Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(14):2712-2726. doi:10.7150/ijbs.48169 This issue Cite
Research Paper
SIRT1/FOXO3a axis plays an important role in the prevention of mandibular bone loss induced by 1,25(OH)2D deficiency
Haiyun Chen1, Xiaoqing Hu1, Renlei Yang1, Guoping Wu1, Qian Tan2, David Goltzman3, Dengshun Miao1 ![]()
1. The Research Center for Aging, Affiliated Friendship Plastic Surgery Hospital of Nanjing Medical University, Nanjing 210029, China.
2. Department of Burns and Plastic Surgery, The Drum Tower Clinical Medical College, Affiliated Drum Tower Hospital, Nanjing Medical University, Nanjing 210008, China.
3. Calcium Research Laboratory, McGill University Health Centre and Department of Medicine, McGill University, Montreal, Quebec H3A 1A1, Canada.
Received 2020-5-14; Accepted 2020-8-11; Published 2020-8-19
Abstract

It has been reported that 1,25 dihydroxyvitamin D [1,25(OH)2D] deficiency leads to the loss of mandibular bone, however the mechanism is unclear. We investigated whether the Sirt1/FOXO3a signaling pathway is involved in this process. Using a 1,25(OH)2D deficiency model induced by genetic deletion in mice of 25-hydroxyvitamin D-1α hydroxylase [1α(OH)ase-/- mice]. We first documented a sharp reduction of expression levels of Sirt1 in the 1α(OH)ase-/- mice in vivo. Next, we demonstrated dose-dependent upregulation of Sirt1 by treatment with exogenous 1,25(OH)2D3 in vitro. We then identified a functional VDR binding site in the Sirt1 promoter. By crossing Prx1-Sirt1 transgenic mice with 1α(OH)ase-/- mice we demonstrated that the overexpression of Sirt1 in mesenchymal stem cells (MSCs) greatly improved the 1α(OH)ase-/- mandibular bone loss phenotype by increasing osteoblastic bone formation and reducing osteoclastic bone resorption. In mechanistic studies, we showed, in 1α(OH)ase-/- mice, decreases of Sirt1 and FoxO3a, an increase in oxidative stress as reflected by a reduction of the antioxidant enzymes peroxiredoxin1 (Prdx1), SOD1 and SOD2 expression, and an increase of markers for osteocyte senescence and senescence associated secretory phenotypes (SASP), including β-galactosidase (β-gal), p16, p53 and p21. The targeted overexpression of Sirt1 in the 1α(OH)ase-/- mice restored the expression levels of these molecules. Finally, we demonstrated that a Sirt1 agonist can upregulate FOXO3a activity by increasing deacetylation and nuclear translocation. Overall, results from this study support the concept that targeted increases in Sirt1/FOXO3a signaling levels can greatly improve the bone loss caused by 1,25(OH)2D deficiency.
Keywords: mandibular bone, Sirt1 promoter, mesenchymal stem cells, antioxidant enzymes peroxiredoxin1
Introduction
Vitamin D plays an essential role in regulating calcium and phosphate metabolism and maintaining a healthy mineralized skeleton. Vitamin D is obtained from sunlight exposure, and/or dietary foods and supplements. Once vitamin D enters the circulation it is converted by 25-hydroxylase in the liver to 25-hydroxyvitamin D [25(OH)D], which is further converted by the 25-hydroxyvitamin D-1α-hydroxylase [1α(OH)ase] in the kidneys and other tissues to the active form, 1,25-dihydroxyvitamin D [1,25(OH)2D]. 1,25(OH)2D binds to its nuclear vitamin D receptor (VDR) to exert its physiologic functions, which include promotion of intestinal calcium and phosphate absorption, renal tubular calcium reabsorption, and calcium mobilization from bone [1, 2]. Vitamin D deficiency is a major global public health problem in all age groups. It is estimated that 1 billion people worldwide have vitamin D deficiency or insufficiency [3]. Vitamin D deficiency has been assumed to be an osteoporotic risk factor, and the vitamin D level of 25-hydroxyvitamin D [25(OH)D] has been reported to be negatively correlated with the occurrence of osteoporosis in Western populations [4, 5]. Our previous studies, using homozygous knockout of 1α(OH)ase (Cyp27b1) (in 1α(OH)ase-/- mice) or double homozygous knockout of 1α(OH)ase and the parathyroid hormone gene (Pth) or double homozygous knockout of 1α(OH)ase and the calcium sensitive receptor gene (Casr), demonstrated that endogenous and exogenous 1,25(OH)2D could directly stimulate osteoblastic bone formation via the VDR [6-10]. Recently we further demonstrated that 1,25(OH)2D plays an anti-osteoporotic role via transcriptionally up-regulating expression of Bmi1 and Ezh2 in osseous cells via the VDR, subsequently, inhibiting oxidative stress and DNA damage, inactivating the p16 and p19 signaling pathways, inducing osseous cell senescence and SASP, facilitating osteoblastic bone formation, and inhibiting osteoclastic bone resorption [11, 12]. These studies focused on long bone or vertebrae, however, the role and mechanism of 1,25(OH)2D in preventing mandibular bone loss [13] is less well investigated.
The bone formation processes in dentin and dental alveolar bone of the mandibles are distinctly regulated in the 1,25(OH)2D-deficient state relative to the processes driving formation in trabecular bone in long bones. The trabecular bone volume and osteoblast numbers are markedly increased in long bones [7, 8, 14], but not in mandibles of 1α(OH)ase-/- mice with increased circulating parathyroid hormone [13]. Significant associations exist between periodontal health and intake of vitamin D and calcium, and dietary supplementation with calcium and vitamin D may improve periodontal health, increase bone mineral density in the mandible and inhibit alveolar bone resorption [15, 16]. Our previous studies had demonstrated that 1,25(OH)2D deficiency resulted in mandibular bone loss in mice [13, 17], however, the underlying mechanism is unclear.
Sirt1 is a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase and is considered as a longevity gene [18]. Global Sirt1 transgenic mice are protected against the metabolic decline related to aging [19-22]. Recent animal studies indicate that longevity-associated Sirt1 may serve as an attractive pharmacological target for the treatment of osteoporosis and other bone related disorders. Pre-clinical studies demonstrated that mice treated with Sirt1 agonists show protection against age-related and post-menopausal models of osteoporosis [23, 24]. Conversely, Sirt1 knockout models display low bone mass phenotypes associated with increased bone resorption and decreased bone formation [25, 26]. To investigate the role in osteogenesis of Sirt1 in mesenchymal stem cells (MSCs), we constructed a transgenic mouse model by overexpressing Sirt1 in MSCs using Prx1 as a promoter (Sirt1Tg) [27, 28]. The transcription factor Prx1, a marker of MSCs, is widely used for mesenchymal lineage-specific knockdown or overexpression of target genes [29]. The osteogenesis of alveolar bone is also under the control of Prx1 [30]. Results from our studies showed that overexpression of Sirt1 in MSCs protected against bone loss induced by Bmi1 deficiency not only in long bone, but also in mandibles [27, 28]. Pre-treatment with 1,25(OH)2D3 attenuated ROS-induced damage and MSC dysfunction by up-regulating the expression levels of Sirt1 in MSCs [31]. However, it is unclear whether 1,25(OH)2D3 can regulate Sirt1 expression at a transcriptional level and whether overexpression of Sirt1 in MSCs protects against bone loss in mandibles induced by 1,25(OH)2D3 deficiency.
To answer the above questions, we examined the effect of 1,25(OH)2D deficiency on Sirt1 expression in mandibular tissue. In view of the fact that the vitamin D receptor (VDR), Prx1 and Sirt1 are all expressed in mandibular mesenchyme [28, 32, 33], we generated 1α(OH)ase-/- mice with Sirt1 overexpression in MSCs (Sirt1Tg1α(OH)ase-/-). We compared mandibular phenotypes of these mice with those of Prx1-driven Sirt1 transgenic (Sirt1Tg), 1α(OH)ase-/-, and WT mice to assess whether overexpression of Sirt1 in MSCs corrected 1,25(OH)2D deficiency-induced mandibular bone loss. We also examined whether the Sirt1 agonist resveratrol promoted osteogenesis of mandibular MSCs by increasing FOXO3a deacetylation and nuclear translocation using in vitro treatment with the Sirt1 agonist resveratrol or the Sirt1 inhibitor Ex527 or with FoxO3a knockdown.
Materials and Methods
Animals
We used three types of mutant mouse models in this study: (1) Sirt1Tg mice are transgenic mice that express highly elevated Sirt1 under the control of the 2.4kb Prx1 promoter and were generated in our laboratory and genotyped as described previously [28]. (2) 1α(OH)ase-/- mice, were generated through the breeding of heterozygous mice, and genotyped as we previously described [34]. (3) Sirt1Tg1α(OH)ase-/-mice were generated by crossing Sirt1Tg1α(OH)ase+/- double-mutant mice. All mice were maintained on a C57BL/6J background. All 1α(OH)ase-/- models were fed a high calcium/phosphate (rescue) diet. Six-month-old male wild-type (WT), Sirt1Tg, 1α(OH)ase-/- and Sirt1Tg/1α(OH)ase-/- littermates were used in this study. All mice used were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University and maintained in the SPF Laboratory Animal Center of Nanjing Medical University.
Radiography and micro-computed tomography (micro-CT)
Mandibles were removed and dissected free of all soft tissues for radiography and micro-computed tomography (micro-CT) as described [13].
Histology and histochemistry
Mandibles were removed and histologically processed as described previously [13, 35]. Paraffin sections were stained with hematoxylin and eosin (H&E), or histochemically for total collagen, alkaline phosphatase (ALP) activity or tartrate-resistant acid phosphatase (TRAP) activity as previously described [13, 28]. The cortical thickness of mandibles corresponding to the apex of the first molar was measured on H&E stained sections.
Immunohistochemical staining
Immunohistochemical staining was performed for osterix, SOD1, SOD2, FoxO3a, β-galactosidase (β-gal), p16 and p21 using the avidin-biotin-peroxidase complex technique as described [12]. Briefly, dewaxed and rehydrated paraffin-embedded sections were incubated with 6% hydrogen peroxide to block endogenous peroxidase activity and then washed in PBS (pH 7.6). The slides were then incubated at 4°C overnight with the primary antibodies to osterix (Abcam), SOD1/2 (Abcam), FOXO3a (Abcam), β-gal (Abcam), p16 (Abcam), and p21 (Santa Cluz). After rinsing with PBS for 15 min, tissues were incubated with secondary antibody (biotinylated goat anti-rabbit IgG and goat anti-mouse IgG, Sigma). Sections were then washed and incubated with the Vectastain Elite ABC reagent (Vector Laboratories) for 30 min. Staining was done using 3,3-diaminobenzidine (2.5mg/ml) followed by counterstaining with Mayer's hematoxylin.
Human mandible-derived bone marrow mesenchymal stem cells (BM-MSCs) cultures
Human mandible-derived BM-MSCs were donated by Dr. Wen Sun of Jiangsu dental hospital. Detailed written informed consent was obtained from all volunteers in accordance with protocols approved by the Human Subjects Institutional Review Board of Nanjing Medical University (Approval ID 2016115) [28]. Human mandible-derived BM-MSCs were cultured in α-MEM containing 10% FBS with 50 μg/ml ascorbic acid and 100nM dexamethasone (Science cell Research Laboratories, Carlsbad, CA, USA) in the absence or presence of 10-9-10-8M or 10 μM resveratrol and 10 μM Sirt1 inhibitor Ex527 for 5-10 days. Cells were then stained cytochemically for ALP or senescence associated β-galactosidase (SA-β-gal) as previously described [12], or by immunofluorescence staining for FOXO3a, or for ethynyl deoxyuridine (EdU) incorporation assay. For immunofluorescence staining, cells were fixed with 4% paraformaldehyde and blocked in 10% normal goat serum containing 0.5% BSA and 0.1% Triton X-100 for 1 h and then stained overnight with anti-FOXO3a antibody (Cell Signaling Technology, Danvers, MA, USA;1:500) at 4°C. After rinsing with PBS for 15 min, cells were incubated with DyLight594 goat anti-rabbit IgG (Multi Sciences, Nanjing, China) at room temperature. Slides were mounted with mounting medium containing DAPI (Sigma-Aldrich, St. Louis, MO), and images were taken with a fluorescence microscope (Leica, Wetzlar, Germany). The EdU incorporation assay is the gold standard for detecting cell proliferation. EdU can be incorporated into newly synthesized DNA strands during DNA replication, and cell proliferation can then be detected by fluorescence intensity. For the EdU incorporation assay, human mandible-derived BM-MSCs were cultured in α-MEM containing 10% FBS and 50 μM EdU for 2 h at 37 °C before fixation. Detection of EdU was achieved with the Cell-Light EdU Apollo 567 (catalog no. C10310-1; RiboBio), according to the manufacturer's protocol.
Immunoprecipitation and Western blot analysis
Whole-cell lysates or cell nuclear lysates were prepared from human mandible-derived BM-MSCs. Immunoprecipitation experiments were extracted by using Pierce™ Crosslink Magnetic IP Kit (Thermo Scientific, Waltham, MA, USA) and a Nuclear and Cytoplasmic Protein Extraction Kit (Thermo Scientific, Waltham, MA, USA). An immunoprecipitation assay was performed as recommended by the supplier. Proteins extracted from 2×106 human mandible-derived BM-MSCs was mixed with 1 μg of antibody and prewashed Protein A/G, then incubated overnight. The bound antigens were eluted from the beads by boiling samples for 10 min. Eluted samples were obtained from SDS-PAGE. Immunoblotting was carried out as previously described [36]. Proteins were extracted from mandibles as previously [13]. Primary antibodies against Sirt1, FOXO3a, acetylated-lysine, Histone H3, and β-actin (Cell Signaling Technology, Danvers, MA, USA) were used. The immunoreactive bands were visualized by ECL chemiluminescence (Amersham) and analyzed by the Scion image Beta 4.02 (Scion, National Institutes of Health).
ChIP-qPCR
Human mandible-derived BM-MSCs were cultured to perform chromatin immunoprecipitation (ChIP) analyses of VDR recruitment by using an anti-VDR antibody (Abcam) and SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology, Danvers, MA, USA). ChIP analyses were performed as recommended by the supplier to identify the VDR recruitment. The primers used to amplify the 153bp fragment of the human FOXO3a gene promoter were 5'-AGCCCGCTTCTACACTCTGA-3' (forward) and 5'-ATTCTACGATCCGTGCCCCA-3' (reverse). The PCR products were electrophoresed on 2% agarose gels, and visualized by ethidium bromide staining.
RNA isolation and real-time RT-PCR
Total RNA was extracted from the cultured BM-MSCs and mandibles using Trizol reagent (Invitrogen) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized using Synthesis SuperMix (Invitrogen). Real-time RT-PCR was carried out using an Agilent Real-time System. Gapdh was amplified at the same time to normalize gene expression. Each experiment was repeated three times to determine relative gene expression differences. The sequence-specific primers of human and mice are displayed in Table 1.
Primers used in this study for real time RT-PCR
| Name | S/AS | sequence | species | Tm (°C) | Length (bp) |
|---|---|---|---|---|---|
| P53 | S | GTCACAGCACATGACGGAGG | mouse | 60 | 129 |
| AS | TCTTCCAGATGCTCGGGATAC | ||||
| P21 | S | CCTGGTGATGTCCGACCTG | mouse | 58 | 103 |
| AS | CCATGAGCGCATCGCAATC | ||||
| P16 | S | CGCAGGTTCTTGGTCACTGT | mouse | 60 | 127 |
| AS | TGTTCACGAAAGCCAGAGCG | ||||
| IL-1a | S | CGAAGACTACAGTTCTGCCATT | mouse | 58 | 126 |
| AS | GACGTTTCAGAGGTTCTCAGAG | ||||
| IL-1b | S | GCAACTGTTCCTGAACTCAACT | mouse | 60 | 89 |
| AS | ATCTTTTGGGGTCCGTCAACT | ||||
| MMP3 | S | ACATGGAGACTTTGTCCCTTTTG | mouse | 60 | 192 |
| AS | TTGGCTGAGTGGTAGAGTCCC | ||||
| MMP13 | S | CTTCTTCTTGTTGAGCTGGACTC | mouse | 60 | 173 |
| AS | CTGTGGAGGTCACTGTAGACT | ||||
| IL-6 | S | TGTATGAACAACGATGATGCACTT | mouse | 60 | 197 |
| AS | ACTCTGGCTTTGTCTTTCTTGTTATCT | ||||
| SOD1 | S | GGTGAACCAGTTGTGTTGTC | mouse | 57 | 203 |
| AS | CCGTCCTTTCCAGCAGTC | ||||
| SOD2 | S | GACCTGCCTTACGACTATG | mouse | 55 | 166 |
| AS | GAAGAGCGACCTGAGTTG | ||||
| FOXO3a | S | CTGGGGGAACCTGTCCTATG | mouse | 60 | 210 |
| AS | TCATTCTGAACGCGCATGAAG | ||||
| Duox1 | S | AAAACACCAGGAACGGATTGT | mouse | 60 | 123 |
| AS | AGAAGACATTGGGCTGTAGGG | ||||
| Duox2 | S | AAGTTCAAGCAGTACAAGCGAT | mouse | 60 | 104 |
| AS | TAGGCACGGTCTGCAAACAG | ||||
| NQO1 | S | AGGATGGGAGGTACTCGAATC | mouse | 58 | 144 |
| AS | AGGCGTCCTTCCTTATATGCTA | ||||
| CAT1 | S | CAGGTGCGGACATTCTAC | mouse | 55 | 202 |
| AS | TTGCGTTCTTAGGCTTCTC | ||||
| GAPDH | S | CTTGCCAGACACAGATGATCG | mouse | 60 | 163 |
| AS | GGGGACAGAAGTTGAGTTTC | ||||
| VDR-Sirt1-chip | S | TATGGAGTCACAGTGTGCCAG | human | 60 | 192 |
| AS | GCGTGAGCTATCTAGCCGT |
Construction of promoter-reporter plasmids and dual-luciferase transient expression assay
Analysis of the transcriptional activity of the VDR was conducted. The full coding sequence of VDR was amplified and cloned into a pCDNA3.1 vector used as an effector vector. The promoters of the Sirt1 gene were cloned into the GV238-LUC reporter vector. The plasmid pGL3-Sirt1 containing -agagtaca- in the promoter region of the human Sirt1 gene linked to the promoterless firefly luciferase gene, and a mutational plasmid pGL3-Sirt1-mut in which -agagtaca- was changed into -ctctccat-, were constructed. Human mandible-derived BM-MSCs cells were plated in 24-well Falcon plates at a density of 100,000 cells/well in α-MEM with 10% FBS 24 hours prior to transient transfection. Plasmids were transfected in individual wells using Opti-MEM and liposome according to the manufacturer`s protocol. Briefly, each well was treated with pcDNA3.0-VDR and pGL3-Sirt1 or pGL3-Sirt1-mut (with or without 10-8M 1,25(OH)2D3), along with 40 ng of Renilla reniformis luciferase. The Renilla reniformis luciferase plasmid allows for constitutive, low-level expression to monitor DNA transfection efficiency. Forty-eight hours post-transfection, the cells were lysed in 1× passive lysis buffer (Promega, Madison, WI) and the lysates were collected. Each lysate was then analyzed sequentially for Firefly and Renilla luciferase activity using a Dual-Luciferase Assay Kit (Promega Corp., Madison, WI). All operating procedures followed the instructions provided by the reagent kit. The mean ratio of Firefly/Renilla for 6 biological replicates (wells) was calculated for each experimental treatment group.
RNA interference and plasmid transfection
Human small interfering RNAs (siRNAs) for FOXO3a and the control siRNAs (siCtrl) were designed and synthesized by Invitrogen (Grand Island, NY, USA). The FOXO3a plasmid and its negative control vectors were purchased from GeneChem (Shanghai, China). Lipofectamine 3000 (L3000015, Invitrogen) and used for transfection according to the manufacturer's instructions.
Statistical analysis
Results are expressed as mean ± S.E.M. Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). Comparisons between two groups were analyzed using a two-tailed unpaired Student's t-test. Comparisons among three or more groups were performed using two-way ANOVA followed by Dunnett's postdoc multiple comparisons. P values <0.05 were considered statistically significant.
Results
1,25(OH)2D3 up-regulates Sirt1 expression via VDR-mediated transcription
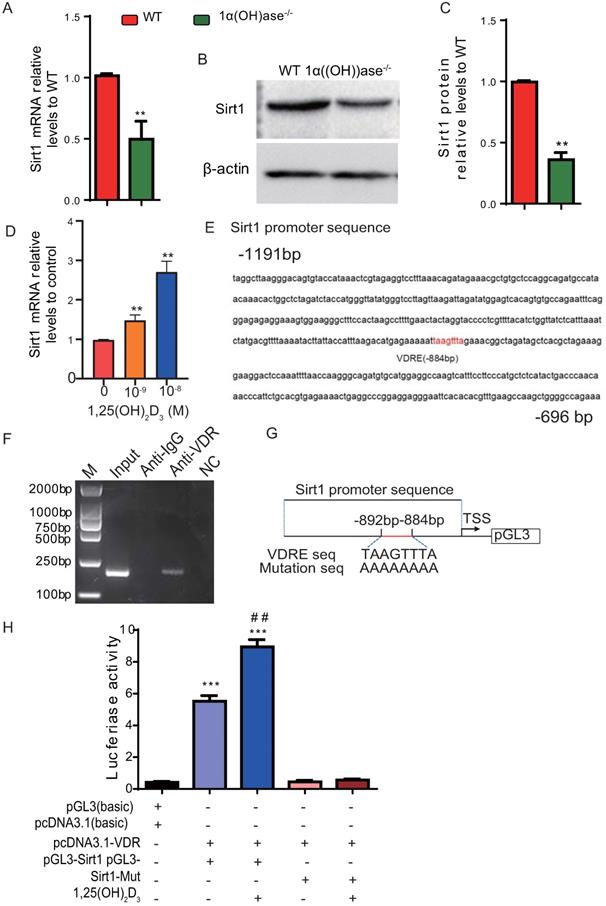
We previously reported that 1,25(OH)2D deficiency in the 1α(OH)ase-/- mice accelerated alveolar bone loss [13, 17]. In this study we assessed whether the alveolar bone loss induced by 1,25(OH)2D deficiency is mediated via the NAD-dependent deacetylase sirtuin-1 (Sirt1). We first examined the alteration of Sirt1 expression at both mRNA and protein levels in mandibular tissue from wild-type and 1α(OH)ase-/- mice. Our results demonstrated that Sirt1 mRNA and protein expression levels in mandibular tissue were down-regulated dramatically in 1α(OH)ase-/- mice compared with their wild-type littermates (Figs. 1A-C). We then examined the effect of exogenous 1,25(OH)2D3 on Sirt1 expression in mandible-derived bone marrow-mesenchymal stem cells (BM-MSCs); we found that 1,25(OH)2D3 significantly up-regulated mRNA expression levels of Sirt1 in a dose-dependent manner in mandible derived BM-MSCs cultured with 10-9-10-8 M 1,25(OH)2D3 for 24 hrs (Fig. 1D). To assess whether 1,25(OH)2D3 regulates Sirt1 via the VDR at a transcriptional level, a VDRE like sequence at position -884 in the 5'-flanking regions of the Sirt1 promoter, retrieved from the NCBI human genome data base, was identified by computer-aided analysis (Fig. 1E). Results of a ChIP-PCR assay demonstrated that the VDR could directly bind to the Sirt1 promoter on the predicted binding site (Fig. 1F). Using human genomic DNA as a template, PCR was used to amplify the whole promoter segment -863 to -1055. The PCR products without and with mutated VDRE were then cloned into pGL3-basic vectors (pGL3-Sirt1, pGL3-Sirt1-mut), which were transiently transfected into mandible derived BM-MSCs (Fig. 1G). Luciferase reporter assays demonstrated that luciferase expression levels were increased significantly in mandible derived BM-MSCs transfected with a Sirt1 binding sequencing plasmid compared with the vehicle, and the luciferase expression levels were augmented by 1,25(OH)2D3; in contrast, luciferase activity was not increased in mandible derived BM-MSCs transfected with a pGL3-Sirt1 mutant plasmid compared with the vehicle (Fig. 1H). Therefore, 1,25(OH)2D3 up-regulated Sirt1 expression via VDR-mediated gene transcription.
1,25(OH)2D3 up-regulates Sirt1 expression through VDR-mediated transcription. (A) Real-time RT-PCR analysis of mandible tissue extracts from 6-month-old WT and 1α(OH)ase-/- mice showing expression of Sirt1. (B) Western blotting was performed on tissue extracts for Sirt1 protein levels and (C) the Sirt1 protein relative levels to β-actin were assessed by densitometric analysis. (D) mRNA expression levels of Sirt1 in mandible derived BM-MSCs cultured with 10-9-10-8 M 1,25(OH)2D3 for 24 hrs, demonstrated by real-time RT-PCR. (E) The VDR-like element (-taagttta-) motif in the Sirt1 promoter was detected by bioinformatics analysis. (F) ChIP analysis was performed using a negative control immunoglobulin G (IgG) or anti-VDR antibody in human mandible derived BM-MSCs cells. (G) Schematic diagram of the structure of the pGL3-Sirt1 promoter reporter plasmid and of the mutant pGL3-Sirt1 promoter reporter plasmid. (H) Sirt1 promoter activity was measured by a dual luciferase reporter assay. Human mandible derived BM-MSCs were transfected with an expression plasmid of the human VDR (pcDNA3.1) or an empty vector (pcDNA3.1) and transfected with the pGL3-Sirt1 promoter, a pGL3-Sirt1 promoter-mutant, or the pGL3 basic vector. A Renilla reniformis luciferase reporter plasmid was co-transfected for 48h. 1,25(OH)2D3 at 10-8 M or vehicle were added following the transfections. Luciferase activity was collected and normalized to Renilla's values at 48 h after transfection. Values are mean ± s. e. m. of 3 determinations per group. *: P < 0.05, **: P < 0.01 compared with WT or control group; ##: P < 0.01 compared with 1,25(OH)2D3 untreated group.
Overexpression of Sirt1 in MSCs prevents 1,25(OH)2D deficiency-induced alveolar bone loss
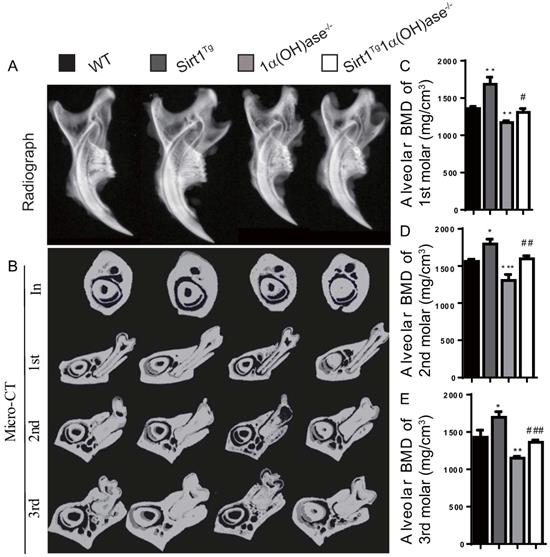
To investigate whether 1,25(OH)2D plays a role in prevention of mandibular bone loss via Sirt1 in vivo, we used Prx1-driven Sirt1 transgenic mice (Sirt1Tg) and generated a mouse model that overexpressed Sirt1 in MSCs on a 1α(OH)ase-/- background (Sirt1Tg1α(OH)ase-/-). We then compared their mandibular phenotypes with 1α(OH)ase-/-, and wild-type littermates at 6 months of age. By radiography and micro-CT scanning analyses, we found that bone mineral density (BMD) was increased significantly in Sirt1Tg mice, but it was decreased significantly in 1α(OH)ase-/- mice and was not altered in Sirt1Tg1α(OH)ase-/- mice at the alveolar bone of 1st, 2nd and 3rd molars compared with their wild-type littermates. Nevertheless, it was increased significantly in Sirt1Tg1α(OH)ase-/- mice compared with 1α(OH)ase-/- littermates (Figs. 2A-E). These results demonstrated that overexpression of Sirt1 in MSCs could prevent 1,25(OH)2D deficiency induced alveolar bone loss in vivo.
Overexpression of Sirt1 in MSCs prevents 1,25(OH)2D deficiency induced mandibular bone loss. (A) Representative radiographs of mandibles from 6-month-old wild-type (WT), Sirt1Tg, 1α(OH)ase-/- and Sirt1Tg1α(OH)ase-/- mice. (B) Representative images of micro-CT-scanned sections through the incisors in front of the first molar (In), the first (1st), second (2nd) and third (3rd) molars. (C-E) Alveolar BMD of 1st, 2nd and 3rd molars. Values are mean ± S.E.M. of 6 determinations per group. *: P <0.05, **: P < 0.01, ***: P < 0.001 compared with WT mice; #: P <0.05, ##: P <0.01, ###: P <0.001 compared with 1α(OH)ase-/- mice.
Overexpression of Sirt1 in MSCs rescues 1,25(OH)2D deficiency-induced alveolar bone turnover defects
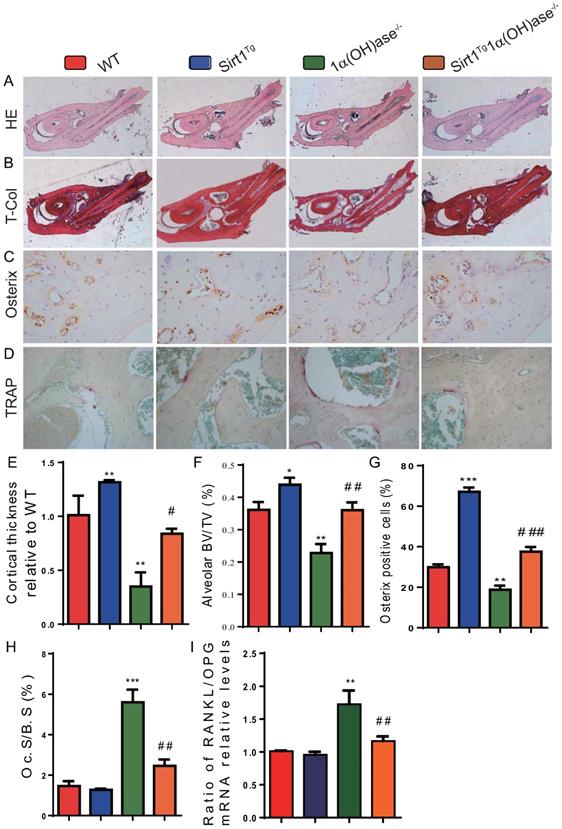
To determine whether 1,25(OH)2D deficiency-induced alveolar bone loss prevented by overexpression of Sirt1 in MSCs was associated with alterations of alveolar bone turnover, alveolar bone formation and resorption were examined using histopathological analyses. The results revealed that the cortical bone volume of mandibles, alveolar bone volume and osterix positive cells were increased significantly in Sirt1Tg mice, but were significantly decreased in 1α(OH)ase-/- mice and were not altered in Sirt1Tg1α(OH)ase-/- mice compared with their wild-type littermates; however, these parameters were increased significantly in Sirt1Tg1α(OH)ase-/- mice compared with 1α(OH)ase-/- littermates (Figs. 3A-C, E-G). In contrast, TRAP positive osteoclastic surface and the ratio of RANKL/OPG mRNA levels were increased dramatically in 1α(OH)ase-/- mice but were not significantly increased in Sirt1Tg1α(OH)ase-/- mice compared with their wild-type littermates; these indices were decreased significantly in Sirt1Tg1α(OH)ase-/- mice compared with 1α(OH)ase-/- littermates (Figs. 3D, H & I). These results implied that overexpression of Sirt1 in MSCs prevented 1,25(OH)2D deficiency induced alveolar bone loss by stimulating osteoblastic alveolar bone formation and inhibiting osteoclastic alveolar bone resorption.
Overexpression of Sirt1 in MSCs rescues 1,25(OH)2D deficiency induced alveolar bone turnover defects. Representative micrographs of decalcified paraffin-embedded sections through the first molars and the incisors from 6-month-old WT, Sirt1Tg, 1α(OH)ase-/- and Sirt1Tg1α(OH)ase-/- mice were stained with (A) hematoxylin and eosin (HE), (B) histochemically for total collagen (T-Col), (C) immunohistochemically for osterix and (D) histochemically for tartrate-resistant acid phosphatase (TRAP). (E) Cortical thickness, (F) alveolar bone volume relative to tissue volume (BV/TV, %). (G) Osterix-positive cells relative to total cells. (H) Osteoclastic surface relative to bone surface (Oc.S/B.S, %). (I) RT-PCR of tissue extracts of mandibles for expression of RANKL and OPG. Messenger RNA expression assessed by real-time RT-PCR is calculated as a ratio relative to Gapdh, and expressed as ratio of RANKL/OPG mRNA relative levels to WT mice. Values are mean ± S.E.M. of 6 determinations per group. *: P <0.05, **: P < 0.01, ***: P < 0.001 compared with WT mice; #: P <0.05, ##: P <0.01, ###: P <0.001 compared with 1α(OH)ase-/- mice.
Overexpression of Sirt1 in MSCs largely corrects 1,25(OH)2D deficiency-induced oxidative stress
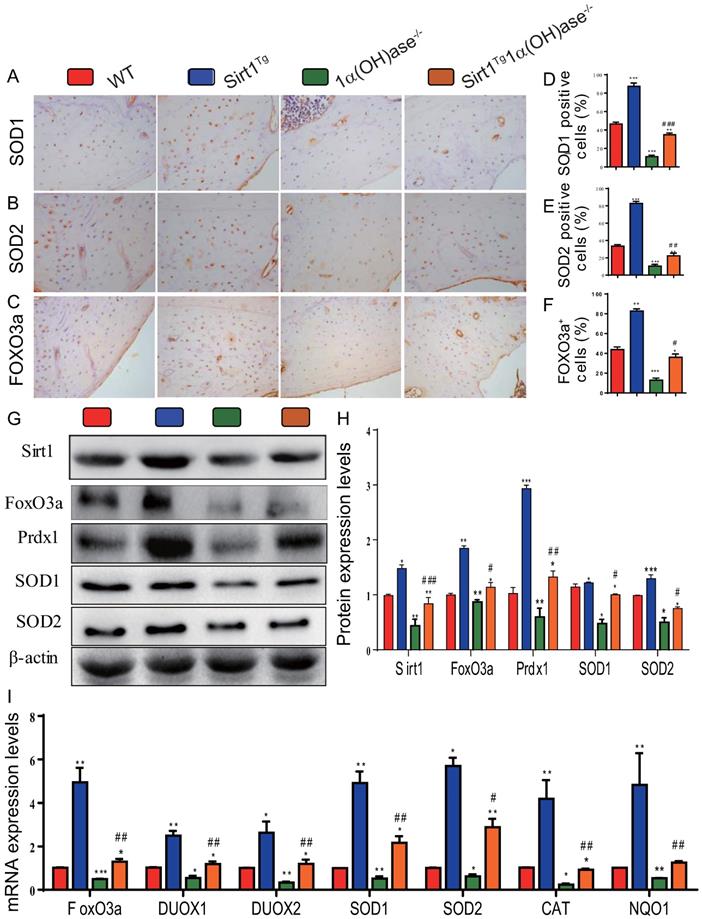
To determine whether 1,25(OH)2D deficiency-induced alveolar bone loss prevented by overexpression of Sirt1 in MSCs was associated with alterations of redox balance, we examined the expression levels of antioxidant enzyme proteins and genes in mandibular tissue using immunohistochemistry, Western blots and real-time RT-PCR. The results showed that the percentage of superoxide dismutase (SOD)1, SOD2 and forkhead box O3 (FOXO3a) positive cells (Figs. 4A-F), the protein expression levels of Sirt1, FOXO3a, peroxiredoxin1 (Prdx1), SOD1 and SOD2 (Figs. 4G & H), and the mRNA expression levels of FOXO3a, dual oxidase 1 (Duox1), dual oxidase 2 (Duox2), SOD1, SOD2, catalase (CAT) and quinone oxidoreductase-like protein 1 (NQO1) (Fig. 4I) were increased dramatically in Sirt1Tg mice, but were decreased in 1α(OH)ase-/- mice compared with their wild-type littermates; they were also increased significantly in Sirt1Tg1α(OH)ase-/- mice compared with 1α(OH)ase-/- littermates. These results indicate that overexpression of Sirt1 in MSCs largely corrected 1,25(OH)2D deficiency-induced oxidative stress by up-regulating the expression levels of antioxidant enzymes.
Overexpression of Sirt1 in MSCs largely corrects 1,25(OH)2D deficiency-induced oxidative stress. Representative micrographs of decalcified paraffin-embedded sections through the first molars and the incisors from 6-month-old WT, Sirt1Tg, 1α(OH)ase-/- and Sirt1Tg/1α(OH)ase-/- stained immunohistochemically for (A) SOD1, (B) SOD2 and (C) FOXO3a. The percentages of (D) SOD1 positive cells, (E) SOD2 positive cells and (F) FOXO3a positive cells. (G) Western blots of mandibular extracts were carried out for expression of Sirt1, FOXO3a, Prdx1, SOD1 and SOD2. (H) Protein levels relative to β-actin were assessed by densitometric analysis and expressed as percentage of the levels of WT mice. (I) RT-PCR of tissue extracts of mandibles for expression of FoxO3, Duox1, Duox2, SOD1, SOD2, CAT and NQO1. Messenger RNA expression assessed by real-time RT-PCR is calculated as a ratio relative to Gapdh, and expressed relative to WT mice. Values are mean ± S.E.M. of 6 determinations per group. *: P <0.05, **: P < 0.01, ***: P < 0.001 compared with WT mice; #: P <0.05, ##: P <0.01, ###: P <0.001 compared with 1α(OH)ase-/- mice.
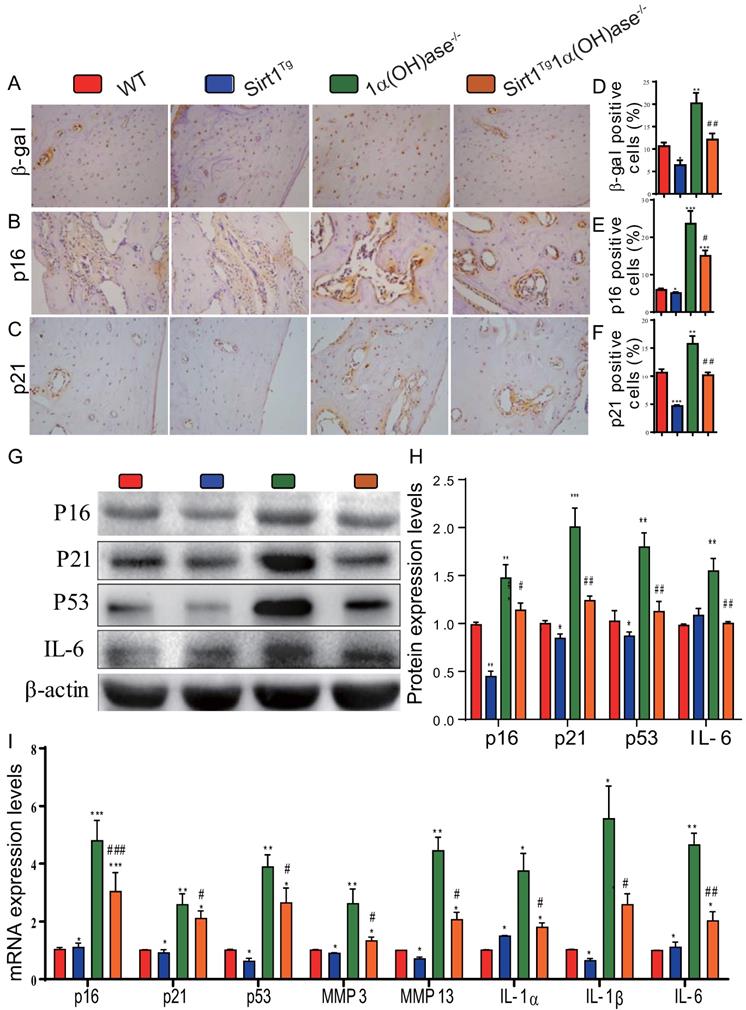
Overexpression of Sirt1 in MSCs largely corrects 1,25(OH)2D deficiency-induced osteocyte senescence and SASP
To determine whether 1,25(OH)2D deficiency-induced alveolar bone loss prevented by overexpression of Sirt1 in MSCs was associated with alterations of osteocyte senescence and senescence associated secretory phenotypes (SASP), we examined the expression levels of cell senescence and SASP related proteins and genes in mandibular tissue using immunohistochemistry, Western blots and real-time RT-PCR. The results showed that the percentage of β-galactosidase (β-gal), p16 and p21 positive osteocytes (Figs. 5A-F), the protein expression levels of p16, p21, p53 and IL-6 (Figs. 5G & H), and the mRNA expression levels of p16, p21, p53, MMP3, MMP13, IL-1α, IL-1β and IL-6 (Fig. 5I) were all increased significantly in 1α(OH)ase-/- mice compared with their wild-type littermates, however, they were decreased significantly in Sirt1Tg1α(OH)ase-/- mice compared with 1α(OH)ase-/- littermates. These results indicate that overexpression of Sirt1 in MSCs largely corrected 1,25(OH)2D deficiency-induced osteocyte senescence and SASP.
Overexpression of Sirt1 in MSCs largely corrects 1,25(OH)2D deficiency-induced osteocyte senescence and SASP. Representative micrographs of decalcified paraffin-embedded sections through the first molars and the incisors from 6-month-old WT, Sirt1Tg, 1α(OH)ase-/- and Sirt1Tg1α(OH)ase-/- stained immunohistochemically for (A) β-gal, (B) p16, and (C) p21. The percentages of (D) β-gal positive cells, (E) p16 positive cells and (F) p21 positive cells. (G) Western blots of mandibular extracts were carried out for expression of p16, p21, p53 and IL-6. (H) Protein levels relative to β-actin were assessed by densitometric analysis and expressed as percentage of the levels of WT mice. (I) RT-PCR of tissue extracts of mandibles for expression of p16, p21, p53, MMP3, MMP13, IL-1α, IL-1β and IL-6. Messenger RNA expression assessed by real-time RT-PCR is calculated as a ratio relative to Gapdh, and expressed relative to WT mice. Values are mean ± S.E.M. of 6 determinations per group. *: P <0.05, **: P < 0.01, ***: P < 0.001 compared with WT mice; #: P <0.05, ##: P <0.01, ###: P <0.001 compared with 1α(OH)ase-/- mice.
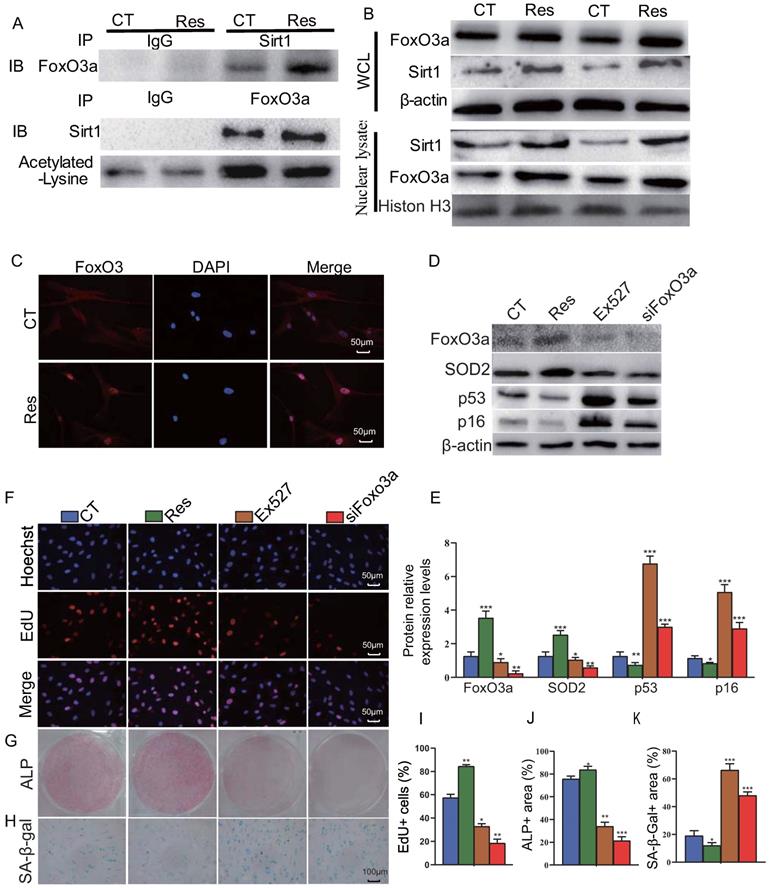
Sirt1 agonist resveratrol promotes osteogenesis of mandible BM-MSCs by increasing FOXO3a deacetylation and nuclear translocation
To further examine the mechanism of Sirt1 action, human mandible-derived BM-MSCs were treated with 10 μM resveratrol for 6 hours, and then whole cell lysates or nuclear lysates were immunoprecipitated with anti-Sirt1 or anti-FOXO3a antibodies or IgG control and blotted with anti-FOXO3a or anti-Sirt1 antibodies or anti-acetylated lysine antibody. The subcellular localization of FOXO3a was detected by immunofluorescence staining. The results of co-immunoprecipitation studies showed that Sirt1 and FOXO3a were able to bind each other, while resveratrol could enhance their binding, and reduce significantly the acetylation level of FOXO3a (Fig. 6A). Results from Western blots also showed that resveratrol treatment could up-regulate the expression levels of Sirt1 and FOXO3a in total cell protein and more dramatically in nuclear protein (Fig. 6B). Immunofluorescence staining revealed that resveratrol treatment significantly increased the nuclear localization of FOXO3a (Fig. 6C). To further explore the role of Sirt1 to mediate the osteogenesis of BM-MSCs via FOXO3a, we used 10 μM resveratrol and 10 μM Sirt1 inhibitor Ex527 to treat human mandible-derived BM-MSCs, or knocked down FOXO3a in these cells. Alterations of BM-MSC proliferation, osteogenic differentiation and senescence were then analyzed by EdU incorporation, and cytochemical staining for ALP or SA-β-gal respectively. The results showed that resveratrol significantly up-regulated FOXO3a and SOD2 expression levels and significantly down-regulated p16 and p53 expression levels, while both Ex527 and FOXO3a knockdown significantly down-regulated FOXO3a and SOD2 expression levels, and significantly up-regulated p16 and p53 expression levels (Figs. 6D & E). The percentage of EdU-positive cells and the areas of ALP-positive cells were increased significantly in the resveratrol-treated group, decreased significantly in the Ex527-treated group, and decreased more significantly in the FOXO3a knockdown group (Figs. 6F, G, I & J). In contrast, the areas of SA-β-gal-positive cells were decreased significantly in the resveratrol-treated group, increased significantly in the Ex527-treated group and the FOXO3a knockdown group (Figs. 6H & K). These results indicate that the Sirt1 agonist resveratrol can increase the interaction between Sirt1 and FOXO3a, reduce the level of FOXO3a acetylation, and promote its nuclear translocation, thereby promoting the proliferation and osteogenic differentiation of BM-MSCs and inhibiting their senescence.
Sirt1 agonist resveratrol promotes osteogenesis of mandible-derived BM-MSCs by increasing FOXO3a deacetylation and nuclear translocation. (A) Human mandible-derived BM-MSCs were cultured in the absence (CT) or presence of 10μM resveratrol (Res) and whole cell lysates (WCL) were subjected to IP with anti-Sirt1 or anti-FOXO3a antibody and blotted with anti-FOXO3a or anti-Sirt1 or acetylated-lysine antibody. IB, immunoblot. (B) Whole cell lysates (WCL) and nuclear lysates were blotted with anti-Sirt1 or anti-FOXO3a antibody. (C) Representative micrographs of immunofluorescence staining for FOXO3a (Red), DAPI (Blue) and merge. (D) Human mandible-derived BM-MSCs were cultured in the absence (CT) or presence of 10μM resveratrol (Res) or 10 μM Sirt1 inhibitor Ex527 or transfected with siFOXO3a plasmid; whole cell lysates were blotted with anti-FOXO3a or anti-SOD2 or anti-p53 or anti-p16 antibody. (E) Protein levels relative to β-actin were assessed by densitometric analysis and expressed as a percentage of the levels of control (CT) cultures. (F) Representative micrographs of immunofluorescence staining for Hoechst (Blue), EdU (Red) and merge. Representative images of cells stained cytochemically for (G) ALP and (H) SA-β-gal. (I) The percentages of: EdU-positive cells, (J) ALP positive area and (K) SA-β-gal-positive area. Values are mean ± S.E.M. of 3 determinations per group. *: P <0.05, **: P < 0.01, ***: P < 0.001 compared with control (CT) cultures.
Discussion
Our previous studies had demonstrated that 1,25(OH)2D deficiency resulted in bone loss in mouse mandibles [13, 17]. Other studies have reported that pre-treatment with 1,25(OH)2D3 attenuated the ROS-induced damage and MSC dysfunction by up-regulating the expression levels of Sirt1 in MSCs [31]. In this study, we investigated the mechanism whereby Sirt1 mediated mandibular bone loss induced by 1,25(OH)2D deficiency. We found that the expression of Sirt1 at both mRNA and protein levels was remarkably downregulated in mandibular tissue of 1α(OH)ase-/- mice compared with that of wild-type littermates. We also demonstrated that the mRNA levels of Sirt1 were up-regulated in mandible-derived BM-MSCs from wild-type mice when exposed to 1,25(OH)2D3 in a dose dependent manner. Next, using bioinformatic analysis, ChIP, and dual luciferase assay, we showed that 1,25(OH)2D3 could regulate Sirt1 expression directly via the VDR at a transcriptional level. These results therefore imply that 1,25(OH)2D up-regulates Sirt1 expression in mandible derived BM-MSCs through VDR-mediated transcription.
Previously studies have shown that Sirt1 haploinsufficient female mice exhibited substantially reduced bone formation and mass with reduced osteogenesis [25] and MSC specific deletion of Sirt1 resulted in lower cortical and trabecular bone volume with reduced MSC osteogenesis [26]. Recently we generated transgenic mice overexpressing Sirt1 in MSCs driven by the Prx1 gene and demonstrated that overexpression of Sirt1 in MSCs stimulated osteogenesis of both long bone- and mandible-derived BM-MSCs and increased both long bone and alveolar bone volume [27, 28]. Furthermore, overexpression of Sirt1 in MSCs protected against long bone loss induced by Bmi1 deficiency and alveolar bone loss induced by Bmi1 or estrogen deficiency [27, 28]. In our current study, we generated 1α(OH)ase-/- mice with Sirt1 overexpression in MSCs. We found that Sirt1 constitutively expressed in MSCs can produce normalization of many of the parameters that were altered in mandibles at 6 months of age in 1α(OH)ase-/- mice, and therefore corrected the mandibular bone loss phenotype caused by 1,25(OH)2D3 deficiency by increasing mandibular BMD and alveolar bone mass through enhancing osteoblastic bone formation and reducing osteoclastic bone resorption. This indicates that providing Sirt1, a downstream target of 1,25(OH)2D, can bypass the requirement for 1,25(OH)2D/VDR and normalize the mandibular phenotype of 1,25(OH)2D deficiency.
Oxidative stress levels of osteon structures are increased with age [37] and mouse models of osteoporosis are usually accompanied by increased oxidative stress [38, 39]. 1,25(OH)2D3 reduced oxidative stress in human prostate epithelial cells [40] and human endothelial cells by activation of Nrf2-antioxidant signals [41]. Cellular senescence is a process in which a cell enters permanent cell cycle arrest, and senescent cells acquire a senescence-associated secretory phenotype (SASP) [42]. SASP includes pro-inflammatory cytokines, growth factors, chemokines, and matrix remodeling enzymes [43]. Senescent cells cause or aggravate the development of aging-related diseases through their growth arrest phenotype and SASP factors. 1,25(OH)2D3 has been reported to delay cellular senescence with increased nuclear translocation of VDR in human BM-MSCs [44]. We recently demonstrated that 1,25(OH)2D3 plays an anti-aging role by suppressing oxidative stress, DNA damage, p16-Rb and p53-p21 pathways, and cell senescence and SASP via upregulating Nrf2 [34]. We also demonstrated that 1,25(OH)2D deficiency accelerated age-related osteoporosis by down-regulating Bmi1 and Ezh2 via VDR-mediated transcription, decreasing H3K27me3 and enhancing p16 and p19 transcription, thus inhibiting the proliferation and osteogenesis of BM-MSCs and osteoblastic bone formation, and inducing osteocyte senescence, SASP and osteoclastic bone resorption. In contrast, supplementation of exogenous 1,25(OH)2D3, overexpression of Bmi1 in MSCs and the deletion of p16 corrected the osteoporotic phenotype caused by 1,25(OH)2D deficiency [11, 12]. In the current study, we demonstrated that 1,25(OH)2D deficiency induced oxidative stress, cellular senescence and SASP in mandibular tissue, by reducing the percentage of SOD1/2 and FOXO3a positive cells, the protein expression levels of Sirt1, FOXO3a, Prdx1 and SOD1/2 and the mRNA expression levels of FOXO3a, Duox1/2, SOD1/2, CAT and NQO1; significant augmentation of the percentage of β-gal, p16 and p21 positive osteocytes, the protein expression levels of p16, p21, p53 and IL-6, and the mRNA expression levels of p16, p21, p53, MMP3, MMP13, IL-1α, IL-1β and IL-6, were also observed. In contrast, these abnormalities induced by 1,25(OH)2D deficiency were largely corrected through Sirt1 overexpression in MSCs. Therefore, our results indicate that 1,25(OH)2D can prevent mandibular bone loss by inhibiting oxidative stress, cellular senescence and SASP via Sirt1 mediation.
Previous studies have reported that Sirt1 caused the deacetylation of histones at the Sost promoter, thus repressing Sost expression and preventing its negative regulation of osteoblast bone anabolic activity. In addition, Sirt1 promotes the activation of Runx2 and suppression of NF-κB signaling, thereby both stimulating osteoblastogenesis and inhibiting osteoclastogenesis [45-47]. It has also been demonstrated that Sirt1 deacetylated FOXO3a resulting in activation of transcriptional activity of FOXO3a and increased expression of its downstream targets, thus regulating cell proliferation, anti-oxidation and apoptosis [48-50]. We previously demonstrated that Sirt1 can deacetylate FOXO3a and up-regulate SOD2 expression, thereby inhibiting oxidative stress in bony tissue, inhibiting the senescence of BM-MSCs and stimulating the differentiation of long bone derived BM-MSCs into osteoblasts [27]. In the current study, we examined possible mechanisms of Sirt1 in stimulating osteogenesis of mandible derived BM-MSCs by the treatment of Sirt1 agonist resveratrol in cultures in vitro. Our results revealed that resveratrol not only up-regulated Sirt1 and FOXO3a expression in total protein and more dramatically in nuclear protein, but also increased the interaction between Sirt1 and FOXO3a, reduced the level of FOXO3a acetylation, and promoted its nuclear translocation. Furthermore, resveratrol promoted the proliferation and osteogenic differentiation of human mandible BM-MSCs and inhibited their senescence, whereas both Sirt1 inhibitor Ex527 and FOXO3a knockdown blocked the above actions of resveratrol. Our results also showed that resveratrol up-regulated the FOXO3a target gene SOD2, down-regulated p53 and p16 expression, whereas Sirt1 inhibitor Ex527 and FOXO3a knockdown down-regulated SOD2 expression, but up-regulated p53 and p16 expression. Previous studies have also shown that the expression of FOXO3a induced significant downregulation of p53 and p16 [51]; the precise mechanisms whereby FOXO3a regulated p53 and p16, however, remains to be investigated. Our results therefore indicate that the Sirt1 agonist resveratrol can increase the interaction between Sirt1 and FOXO3a, reduce the level of FOXO3a acetylation, and promote its nuclear translocation, and down-regulation of p53 and p16, thereby promoting the proliferation and osteogenic differentiation of BM-MSCs and inhibiting their senescence.
Overall, therefore, the results of this study provide a model that suggests that 1,25(OH)2D deficiency induced mandibular bone loss by down-regulating Sirt1 expression through VDR-mediated transcription, increasing FOXO3a acetylation and oxidative stress, activating p16 and p53 signaling, inducing cellular senescence and SASP, and inhibiting osteogenic differentiation of mandibular BM-MSCs with decreased osteoblastic bone formation, and increased osteoclastic bone resorption; These processes can be rescued by overexpression of Sirt1 in MSCs. Our findings have potential translational significance, in that modification of MSCs genetically to overexpress Sirt1, or administration of resveratrol to activate Sirt1, might be novel approaches for the treatment of mandibular bone loss.
Acknowledgements
This work was supported by grants from National Key R&D Program of China (2018YFA0800800 to DM) and from the National Natural Science Foundation of China (81230009 and 81730066 to DM) and from the Canadian Institutes of Health Research (CIHR) (PJT-152963 to DG).
Author contributions
D.M. and D.G. conceived the project. H.C. and X.H. performed most of the experiments, analyzed and compiled the data. R.Y., G.W. and Q.T. helped with experiments and data analysis. H.C., D.M. and D.G. participated in writing or editing the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Feldman D, P JM. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. BoneKEy reports. 2014;3:510
2. Glorieux FH, St-Arnaud R. Molecular cloning of (25-OH D)-1 alpha-hydroxylase: an approach to the understanding of vitamin D pseudo-deficiency. Recent progress in hormone research. 1998;53:341-9 discussion 50
3. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-81
4. Jagelaviciene E, Vaitkeviciene I, Silingaite D, Sinkunaite E, Daugelaite G. The Relationship between Vitamin D and Periodontal Pathology. Medicina. 2018 54
5. van Schoor NM, Visser M, Pluijm SM, Kuchuk N, Smit JH, Lips P. Vitamin D deficiency as a risk factor for osteoporotic fractures. Bone. 2008;42:260-6
6. Liu J, Lv F, Sun W, Tao C, Ding G, Karaplis A. et al. The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and PTH. PLoS Genet. 2011;7:e1002294
7. Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN. et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754-66
8. Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN. et al. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498-503
9. Shu L, Ji J, Zhu Q, Cao G, Karaplis A, Pollak MR. et al. The calcium-sensing receptor mediates bone turnover induced by dietary calcium and parathyroid hormone in neonates. J Bone Miner Res. 2011;26:1057-71
10. Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous 1,25-dihydroxyvitamin D3 exerts a skeletal anabolic effect and improves mineral ion homeostasis in mice that are homozygous for both the 1alpha-hydroxylase and parathyroid hormone null alleles. Endocrinology. 2006;147:4801-10
11. Sun H, Qiao W, Cui M, Yang C, Wang R, Goltzman D. et al. The Polycomb Protein Bmi1 Plays a Crucial Role in the Prevention of 1,25(OH)2 D Deficiency-Induced Bone Loss. J Bone Miner Res. 2019
12. Yang R, Chen J, Zhang J, Qin R, Wang R, Qiu Y. et al. 1,25-Dihydroxyvitamin D protects against age-related osteoporosis by a novel VDR-Ezh2-p16 signal axis. Aging Cell. 2019: e13095.
13. Liu H, Guo J, Wang L, Chen N, Karaplis A, Goltzman D. et al. Distinctive anabolic roles of 1,25-dihydroxyvitamin D(3) and parathyroid hormone in teeth and mandible versus long bones. J Endocrinol. 2009;203:203-13
14. Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135-41
15. Cozzolino M, Lu Y, Finch J, Slatopolsky E, Dusso AS. p21WAF1 and TGF-alpha mediate parathyroid growth arrest by vitamin D and high calcium. Kidney Int. 2001;60:2109-17
16. Zittermann A. Vitamin D in preventive medicine: are we ignoring the evidence? Br J Nutr. 2003;89:552-72
17. Gong A, Chen J, Wu J, Li J, Wang L, Goltzman D. et al. 1,25-Dihydroxyvitamin D deficiency accelerates alveolar bone loss independent of aging and extracellular calcium and phosphorus. J Periodontol. 2018
18. Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED. et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416-30
19. Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L. et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333-41
20. Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A. et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759-67
21. Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O. et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3
22. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
23. Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N. et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157-68
24. Feng J, Liu S, Ma S, Zhao J, Zhang W, Qi W. et al. Protective effects of resveratrol on postmenopausal osteoporosis: regulation of SIRT1-NF-kappaB signaling pathway. Acta Biochim Biophys Sin (Shanghai). 2014;46:1024-33
25. Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I. et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology. 2011;152:4514-24
26. Simic P, Zainabadi K, Bell E, Sykes DB, Saez B, Lotinun S. et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med. 2013;5:430-40
27. Sun W, Qiao W, Zhou B, Hu Z, Yan Q, Wu J. et al. Overexpression of Sirt1 in mesenchymal stem cells protects against bone loss in mice by FOXO3a deacetylation and oxidative stress inhibition. Metabolism. 2018;88:61-71
28. Wang H, Hu Z, Wu J, Mei Y, Zhang Q, Zhang H. et al. Sirt1 Promotes Osteogenic Differentiation and Increases Alveolar Bone Mass via Bmi1 Activation in Mice. J Bone Miner Res. 2019;34:1169-81
29. Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T. et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008;14:306-14
30. ten Berge D, Brouwer A, Korving J, Martin JF, Meijlink F. Prx1 and Prx2 in skeletogenesis: roles in the craniofacial region, inner ear and limbs. Development. 1998;125:3831-42
31. Zhou J, Wang F, Ma Y, Wei F. Vitamin D3 contributes to enhanced osteogenic differentiation of MSCs under oxidative stress condition via activating the endogenous antioxidant system. Osteoporos Int. 2018;29:1917-26
32. Bailleul-Forestier I, Davideau JL, Papagerakis P, Noble I, Nessmann C, Peuchmaur M. et al. Immunolocalization of vitamin D receptor and calbindin-D28k in human tooth germ. Pediatr Res. 1996;39:636-42
33. Doufexi AE, Mina M. Signaling pathways regulating the expression of Prx1 and Prx2 in the chick mandibular mesenchyme. Dev Dyn. 2008;237:3115-27
34. Chen L, Yang R, Qiao W, Zhang W, Chen J, Mao L. et al. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell. 2019: e12951.
35. Wang H, Lv C, Gu Y, Li Q, Xie L, Zhang H. et al. Overexpressed Sirt1 in MSCs Promotes Dentin Formation in Bmi1-Deficient Mice. J Dent Res. 2018;97:1365-73
36. Chen L, Yang R, Qiao W, Yuan X, Wang S, Goltzman D. et al. 1,25-Dihydroxy vitamin D prevents tumorigenesis by inhibiting oxidative stress and inducing tumor cellular senescence in mice. Int J Cancer. 2018;143:368-82
37. Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free radical biology & medicine. 2007;43:477-503
38. de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H. et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276-9
39. Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H. et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45-53
40. Bao BY, Ting HJ, Hsu JW, Lee YF. Protective role of 1 alpha, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. Int J Cancer. 2008;122:2699-706
41. Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926-30
42. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS biology. 2008;6:2853-68
43. Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology. 2014;15:627-42
44. Klotz B, Mentrup B, Regensburger M, Zeck S, Schneidereit J, Schupp N. et al. 1,25-dihydroxyvitamin D3 treatment delays cellular aging in human mesenchymal stem cells while maintaining their multipotent capacity. PloS one. 2012;7:e29959
45. Edwards JR, Perrien DS, Fleming N, Nyman JS, Ono K, Connelly L. et al. Silent information regulator (Sir)T1 inhibits NF-kappaB signaling to maintain normal skeletal remodeling. J Bone Miner Res. 2013;28:960-9
46. Shakibaei M, Buhrmann C, Mobasheri A. Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-kappaB ligand (RANKL) activation of NF-kappaB signaling and inhibit osteoclastogenesis in bone-derived cells. J Biol Chem. 2011;286:11492-505
47. Tseng PC, Hou SM, Chen RJ, Peng HW, Hsieh CF, Kuo ML. et al. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J Bone Miner Res. 2011;26:2552-63
48. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011-5
49. Ferguson D, Shao N, Heller E, Feng J, Neve R, Kim HD. et al. SIRT1-FOXO3a regulate cocaine actions in the nucleus accumbens. J Neurosci. 2015;35:3100-11
50. Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A. et al. Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation. 2013;127:1692-701
51. Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C. et al. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem. 2008;283:25692-705
Author contact
![]() Corresponding author: Dengshun Miao, M.D., Ph.D. The Research Center for Aging, Affiliated Friendship Plastic Surgery Hospital of Nanjing Medical University, Nanjing Medical University, Nanjing, Jiangsu, 210029, The People's Republic of China. Tel. & Fax: 011-86-25-8686-9377; E-mail: dsmiaoedu.cn.
Corresponding author: Dengshun Miao, M.D., Ph.D. The Research Center for Aging, Affiliated Friendship Plastic Surgery Hospital of Nanjing Medical University, Nanjing Medical University, Nanjing, Jiangsu, 210029, The People's Republic of China. Tel. & Fax: 011-86-25-8686-9377; E-mail: dsmiaoedu.cn.