Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The Diabetogenic mechanisms of...
Statin therapy for nervous...
Statins for the treatment of...
The anti-inflammatory effects of...
Statins are potential Anticancer...
Discussion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(14):2704-2711. doi:10.7150/ijbs.42965 This issue Cite
Review
Pleiotropic use of Statins as non-lipid-lowering drugs
Qijia Zhang1, Jianlong Dong2, Ze Yu3 ![]()
1. Digestive internal medicine and Department of infectious diseases, Zhuhai Hospital of Integrated Traditional Chinese and Western Medicine, Zhuhai, China.
2. College of Life Science, Northeast Agricultural University, Harbin, China.
3. Affiliated Cancer Hospital and Institute of Guangzhou Medical University, State Key Laboratory of Respiratory Disease, Guangzhou, China.
Received 2019-12-11; Accepted 2020-8-1; Published 2020-8-13
Abstract

Statins, known as HMG-CoA reductase (HMGCR) inhibitors, have primarily been utilized for metabolic and angiographic medical applications because of their cholesterol-lowering effects. Similar to other drugs, statins may also induce a series of potential side effects. Statins inhibit the HMGCR (rate-limiting enzyme) activity in early stages of mevalonate pathway and then indirectly affect a number of intermediate products, including non-sterol isoprenoids (coenzyme Q10, dolichol etc.), which can result in impaired functions of body organs. Recently, scores of studies have uncovered additional functional mechanisms of statins in other diseases, such as diabetes mellitus, nervous system diseases, coronary heart disease, inflammation and cancers. This review aims to summarize the positive and adverse mechanisms of statin therapy. Statin care should be taken in the treatment of many diseases including cancers. Since the underlying mechanisms are not fully elucidated, future studies should spend more time and efforts on basic research to explore the mechanisms of statins.
Keywords: Statins, mevalonate pathway, non-lipid-lowering function, anticancer agents
Introduction
Statins are a class of drugs that contain a naphthyl ring skeleton structure and are known as HMGCR inhibitors, which regulate the concentration of plasma lipoproteins by inhibiting cholesterol synthesis. Statins could reduce the synthesis of cholesterol, thus increasing the number and activity of low-density lipoprotein (LDL) receptors on the membrane of liver cells and enhancing the clearance of plasma cholesterol [1, 2]. Since the hydrophilic group of statins is similar to the HMG portion of the substrate [3], statins can be used as competitive inhibitors of HMG-CoA. Therefore, statins can reduce cholesterol synthesis to prevent cardiovascular diseases such as atherosclerosis [4-6].
Mevastatin was the first discovered HMG-CoA reductase inhibitor and purified by Akira Endo from the mold fermentation nutrient medium [7]. Then, an analogue of lovastatin was isolated from the broth of Aspergillus terreus in 1980 and marketed by Merck in 1987. Subsequently, other statin, including lovastatin and simvastatin, were discovered and studied [8, 9]. Such natural micro-molecules were recognized as the first generation of HMG-CoA reductase inhibitors, retaining the highly absorptive, fat-soluble and hypo-toxic characteristics [10]. However, complicated synthesis processes and expensive costs had restricted their applications. Thus, artificial modifications of statin structure were required to reduce the costs. The subsequent second generation of statins, such as pravastatin and fluvastatin, had higher solubility in water, lower lipid solubility and faster oral absorption than the first generation [9].
As an important component in the electron transport chain during cellular respiration, coenzyme Q10 (CoQ10) (ubiquinone) mediates electron transfer and results in the shifting of hydrogen ions into the mitochondrial intermembrane space to form a hydrogen ion concentration gradient, ultimately required for the formation of ATP [11]. Statins, along with inhibiting the activity of HMG-CoA reductase enzyme, also block the pathway leading to the production of CoQ10 [12, 13]. The lack of CoQ10 resulting from statins may block normal cellular aerobic respiration and produce an abundance of free radicals, which are toxic to cells [12, 14].
The Diabetogenic mechanisms of Statins and clinical implications
Recently, clinical trials and observational studies have indicated an approximate 10% increase in new-onset diabetes mellitus among patients taking statins, but the mechanisms underlying these clinical effects are not yet fully understood. Clinical and basic studies have shown that statins can potentially increase plasma glucose levels and diabetes risk by enhancing insulin resistance or impairing β-cell function. Here, we describe the potential mechanisms underlying how statins affect glucose homeostasis. Many studies have indicated that the effects of statins in diabetes are related to the inhibition of HMG-CoA reductase activity [15].
Statin-induced cholesterol depletion can decrease GLUT4 gene expression and increase GLUT1 protein levels [16]. Moreover, membrane cholesterol depletion results in a remarkable decline in the tyrosine-specific phosphorylation of insulin receptor substrate (IRS) and the serine-threonine phosphorylation of AKT in response to insulin [17]. IRS1/2 mediate the control of various cellular processes by insulin. When phosphorylated by the insulin receptor, IRS1/2 bind specifically to various cellular proteins containing SH2 domains. Statins can downregulate GLUT4 gene expression and reduce insulin-stimulated glucose transport in adipocytes, while adding mevalonate can reverse these effects; HMG-CoA reductase catalyzes this process [16]. These results indicate that statins regulate the GLUT4 expression via influencing mevalonate pathway and some intermediate metabolites in mevalonate pathway play an important role in regulating transcription levels of GLUT4. Besides, statins also reduce expression level of the small GTP-binding protein RhoA, which is a cell membrane-anchored protein and requires isoprenylation to activate it. Isoprenylation of RhoA is crucial to support cholesterol homeostasis [18]. These results suggest that the reduction of isoprenoid intermediates in the cholesterol biosynthetic pathway is related to insulin resistance.
Isoprene residues are among the important intermediate molecules of the mevalonate synthesis pathway and are also required for the synthesis of CoQ10 [19]. CoQ10 participates in mitochondrial oxidative phosphorylation and the generation of ATP (Figure 1). Both in vivo and in vitro experiments have proved that simvastatin reduce ATP production in a dose-dependent manner and that insulin secretion requires ATP [20]. Depressed coenzyme Q10 levels are likely to impact β-cell function, decrease insulin sensitivity, disturb glucose tolerance and inhibit oxidative phosphorylation in mitochondria [21]. Additionally, the accumulation of small G-proteins is paralleled by a reduction in insulin levels. It is known that lovastatin can decrease glucose-induced insulin secretion from pancreatic islets by 50%, while mevalonate can rescue this effect [22].
The diabetogenic mechanisms of statins. Extracellular insulin can activate Insulin receptor tyrosine kinase, which results in the phosphorylation of IRS1, and AKT is subsequently activated. Statins could facilitate GLUT1 transcription, while repress GLUT4. Statins prevent RhoA from transferring to the cell membrane via inhibiting its translation. Statins reduce CoQ10 levels via depressing the HMG-CoA activity to inhibition of the mevalonate pathway impairing mitochondrial electron transport chain and ATP production.
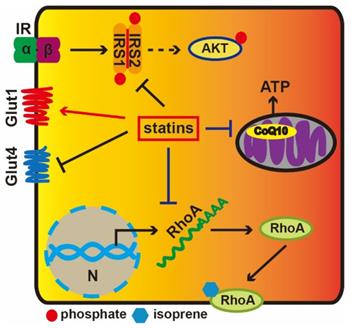
Our purpose is to explore possible risks underlying the effects of statins in diabetogenic clinical treatment. Relevant risk factors related to diabetes during the administration of statins, such as statin variety, treatment dose and hazards for patients with type II diabetes, are very important to be considered in the treatment of diabetic patients. Some studies have found that patients with diabetes receiving statin therapy appear to suffer more risk factors for type II diabetes, particularly in elderly age (age >70 years) [23].
Statin therapy for nervous system diseases
Clinically, a great body of evidence indicates statins are employed to treat atherosclerosis in patients with cerebral thrombosis and Alzheimer's disease as it can significantly reduce the incidence of ischemic and hemorrhagic stroke [24]. Despite that numerous clinical studies seek to confirm the therapeutic potential of statins in various central nervous system disorders, including multiple sclerosis, epilepsy, dementia, depression and stroke, there still has been a gap in our understanding of mechanisms underlying the neurological effects of statins. In 2006, it was announced that lower cholesterol levels and stroke prevention studies represented the first milestone for the prevention of recurrent stroke in patients administered statins after stroke. These results suggest that despite the simultaneous reduction of cardiovascular events in the statin intervention group, the incidence of hemorrhagic stroke increased slightly, although bleeding in the two groups did not increase the number of deaths caused by stroke, and the incidence of hemorrhagic stroke was extremely low (2.3%; 1.4% in the control group) [25].
Studies from Yousself et al. have shown that statins decrease the infiltration of the central nervous system (TH1 cell) with inflammatory cells, reduce the expression of major histocompatibility complex II (MHC-II) and inhibit CD40, CD80, and CD86. Oral atorvastatin can prevent and reverse chronic relapses of multiple sclerosis in patients with clinical symptoms of paralysis. Atorvastatin can increase the secretion of TH2 cytokines (IL-4, IL-5, IL-10 and TNF-β) (Figure 2). Mevalonate can reverse the effects of atorvastatin in T cells and antigen-presenting cells and inhibit isoprenoid synthesis, thereby preventing intracellular signaling [26].
Statin therapy for nervous system diseases. In the central nervous system, statins decrease the infiltration of inflammatory cells (TH1 and TH2 cell). In the TH1 cell, statins depress MHC-II, CD40, CD80, and CD86 marker proteins expression. Meanwhile, conversely, in TH2 cell, statins increase the secretion of TH2 cytokines (IL-4, IL-5, IL-10, and TNF-β).
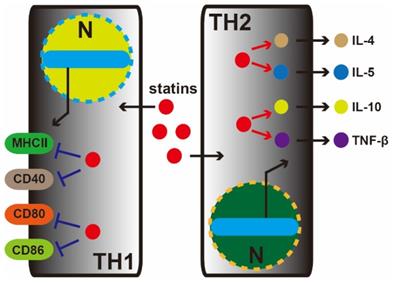
Recent studies also suggest that statins can mitigate Alzheimer's disease (AD). One study found that among 845 cases of elderly patients (average: 80.5 years old), 20.1% had dementia, while in the 10.9% of people without dementia taking lipid-lowering medication, only 3.5% developed dementia. AD is one of the most common causes of dementia, and taking statins may reduce morbidity, improve cognitive function and delay the progression of AD; thus, statin treatment brings new hope to AD therapy [27].
The association between long-term statin use and adverse cognitive effects has not been well established. A recent review analyzed the MedWatch drug surveillance system and found statins could also cause memory loss in some patients. It was reported that 60 cases of memory loss were associated with statin use, of which 36 cases took simvastatin, 23 cases took atorvastatin, 1 took pravastatin. Memory loss occurred within 2 months after taking statins in 50% of patients. Among 25 cases taking simvastatin, 14 patients developed improved memory after stopping the drug. However, many of the studies evaluating the effects of statins on cognitive dysfunction have a relatively short follow-up, limiting the ability to assess whether there is a true caused relationship between long-term statin use and cognitive function [28]. Consequently, we cannot evaluate whether or not the risk of statin-associated cognitive side effects increases with time.
Statins for the treatment of Coronary Heart Disease
Statins can be used to treat coronary heart disease by alleviating endothelial dysfunction, inhibiting adhesion between leukocytes and endothelial cells, resisting oxidation, stabilizing plaques, affecting blood flow, suppressing smooth muscle cell proliferation and exerting other effects [29, 30]. Research has shown that statins affect vascular smooth muscle cell proliferation that is related to Rho protein isoprenylation. Rho protein not only plays a key role in maintaining cell morphology but also is responsible for degrading cell cycle inhibitory p27 [31]. Additionally, statins could reduce the number of Rho proteins attached to cell membrane and decrease Cdk2 expression level in vascular smooth muscle cells, ultimately inhibiting cells from transitioning between G1 to S phase and causing cell cycle arrest [32].
To study whether statins can suppress cell proliferation via affecting DNA replication in rat aorta vascular smooth muscle cells, Bruemmer et al. selected two chromosomal proteins, MCM6 and MCM7, which played important roles in DNA replication initiation stage. Results showed that MCM6 and MCM7 expression was suppressed by atorvastatin in a dose-dependent manner, and mevalonate could completely reverse the effect. And their study also found that atorvastatin may inhibit MCM6 and MCM7 enhancer activity. Meanwhile, since the MCM6/7 enhancer contains several E2F transcription sites, adenovirus-mediated overexpression of E2F could reverse the inhibition exerted on MCM6 and MCM7 by atorvastatin. These results suggested that atorvastatin was associated with MCM6 and MCM7 mRNA transcription [33].
Recent advances have shown that Rac1 GTPase is a major master regulator of cell motility through regulating the generation of reactive oxygen species and cortical actin reorganization, which are induced by regulating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity [34]. Statins can inhibit Rac1 activity (Figure 3). One critical issue is to evaluate the activation of Rac1 GTPase, which is a key component of cardiovascular pathologies, including cardiac hypertrophy, fibrosis and atrial fibrillation [35]. However, the signal transduction involved in these processes has yet to be deciphered. An improved understanding of the Rac1-mediated effects of statins may help us to identify novel therapeutic strategies and targets.
Statin therapy for coronary heart disease. In the vascular epithelial cells, the key effectors are Rac1 and RhoA protein in the mevalonate pathway, Rac1 can maintain the stability of the cytoskeleton and promote cell migration via regulating NOX, and RhoA protein can keep from the ubiquitin degradation of p27, inhibit cell cycle progression. As the inhibitor of the mevalonate pathway, statins are able to inhibiting the formation of Rac1 and RhoA. Meanwhile, statins can also prevent the transition of G1-S phase through inhibiting CDK2. Moreover, statins inhibit cell proliferation by depressing the transcription of MCM6 and MCM7.
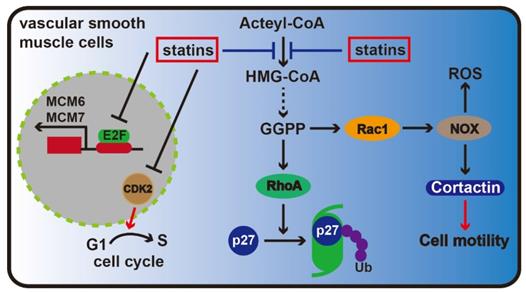
The anti-inflammatory effects of Statins
Basic and clinical studies have demonstrated statins' anti-inflammatory effects [36, 37]. The primary mechanism involves in cell proliferation inhibition, aggregation of inflammatory cells, and the secretion of cellular factors, increasing endothelial nitric oxide production, and protecting endothelial function [38].
Nuclear factor κB (NF-κB) is an important transcription regulatory protein and it is a homo- or heterodimeric complex formed by the Rel-like domain-containing proteins. It is involved in activation of immune cells, T and B lymphocyte development, the stress response, apoptosis and other cellular activities [39]. Its abnormal activation can exacerbate inflammatory reactions. Chemokines, adhesion molecules and other inflammation-related gene promoters all contain nuclear factor NF-κB recognition sites. Statins not only inhibit the activity of nuclear factor κB expression, but also prevent NF-κB from entering the nucleus, and ultimately reduce the expression of adhesion molecules and inflammatory factors [40].
Nitric oxide (NO) synthesis is catalyzed by nitric oxide synthase (NOS) with L-arginine, NADPH and molecular oxygen. It is released from the endothelium and present in a sustained form in the vascular system and also acts as an important protective factor. NO could inhibit platelet aggregation and leukocyte adhesion and infiltration, eliminate free oxygen radicals, dilate blood vessels and affect inflammatory reactions in many other ways [41]. The catalysis pathway of NO mediated by NOS is a highly potentially anti-inflammatory pathway. Statins can employ a variety of mechanisms to increase the synthesis of NO through NOS. Allan et al. found that simvastatin and lovastatin exerted dose-dependent protective effects in experimental stroke models by increasing NOS levels [42]. Other studies have confirmed statin can activate NOS gene transcription, extend the half-life of NOS mRNA in skin cells and inhibit mevalonate- mediated isoprenoid synthesis to increase endothelial NOS activity [43] (Figure 4).
The anti-inflammatory effects of statins. Statins can inhibit the activity of NF-κB, and NF-κB is an important transcription regulatory protein in inflammatory response. In addition, statins are able to activating NOS gene transcription, and stimulate the production of NO.
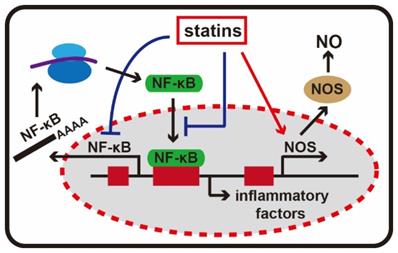
Statins are potential Anticancer Agents
Geranylgeranyl pyrophosphate and isoprenoid farnesyl pyrophosphate produced by the mevalonate pathway are able to make proteins lipidation with C-terminal motifs [44]. The process of isoprenylation is essential for the function and localization of small guanosine triphosphatases (GTPases), such as Ras protein, which is a key point in cancer progression [45].
Unlimited proliferation is one of the important characteristics of cancer cells and is an important reason why cancer is difficult to cure. Ras, RhoA and other small G-proteins play critical roles in cell proliferation, metastasis and apoptosis [46, 47]. As these small G-proteins require isoprenylation to be activated, their activity is fundamentally dependent on HMGCR. Statins inhibit cancer cell proliferation by disturbing the prenylation of several major small G-proteins. Different types of drugs exert various effects on the proliferation of different cancer cells. In a breast cancer mouse model, lipophilic statins, such as simvastatin, inhibit tumor growth, while the hydrophilic pravastatin did not demonstrate inhibition [48]. Notably, it has been shown that the disruption of isoprenylated proteins, such as Ras and RhoA, is unlikely to mediate the anticancer activities of statins alone. This finding indicates that the loss of multiple isoprenylated proteins immediately leads to statin-induced apoptosis [49].
Signal transduction process is closely linked to cell proliferation and survival. Vosper et al. found lovastatin blocked the cell cycle in prostate cancer cells, causing cells to stay in the G1 phase, and inhibited cancer cell proliferation. Downregulation of CDKs or upregulation of cell cycle blocking factor occurred during this process. However, the addition of farnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate (GGPP) recovered cell cycle [50]. Gbelcová et al. studied the anticancer effects of different statins in pancreatic cancer and found that the effects of mevastatin on pancreatic cancer cell were superior, while in animal models, rosuvastatin was found to be better [51]. These effects are associated with blocking of the Ras protein-mediated cell signal transduction.
A large number of studies have shown that cessation of the cell cycle does not depend on the lipid-lowering effects of statins, but statins inhibit cancer cell mitosis in a protease-associated manner and exert anti-proliferation effects involving the downregulation of CDK2 expression and upregulation of p21 and p27 activity [52, 53]. Researchers have used proteomics methods to study the mechanisms of lovastatin resistance of breast cancer and demonstrated that the inhibition of breast cancer cells occurred through the regulation of Ras and AKT signaling molecules [54, 55].
Apoptosis is a hallmark of cancer cells and induction of cancer cell apoptosis is a hotspot for anti-cancer drug researches. It has been confirmed both in vivo and in vitro that statins can induce cancer cell apoptosis, including lovastatin and simvastatin. [56, 57]. In recent years, studies have shown that statin-induced cancer cell apoptosis is involved in regulating many apoptosis signaling pathways. Jung et al. found that simvastatin activated caspase-8, caspase-3 and caspase-9 in prostate cancer cells and induced apoptosis [58]. Studies from Wang et al. showed that simvastatin induced apoptosis of breast cancer cells via reducing the activity of PI3K/AKT [59]. In addition, Chang et al. found simvastatin could induce apoptosis of colorectal cancer cells by activating MAPK-p53-survivin cascade [60]. In short, various studies have shown that different statins may act on different tumor cells to induce apoptosis through different signaling pathways. Recently, there has also been a large number of reports indicating that statins inhibit the expansion and metastasis of cancer via the Hippo pathway. Statins regulate YAP protein entry into the nucleus by influencing the conformation of cytoskeleton [61, 62].
In many cancers, angiogenesis is induced to provide nutrients and oxygen for cancer cell growth through the transfer of vessels providing transport to other organs and parts of the body, leading to tumor metastasis. A large number of experiments have shown that statins can inhibit tumor angiogenesis. In Ras-3T3 transgenic mouse tumor models, lovastatin suppresses tumor growth and angiogenesis by inhibiting tumor necrosis factor (TNF-α) [63] (Figure 5). Researchers found low concentrations of cerivastatin and lovastatin enhanced endothelial cell proliferation, whereas high concentrations inhibited angiogenesis. Furthermore, these phenotypic variations are related to the prenylation of small G-proteins. Accumulating data show statins induce angiogenesis in a dose-dependent manner, that is, low concentration stimulating angiogenesis and high concentration suppressing angiogenesis [64].
Statin therapy for cancer. When statins enter cells and block HMG-CoA reductase activity, the mevalonate pathway end product, RhoA is depleted. Depression of RhoA can affect the stability of the cytoskeleton, further prevent the YAP entering cell nucleus, here, YAP will drive transcription of target genes with TEAD to promote the tumor tissue growth. At the same time, KRAS is another important downstream molecule in the mevalonate pathway, which can activate the MAPK signal to accelerate cell proliferation, thus restraining KRAS will delay cancer cell proliferation. In addition to the mevalonate pathway, statins could directly depress the AKT signal, effect the synthesis and degradation p21 and p27, at last block the cell cycle progression. Besides, statin can activate caspase-8, caspase-3 and caspase-9 to induce apoptosis, and suppress angiogenesis by inhibiting TNF-α.
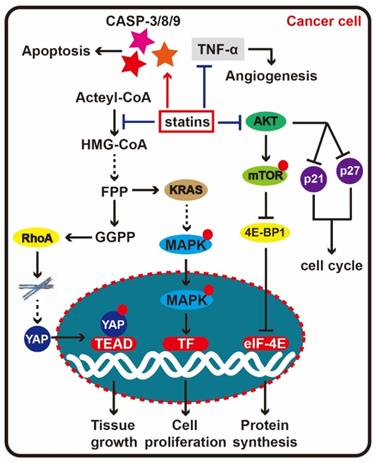
Discussion
Statins can cause many side effects while treating various diseases. The side effects of statins include liver toxicity [65], muscle toxicity [66] and neurotoxicity [67], as well as the following: effects on coenzyme Q10 and the mitochondrial membrane [68, 69]; effects on isopentenyl and glycosylase, and effects on ion channel signaling [70]. The dysfunction of cellular metabolism further leads to various adverse effects.
Statins are primarily metabolized by the liver and excreted by the kidneys. Therefore, if there is liver and renal failure, statins should not be administered. Alcoholics should be closely followed after using statins. Cholestasis can delay the excretion of statins, and increased plasma concentrations of statins may increase the risks of statin-induced myopathy (SIM). In addition, a history of statin drug allergy in SIM patients should also preclude the use of statins [71]. Thus, we must pay more attention to their application. In the meantime, some SIM symptoms can be overcome with concomitant use of non-sterol isoprenoids.
Despite being originally developed to address the escalating problem of high cholesterol in cardiovascular disease, statins appear to have extensive untapped potential. What we need to do is to maximize the benefits of statins and try to eliminate the potential side effects. Based on clinical experience, the side effects of various statins are not identical. By comparing the toxicities of various statins in liver cancer cells, it was found that the rank order of cytotoxicity was cerivastatin > simvastatin > atorvastatin > lovastatin > pravastatin [72]. Therefore, we should choose statins displaying less toxicity and fewer side effects. Moreover, Statin-induced myopathy is caused by the long-term use of higher doses, when certain drugs have been used previously (cyclosporine, erythromycin and clarithromycin), we should be fully aware of toxic side effects and other drug interactions and monitor for adverse reactions. Additionally, care should be taken in the treatment of many diseases including cancer because the underlying mechanisms are not fully clear and because the experimental results cannot necessarily be repeated. Future studies should spend more time and efforts on basic research to explore the mechanisms of statins.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Iannelli F, Lombardi R, Milone MR, ucci B, De Rienzo S, Budillon A. et al. Targeting Mevalonate Pathway in Cancer Treatment: Repurposing of Statins. Recent Pat Anticancer Drug Discov. 2018;13:184-200
2. Smit RAJ, Trompet S, Leong A, Goodarzi MO, Postmus I, Warren H. et al. Statin-induced LDL cholesterol response and type 2 diabetes: a bidirectional two-sample Mendelian randomization study. Pharmacogenomics J. 2020;20:462-470
3. Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Statins: pros and cons. Med Clin (Barc). 2018;150:398-402
4. Kazi DS, Penko JM, Bibbins-Domingo K. Statins for Primary Prevention of Cardiovascular Disease: Review of Evidence and Recommendations for Clinical Practice. Med Clin North Am. 2017;101:689-699
5. Yang Z, Wang H, Edwards D, Ding C, Yan L, Brayne C. et al. Association of blood lipids, atherosclerosis and statin use with dementia and cognitive impairment after stroke: A systematic review and meta-analysis. Ageing Res Rev. 2020;57:100962
6. Chung JW, Cha J, Lee MJ, Yu IW, Park MS, Seo WK. et al. Intensive Statin Treatment in Acute Ischaemic Stroke Patients with Intracranial Atherosclerosis: a High-Resolution Magnetic Resonance Imaging study (STAMINA-MRI Study). J Neurol Neurosurg Psychiatry. 2020;91:204-211
7. Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569-1582
8. Kostner GM. Pharmacology of HMG CoA reductase inhibitors (statins). Wien Med Wochenschr. 1999;149:120-124
9. Blum CB. Comparison of properties of four inhibitors of 3-hydroxy-3- methylglutaryl-coenzyme A reductase. Am J Cardiol. 1994;73:3D-11D
10. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160-1164
11. Hargreaves IP, Mantle D. Coenzyme Q10 Supplementation in Fibrosis and Aging. Adv Exp Med Biol. 2019;1178:103-112
12. Littarru GP, Langsjoen P. Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion. 2007;7(Suppl):S168-74
13. McGregor GH, Campbell AD, Fey SK, Tumanov S, Sumpton D, Blanco GR. et al. Targeting the Metabolic Response to Statin-Mediated Oxidative Stress Produces a Synergistic Antitumor Response. Cancer Res. 2020;80:175-188
14. Cordes T, Metallo CM. Statins Limit Coenzyme Q Synthesis and Metabolically Synergize with MEK Inhibition in Pancreatic Tumors. Cancer Res. 2020;80:151-152
15. Chrysant SG. New onset diabetes mellitus induced by statins: current evidence. Postgrad Med. 2017;129:430-435
16. Zhao W, Zhao SP. Different effects of statins on induction of diabetes mellitus: an experimental study. Drug Des Devel Ther. 2015;9:6211-6123
17. Parpal S, Karlsson M, Thorn H, Strålfors P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem. 2001;276:9670-9678
18. Medina MW, Theusch E, Naidoo D, Bauzon F, Stevens K, Mangravite LM. et al. RHOA is a modulator of the cholesterol-lowering effects of statin. PLoS Genet. 2012;8:e1003058
19. Nawarskas JJ. HMG-CoA reductase inhibitors and coenzyme Q10. Cardiol Rev. 2005;13:76-79
20. Koster JC, Permutt MA, Nichols CG. Diabetes and insulin secretion: the ATP-sensitive K+ channel (K ATP) connection. Diabetes. 2005;54:3065-3072
21. Sumi K, Okura T, Fujioka Y, Kato M, Imamura T, Taniguchi SI. et al. Coenzyme Q10 suppresses apoptosis of mouse pancreatic β-cell line MIN6. Diabetol Metab Syndr. 2018;10:47
22. Tsuchiya M, Hosaka M, Moriguchi T, Zhang S, Suda M, Yokota-Hashimoto H. et al. Cholesterol biosynthesis pathway intermediates and inhibitors regulate glucose-stimulated insulin secretion and secretory granule formation in pancreatic beta-cells. Endocrinology. 2010;151:4705-4716
23. Sattar NA, Ginsberg H, Ray K, Chapman MJ, Arca M, Averna M. et al. The use of statins in people at risk of developing diabetes mellitus: evidence and guidance for clinical practice. Atheroscler Suppl. 2014;15:1-15
24. Castilla-Guerra L, Del Carmen Fernandez-Moreno M, Colmenero-Camacho MA. Statins in Stroke Prevention: Present and Future. Curr Pharm Des. 2016;22:4638-4644
25. Ní Chróinín D1, Asplund K, Åsberg S, Callaly E, Cuadrado-Godia E, Díez-Tejedor E. et al. Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke. 2013;44:448-456
26. Youssef S1, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM. et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78-84
27. Mendoza-Oliva A, Zepeda A, Arias C. The complex actions of statins in brain and their relevance for Alzheimer's disease treatment: an analytical review. Curr Alzheimer Res. 2014;11:817-833
28. Huddy K, Dhesi P, Thompson PD. Do the frequencies of adverse events increase, decrease, or stay the same with long-term use of statins? Curr Atheroscler Rep. 2013;15:301
29. Tsujimoto T, Kajio H. Favorable effects of statins in the treatment of heart failure with preserved ejection fraction in patients without ischemic heart disease. Int J Cardiol. 2018;255:111-117
30. Ray KK, Corral P, Morales E, Nicholls SJ. Pharmacological lipid-modification therapies for prevention of ischaemic heart disease: current and future options. Lancet. 2019;394:697-708
31. Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8:e2626
32. Chan KC, Wu CH, Huang CN, Lan KP, Chang WC, Wang CJ. Simvastatin inhibits glucose-stimulated vascular smooth muscle cell migration involving increased expression of RhoB and a block of Ras/Akt signal. Cardiovasc Ther. 2012;30:75-84
33. Bruemmer D, Yin F, Liu J, Kiyono T, Fleck E, Van Herle A. et al. Atorvastatin inhibits expression of minichromosome maintenance proteins in vascular smooth muscle cells. Eur J Pharmacol. 2003;462:15-23
34. Adam O, Laufs U. Rac1-mediated effects of HMG-CoA reductase inhibitors (statins) in cardiovascular disease. Antioxid Redox Signal. 2014;20:1238-1250
35. Sawada N, Liao JK. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid Redox Signal. 2014;20:1251-167
36. Li GM, Zhao J, Li B, Zhang XF, Ma JX, Ma XL. et al. The anti-inflammatory effects of statins on patients with rheumatoid arthritis: A systemic review and meta-analysis of 15 randomized controlled trials. Autoimmun Rev. 2018;17:215-225
37. Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C. Statins as anti-inflammatory agents in atherogenesis: molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des. 2012;18:1519-1530
38. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977-987
39. Korbecki J, Bobiński R, Dutka M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm Res. 2019;68:443-458
40. Chang CH, Hsu YM, Chen YC, Lin FH, Sadhasivam S, Loo ST. et al. Anti-inflammatory effects of hydrophilic and lipophilic statins with hyaluronic acid against LPS-induced inflammation in porcine articular chondrocytes. J Orthop Res. 2014;32:557-565
41. Bailey JD, Diotallevi M, Nicol T, McNeill E, Shaw A, Chuaiphichai S. et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019;28:218-230
42. Ma S, Ma CC. Recent developments in the effects of nitric oxide-donating statins on cardiovascular disease through regulation of tetrahydrobiopterin and nitric oxide. Vascul Pharmacol. 2014;63:63-70
43. Datar R, Kaesemeyer WH, Chandra S, Fulton DJ, Caldwell RW. Acute activation of eNOS by statins involves scavenger receptor-B1, G protein subunit Gi, phospholipase C and calcium influx. Br J Pharmacol. 2010;160:1765-1772
44. Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241-269
45. Adderley H, Blackhall FH, Lindsay CR. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711-716
46. Nam S, Kim JH, Lee DH. RHOA in Gastric Cancer: Functional Roles and Therapeutic Potential. Front Genet. 2019;10:438
47. Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG. et al. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell. 2019;73:830-844
48. Pocobelli G, Newcomb PA, Trentham-Dietz A, Titus-Ernstoff L, Hampton JM, Egan KM. Statin use and risk of breast cancer. Cancer. 2008;112:27-33
49. Tsubaki M, Fujiwara D, Takeda T, Kino T, Tomonari Y, Itoh T. et al. The sensitivity of head and neck carcinoma cells to statins is related to the expression of their Ras expression status, and statin-induced apoptosis is mediated via suppression of the Ras/ERK and Ras/mTOR pathways. Clin Exp Pharmacol Physiol. 2017;44:222-234
50. Vosper J, Masuccio A, Kullmann M, Ploner C, Geley S, Hengst L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget. 2015;6:2889-2902
51. Gbelcová H, Lenícek M, Zelenka J, Knejzlík Z, Dvoráková G, Zadinová M. et al. Differences in antitumor effects of various statins on human pancreatic cancer. Int J Cancer. 2008;122:1214-1221
52. Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH, Chen CC. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter- associated HDAC1/2. Cancer Res. 2008;68:2375-2383
53. Feldt M, Bjarnadottir O, Kimbung S, Jirström K, Bendahl PO, Veerla S. et al. Statin-induced anti-proliferative effects via cyclin D1 and p27 in a window-of-opportunity breast cancer trial. J Transl Med. 2015;13:133
54. Vásquez-Bochm LX, Velázquez-Paniagua M, Castro-Vázquez SS, Guerrero- Rodríguez SL, Mondragon-Peralta A, De La Fuente-Granada M. et al. Transcriptome-based identification of lovastatin as a breast cancer stem cell-targeting drug. Pharmacol Rep. 2019;71:535-544
55. Yang T, Yao H, He G, Song L, Liu N, Wang Y. et al. Effects of Lovastatin on MDA-MB-231 Breast Cancer Cells: An Antibody Microarray Analysis. J Cancer. 2016;7:192-199
56. Wang F, Liu W, Ning J, Wang J, Lang Y, Jin X. et al. Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis. Int J Biol Sci. 2018;14:406-417
57. Lee HY, Kim IK, Lee HI, Mo JY, Yeo CD, Kang HH. et al. The apoptotic effect of simvastatin via the upregulation of BIM in nonsmall cell lung cancer cells. Exp Lung Res. 2016;42:14-23
58. Jung EJ, Chung KH, Kim CW. Identification of simvastatin-regulated targets associated with JNK activation in DU145 human prostate cancer cell death signaling. BMB Rep. 2017;50:466-471
59. Wang T, Seah S, Loh X, Chan CW, Hartman M, Goh BC. et al. Simvastatin- induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget. 2016;7:2532-2544
60. Chang HL, Chen CY, Hsu YF, Kuo WS, Ou G, Chiu PT. et al. Simvastatin induced HCT116 colorectal cancer cell apoptosis through p38MAPK-p53-survivin signaling cascade. Biochim Biophys Acta. 2013;1830:4053-4064
61. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357-366
62. Hao F, Xu Q, Wang J, Yu S, Chang HH, Sinnett-Smith J. et al. Lipophilic statins inhibit YAP nuclear localization, co-activator activity and colony formation in pancreatic cancer cells and prevent the initial stages of pancreatic ductal adenocarcinoma in KrasG12D mice. PLoS One. 2019;14:e0216603
63. Feleszko W, Bałkowiec EZ, Sieberth E, Marczak M, Dabrowska A, Giermasz A. et al. Lovastatin and tumor necrosis factor-alpha exhibit potentiated antitumor effects against Ha-ras-transformed murine tumor via inhibition of tumor-induced angiogenesis. Int J Cancer. 1999;81:560-567
64. Cho SJ, Kim JS, Kim JM, Lee JY, Jung HC, Song IS. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer. 2008;123:951-957
65. Millar DA, Bowles S, Windvogel SL, Louw J, Muller CJF. Effect of Rooibos (Aspalathus linearis) extract on atorvastatin-induced toxicity in C3A liver cells. J Cell Physiol. 2020 p: 27
66. Attalah Nee Rezkallah C, Thongkum A, Zhu C, Chen QM. Resveratrol for protection against statin toxicity in C2C12 and H9c2 cells. J Biochem Mol Toxicol. 2020;34:e22484
67. Li HH, Lin CL, Huang CN. Neuroprotective effects of statins against amyloid β-induced neurotoxicity. Neural Regen Res. 2018;13:198-206
68. Cirigliano A, Amelina A, Biferali B, Macone A, Mozzetta C, Bianchi MM. et al. Statins interfere with the attachment of S. cerevisiae mtDNA to the inner mitochondrial membrane. J Enzyme Inhib Med Chem. 2020;35:129-137
69. Ghadge GA, Gourishetti K, Chamallamudi MR, Nampurath GK, Nandakumar K, Kumar N. Sesamol protects MIN6 pancreatic beta cells against simvastatin-induced toxicity by restoring mitochondrial membrane potentials. 3 Biotech. 2020 10: 149
70. Paseban M, Butler AE, Sahebkar A. Mechanisms of statin-induced new-onset diabetes. J Cell Physiol. 2019;234:12551-12561
71. Bellosta S, Corsini A. Statin drug interactions and related adverse reactions: an update. Expert Opin Drug Saf. 2018;17:25-37
72. Kobayashi M, Chisaki I, Narumi K, Hidaka K, Kagawa T, Itagaki S. et al. Association between risk of myopathy and cholesterol-lowering effect: a comparison of all statins. Life Sci. 2008;82:969-975
Author contact
![]() Corresponding author: Ze Yu, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Heng Zhi Gang Road 78, Guangzhou, 510095, China, E-mail: gjw94edu.cn; zeyunfucom.
Corresponding author: Ze Yu, Affiliated Cancer Hospital and Institute of Guangzhou Medical University, Heng Zhi Gang Road 78, Guangzhou, 510095, China, E-mail: gjw94edu.cn; zeyunfucom.