicate that 80% of COVID-19 infections are mild or asymptomatic, while approximately 15% are severe and require oxygen supplementation, and 5-10% are critical, characterized by SARS with acute respiratory distress syndrome (ARDS) and requiring mechanical ventilation in an intensive care unit [1-4]. These subgroups of severe and critically infected patients are larger than those observed for influenza infection [5]. Due to the rapid spread of COVID-19, affecting almost 200 countries, the World Health Organization (WHO) announced on March 11th the elevation of COVID-19 from an epidemic to a pandemic, raising this infection to a global health priority [6, 7]. Accordingly and to avoid the spread of the disease, several governments have implemented extraordinary measures, such as declaring states of emergency and instituting quarantines [8].
According to data obtained from numerous cohorts, the main causes of death by COVID-19 include respiratory failure and the onset of sepsis. In fact, sepsis has been observed in nearly all deceased patients in many of the reported cohorts [9-12]. However, the sepsis figures were not always related to the bacterial findings in the microbiological work-up, suggesting SARS-CoV-2 as the etiological agent causative of this systemic condition [13]. Additionally, comorbidities such as diabetes, hypertension and coronary disease and factors such as age, procalcitonin and interleukin (IL)-6 levels, leukocytosis and lymphocytopenia have been included as associated with mortality in patients with COVID-19 [9, 14]. Abnormalities in these factors in patients with unfavorable progression, combined with the high incidence of sepsis, strongly suggest the involvement of significant changes in the host's immune response. Additionally, evidence suggests the potential role of immune receptors such as Toll-like receptors (TLRs) and dipeptidyl peptidase 4 in the hijacking and virulence of the infection [15-17].
Current treatments employed against SARS-CoV-2 include a number of antiretroviral therapies recycled from other infections such as ribavirin, lopinavir/ritonavir, oseltamivir, hydroxychloroquine and remdesivir [18, 19], inflammation modulators and anticoagulants [20]. Their usefulness, however, has led to major public debate. According to a randomized trial on hydroxychloroquine as post-exposure prophylaxis for COVID-19, the compound did not prevent the disease when administered within 4 days after exposure to the virus [21]. Although remdesivir has shown significant activity both in vitro and in a primate model against SARS-CoV-2 [22-24], the drug has only provided moderate clinical benefit for the treatment of patients with COVID-19 [25, 26]. Thus, the lack of a successful treatment and the absence of vaccines has prompted the scientific community to explore other avenues. Host-directed therapies such as immunomodulators could be an interesting alternative for treating patients with COVID-19.
In this review, we summarize the immune changes observed in SARS-CoV-2 infection, with an emphasis on the similarities with sepsis, and the role of immune compounds as severity markers and therapeutic targets.
Coronavirus heterogeneity
SARS-CoV-2 is a β-coronavirus included in the sarbecovirus subgenus, orthocoronavirinae subfamily, which is broadly distributed in humans and other mammals [7]. The first human coronaviruses (HCoV-229E and HCoV-OC43) were described in the 1960s, while HCoV-NL63 and HCoV-HKU1 (which cause mild infections in immunocompetent individuals) were described in the early 2000s [27]. However, SARS-CoV1 (SARS1) and MERS-CoV (MERS), the other two members of the family that have affected humans in this century, are highly transmissible and pathogenic. In fact, the two pathogens have caused more than 700 deaths in 27 countries during their respective outbreaks in 2002 and 2012 [28].
SARS-CoV-2 shows close similarities with SARS1 and MERS in terms of pathogenicity and has largely overtaken the death toll of the latter two. As with SARS1 and MERS [29], SARS-CoV-2 presents a serious risk of infection for healthcare workers on the frontlines [30], which is likely due to nosocomial transmission [31] given the substantial virus shedding that only occurs after the onset of symptoms (i.e., when patients are already hospitalized for days) and the fact that the viruses can remain on hospital surfaces for several days after patients no longer test positive [32-34].
Since they belong to the order Nidovirales, the viruses' molecular structures show similarities, although there are slight differences, which will be discussed below. Basically, SARS-CoV-2 is an enveloped, non-segmented, positive-sense RNA virus that has four main structural proteins: the spike (S) glycoprotein, the small envelope (E) glycoprotein, the membrane (M) glycoprotein, and the nucleocapsid (N) protein. In addition, SARS-CoV-2 has several accessory proteins that are relevant to the host immune process, as will be discussed below.
SARS-CoV-2 entry points
According to reported data, angiotensin-converting enzyme 2 (ACE2) has been identified as a functional receptor for SARS1 [35]. ACE2 is highly expressed in several tissues including myocardial cells, kidney proximal tubule cells, bladder urothelial cells, testis cells and lung cells [36-38]. In lung tissue, ACE2 is mainly located on the apical side of the epithelial cells in the alveolar space [39, 40]. SARS1 primarily infects ciliated bronchial epithelial cells, unlike MERS, which primarily infects unciliated bronchial epithelial cells by binding to dipeptidyl peptidase 4 (CD26) receptors [27]. This difference is due to each virus' differing structure for the receptor-binding domains (RBD) of S proteins. While the SARS1 RBD contains a receptor-binding motif (RBM) rich in loops, MERS-RBD has an RBM that contains a four stranded β-sheet [27].
Structural and functional analyses have shown that SARS-CoV-2 also binds to ACE2 [41-43]. Given that the SARS-CoV-2 protein S interacts with ACE2 expressed on type II pneumocytes in the lungs [44], ACE2 has been postulated as the entry point of this virus into pneumocytes. Dipeptidyl peptidase 4 [15] and CD147 [45] have also been reported as entry points for SARS-CoV-2.
Lung-resident macrophages are located on the apical side of the epithelium and, together with dendritic cells, act as innate immune cells to attack viruses until adaptive immunity is involved. It is still an open question as to how SARS-CoV-2 interacts with and eventually enters immune cells. Both ACE2 and dipeptidyl peptidase 4 are expressed in these cells but to a limited extent [46-50]. However, phagocytosis of virus-infected apoptotic epithelial cells by resident macrophages and the presence of other proteins binding to SARS-CoV-2 could be pathways of interaction between the cells and SARS-CoV-2.
Successful virus-host cell fusion requires other molecules. Several research groups have attempted to inhibit host proteases, such as cathepsins and TMPRSS2, a protease that processes S protein and favors virus entry into cells [51]. Other authors have demonstrated that camostat mesylate, an inhibitor of TMPRSS2 protease activity, partially inhibited the entry of SARS-CoV-2 into primary lung epithelial cells [44].
The immune response in COVID-19
In terms of the circulatory system, several reports have demonstrated a direct association between proinflammatory cytokine levels in plasma and lung injury during infection by coronaviruses such as SARS1 [52] and MERS [53]. Changes in the inflammatory response have explained the high impact of MERS on infected patients with diabetes [9]. SARS-CoV-2 has also been shown to severely affect cytokine levels [9, 14].
Innate immune system (IIS) cells, mainly monocytes and macrophages, are the principal players in orchestrating the host's inflammatory response by activating the nuclear factor kappa B (NF-κB) and interferon regulatory factor (IRF) pathways [54, 55]. As previously reported for sepsis, patients with severe COVID-19 infections show excessive inflammation and cytokine storms, including overexpression of interleukin (IL)-1β, IL-2, IL-6, and tumor necrosis factor alpha, in the early phase of the disease (Table 1) [56]. These hallmarks of sepsis have been widely explained by an exacerbation of macrophage activation [57].
Pathological hallmarks of COVID-19 and associated biomarkers
| Immune System | COVID-19 Hallmark | Phenotype associated to severe patients |
|---|---|---|
| Innate | Cytokine storm and over inflammation | Neutrophilia |
| ↑ Acute phase reactants: Reactive C protein, fibrinogen, procalcitonin and haptoglobin | ||
| ↑ Basal IL-6 | ||
| Reduced antiviral cytotoxicity | ↓ NKs Frequency | |
| Coagulopathy | ↑ D-Dimer | |
| ↑ Platelets | ||
| Adaptative | T cell exhaustion and reduced humoral response | Lymphocytopenia |
| ↓ T Lymphocyte counts | ||
| ↓ B Lymphocyte counts |
According to several studies, the inflammatory phase for patients with severe COVID-19 is limited to the initial period of the disease [7]. The subsequent chronic basal inflammation, which lasts several days, leads the immune system towards a refractory state, which is also observed in protracted sepsis. A comparative study of patients with severe and mild COVID-19 demonstrated that all cytokines, except IL-6 and IL-10, reached their peak serum levels 3-6 days after disease onset. IL-6 levels began to drop approximately 16 days later, and IL-10 levels were at their lowest 13 days after disease onset. Interestingly, the cytokine levels reached similar points for all patients with severe and mild disease 16 days after disease onset [56]. This phenomenon mirrors the most advanced phases of sepsis, when macrophages develop a refractory state characterized by strong inhibition of the NF-κB and IRF pathways in response to pathogens [54, 55, 57]. However, little is known about the role of the TLR family in this context. Authors have postulated that SARS1 infection regulates immune-related genes in myeloid cells by TLRs [58], which might be important to the pathogenesis of SARS. Other authors have demonstrated that mice deficient in the TLR3/TLR4 TIR-domain-containing adapter-inducing interferon-β (TRIF) were highly susceptible to SARS1 infection [59]. Both SARS1 and SARS-CoV-2 require acidification of endosomes and lysosomes to infect cells [24, 60]. A number of authors have suggested that the ssRNA of SARS-CoV-2 could bind TLR3/7/8 resulting in the induction of type I interferon (IFN-I) [61]. Therefore, the overactivation of inflammatory signaling points to the important role of TLRs in SARS-CoV-2.
Various clinical trials have included IL-6 and IL-6R-blocking antibodies to prevent this anaphylactic toxicity observed in patients with COVID-19 [5, 56]. In this context, however, anticytokine-based therapies might control only the cytokine storm without deleterious effects on virus replication. In addition, colony stimulation factors (CSFs) show significantly aberrant overexpression in patients with COVID-19. High granulocyte-macrophage (GM)-CSF levels have been detected in circulating lymphocyte populations, excluding natural killers (NKs) and B cells, from patients with COVID-19 admitted to intensive care units [62]. Similarly, a number of studies on sepsis have shown that GM-CSF deficiency protects mice in models of lethal endotoxemia [63]. Thus, the potential effect of GM-CSF-blocking antibodies as a treatment for SARS-CoV-2 is being evaluated by research groups and pharmaceutical companies (NCT04351152 and NCT04341116) [1, 64, 65]. Other current strategies undergoing clinical investigation for reducing macrophage activation include the blockade of certain cytokines, inhibition of CCR5-mediated migration and CD14 blockade by monoclonal antibodies (NCT04348448, NCT04324021, NCT04345445, NCT04347239 and NCT04346277) [66, 67].
An increasing number of reports have indicated that not only the IIS but also the adaptive system becomes deregulated during COVID-19 infection. The approximately 83% of patients who have shown lymphocytopenia at admission illustrates this point [10, 56]. Although macrophages appear to play a major role in the early phase of the pathophysiology of SARS-CoV-2, the adaptive immune system likely emerges as a crucial factor in the late phase. These observations however apply only to those patients who require hospitalization and clinical care. Approximately 80% of individuals infected with SARS-CoV-2 are asymptomatic or experience mild symptoms [1-4].
A number of analyzed cohorts have confirmed that the severe phase appears in these patients approximately 8-11 days after the onset of the disease, when proinflammatory cytokines reach their peak expression, probably due to an exacerbated innate immune response [56]. However, the total number of lymphocytes is significantly reduced in those patients with a poor prognosis [56]. A multicenter retrospective study showed that the lymphocyte count was an independent high-risk factor associated with COVID-19 progression [68]. Other researchers have found an inverse correlation between lymphocyte counts and time to symptom reappearance in a cohort of patients with COVID-19 discharged from hospital [69].
Similarly, an obvious depletion of lymphocytes during sepsis has been demonstrated, which compromises the adaptive response in the second phase of the disease [70]. Several researchers have studied this phenomenon, indicating a strong implication of immune checkpoints (ICs) and their ligands, such as the programmed death-1 (PD-1) and PD-L1 axis [71]. Furthermore, PD-1/PD-L1 interaction during sepsis induces not only apoptosis but also lymphocyte “exhaustion”, an effect that can be reversed by blocking monoclonal antibodies against either PD-1 or PD-L1 [72, 73]. SARS-CoV-2 has observed to induce apoptosis of peripheral blood lymphocytes via P53 activation [74], and PD-1 [75] has been found to be upregulated in the late phase of COVID-19 infection. Interestingly, PD-1 overexpression has also been reported in other infections by retroviruses such as the lymphocytic choriomeningitis virus and human immunodeficiency virus. Moreover, there are a number similarities between HIV and SARS-CoV-2 in terms of origin, aggressiveness to human hosts but not animal hosts, etc., which might help in finding new ways to combat SARS-CoV-2 [76]. Other ICs, such as PSGL-1, play an important role in the pathophysiology of chronic viral infections [77].
The collective data available point to an exaggerated innate response followed by an inappropriate switch to the adaptive response, which leads to immune system exhaustion during SARS-CoV-2 infection (Fig. 1). Likewise, the potential role of ICs and their ligands in lymphocytopenia observed in patients with severe COVID-19 should be a focus of research on SARS-CoV-2 infection. There is a crucial need for thorough studies on the development of clinical tools for personalized medicine for this infection. IC levels and IC-ligand expression would not only help physicians stratify patients at admission but also serve as pharmacological targets for those patients with a poor prognosis. Due to the impact of IC inhibitors in cancer therapies, several antibodies against ICs have been approved by the US Food and Drug Administration for use in certain malignancies [78, 79]. However, only camrelizumab (a PD-1 monoclonal antibody) and thymosin have so far been included in a clinical trial (NCT04268537) for COVID-19 treatment [80, 81].
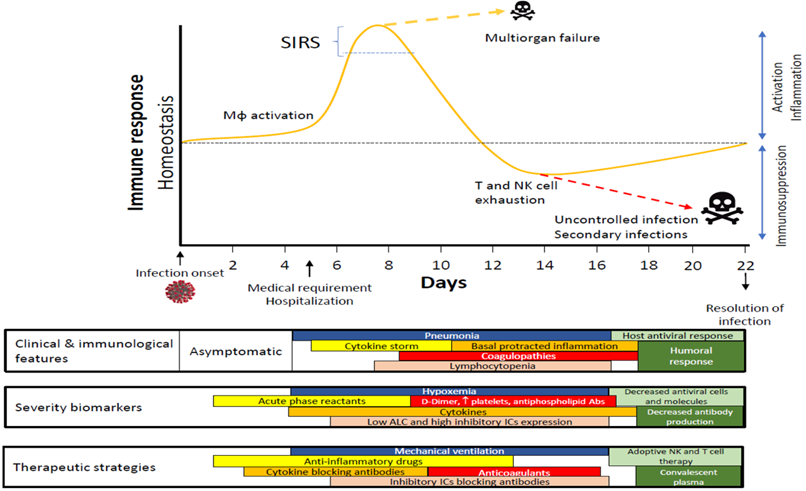
Time course proposed for immune response in COVID-19. Abbreviations: Abs, antibodies; ALC, absolute lymphocyte counts; Mφ, Macrophage/Monocyte; SIRS, systemic inflammatory response syndrome.
Interferons and SARS-CoV-2
Previous data from animal models of SARS1 and MERS infection have shown a significant delay in IFN-I production by macrophages, which leads to lethal pneumonia in mice [82, 83]. SARS1 and MERS encode at least 8 proteins that interact with the signaling cascades downstream of pattern recognition receptors, some of which suppress IFN signaling [28, 76]. Several MERS proteins can inhibit NF-κB and IRF pathways, promoting an evasion of the innate immune response [84]. Nevertheless, other authors have published conflicting results. While IFNβ-1b was shown to reduce disease severity and mean viral loads in necropsied lungs and extrapulmonary tissue in MERS [85], IFNα-2b neither decreased mortality nor accelerated viral clearance in a retrospective observational clinical study [86]. IFNγ treatment, however, showed a protective role in lethal respiratory disease in mice infected by SARS1 [87]. Other authors have demonstrated that the beneficial or detrimental role of IFN-I depends on the timing of the SARS1 infection, indicating that IFN-I and inflammatory macrophages (IMMs) promote lethal SARS-CoV1 infection and identifying IFN-I and IMMs as potential therapeutic targets in patients infected with pathogenic coronavirus [82]. Several combinations of antiretrovirals and human recombinant IFNs (such as IFNβ-1b and IFNα-2b) are currently employed for managing COVID-19 [88, 89]. Further research on IFNs for managing COVID-19 is therefore warranted.
The IFN-I response also has a key role in NK-cell activation and, subsequently, on triggering their antitumoral abilities. NK cell levels have been found to be reduced and to present an anergic status in patients with COVID-19 [90]. Chimeric antigen receptor-engineered NK cells are also being tested for treating COVID-19 (NCT04344548 and NCT04324996) [91, 92].
Antibody and plasma therapy
Similar to common acute viral infections, the antibody profile against SARS1 has a typical pattern of immunoglobulin M (IgM) and IgG production. The kinetics reported for SARS1-specific IgM antibodies indicate that IgMs disappear after 12 weeks of disease onset. However, IgGs can last a long time and play a protective role [93]. Little is known about this scenario for SARS-CoV-2. There have been several convalescent patients who donated plasma against SARS-CoV-2, as was the case for both SARS1 [94] and MERS [95] at the start of their respective outbreaks. A study conducted with 10 adult patients with severe COVID-19 showed that one 200-mL dose of plasma was well tolerated by the patients, leading to the disappearance of viremia in 7 days, while the clinical symptoms and paraclinical criteria rapidly improved within 3 days [96]. Cross-reactivity between antibodies generated against SARS1 and SARS-CoV-2 has also been reported [42, 44, 97]. Although this cross-reactivity might jeopardize the detection by serological tests of patients with COVID-19, it could favor the resolution of the infection in certain individuals.
Generating recombinant human monoclonal antibodies could be another method for neutralizing SARS-CoV-2. CR3022, a SARS coronavirus-specific human monoclonal antibody, has been reported capable of binding to SARS-CoV-2-RBD [98]. Monoclonal antibodies that neutralize SARS1, such as CR3014, could be a treatment option for SARS-CoV-2 infection [99]. S309, an antibody previously identified in a patient who contracted SARS1 in 2003, appears to be promising candidate for neutralizing SARS-CoV-2 [100].
Coagulation abnormalities: another effect of dysregulation of the innate immune response with a mirror image in sepsis
Since the 1993 report on tissue factor (TF) synthesis by activated macrophages [101], several studies have demonstrated the association between inflammation and coagulation [102]. During inflammation, macrophages contribute to disseminated intravascular coagulation (DIC) in numerous clinical contexts. Aberrant in vivo TF expression plays a pivotal role in the activation of blood coagulation in the setting of sepsis and endotoxemia [103], and the high incidence of sepsis-associated DIC is well known. TF expression provokes widespread thrombosis in the microcirculation of various organs, contributing to multiple organ dysfunction, a major determinant of mortality in sepsis [104-106]. Peripheral blood mononuclear cells have shown obvious overexpression of coagulation-related genes in COVID-19 infection. In fact, a DIC score ≥5 points has been associated with mortality in patients with COVID-19, and more than 70% patients who die from the infection meet the International Society of Thrombosis and Hemostasis criteria for a DIC [107]. Furthermore, anticoagulant treatment has been associated with hospital survival in hospitalized patients with COVID-19 [108].
Proinflammatory cytokines induce the production of C-reactive protein (CRP) and haptoglobin by hepatocytes. A number of reports have indicated high CRP and haptoglobin levels in patients with COVID-19, with higher levels in those patients with a severe condition [10, 109-111]. The presence of these acute-phase proteins has been associated with D-dimer levels, a fibrin cleavage product that serves as a biomarker of pulmonary embolism [112]. A study indicated that patients with COVID-19 who died showed higher D-dimer levels and lower platelet counts than those who survived [9]. Longer prothrombin and activated partial thromboplastin times have also been associated with poorer outcomes in COVID-19 infection [107]. To prevent these abnormalities, anticoagulant heparin, tinzaparin and enoxaparin are being tested in patients with COVID-19 (NCT04344756 and NCT04345848).
Outlook
COVID-19 has emerged as a complex disease that shares clinical characteristics with sepsis. Figure 1 summarizes the progression of infected patients who usually require hospitalization from days 5 to 6 since the start of their infection. Depending on their immune response, patients might experience an exacerbated cytokine production that compromises their vital functions and leads to a state of exhaustion that hinders the activation of the adaptive response.
A number of published studies have reported the onset of symptoms compatible with pneumonia accompanied by coagulopathy and lymphocytopenia during COVID-19 infection. However, there are still no solid markers to predict these patients' progression. Cytokine elevation rates, the presence of acute phase reactants such as D-dimer, platelet activation and IC expression are some of the biomarkers proposed in this context. Given the similarity of COVID-19 infection to sepsis, it is possible that testing for the early expression of ICs and their ligands on innate immune cells such as monocytes and macrophages could serve as a tool for classifying patients on admission, thereby opening up new avenues for treatment. As has been observed in prolonged sepsis, T-cell exhaustion due to SARS-CoV-2 and even reduced T-cell counts due to apoptosis hinder the host's response to infection. In such a scenario, new infections might emerge, increasing the risk of mortality.
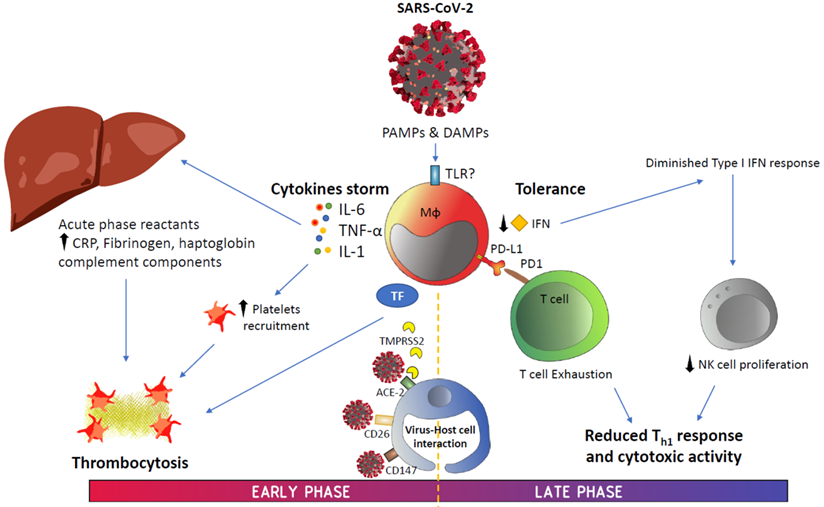
According to data generated on COVID-19 since its emergence, this disease has shown two potentially overlapping phases (Figure 2). The first phase is strongly characterized by a disproportionate IIS reaction that causes a cytokine storm and, subsequently, generates significant damage to the body. In addition, inflammation induces the production of a significant number of factors related to the coagulation cascade, resulting in the onset of thrombi and associated DIC, a condition that highly resembles that observed during sepsis.
Graphical summary. Abbreviations: ACE-2, angiotensin-converting enzyme 2; CRP, C-reactive protein; DAMPS, danger-associated molecular patterns; IFN, interferon; IL, interleukin; PAMPS, pathogen-associated molecular patterns; PD-1, programmed cell death protein-1; PD-L1, programmed death ligand-1; TF, tissue factor; TMPRSS2, transmembrane serine protease 2; TNF-α, tumor necrosis factor alpha.
The urgency and severity of COVID-related events in various countries is facilitating a significant number of clinical trials (Table 2). To date, we have only partial results, and exact answers have not been found in the clinics. In fact, there have been cases of conflicting results from clinical trials and on the use of drugs that have had deleterious effects [86, 113-117].
Proposed host-directed therapies against the most relevant pathological hallmarks of COVID-19
| Pathological phenomenon | Treatment | Mechanism of action | Clinical trial | Toxicity and pharmacokinetic studies in humans |
|---|---|---|---|---|
| Cytokine storm and Mφ | Corticosteroids | Anti-inflammatory steroid hormones | Dexamethasone: NCT04327401 and NCT04325061. Methylprednisolone: NCT04329650 and NCT04343729. Prednisone: NCT04344288. | Yes. Approved for several pathologies |
| Sirolimus | mTOR inhibitor, immunosuppressant | NCT04341675 | Yes. Approved for preventing of transplant rejection and lyphangioleiomyomatosis | |
| MSCs infusion | BM, DP and UC and NestCell® MSCs for increasing anti-inflammatory environment | NCT04346368, NCT0402519, NCT04339660 and NCT04315987 | No. Biological treatment. Phase I and II trials are currently recruiting patients | |
| Pyridostigmine | Acetylcholinesterase inhibitor. Enhances anti-inflammatory activity of α7-nAChR | NCT04343963 | Yes. Approved for myasthenia gravis | |
| Canakinumab | IL-1β blocking antibody | NCT04348448 | Yes. Approved for cryopyrin-associated periodic syndromes | |
| Anakinra | IR-1R blocking antibody | NCT04324021 | Yes. Approved for rheumatoid arthritis | |
| Tocilizumab, Sarilumab | IL-6 blocking antibodies | NCT04345445 and ChiCTR2000030894 | Yes. Approved for rheumatoid arthritis | |
| Emapalumab | IFN-γ blocking antibody | NCT04324021 | Yes. Approved for haemophagocytic lymphohistiocytosis | |
| Eculizumab, IFX-1 | C5 blocking antibodies | NCT04288713 and NCT04341116 | Yes. Eculizumab approved for paroxysmal nocturnal hemoglobinuria. IFX-1 texted for hijdradenitis suppurativa | |
| Lenzilumab, TJ003234 | GM-CSF blocking antibody | NCT04351152 and NCT04341116 | Yes. Phase I trial in healthy volunteers | |
| IC14 | CD14 blocking antibody | NCT04346277 | Yes. Tested for sepsis | |
| Immune tolerance and exhaustion | Ruxolitinib, Baricitinib | JAK inhibitors | NCT04348071, NCT04337359, NCT04338958 and NCT04320277 | Yes. Ruxolitinib approved for myelofibrosis, polycythaemia vera and graft-versus-host disease. Baricitinib approved for rheuamatoid arthritis |
| Leronlimab | CCR5 blocking antibody | NCT04347239 | Yes. Phase I trial in HIV | |
| Camrelizumab | PD-1 blocking antibody | ChiCTR2000029806 and NCT04268537 | Yes. Tested for relapsed/refractory classic Hodgkin lymphoma | |
| Thymosin α1 | Recombinant Human Thymosin α1 (rhTh) | ChiCTR2000029806 and NCT04268537 | Yes. Tested for hepatitis B and C and various types of cancer | |
| Expanded NK cell infusion | Allogenic and modified CAR NK cells | NCT04344548 and NCT04324996 | No. Biological treatment. Phase I and II trials are currently recruiting patients | |
| APCs infusion | Lentiviral transfected DCs and APCs with 2019-nCoV for improving antigen presentation | NCT04276896 and NCT04299724 | No. Biological treatment. Phase I and II trials are currently recruiting patients | |
| IFNα-2b | Immunomodulator | ChiCTR2000029308 | Yes. Approved for hepatitis and hematological tumors | |
| IFNβ-1b | Immunomodulator | MIRACLE Study: NCT 02845843 | Yes. Approved for multiple sclerosis | |
| IFN-γ | Immunomodulator | MIRACLE Study: NCT02845843 | Yes. Approved for infections associated with chronic granulomatous disease and malignant osteopetrosis | |
| Convalescent plasma | Hyperimmune antibodies plasma | NCT04343755, NCT04347681 and NCT04345523 | No. Biological treatment. Phase I and II trials are currently recruiting patients | |
| Coagulopathy | Enoxaparin | Anticoagulant. Heparin derivative | NCT04345848 | Yes. Approved for deep vein thrombosis and pulmonary embolism |
| Tinzaparin | Anticoagulant. Low molecular weight heparin | NCT04344756 | Yes. Approved for deep vein thrombosis | |
| Heparin | Unfractionated heparin | NCT04344756 | Yes. Approved for several diseases | |
| Host viral entry | Camostat | TMPRSS-2 inhibitor | NCT04321096 | No. Phase I and II trials are currently recruiting patients |
| Linagliptin | DPP4 inhibitor | NCT04341935 | Yes. Approved for type 2 diabetes mellitus | |
| APNO1 | Recombinant Human Angiotensin 2 Converting Enzyme Analogue (rhACE2) | NCT04287686 | Yes. Phase I safety trial in healthy volunteers |
Abbreviations: α7-nAChR, alpha-7 nicotinic receptor; APCs, antigen-presenting cells; BM, bone marrow; CAR, chimeric antigen receptor; CCR-5, C-C chemokine receptor type 5; DCs, dendritic cells; DP, dental pulp; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; MSCs, mesenchymal stem cells; mTOR, mammalian target of rapamycin; PD-1, programmed cell death protein-1; TMPRSS2, transmembrane serine protease 2; UC, umbilical cord.
An in-depth study of the interaction between the virus and the immune system is warranted to identify appropriate therapeutic targets. We need to achieve a finely tuned balance between regulating the first wave of cytokines and reactivating an appropriate adaptive response, a delicate balance struck between simultaneously blocking and unblocking the immune response. This balancing act will require biomarkers that clearly show the proper approach. It is likely that IC ligands (already known or to be discovered) are overexpressed from the onset of COVID-19 infection. As mentioned previously, IC ligand levels could help stratify patients during admission and prevent the disease from progressing. A massive international study approach could be useful for developing this tool. Given that COVID-19 is a dynamic disease, however, we will need to study patients at different stages of the disease to identify predictive factors and appropriate targets. A single picture of cytokine levels, cell population distribution, and other markers of interest would only give us a partial idea of an evolving disease.
Conclusion
Based on the evidence presented in this article, the study of the immune system and the coagulation cascade during COVID-19 infection could provide valuable information for approaching the diagnosis and treatment of this disease. There are many questions that remain unanswered in the context of COVID-19, including “Can we predict the progression of patients with COVID-19 by establishing their immune profiles on admission?”, “Could IC expression regulate the second phase of COVID-19?” and “Can IC inhibitors and their ligands be useful for COVID-19 therapy as they have been in many types of cancers and as they have been postulated in sepsis?” Identifying those patients with poorer prognoses will therefore facilitate the development of accurate host-directed therapies.
Acknowledgements
We would like to thank the Biomedical Research Foundation of Hospital La Paz, the Alonso Family Foundation and Ayuntamiento de Madrid for their support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Borobia AM, Carcas AJ, Arnalich F, Alvarez-Sala R, Monserrat-Villatoro J, Quintana M. et al. A Cohort of Patients with COVID-19 in a Major Teaching Hospital in Europe. J Clin Med. 2020 9
2. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H. et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475-81
3. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020
4. Verity R, Okell LC, Dorigatti I, Winskill P, Whittaker C, Imai N. et al. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect Dis. 2020;20:669-77
5. Buonaguro FM, Puzanov I, Ascierto PA. Anti-IL6R role in treatment of COVID-19-related ARDS. J Transl Med. 2020;18:165
6. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020
7. Astuti I, Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab Syndr. 2020;14:407-12
8. Coronavirus latest. WHO says Europe now epicentre of pandemic. Nature. 2020 https://www.bbc.com/news/world-europe-51876784
9. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054-62
10. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX. et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020
11. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G. et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091
12. Chao JY, Derespina KR, Herold BC, Goldman DL, Aldrich M, Weingarten J. et al. Clinical Characteristics and Outcomes of Hospitalized and Critically Ill Children and Adolescents with Coronavirus Disease 2019 (COVID-19) at a Tertiary Care Medical Center in New York City. J Pediatr. 2020
13. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N. et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020;395:1517-20
14. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020
15. Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9:601-4
16. Conti P, Gallenga CE, Tete G, Caraffa A, Ronconi G, Younes A. et al. How to reduce the likelihood of coronavirus-19 (CoV-19 or SARS-CoV-2) infection and lung inflammation mediated by IL-1. J Biol Regul Homeost Agents. 2020 34
17. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I. et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020 34
18. Khalili JS, Zhu H, Mak NSA, Yan Y, Zhu Y. Novel coronavirus treatment with ribavirin: Groundwork for an evaluation concerning COVID-19. J Med Virol. 2020
19. Martinez MA. Compounds with Therapeutic Potential against Novel Respiratory 2019 Coronavirus. Antimicrob Agents Chemother. 2020 64
20. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020
21. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC. et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med. 2020
22. Pizzorno A, Padey B, Julien T, Trouillet-Assant S, Traversier A, Errazuriz-Cerda E. et al. Characterization and treatment of SARS-CoV-2 in nasal and bronchial human airway epithelia. BioRxiv. 2020
23. Williamson BN, Feldmann F, Schwarz B, Meade-White K, Porter DP, Schulz J. et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature. 2020
24. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269-71
25. Dolin R, Hirsch MS. Remdesivir - An Important First Step. N Engl J Med. 2020
26. Goldman JD, Lye DCB, Hui DS, Marks KM, Bruno R, Montejano R. et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N Engl J Med. 2020
27. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181-92
28. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523-34
29. Graham CA. Outbreaks, Middle East respiratory syndrome and sepsis in emergency care. Eur J Emerg Med. 2015;22:297
30. Black JRM, Bailey C, Przewrocka J, Dijkstra KK. COVID-19: the case for health-care worker screening to prevent hospital transmission. The Lancet. 2020
31. Luong-Nguyen M, Hermand H, Abdalla S, Cabrit N, Hobeika C, Brouquet A. et al. Nosocomial infection with SARS-Cov-2 within Departments of Digestive Surgery. J Visc Surg. 2020
32. Ryu BH, Cho Y, Cho OH, Hong SI, Kim S, Lee S. Environmental contamination of SARS-CoV-2 during the COVID-19 outbreak in South Korea. Am J Infect Control. 2020
33. Wang J, Feng H, Zhang S, Ni Z, Ni L, Chen Y. et al. SARS-CoV-2 RNA detection of hospital isolation wards hygiene monitoring during the Coronavirus Disease 2019 outbreak in a Chinese hospital. Int J Infect Dis. 2020;94:103-6
34. Chia PY, Coleman KK, Tan YK, Ong SWX, Gum M, Lau SK. et al. Detection of air and surface contamination by SARS-CoV-2 in hospital rooms of infected patients. Nat Commun. 2020;11:2800
35. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450-4
36. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020
37. Sun P, Lu X, Xu C, Wang Y, Sun W, Xi J. CD-sACE2 inclusion compounds: An effective treatment for coronavirus disease 2019 (COVID-19). J Med Virol. 2020
38. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259-60
39. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631-7
40. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J. et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79:14614-21
41. Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020
42. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281-92 e6
43. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562-9
44. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271-80 e8
45. Wang K, Chen W, Zhou Y-S, Lian J-Q, Zhang Z, Du P. et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv. 2020
46. Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J. et al. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes. 2013;62:149-57
47. Gliddon DR, Howard CJ. CD26 is expressed on a restricted subpopulation of dendritic cells in vivo. Eur J Immunol. 2002;32:1472-81
48. Epardaud M, Bonneau M, Payot F, Cordier C, Megret J, Howard C. et al. Enrichment for a CD26hi SIRP- subset in lymph dendritic cells from the upper aero-digestive tract. J Leukoc Biol. 2004;76:553-61
49. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9:45
50. Ellingsen T, Hornung N, Moller BK, Hjelm-Poulsen J, Stengaard-Pedersen K. In active chronic rheumatoid arthritis, dipeptidyl peptidase IV density is increased on monocytes and CD4(+) T lymphocytes. Scand J Immunol. 2007;66:451-7
51. Totura AL, Bavari S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin Drug Discov. 2019;14:397-412
52. Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH. et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95-103
53. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104:8-13
54. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475-87
55. Lopez-Collazo E, del Fresno C. Pathophysiology of endotoxin tolerance: mechanisms and clinical consequences. Crit Care. 2013;17:242
56. Liu J, Li S, Liu J, Liang B, Wang X, Wang H. et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020;55:102763
57. Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez-Jimenez E. et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity. 2015;42:484-98
58. Hu W, Yen YT, Singh S, Kao CL, Wu-Hsieh BA. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol. 2012;25:277-88
59. Totura AL, Whitmore A, Agnihothram S, Schafer A, Katze MG, Heise MT. et al. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio. 2015;6:e00638-15
60. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101:4240-5
61. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020
62. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y. et al. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. National Science Review. 2020
63. Basu S, Dunn AR, Marino MW, Savoia H, Hodgson G, Lieschke GJ. et al. Increased tolerance to endotoxin by granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol. 1997;159:1412-7
64. NCT04351152. Phase 3 Study to Evaluate Efficacy and Safety of Lenzilumab in Hospitalized Patients With COVID-19 Pneumonia. https://ClinicalTrials.gov/show/NCT04351152.
65. NCT04341116. Study of TJ003234 (Anti-GM-CSF Monoclonal Antibody) in Subjects With Severe Coronavirus Disease 2019 (COVID-19). https://ClinicalTrials.gov/show/NCT04341116.
66. Saghazadeh A, Rezaei N. Towards treatment planning of COVID-19: Rationale and hypothesis for the use of multiple immunosuppressive agents: Anti-antibodies, immunoglobulins, and corticosteroids. Int Immunopharmacol. 2020;84:106560
67. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents. 2020;55:105954
68. Wu J, Liu J, Zhao X, Liu C, Wang W, Wang D. et al. Clinical Characteristics of Imported Cases of COVID-19 in Jiangsu Province: A Multicenter Descriptive Study. Clin Infect Dis. 2020
69. Yuan J, Kou S, Liang Y, Zeng J, Pan Y, Liu L. PCR Assays Turned Positive in 25 Discharged COVID-19 Patients. Clin Infect Dis. 2020
70. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC. et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166:6952-63
71. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S. et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. 2014;18:R3
72. Avendano-Ortiz J, Maroun-Eid C, Martin-Quiros A, Lozano-Rodriguez R, Llanos-Gonzalez E, Toledano V. et al. Oxygen Saturation on Admission Is a Predictive Biomarker for PD-L1 Expression on Circulating Monocytes and Impaired Immune Response in Patients With Sepsis. Front Immunol. 2018;9:2008
73. Avendano-Ortiz J, Maroun-Eid C, Martin-Quiros A, Toledano V, Cubillos-Zapata C, Gomez-Campelo P. et al. PD-L1 Overexpression During Endotoxin Tolerance Impairs the Adaptive Immune Response in Septic Patients via HIF1alpha. J Infect Dis. 2018;217:393-404
74. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A. et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020;9:761-70
75. Thevarajan I, Nguyen THO, Koutsakos M, Druce J, Caly L, van de Sandt CE. et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. 2020;26:453-5
76. Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. 2020;9:558-70
77. Tinoco R, Carrette F, Barraza ML, Otero DC, Magana J, Bosenberg MW. et al. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity. 2016;44:1470
78. Abdou Y, Pandey M, Sarma M, Shah S, Baron J, Ernstoff MS. Mechanism based treatment of cancer with immune-checkpoint inhibitor therapies. Br J Clin Pharmacol. 2020
79. Wieder T, Eigentler T, Brenner E, Rocken M. Immune checkpoint blockade therapy. J Allergy Clin Immunol. 2018;142:1403-14
80. NCT04268537. Immunoregulatory Therapy for 2019-nCoV. https://ClinicalTrials.gov/show/NCT04268537.
81. Lythgoe MP, Middleton P. Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol Sci. 2020
82. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK. et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181-93
83. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M. et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129:3625-39
84. Shokri S, Mahmoudvand S, Taherkhani R, Farshadpour F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J Cell Physiol. 2019;234:2143-51
85. Chan JF, Yao Y, Yeung ML, Deng W, Bao L, Jia L. et al. Treatment With Lopinavir/Ritonavir or Interferon-beta1b Improves Outcome of MERS-CoV Infection in a Nonhuman Primate Model of Common Marmoset. J Infect Dis. 2015;212:1904-13
86. Arabi YM, Shalhoub S, Mandourah Y, Al-Hameed F, Al-Omari A, Al Qasim E. et al. Ribavirin and Interferon Therapy for Critically Ill Patients With Middle East Respiratory Syndrome: A Multicenter Observational Study. Clin Infect Dis. 2020;70:1837-44
87. Nagata N, Iwata N, Hasegawa H, Fukushi S, Harashima A, Sato Y. et al. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am J Pathol. 2008;172:1625-37
88. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 2020;178:104791
89. Sheahan TP, Sims AC, Leist SR, Schafer A, Won J, Brown AJ. et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun. 2020;11:222
90. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D. et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020
91. NCT04344548. Phase I II Clinical Study of Immunotherapy Based on Adoptive Cell Transfer as a Therapeutic Alternative for Patients With COVID-19 in Colombia. https://ClinicalTrials.gov/show/NCT04344548.
92. NCT04324996. A Phase I/II Study of Universal Off-the-shelf NKG2D-ACE2 CAR-NK Cells for Therapy of COVID-19. https://ClinicalTrials.gov/show/NCT04324996.
93. Li G, Chen X, Xu A. Profile of specific antibodies to the SARS-associated coronavirus. N Engl J Med. 2003;349:508-9
94. Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw FM, Lim WS. et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J Infect Dis. 2015;211:80-90
95. Koenig KL. Identify-Isolate-Inform: A Modified Tool for Initial Detection and Management of Middle East Respiratory Syndrome Patients in the Emergency Department. West J Emerg Med. 2015;16:619-24
96. Duan K, Liu B, Li C, Zhang H, Yu T, Qu J. et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A. 2020
97. Lv H, Wu NC, Tsang OT, Yuan M, Perera R, Leung WS. et al. Cross-reactive Antibody Response between SARS-CoV-2 and SARS-CoV Infections. Cell Rep. 2020;31:107725
98. Tian X, Li C, Huang A, Xia S, Lu S, Shi Z. et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9:382-5
99. Zhang L, Liu Y. Potential interventions for novel coronavirus in China: A systematic review. J Med Virol. 2020;92:479-90
100. Pinto D, Park YJ, Beltramello M, Walls AC, Tortorici MA, Bianchi S. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020
101. Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood. 1993;82:513-20
102. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26-34
103. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. 2010;2:e2010024
104. Levi M, Schultz M, van der Poll T. Disseminated intravascular coagulation in infectious disease. Semin Thromb Hemost. 2010;36:367-77
105. Levi M. Current understanding of disseminated intravascular coagulation. Br J Haematol. 2004;124:567-76
106. van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW. et al. Review: infectious diseases and coagulation disorders. J Infect Dis. 1999;180:176-86
107. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844-7
108. Paranjpe I, Fuster V, Lala A, Russak A, Glicksberg BS, Levin MA. et al. Association of Treatment Dose Anticoagulation with In-Hospital Survival Among Hospitalized Patients with COVID-19. J Am Coll Cardiol. 2020
109. Tan C, Huang Y, Shi F, Tan K, Ma Q, Chen Y. et al. C-reactive protein correlates with computed tomographic findings and predicts severe COVID-19 early. J Med Virol. 2020
110. Yang Z, Shi J, He Z, Lu Y, Xu Q, Ye C. et al. Predictors for imaging progression on chest CT from coronavirus disease 2019 (COVID-19) patients. Aging (Albany NY). 2020;12:6037-48
111. Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A. et al. Clinical Characteristics of Covid-19 in New York City. N Engl J Med. 2020
112. Wells PS, Anderson DR, Rodger M, Forgie M, Kearon C, Dreyer J. et al. Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med. 2003;349:1227-35
113. O'Brien TR, Thomas DL, Jackson SS, Prokunina-Olsson L, Donnelly RP, Hartmann R. Weak Induction of Interferon Expression by SARS-CoV-2 Supports Clinical Trials of Interferon Lambda to Treat Early COVID-19. Clin Infect Dis. 2020
114. Lu CC, Chen MY, Chang YL. Potential therapeutic agents against COVID-19: What we know so far. J Chin Med Assoc. 2020
115. Prokunina-Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R. et al. COVID-19 and emerging viral infections: The case for interferon lambda. J Exp Med. 2020 217
116. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G. et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med. 2020
117. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y. et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. The Lancet. 2020
Author contact
![]() Corresponding author: E-mails: elopezcmadrid.org (E.L-C.) and luis.augusto.aguirrees (L.A.A.).
Corresponding author: E-mails: elopezcmadrid.org (E.L-C.) and luis.augusto.aguirrees (L.A.A.).