Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
NEDD4-1
NEDD4L
ITCH
WWP1
WWP2
SMURF1
SMURF2
Conclusions and Prospects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(14):2727-2740. doi:10.7150/ijbs.48437 This issue Cite
Review
E3 Ubiquitin ligase NEDD4 family‑regulatory network in cardiovascular disease
Ying Zhang1, Hao Qian1, Boquan Wu1, Shilong You1, Shaojun Wu1, Saien Lu1, Pingyuan Wang2, Liu Cao3 ![]() , Naijin Zhang1
, Naijin Zhang1 ![]() , Yingxian Sun1
, Yingxian Sun1 ![]()
1. Department of Cardiology, the First Hospital of China Medical University, Shenyang, Liaoning, P.R. China.
2. Staff scientist, Center for Molecular Medicine National Heart Lung and Blood Institute, National Institutes of Health, the United States.
3. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning, China.
Received 2020-5-20; Accepted 2020-8-6; Published 2020-8-21
Abstract

Protein ubiquitination represents a critical modification occurring after translation. E3 ligase catalyzes the covalent binding of ubiquitin to the protein substrate, which could be degraded. Ubiquitination as an important protein post-translational modification is closely related to cardiovascular disease. The NEDD4 family, belonging to HECT class of E3 ubiquitin ligases can recognize different substrate proteins, including PTEN, ENaC, Nav1.5, SMAD2, PARP1, Septin4, ALK1, SERCA2a, TGFβR3 and so on, via the WW domain to catalyze ubiquitination, thus participating in multiple cardiovascular-related disease such as hypertension, arrhythmia, myocardial infarction, heart failure, cardiotoxicity, cardiac hypertrophy, myocardial fibrosis, cardiac remodeling, atherosclerosis, pulmonary hypertension and heart valve disease. However, there is currently no review comprehensively clarifying the important role of NEDD4 family proteins in the cardiovascular system. Therefore, the present review summarized recent studies about NEDD4 family members in cardiovascular disease, providing novel insights into the prevention and treatment of cardiovascular disease. In addition, assessing transgenic animals and performing gene silencing would further identify the ubiquitination targets of NEDD4. NEDD4 quantification in clinical samples would also constitute an important method for determining NEDD4 significance in cardiovascular disease.
Keywords: NEDD4 E3 ligases, post translation modification, ubiquitin proteasome system, cardiovascular disease
Introduction
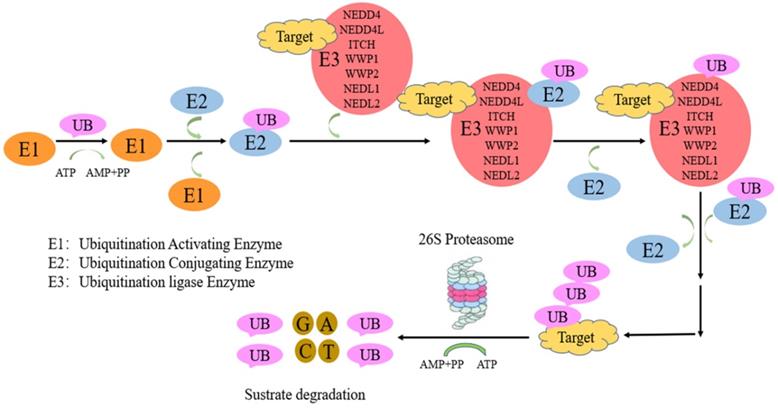
Protein ubiquitination, a critical post translation modification, is necessary for ubiquitin-associated protein degradation by the ubiquitin proteasome system (UPS). UPS system comprise ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3). First, ubiquitin activation is performed by E1. Then, E2 transfers ubiquitin to E3. Finally, E3 catalyses the covalent binding of ubiquitin to the target protein [1]. E3 ligases are divided into three main families: zinc-binding RING finger adaptor (a recently discovered gene) [2], HECT (homologous with the carboxyl end of E6AP) catalytic [3] and U-box (a modified cyclic motif) families [4]. E3 enzymes involve in substrate recognition and ubiquitin transfer to a single, multiple Lys (mono-/multi-ubiquitination) or as a poly-Ub chain (poly-ubiquitination) residues of the substrate (HECT family) [5] or easing ubiquitin transfer from E2 to the target protein (RING family). All seven lysine (K6, K11, K27, K29, K33, K48 and K63) and the N-terminal M1 residues constitute linkage points during chain elongation [6], although lys48 and lys63 are mostly involved. K48-, K11-, K29 linked poly-Ub chains direct substrates to proteasomal degradation [7, 8], while mono-ubiquitination and K63-linked poly-Ub chains have non-proteolytic functions [9]. In contrast, K63-linked chains control “proteasome-independent” events, including inflammation-related signaling pathways, DNA repair, endocytosis, and selective autophagy [10,11]. Nedd4-family E3 members mostly synthesize K63-bound poly-Ub chains [12, 13], unlike other HECT ligases. This is followed by protein recognition and degradation by the 26S proteasome [5, 14]. The neuronal precursor cell-expressed developmentally downregulated 4 (NEDD4) family constitutes an important group in the HECT group [15, 16].
NEDD4-1 and NEDD4-like (NEDD4L or NEDD4-2) enzymes represent E3 ubiquitin-protein ligases of the HECT family [17, 18]. NEDD4 family enzymes are conserved from yeast to humans [19]. As shown in Figure 1, the NEDD4 family contains nine members in humans: NEDD4-1 (RPF1), NEDD4L (NEDD4-2), ITCH/atropine-1 interaction protein 4 (AIP4), WW domain-containing E3 ubiquitin protein ligase 1 (WWP1), WWP2/Atropine-1-interacting protein 2 (AIP2), NEDL1 (HECW1), NEDL2 (HECW2), SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1) and SMURF2 [20]. NEDD4 family enzymes structure consists of N-terminal C2 domain (Ca2+/phosphorlipase and membrane-binding; mediating Ca2+-associated targeting to the cell membrane substrate recognition [21]), 2-4 WW domain and catalytic C-terminal domain (HECT; 350 residues controlling ubiquitin binding to e-NH2 groups of lysine residues on protein substrates [5, 18, 22]). The WW domain catalyzes protein ubiquitination and catabolism (or endocytosis) [15]. The WW domain recognizes and binds to predominantly proline-rich sequences on substrate proteins [23], e.g., PPxY (where x represents any amino acid) [24], PPLP [25], PR [26] and phosphoserine/threonine (pS/pT) residues preceding proline [27]. The catalytic HECT domain consists of an "n-leaf" and a catalytic cysteine residue in a "c-leaf" which confers the ligase its catalytic activity [28]. NEDD4 E3 ligases differ in function because of distinct WW domains [29] and different substrates [16]. In the past few years, the ubiquitin proteasome system has been attributed key roles in regulating diverse cardiovascular disease [30, 31], including atherosclerosis, familial cardiopathy, idiopathic dilated cardiomyopathy and myocardial ischemia [32]. Here we review the current evidence accumulated concerning the cardiovascular role of NEDD4 family members in cardiovascular disease.
The ubiquitination-proteasome of NEDD4 E3 ligases pathway. Proteins targeted for UPS-induced degradation by NEDD4 E3 ligases comprise polyubiquitin chain by a process involving three proteins, including ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3). First, ubiquitin activation is performed by E1. Then, E2 transfers ubiquitin to E3. Finally, NEDD4 E3 ligases catalyses the covalent binding of ubiquitin to the target protein.
NEDD4-1
NEDD4-1 function
NEDD4-1 was first isolated from mouse neural progenitor cells in 1992, with reduced mRNA levels during mouse brain development. NEDD4-1 is widely expressed in the heart, lung, brain, somite, kidney, and other tissues. It may be involved in many human cell functions [33]. NEDD4-1 catalyses the degradation of its substrate by polyubiquitination at K48 and K63 [34-36] or by single ubiquitination of K6 or K27 [37-38], indicating that NEDD4-1 plays a variety of regulatory roles through single/multiple ubiquitination.
The role of NEDD4-1 in myocardial reperfusion injury
Myocardial reperfusion injury involves tissue damage occurring with blood supply return to the cardiac tissue following ischemia or lack of oxygen, causing inflammation [39]. NEDD4-1 expression is reduced in the late stage of ischemia/reperfusion (I/R), thereby attenuating its protective effects against cell death and cardiac I/R injury. In addition, activation of the AKT serine/threonine kinase (AKT) pathway protects the heart from I/R injury [40]. NEDD4-1 promotes nuclear trafficking of active AKT. Phosphatase and Tensin Homolog (PTEN) represents a critical suppressor of Phosphatidylinositol-3-Kinase (PI3K) signaling and is controlled by NEDD4-1 via polyubiquitination [41-42]. PTEN-associated AKT inhibition is suppressed by NEDD4-1, while AKT signaling is activated to protect against I/R-induced cell damage and apoptotic cell death, as demonstrated by decreased BAX and cleaved-caspase3/7 levels and elevated BCL2 amounts. Therefore, NEDD4-1 protects the myocardium from I/R induced apoptosis by activating PI3K/Akt pathway. This regulatory pathway provides a novel insight into reducing the intraoperative injury in patients with myocardial infarction.
The role of NEDD4-1 in heart development
In embryonic day 10.5 (E10.5) mice, NEDD4-1 is expressed in the pharyngeal and gill arches near the heart. Nedd4 knockout mice show severe heart and vascular defects in the second of pregnancy, leading to fetal death, accompanied by significant heart defects and vascular system abnormalities. In particular, outflow tract defects in knockout animals display a double outlet right ventricle [43]. Nedd4 knockout mice also have endocardial cushion defects. Abnormalities are also present in the vascular system of Nedd4 knockout mice. In particular, the cephalic plexus vein in some knockout embryos is abnormal. In addition to these significant cardiovascular defects, Nedd4 knockout embryos exhibit delayed maturation of the lungs, where Nedd4 is highly expressed [44].
The role of NEDD4-1 in Vascular Calcification
Vascular calcification represents a major complication of atherosclerosis, chronic renal failure, diabetes, cardiovascular disease and other pathological conditions. More and more evidences show that vascular calcification likens osteogenesis [45-47]. Among TGFβ superfamily proteins, BMP2 and TGFβ1 can be targeted for the treatment of multiple bone disorders. In previous study, NEDD4-1 suppression in SM22α+ mouse tissues resulted in deformed aortic structures. Meanwhile, Vitamin D-associated aorta vascular calcification was markedly elevated in Nedd4-null animals compared with wild type littermates. In addition, methylation of human NEDD4 gene promoter is remarkably enhanced in individuals with atherosclerosis. Further research indicated that NEDD4-1 E3 ligase is an important BMP/Smad signaling inhibitor, via polyubiquitination-associated degradation of C-terminal phosphorylated Smad1 (pSmad1) activated by TGF-β. Thus, dysregulated or dysfunctional NEDD4-1 E3 ligase could contribute to vascular calcification in VSMCs through induction of bone generating signals in the process of atherosclerosis progression [48].
NEDD4L
NEDD4L function
NEDD4L on human chromosome 18q21 contains the WW and HECT domains as similar to other NEDD4 genes. In mice, Nedd4L shows homology with Nedd4-1, but has no C2 domain in the N-terminus region. In addition, human NEDD4L and mouse nedd4-2 are homologous [49,50]. Nedd4L is widely expressed during mouse development and in adult tissues, especially in the liver, kidney, heart, brain and lung [51,52]. Furthermore, NEDD4L plays a vital role in hypertension and arrhythmia.
The role of NEDD4L in hypertension
Hypertension represents an important risk factor for cardiovascular ailments, including myocardial infarction, stroke, heart failure and kidney disease. It reflects salt-sensitivity or resistance when blood pressure response to high- or low-salt diets intake varies substantially [53-54]. Epithelial sodium channel (ENaC) suppression in the kidney, lung via the NEDD4 family is important in fluid and electrolyte homeostasis. The ENaC significantly affects kidney Na+ reabsorption [55]. It is a hetero-multimeric membrane protein consisting of the homologous subunits α, β, and γ, each comprising intracellular N and C terminal [55]. NEDD4L was originally identified as a ligand of the ENaC; NEDD4L binds to β- and γ-subunits of the ENaC through the ENaC proline-tyrosine (PY) motif in the C-terminal region, interacting with the WW domain in NEDD4L, followed by ubiquitination and degradation [56-58]. In Liddle's syndrome, an ENaC incorporating variant β or γ subunits that lack the PY motif leads to deficient interaction with the WW domain of NEDD4L, resulting in excessive Na+ reabsorption and hypertension, with the characteristics of salt-sensitivity, hypokalaemia, metabolic alkalosis, and reduced renin activity and aldosterone amounts [22, 59-61]. Nedd4 knockout mice display high blood pressure even with normal diets, an aliment exacerbated with high-salt diets [62]. The IL17A-SGK1/NEDD4L-dependent pathway modulates renal sodium transport by improving renal function in hypertension and other autoimmune disorders. Glucocorticoid Regulated Kinase 1 (SGK1) induced by serum and glucocorticoids triggers a cascade that leads to hypertension by ENaC activation [61-62]. SGK1 phosphorylation of NEDD4L promotes its interaction with the chaperone 14-3-3, thus eliminates the ubiquitination of its target substrates [63-64]. SGK1 phosphorylates NEDD4L on serine 444 [65], which is necessary for the binding of NEDD4L to 14-3-3, and results in a reduction of ENaC ubiquitination and an enhancement of ENaC activity mediated by NEDD4L. SGK1 is activated by WNK1 (WNK Lysine Deficient Protein Kinase 1), which is involved in pseudo-hypoaldosteronism type II hypertension. WNK1 activates SGK1, inhibits NEDD4L, enhances ENaC activity and leads to hypertension [66]. ENaC activity was also observed during the inhibition of NEDD4L by AMP-activated Protein Kinase (AMPK) [65]. These results indicate that IL17A induces an increase in NCC activity through SGK1 phosphorylation and the inhibition of NEDD4L-mediated ubiquitination and NCC degradation. These studies provide a mechanistic link by which the IL17A-SGK1/NEDD4L-dependent pathway modulates renal sodium transport, which may improve renal function in hypertension and other autoimmune disorders [67].
The role of NEDD4L in arrhythmia
Reduced voltage-gated sodium channel (Nav1.5) function and expression supply a slowed conduction substrate for heart arrhythmias. Calcium-associated increases in NEDD4L reduces Nav1.5 levels by ubiquitination. Nav1.5 co-localizes with NEDD4L, and ubiquitin is downregulated in the failing heart in rats. The above findings indicate an important role for NEDD4L in Nav1.5 suppression in heart failure (HF) [68]. In addition, defection of the NEDD4L C2 isoform changes cardiac conduction in the resting state as well as pro-arrhythmic alterations upon acute myocardial infarction (MI). Studies indicated that reduced NEDD4L function results in serious heart arrhythmia via modifications of cardiac ion-channels after transcription. Patients with salt sensitivity and hypertension because of NEDD4L anomalies may be more prone to severe cardiovascular ailments compared with individuals without NEDD4L anomalies. MicroRNA-1(miR-1) regulates genes in the heart and skeletal muscle. Analysis of potential miR-1 target genes identified miR-1 as a direct Nedd4 regulator; in addition, NEDD4L modulates cardiac development in Drosophila [69]. Furthermore, miR-1-associated Nedd4 regulation might help control the trafficking and catabolism of cardiac NEDD4L substrates in the cardiac tissue. However, further study is needed to clarify which substrate ubiquitinated by NEDD4L is involved in arrhythmia.
The role of NEDD4L in cardiac regenerative repair
CircRNAs (circular RNAs) represent potent modulators of cardiac development and disease. Studies have found a new circRNA termed Nfix circRNA (circNfix). Down-regulation of circNfix can promote proliferation and angiogenesis in cardiomyocytes, inhibit the apoptosis of cardiomyocytes after myocardial infarction, alleviate cardiac insufficiency and improve patient prognosis. It was found that circNfix enhances Ybx1 (Y-box binding protein 1) interaction with NEDD4L, and induces Ybx1 degradation by ubiquitination via NEDD4L, which inhibits the expression of cyclin A2 and cyclin B1. In addition, suppression of superenhancer modulated circNfix promotes cardiac regeneration and enhances heart function following myocardial infarction via degradation of Ybx1, which may provide a promising strategy to improve prognosis after MI [70].
ITCH
ITCH function
The ubiquitin E3 ligase ITCH is a 113 kDa protein that contains N-terminal C2 (approximately 116 amino acids) domain, 2-4 protein-protein interaction WW (about 40 amino acid) which recognizes PPxY-(PY)-rich sequences, and C-terminal catalytic HECT domains [24, 71-72]. The seven internal lysine moieties in ubiquitin may serve as chain extension sites [73]. The polyubiquitin chain linked to lys48 is a proteolysis marker for 26S proteasome-induced degradation [74]. ITCH is involved in multiple cell processes by targeting modulators of diverse pathways [75].
The role of ITCH in myocardial infarction and cardiotoxicity
The occurrence of heart failure caused by MI or doxorubicin (DOX) is closely related to progressive left ventricular remodeling caused by oxidative stress [76]. The main cause of excessive oxidative stress in HF cases is believed to be elevated intracellular reactivity related to the antioxidant defence against reactive oxygen free radicals (ROS) [77]. Thioredoxin can be activated by inhibiting Thioredoxin Interacting Protein (TXNIP), which plays an important role in antagonizing ROS in cardiomyocytes [78]. Txnip gene knockout in mice protects the animal's heart, and TXNIP degradation protects the heart of rats with ischemic heart disease [79]. The WW domain recognizes a proxy PPxY sequence of TXNIP. The ITCH-dependent TXNIP UPS is essential for intracellular homeostasis [80]. Overexpression of ITCH by enhancing degradation of TXNIP increases thioredoxin activity, thereby inhibiting the production of ROS, p38MAPK and p53, and suppressing the apoptosis of cardiomyocytes. Cytology confirmed that ITCH can maintain ROS homeostasis in cardiomyocytes [81]. Meanwhile, cardiomyocyte-specific ITCH overexpression and transgenic mouse assays showed that ITCH plays a protective role in DOX and hydrogen peroxide-induced apoptosis and cardiotoxicity by degradation TXNIP. Therefore, ITCH plays a protective role in cardiotoxicity and provides a theoretical basis for MI-targeted therapy [82].
The role of ITCH in diabetic cardiomyopathy
The main pathological changes of diabetic cardiomyopathy are cardiac hypertrophy, apoptosis and myocardial interstitial fibrosis [83]. Calcium Sensitive Receptor (CaSR) belongs to the G protein-coupled receptor (GPCR) C family. Activation of CaSR increases intracellular Ca2+ concentration, upregulates ITCH, increases the ubiquitination level of SMAD7, and augments p-SMAD2 and p-SMAD3 amounts. Calhex 231, a CaSR inhibitor, suppresses the ITCH-ubiquitin proteasome and TGFβ1/SMADs pathway, thus inhibiting the proliferation of cardiac fibroblasts, reducing collagen deposition, and decreasing glucose-induced myocardial fibrosis. This regulatory pathway reveals a novel mechanism for ITCH in myocardial fibrosis, indicating that Calhex 231 may represent a novel drug for treating dilated cardiomyopathy [84].
WWP1
WWP1 function
WWP1 is also referred to as TIUL1 (TGIF interaction ubiquitin ligase 1) or AIP5 (atropine-1-interaction protein 5) [85]. In humans, WWP1 comprises 922 amino acids, with a molecular weight of 110 kDa. WWP1 contains N-terminal C2, 4 tandem WW and C-terminal catalytic HECT domains. WW domains bind to precursor-rich peptide ligands that contain PPxY (PY), PPLP, PPR, and P(S/T) P motifs, respectively [86]. WW domains 1 and 3 in WWP1 interact with the PY motif [87]. The C-terminal has a HECT domain interacting with UbcH5 or UbcH7 (ubiquitin binding enzyme) and controls ubiquitin E3 ligase activity [88]. HECT domain contains a catalytic cysteine with the capability of forming a covalent isopeptide bond with ubiquitin.
The role of WWP1 in cardiac hypertrophy
WWP1 is found at high levels in the myocardium and skeletal muscle. Circular RNAs (circRNAs) may mediate the development of cardiac hypertrophy and the reprogramming of fetal genes [89]. Total RNA from the left ventricular of mice with myocardial hypertrophy induced by isoprenaline hydrochloride was sequenced and the results were assessed by Gene Ontology and Kyoto Encyclopedia analysis. It was determined that ANF and mir-23a are the downstream targets of circRNA-WWP1, indicating that circRNA-WWP1 could inhibit cardiac hypertrophy by downregulating ANF and mir-23a, and that circRNA-WWP1 is a potential new therapeutic target for cardiac hypertrophy [90].
The role of WWP1 in atrial fibrillation
Patients with atrial fibrillation (AF) have a high risk of cardiogenic stroke and further complications [91-92]. Atrial fibrosis represents a critical regulator of integrity in atrial fibrillation, both structurally and functionally. Cardiac fibroblast proliferation is essential for atrial fibrosis and structural remodeling in individuals with atrial fibrillation [93-94]. Transforming growth factor-B1 (TGF-b1) regulates cell proliferation in atrial fibrosis during AF. In addition, miR-21 suppresses endothelial progenitor cell proliferation via WWP1 targeting [95]. WWP1 binds to Smad7 and enhances Smad7 interaction with TGF-b1 receptor, which is degraded [96]. WWP-1 suppresses TGF-b-induced p-Smad2 [97]. Eventually, miR-21 suppresses cardiac fibroblast growth by inhibiting TGF-b1/Smad2 signaling through WWP1 upregulation [98].
WWP2
WWP2 function
WWP2 (about 870 amino acids) consists of N-terminal C2 domain, 4 tandem WW domain, and C-terminal HECT domain [99-100]. WWP2 contains three subtypes, namely, the full-length (WWP2-FL, 870 amino acids), N-terminal subtype (WWP2-N, 336 amino acids), and C-terminal subtype (WWP2-C 440 amino acids) molecules. WWP2-N comprises the C2 and first WW (WW1) domains; WWP2-C encompasses the 4th WW (WW4) and HECT domains, and WWP2-FL has all 3 domains [101]. The HECT domain is structurally bilobal [102], and confers intrinsic ubiquitin ligase activity to WWP2. The WW domain of WWP2 recognizes the target protein through residues in the PY motif or substrate [103-104]. WWP2 forms a self-inhibiting structure via intramolecular interactions of its C2 and HECT domains [105]. WWP2 dose-dependently controls its ligase activity through polyubiquitination via K63 linkage [106].
WWP2 function in myocardial fibrosis
The natural myocardial ischemia injury of dilated cardiomyopathy can cause myocardial fibrosis and eventually lead to heart failure [107-108]. The development of tissue fibrosis involves the transforming growth factor β (TGFβ) superfamily of proteins [109]. TGFβ1 binds directly to its receptor via the downstream effector protein SMADS [110]. Through detailed genetic analysis of diabetic cardiomyopathy, WWP2 and WWP2 mRNA expression in fibroid heart disease is only slightly increased [111]. In primary cardiac fibroblasts, TGFβ1 stimulates the WWP2 N-terminal subtype to enter the nucleus, subsequently enhancing the activity of WWP2-FL to promote interaction with SMAD2, and promoting its mono-ubiquitination, which activates the downstream pro-fibrogenic gene program [112]. Myocardial fibrosis is an important therapeutic target in HF cases [113], and WWP2 represents a regulator of TGFβ/SMAD signalling, which has critical functions in the activation and fibrosis of cardiac fibroblasts. Therefore, WWP2, as a new drug target, is of great significance to control pathological cardiac fibrosis and heart failure, and to improve the clinical prognosis of patients with heart disease. However, more studies are warranted to assess whether WWP2-mediated SMAD2 mono-ubiquitination interferes with or directly regulates SMAD2 complex activity [114], and to explain how WWP2 mono-ubiquitination affects SMAD2 nuclear output and regulation from a mechanical point of view [115].
The role of WWP2 in cardiac remodeling
Cardiac remodeling, including cardiac hypertrophy and fibrosis, underlies HF development. Poly (ADP-Ribose) Polymerase 1 (PARP1) is an important damaging factor in cardiovascular disease, especially cardiac remodeling caused by various factors [77-79]. In cardiac remodeling, cleaved-PARP1 amounts increase, which leads to extreme energy utilization by cardiomyocytes and cell injury [116-118]. WWP2 mostly binds to the BRCT domain of PARP1, and can ubiquitinate its K249 and K418 residues. The ubiquitination level of PARP1 is decreased in MycCre+; WWP2Fl/Fl mice, while the expression level of PARP1 induced by ISO is increased. Therefore, WWP2 can degrade PARP1 and protect from ISO-triggered cardiac remodeling. This provides a foundation for investigating novel approaches for the treatment of cardiac remodeling-associated disease [119].
WWP2 function in vascular endothelial injury
Oxidative stress is important in the pathology of endothelial damage, initiating cardiovascular ailments, including atherosclerosis and hypertension [120-122]. Endothelial/myeloid-specific Wwp2 gene silencing markedly increases endothelial damage and vascular remodeling induced by angiotensin II. WWP2 promotes the degradation of the endothelial damage factor Septin4 via lysine residue 174 (K174), thus inhibiting formation of the Septin4-PARP1 endothelial damage complex [123]. This study showed that the WWP2-septin4 pathway might represent a new target for preventing and treating atherosclerosis and hypertension. This was the first report to confirm WWP2 as the first NEDD4 family member to inhibit endothelial damage and vascular remodeling, providing an insight into IOS-related atherosclerosis and hypertension [124].
SMURF1
SMURF1 function
SMURF1 comprises N-terminal C2, tandem WW and C-terminal HECT domains [15]. SMURF1 is localized intracellularly and recruited substrates, with the HECT domain containing the active site cysteine and interacting with E2 proteins, to form ubiquitin thioesters and promote substrate ubiquitination.
SMURF1's role in pulmonary hypertension
Pulmonary hypertension (PAH) is characterized by thickening of the middle layer of the pulmonary artery, eventually causing HF and death [125-126]. PAH is related to bone morphogenetic protein receptor 2 (BMPR2) gene mutation [127]. When bone morphogenesis proteins interact with BMPR2, the cytoplasmic proteins R-SMAD1/5/8 are phosphorylated. R-SMADs interact with the cofactor SMAD4 and undergo nuclear translocation, further regulating cell growth, proliferation and differentiation [127]. The degradation of R-SMADs by SMURF1 blocks this pathway [128]. Damage to the BMPR2 pathway leads to pulmonary artery occlusion and right ventricle afterload increase [129]. MiR-424(322), as a SMURF1 target can maintain the signal transduction of BMPR2. However, hypoxia-induced miR-424(322) secretion results in reduced SMURF1 expression, increased right ventricular hypertrophy and heart failure. In PAH rats, the level of miR-424(322) shows a negative correlation with SMURF1 amounts in the hypertrophic right ventricle. Therefore, the miR-424(322)-SMURF1 regulatory pathway could be explored for diagnosing pulmonary hypertension by measuring peripheral blood miR-424(322) [130].
Another study revealed that mir-140-5p with the target of SMURF1 mRNA, are decreased in individuals with PAH. Indeed, mir-140-5p and SMURF1 amounts are negatively correlated. As a BMPR2 signaling suppressor, Smurf1 knockout mice show right ventricular hypertrophy and decreased pulmonary microvascular remodeling. As mir-140-5p is cytotoxic, a SMURF1 inhibitor for the treatment of PAH, could replace mir-140-5p for treating PAH by inhibiting the degradation of BMPR2 and reducing the therapeutic toxicity [131-132].
The role of SMURF1 in angiogenesis
Angiogenesis represents a multi-step event that involves endothelial cell activation, proliferation and migration. TGFβ family members have critical functions in the development of the vascular system [133]. SMASD7 and SMURF1 exert negative effects on TGFβ1-induced VEGF expression and SMAD3/4-mediated VEGF expression. VEGF is effective for treating pathological angiogenesis, therefore, SMURF1 has the function of regulating angiogenesis [134]. Receptor-Like Kinase 1 (ALK1) is necessary for vascular development, remodeling and abnormal angiogenesis [135]. Bone morphogenic 9 (BMP9) and BMP10 specifically activate endothelial cells [136]. Human umbilical vein endothelial cell (HUVEC) treatment with metformin and AMPK inducers activate AMPK, increasing the expression of SMURF1, which leads to ALK1 degradation and the inhibition of BMP9-induced SMAD1/5 phosphorylation and angiogenesis. The role of SMURF1 in this regulatory pathway indicate a new clinical application of AMPK activators based on metformin and other drugs, suggesting a combination with other strategies to improve therapeutic effects in anti-VEGF resistance-related disease [137].
The role of SMURF1 in heart failure
With the aging of the world population, HF incidence is increasing [138]. Activin A (ActRI), a ligand of ActRII, is associated with aging and muscle atrophy in several disease [139]. The expression of ActRI signaling in the heart is increased, and heart function is damaged with aging. Moreover, SMURF1 is considered a key downstream effector of the ActRI signaling pathway, which promotes the degradation of sarcoplasmic Ca2+ ATPase (SERCA2a), a key factor determining the function of cardiomyocytes. The catabolic pathway mediated by SMURF1 inhibits ActRI signaling, thus can serve for targeted treatment of HF [140].
The role of SMURF1 in myocardial fibrosis
The therapeutic potential of endothelial colony forming cells (ECFCs) in the ischemic environment may be impaired. Normoxia significantly improves the activation of cultured cardiac fibroblasts. These effects were weakened in the hypoxia group. Mir-10b-5p was enriched in normoxia, but not in hypoxia. Mir-10b-5p inhibits the expression of Smurf1 mRNA. Therefore, Smurf1 downregulation by mir-10b-5p could blunt the anti-fibrotic effect of hypoxia, and SMURF1 has a potential function in alleviating myocardial fibrosis [141]. However, it needs to make a profound study how SMURF 1 participates in myocardial fibrosis through ubiquitination of downstream substrates.
The role of SMURF1 in heart valve disease
Heart valve disease represents a main mortality factor and morbidity. Epithelial-to-mesenchymal transition (EMT) of the inner membrane cell subpopulation in the atrioventricular septum (AVC) represents a key step of heart valve development [142]. In accordance with the activation of PAR6/SMURF1 signaling downstream of TGFβR3, targeting ALK5, PAR6 or SMURF1 markedly inhibits the EMT response to TGFβ2 or BMP2. The need for ALK5 activity, PAR6, and SMURF1 in TGFβR3-associated EMT of endocardial cells corroborates the pathway's involvement with tight junctions. Therefore, SMURF1 is involved in heart valve disease [143].
The role of SMURF1 in Heart Development
Heart development comprises complex developmental events, involving the deployment of multiple cell lines [144]. The proepicardium (PE) represents an important transitory cauliflower-like structure between the heart and liver primordia. MicroRNAs highly contribute to the development of the cardiovascular system. It was found that mir-125, miR-146, miR-195 and mir-223 specifically promote chicken cardiac myogenesis of PE/ST explants and also contribute to embryonic epicardium, a process controlled by Smurf1- and Foxp1 [145].
Additionally, heart SMURF1 amounts are high in mice during development [146], mainly distributing in ectodermal tissues. SMURF1 affects many processes in the developing heart, e.g., OFT septation and SMC generation in the main and coronary arteries. Smurf1 plays a role in CNC-mediated OFT septation by regulating the steady state levels of BMP and TGFβ signaling effectors to induce EndoMT, CNC delamination and migration [147-148]. In addition, SMURF1 highly contributes to SMC differentiation from the epicardium by regulating BMP-dependent SMAD signaling, together with RHOA, PAR6 and TGFβ-RIII18 [149]. Furthermore, SMURF1 exerts effects at the cilium to control baseline SMAD1/5 activation to regulate BMP signaling in the generation of cardiomyocytes, substantially contributing to cell-type specification [147]. The mechanisms of SMURF1 participation in heart development have provided insights for improving in vitro differentiation of cardiomyocytes to treat cardiovascular disorders.
SMURF2
SMURF2 function
SMURF2 contains an N-terminal C2 [150], two or more central WW domains interacting with the PY motif of the target protein or the adaptor protein, and a HECT domain at the C-terminus. SMURF2 receives ubiquitin from E2 at its cysteine 716 (Cys 716) active site and transfers it to the target protein to control its stability, localization and function [150]. SMURFs were originally considered negative regulators of TGFβ/BMP signaling. Through interaction with PY motifs in most R-SMADs and SMAD inhibitors, SMURFs regulate SMADs or TGFβ/BMPs, or other downstream components of the targeted pathway [128, 151-152]. After TGFβ-induced expression, the inhibitor of the adaptor protein SMAD7 forms a complex with SMURF2 in the nucleus [126]. SMURF2 then down-regulates TGFβ signal transduction by targeting itself, SMAD7 and TGFβ receptor kinase. In the presence of active TGFβ signaling, SMAD2 interacts with the tryptophan-rich WW domain of SMURF2 through the proline-rich PPxY motif [153].
The role of SMURF2 in calcified aortic valve disease
Calcified aortic valve disease incidence is very high in elderly individuals. In humans, TGFβ1 and BMP2 promote the pre-osteogenic induction of aortic valve interstitial cells (AVICS), which have a critical function in valve calcification [154-155]. TGFβ1 and BMP2 upregulate mir-486 and downregulate mir-204 for promoting osteogenic activity, while mir-486 downregulates Smurf2 to enhance mir-204 downregulation. Smurf2 gene knockout enhances TGFβ1 or BMP2-induced downregulation of mir-204 and results in elevated amounts of the osteoblast markers OSX and RUNX2. Therefore, the mir-486-SMURF2-SMAD regulatory pathway has a critical function in modulating the osteogenic effects of TGFβ1 and BMP2. Targeting this modulatory circuit might have therapeutic potential in inhibiting aortic valve calcification [156].
The role of SMURF2 in myxomatous mitral disease (MMVD)
Myxomatous mitral disease (MMVD) represents a commonly detected heart disease [157]. Increased phosphorylation of SMAD2/3 [158] and SMURF2 expression indicate that TGFβ1 signaling occurs through a typical signaling cascade. The levels of CTGF, MMP2, NOX2 and NOX4 [159-160] in mitral valves of SOD1-deficient mice were shown to be significantly increased. In addition, treatment of mouse valve stromal cells with cell permeability antioxidants resulted in reduced expression of TGFβ1-induced SMURF2 involved in promoting fibrosis and matrix remodeling genes. The study of this regulatory pathway has potential guidance for targeted therapy of MMVD.
The role of SMURF2 in vascular smooth muscle proliferation
Platelet-derived growth factor BB (PDGF-BB) and TGFβ1 are important growth factors regulating vascular smooth muscle cell (VSMC) proliferation. PDGF-BB increases the levels of ALK5, SMURF2, pSMAD2/3 and SMAD4, but decreases those of SMAD2 and SMAD7; these changes are partially reversed by neutralizing anti-TGFβ1 antibodies. The TGFβ signaling pathway mediates PDGF-BB effect in VSMCs by regulating downstream proteins such as SMURF2 [161]. This study revealed the molecular mechanism of SMURF2 in vascular smooth muscle and laid a theoretical foundation for the study of vascular disease.
The role of SMURF2 in Vascular endothelial injury
ROS are important signaling molecules that maintain the homeostasis of the cardiovascular system [162]. However, significant increases in ROS levels are closely related to the occurrence and development of cardiovascular ailments, including HF [163], atherosclerosis [164], hypertension [165] and ischemic heart disease [166], resulting in inflammation, fibrosis and apoptosis [167-168]. Oxidative stress-associated injury is closely related to ubiquitination. In the cardiovascular system, activation of PARP1 could cause vascular endothelial cell damage [168]. Decreasing PARP1 activity could reduce the development of atherosclerotic plaques [169]. The interaction between SMURF2 and PARP1 is enhanced under oxidative stress. In H2O2-treated human umbilical vein endothelial cells, SMURF2 was shown to degrade PARP1 to reduce ROS production and apoptosis, thereby alleviating oxidative stress injury [170]. The molecular mechanism of SMURF2 in atherosclerosis provides an insight into targeted treatment of the disease.
The role of SMURF2 in ischemia-reperfusion
I/R represents a commonly diagnosed and lethal ailment. Studies found that overexpression of mir-322/503 can enhance the expression of p-Akt and p-GSK3β, reduce infarct area, inhibit apoptosis and promote cell proliferation. It was also found that mir-322/503 could directly bind to Smurf2 gene for downregulation at the translational level, reducing the ubiquitination and degradation of EZH2 by Smurf2, activating Akt/GSK3 β signal transduction and protecting cells from I/R injury [171].
The role of SMURF2 in myocardial fibrosis
After myocardial infarction, the heart is severely remolded, and a structured collagen network is generated to resist circumferential deformation [172] for preventing scar rupture and promoting survival. However, abnormal biosynthesis of matrix proteins eventually results in cardiac fibrosis and heart failure [173]. TGF-β plays a role in heart failure development by promoting cardiac fibrosis [174]. Smurf2 is considered a critical TGF-β pathway suppressor via Smad2 degradation [175]. In addition, Smurf2 and Smad7 jointly repress TGF-β signaling [176]. Smurf2's function is therefore paradoxical. This contradictory behavior of Smurf2 is because nuclear Smurf2 induces the TGF-β pathway, while cytosolic Smurf2 exerts opposite effects. As cardiac fibrosis appears to show altered TGF-β signaling, investigating Smad-regulating proteins may help identify an effective way to treat myocardial fibrosis [177].
Conclusions and Prospects
A major HECT group of ubiquitin ligases is the NEDD4 family [5, 6]. NEDD4 proteins identify different substrate proteins to perform ubiquitination catalysis, thus participating in various cardiovascular diseases. The present review investigated the functions and regulation of the NEDD4 family in association with cardiovascular disease (Table 1).
The NEDD4 family functions and regulation
| Enzymes | Mechanisms | Biological response | References |
|---|---|---|---|
| NEDD4-1 (RPF1) | Activate Akt/PI3K signaling pathway and degradate PTEN by binding to its N-terminal | myocardial reperfusion injury (I/R) | [40-42] |
| NEDD4-1 KO mice show a double outlet right ventricle, an endocardial cushion defect and abnormalityof the cephalic plexus vein of embryos | abnormality of heart development | [43,44] | |
| NEDD4-1 deletion in mouse tissues showed deformed aortic structures. Suppress BMP/Smad pathway via degradation of C-terminal pSmad1 activated by TGF-β. | Vascular Calcification | [45-48] | |
| NEDD4L (NEDD4-like or NEDD4-2) | WW domain of NEDD4L recognizes β- and γ-subunits of PY motif of ENaC in C domain | hypertension | [22,56-60] |
| WNK1 activates SGK1 phosphorylating NEDD4L on serine 444 by interaction with the chaperone 14-3-3 and results in a reduction of ENaC ubiquitination. IL17A increased NCC activity in an SGK1/NEDD4L dependent pathway. | hypertension | [61-67] | |
| downregulate Nav1.5 by ubiquitination; defect the NEDD4L C2 isoform | heart failure (HF); myocardial infarction (MI) | [68] | |
| miR-1 regulates the 3′-UTR of Nedd4L | heart development | [69] | |
| circnfix enhanced the interaction between Ybx1 and NEDD4L, and induced Ybx1 degradation by NEDD4L, which inhibited cyclin A2 and cyclin B1. | cardiac regenerative repair | [70] | |
| ITCH (AIP4) | TXNIP binds to HECT domain of ITCH and WW domain recognizes PPxY motif of TXNIP. | myocardial infarction (MI) or doxorubicin (DOX) | [78-82] |
| ITCH which is upregulated by increasing intracellular Ca2+ via CaSR, increases the ubiquitination level of SMAD7, and upregulates the levels of p-SMAD2 and p-SMAD3. | diabetic cardiomyopathy | [83-84] | |
| WWP1 (TIUL1 or AIP5) | circRNA-WWP1 down-regulates ANF and mir-23a | cardiac hypertrophy | [89-90] |
| miR-21 inhibits cardiac fibroblasts proliferation by inactivating the TGF-b1/Smad2 via up-regulation of WWP1 | atrial fibrillation | [95-98] | |
| WWP2 (AIP2) | TGFβ1 stimulates the WWP2 N-terminal subtype to enter the nucleus. The WWP2-N subtype enhances the activity of WWP2-FL to promote interaction with SMAD2, and promote its monoubiquitination. | myocardial fibrosis | [110-115] |
| WWP2 interacts with PARP1 in its BRCT domain and ubiquitinate K249 and K418 of PARP1. | cardiac remodelling | [116-119] | |
| WWP2 promoted the degradation of Septin4-K174, thus inhibiting formation of the Septin4-PARP1 complex. | Vascular endothelial injury | [123-124] | |
| SMURF1 | Secretion of miR-424 (322) by PAECs results in down-regulation of SMURF1, thus blocking the degradation of R-SMADs and increasing BMPR2 pathway activity. | pulmonary hypertension | [127-130] |
| The binding target of mir-140-5p was SMURF1 mRNA. A SMURF1 inhibitor can replace mir-140-5p by inhibiting ubiquitination of BMPR2. | pulmonary hypertension | [131-132] | |
| SMURF1 have negative effects on TGFβ1-induced VEGF expression and SMAD3/4-mediated VEGF expression. | angiogenesis | [134] | |
| Metformin and AMPK activators activate AMPK and SMURF1 is up-regulated, leading to the degradation of ALK1, and inhibition of BMP9-induced SMAD1/5 phosphorylation and angiogenesis. | angiogenesis | [135-137] | |
| SMURF1 promotes degradation of (SERCA2a) therefore inhibit actri signalling. | heart failure | [139-140] | |
| Mir-10b-5p inhibits the expression of Smurf1 | heart failure | [141] | |
| PAR6/SMURF1 pathway downstream of TGFβR3 actives, targeting ALK5, PAR6 or SMURF1 significantly inhibited the EMT response to TGFβ2 or BMP2. | heart valve disease | [143] | |
| SMURF1 regulates BMP and TGFβ signaling to induct EndoMT in CNC-mediated OFT septation, CNC delamination, migration and in SMC differentiation from the epicardium, together with RHOA, PAR6 and TGFβ-RIII18. SMURF1 modulates SMAD1/5 activation to regulate BMP signaling during cardiomyogenesis at the cilium toand. | Heart Development | [147-149] | |
| SMURF2 | Mir-486, up-regulated by TGFβ1 and BMP2, inhibits SMURF2-SMAD regulatory pathway leading to enhance mir-204 down regulation and results in increased of OSX and RUNX2. | valve disease | [154-156] |
| TGFβ1 signalling increases phosphorylation of SMAD2/3 and SMURF2 expression leading to promote fibrosis and matrix remodelling genes. | myxomatous mitral disease | [158-160] | |
| PDGF-BB increases expression of ALK5, SMURF2, pSMAD2/3 and SMAD4, but decreases expression of SMAD2 and SMAD7 | vascular smooth muscle proliferation | [161] | |
| SMURF2 degrades PARP1 through the ubiquitin proteasome pathway | Vascular endothelial injury | [168-170] | |
| mir-322 / 503 binds to Smurf2 gene and inhibits its translation to reduce the degradation of EZH2, to activate Akt/GSK3 β signal transduction and protect cells from I/R injury | ischemia-reperfusion | [171] | |
| Smurf2 inhibits TGF-β pathway via degradation of Smad2. In addition, Smurf2 conjunction with Smad7 to halt TGF-β signal transduction. | myocardial fibrosis | [172-177] | |
| NEDL1 (HECW1) | None | None | None |
| NEDL2 (HECW2) | None | None | None |
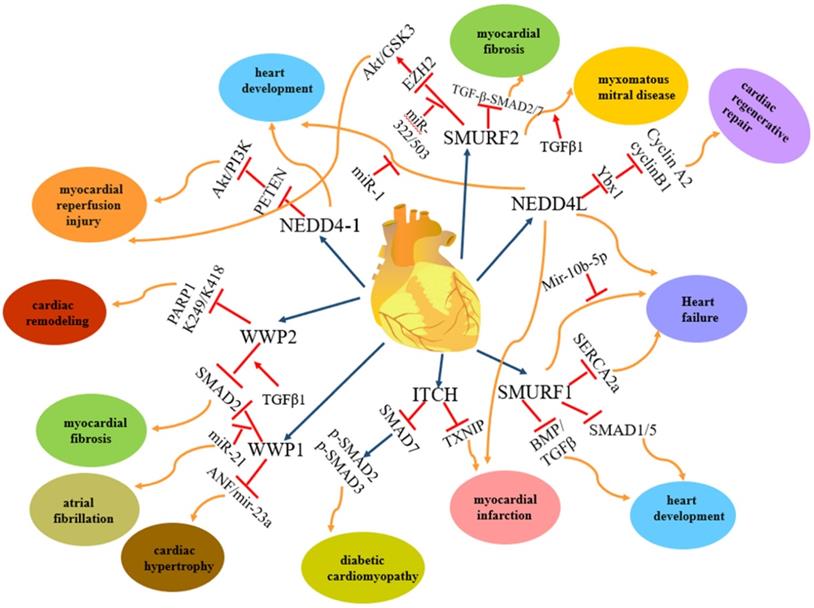
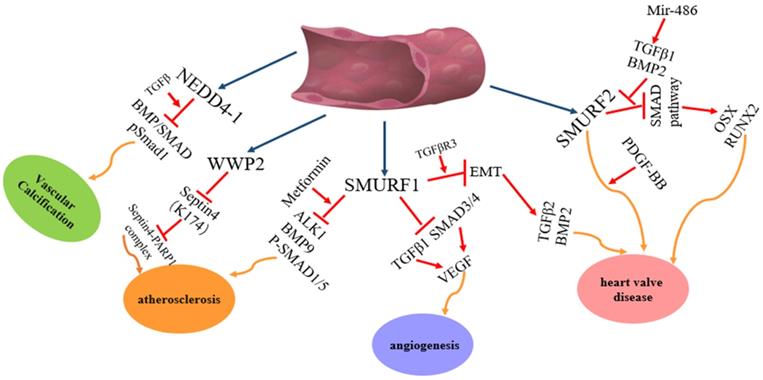
Among the nine members of the NEDD4 family, NEDD4-1, NEDD4L, ITCH, WWP1, WWP2, SMURF1 and SMURF2 are involved in heart diseases. However, each of them plays different roles in heart diseases (Figure 2): NEDD4-1 participates in myocardial ischemia-reperfusion injury [43] and abnormality of heart development [44]; NEDD4L and SMURF1 are involved in heart failure [68], myocardial infarction [68] and heart development [69, 139-141, 147-149]; WWP1 participates in cardiac hypertrophy [89-90] and atrial fibrillation [95-98]; WWP2 is involved in myocardial fibrosis [110-115] and cardiac remodelling [116-119]; ITCH participates in myocardial infarction [78-82] and diabetic cardiomyopathy [83-84] and SMURF2 participates in myocardial fibrosis [172-177] and myocardial ischemia-reperfusion injury [171]. In addition, NEDD4-1, NEDD4L, WWP2, SMURF1 and SMURF2 are involved in vascular disease (Figure 3). NEDD4-1 participates in vascular calcification [45-48] and NEDD4L participates in hypertension [22, 56-67]. WWP2 is involved in vascular endothelial injury [123-124], SMURF1 in atherosclerosis [135-137] and pulmonary hypertension [127-132], and SMURF2 in myxomatous mitral disease [158-160] and vascular endothelial injury [168-170]. However, whether NEDL1 and NEDL2 play biological functions in cardiovascular related disease needs further study.
The regulation and functions of NEDD4 E3 ligases in cardiac disease. The regulatory pathway of NEDD4 E3 ligases including NEDD4-1, NEDD4L, ITCH, WWP1, WWP2, Smurf1 and Smurf2 in cardiac disease, such as myocardial reperfusion injury, heart development, heart failure, myocardial infarction, diabetic cardiomyopathy, cardiac hypertrophy, myocardial fibrosis, cardiac remodelling, myxomatous mitral disease.
The regulation and functions of NEDD4 E3 ligases in vascular disease. The regulatory pathway of NEDD4 E3 ligases including WWP2, Smurf1 and Smurf2 in vascular disease, such atherosclerosis, angiogenesis, heart valve disease.
The research indicated that suppression of circNfix can promote cardiac regeneration and enhance heart function following myocardial infarction via degradation of Ybx1 by NEDD4L, which may provide a promising strategy to improve prognosis after MI [70]. It reveals the role that ITCH/ TGFβ1/SMADs pathway inhibits myocardial fibrosis, indicating that Calhex 231 may represent a novel drug for treating dilated cardiomyopathy [84]. It was indicated that circRNA-WWP1 could inhibit cardiac hypertrophy by downregulating ANF and mir-23a by Gene Ontology and Kyoto Encyclopedia analysis, and that circRNA-WWP1 is a potential new therapeutic target for cardiac hypertrophy [90]. Metformin and AMPK activators activate AMPK and SMURF1, subsequently, degradation of ALK1, and inhibition of BMP9-induced SMAD1/5. It indicates a new clinical application of AMPK activators combination with metformin to improve therapeutic effects in anti-VEGF resistance-related disease [137]. The mir-486-SMURF2-SMAD regulatory pathway in modulating the osteogenic effects might have therapeutic potential in inhibiting aortic valve calcification [156]. Thus, it will play an important role in revealing multiple various regulatory mechanisms and functions of NEDD4 E3 ligase family to explore the above issues deeply, and provide new insights for the development of cardiovascular disease.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China [81900372, 81900355]; and the China Postdoctoral Science Foundation [2019M661175, 2018M641750, 2019M661159, 2018M641730]; and Natural Foundation of Department of education of Liaoning Province [QN2019002].
Authors' contributions
NZ, SY and LC designed the review; YZ searched for literatures and wrote the manuscript; SL and HQ organized literaturs; BW, SY, SW drew the pattern charts. All authors have read and approved the manuscript and agree with publication in this journal.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13-21
2. Joazeiro CA, Weissman AM. RING Finger Proteins: Mediators of Ubiquitin Ligase Activity. Cell. 2000;102:549-52
3. Huibregtse JM, Scheffner M, Beaudenon S. et al. A Family of Proteins Structurally and Functionally Related to the E6-AP Ubiquitin-Protein Ligase. Proc Natl Acad Sci USA. 1995;92:2563-67
4. Andrew Paul Hutchins, Shaq Liu, Diego Diez. et al. The Repertoires of Ubiquitinating and Deubiquitinating Enzymes in Eukaryotic Genomes. Mol Biol Evol. 2013;30:1172-87
5. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373-428
6. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep. 2008;9:536-542
7. Jin L, Williamson A, Banerjee S. et al. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653-665
8. Chau V, Tobias JW, Bachmair A. et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576-1583
9. Tanno H, Komada M. The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem. 2013;153:497-504
10. Husnjak K, Dikic I. Ubiquitin-binding proteins: Decoders of ubiquitin mediated cellular functions. Annu Rev Biochem. 2012;81:291-322
11. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203-229
12. Maspero E, Valentini E, Mari S. et al. Structure of a ubiquitin-loaded HECT ligase reveals the molecular basis for catalytic priming. Nat Struct Mol Biol. 2013;20:696-701
13. Kristariyanto YA, Choi SY, Rehman SA. et al. Assembly and structure of Lys33-linked polyubiquitin reveals distinct conformations. J Biochem. 2015;467:345-352
14. Glickman MH, Adir N. The proteasome and the delicate balance between destruction and rescue. PLoS Biol. 2004;2:E13
15. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398-409
16. Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17:68-77
17. Harvey KF, Dinudom A, Komwatana P. et al. All Three WW Domains of Murine Nedd4 Are Involved in the Regulation of Epithelial Sodium Channels by Intracellular Na+. J Biol Chem. 1999;274:12525-30
18. Rotin D, Staub O, Haguenauer-Tsapis R. Ubiquitination and Endocytosis of Plasma Membrane Proteins: Role of Nedd4/Rsp5p Family of Ubiquitin-Protein Ligases. J Membr Biol. 2000;176:1-17
19. Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004;23:1972-84
20. Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta. 2014;1843:61-74
21. Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879-82
22. Staub O, Dho S, Henry P. et al. WW Domains of Nedd4 Bind to the Proline-Rich PY Motifs in the Epithelial Na+ Channel Deleted in Liddle's Syndrome. EMBO J. 1996;15:2371-80
23. Staub O, Rotin D. WW domains. Structure. 1996;4:495-9
24. Macias MJ, Wiesner S, Sudol M. WW and SH3 Domains, Two Different Scaffolds to Recognize Proline-Rich Ligands. FEBS Lett. 2002;513:30-7
25. Bedford MT, Chan DC, Leder P. FBP WW Domains and the Abl SH3 Domain Bind to a Specific Class of Proline-Rich Ligands. EMBO J. 1997;16:2376-83
26. Bedford MT, Reed R, Leder P. WW Domain-Mediated Interactions Reveal a Spliceosome-Associated Protein That Binds a Third Class of Proline-Rich Motif: The Proline Glycine and Methionine-Rich Motif. Proc Natl Acad Sci USA. 1998;95:10602-7
27. Yaffe MB, Schutkowski M, Shen M. et al. Sequence-specific and Phosphorylation-Dependent Proline Isomerization: A Potential Mitotic Regulatory Mechanism. Science. 1997;278:1957-60
28. Huang L, Kinnucan E, Wang G. et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321-6
29. Dodson EJ, Fishbain YV, Rotem BS. et al. Versatile communication strategies among tandem WW domain repeats. Exp Biol Med (Maywood). 2015;240:351-60
30. Willis MS, Townley-Tilson WH, Kang EY. et al. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463-78
31. Patterson C, Ike C, Willis PW IV. et al. The bitter end: the ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456-63
32. Powell SR, Herrmann J, Lerman A. et al. The ubiquitin-proteasome system and cardiovascular disease. Prog Mol Biol Transl Sci. 2012;109:295-346
33. Fagerberg L, Hallström BM, Oksvold P. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteom. 2014;13:397-406
34. Xu C, Fan CD, Wang X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene. 2015;34:281-9
35. French ME, Klosowiak JL, Aslanian A. et al. Mechanism of ubiquitin chain synthesis employed by a HECT domain ubiquitin ligase. J Biol Chem. 2017;292:10398-413
36. Sluimer J, Distel B. Regulating the human HECT E3 ligases. Cell Mol Life Sci. 2018;75:3121-41
37. FukushimaT Yoshihara H, Furuta H et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat Commun. 2015;6:6780
38. Murillas R, Simms KS, Hatakeyama S. et al. Identification of developmentally expressed proteins that functionally interact with Nedd4 ubiquitin ligase. J Biol Chem. 2002;277:2897-907
39. Xiong J, Xue FS, Yuan YJ. et al. Cholinergic anti-inflammatory pathway: a possible approach to protect against myocardial ischemia reperfusion injury. Chin Med J (Engl). 2010;123:2720-6
40. Fujio Y, Nguyen T, Wencker D. et al. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660-7
41. Wang X, Trotman LC, Koppie T. et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129-39
42. Jovana Drinjakovic, Hosung Jung, Douglas S. Campbell. E3 Ligase Nedd4 Promotes Axon Branching by Downregulating PTEN. Neuron. 2010;65:341-57
43. Obler D, Juraszek AL, Smoot LB. et al. Double Outlet Right Ventricle: Aetiologies and Associations. J Med Genet. 2008;45:481-97
44. Fouladkou F, Lu C, Jiang C. et al. The Ubiquitin Ligase Nedd4-1 Is Required for Heart Development and Is a Suppressor of Thrombospondin -1*. J Biol Chem. 2010;285:6670-80
45. Doherty TM, Asotra K, Fitzpatrick LA. et al. Calcification in atherosclerosis: bone biology and chronic inflammation at the arterial crossroads. Proc Natl Acad Sci U S A. 2003;100:11201-6
46. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044-59
47. Bostrom K, Watson KE, Horn S. et al. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800-9
48. Lee JH, Jeon SA, Kim BG. et al. Nedd4 Deficiency in Vascular Smooth Muscle Promotes Vascular Calcification by Stabilizing pSmad1. J Bone Miner Res. 2017;32:927-38
49. Kamynina E, Debonneville C, Bens M. et al. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 2001;15:204-14
50. Chen H, Ross CA, Wang N. et al. NEDD4L on human chromosome 18q21 has multiple forms of transcripts and is a homologue of the mouse Nedd4-2 gene. Eur J Hum Genet. 2001;9:922-30
51. Harvey KF, Dinudom A, Cook DI. et al. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem. 2001;276:8597-601
52. Araki N, Umemura M, Miyagi Y. et al. Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: implications for essential hypertension. Hypertension. 2008;51:773-7
53. Dahl LK. Possible role of chronic excess salt consumption in the pathogenesis of essential hypertension. Am J Cardiol. 1961;8:571-5
54. Weinberger MH, Miller JZ, Luft FC. et al. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8:II127-II134
55. Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol. 2014;15:33-46
56. Abriel H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007;76:381-9
57. Shi YQ, Yan CC, Zhang X. et al. Mechanisms underlying probucol-induced hERG-channel deficiency. Drug Des Dev Ther. 2015;9:3695-704
58. Kang Y, Guo J, Yang T. et al. Regulation of the human ether-a-go-go-related gene (hERG) potassium channel by Nedd4 family interacting proteins (Ndfips). Biochem J. 2015;472:71-82
59. Schild L, Lu Y, Gautschi I. et al. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J. 1996;15:2381-7
60. Shi PP, Cao XR, Sweezer EM. et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am j Physiol Renal Physiol. 2008;295:F462-70
61. Busjahn A, Aydin A, Uhlmann R. et al. Serum- And Glucocorticoid-Regulated Kinase (SGK1) Gene and Blood Pressure. Hypertension. 2002;40:256-60
62. Pearce D. The Role of SGK1 in Hormone-Regulated Sodium Transport. Trends Endocrinol Metab. 2001;12:341-7
63. Hallows KR, Bhalla V, Oyster NM. et al. Phosphopeptide screen uncovers novel phosphorylation sites of Nedd4-2 that potentiate its inhibition of the epithelial Na+ channel. J Biol Chem. 2010;285:21671-8
64. Lang F, Stournaras C. Serum and glucocorticoid inducible kinase, metabolic syndrome, inflammation, and tumor growth. Hormones (Athens). 2013;12:160-71
65. Bhalla V, Daidie D, Li H. et al. Serum- And Glucocorticoid-Regulated Kinase 1 Regulates Ubiquitin Ligase Neural Precursor Cell-Expressed, Developmentally Down-Regulated Protein 4-2 by Inducing Interaction With 14-3-3. Mol Endocrinol. 2005;19:3073-84
66. Xu BE, Stippec S, Chu PY. et al. WNK1 Activates SGK1 to Regulate the Epithelial Sodium Channel. Proc Natl Acad Sci USA. 2005;102:10315-20
67. Norlander AE, Saleh MA, Kamat NV. et al. Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II-Induced Hypertension. Hypertension. 2016;68:167-74
68. Luo L, Ning F, Du Y. et al. Calcium-dependent Nedd4-2 upregulation mediates degradation of the cardiac sodium channel Nav1.5: implications for heart failure. Acta Physiol (Oxf). 2017;221:44-58
69. Zhu JY, Heidersbach A, Kathiriya IS. et al. The E3 ubiquitin ligase Nedd4/Nedd4L is directly regulated by microRNA 1. Development. 2017;144:866-75
70. Huang SL, Li XZ, Zheng H. et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation. 2019;139:2857-76
71. Schwarz SE, Rosa JL, Scheffner M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J Biol Chem. 1998;273:12148-54
72. Pickart CM. Mechanisms Underlying Ubiquitination. Annu Rev Biochem. 2001;70:503-33
73. Peng J, Schwartz D, Elias JE. et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921-6
74. Deng L, Wang C, Spencer E. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351-61
75. Melino G, Gallagher E, Aqeilan RI. et al. Itch: a HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008;15:1103-12
76. Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198-200
77. Li JM, Gall NP, Grieve DJ. et al. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477-84
78. Junn E, Han SH, Im JY. et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287-95
79. Yoshioka J, Chutkow WA, Lee S. et al. Deletion of thioredoxininteracting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122:267-79
80. Zhang P, Wang C, Gao K. et al. The ubiquitin ligase ITCH regulates apoptosis by targeting thioredoxininteracting protein for ubiquitin-dependent degradation. J Biol Chem. 2010;285:8869-79
81. Hatakeyama S, Nakayama KI. Ubiquitylation as a quality control system for intracellular proteins. J Biochem. 2003;134:1-8
82. Takahashi H, Takeishi Y, Seidler T. et al. Adenovirus-mediated overexpression of diacylglycerol kinase-zeta inhibits endothelin-1-induced cardiomyocyte hypertrophy. Circulation. 2005;111:1510-6
83. Francisco W, Riquelme JA, Mario P. et al. New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Frontiers in Physiology. 2016;7:125
84. Tharmalingam S, Hampson DR. The Calcium-Sensing Receptor and Integrins in Cellular Differentiation and Migration. Frontiers in Physiology. 2016;7:190
85. Wood JD, Yuan J, Margolis RL. et al. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci. 1998;11:149-60
86. Sudol M, Hunter T. NeW wrinkles for an old domain. Cell. 2000;103:1001-4
87. Li Y, Zhou Z, Alimandi M. et al. WW domain containing E3 ubiquitin protein ligase 1 targets the full-length ErbB4 for ubiquitin-mediated degradation in breast cancer. Oncogene. 2009;28:2948-58
88. Verdecia MA, Joazeiro CA, Wells NJ. et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell. 2003;11:249-59
89. Li M, Ding W, Sun T. et al. Biogenesis of circular RNAs and their roles in cardiovascular development and pathology. FEBS J. 2018;285:220-32
90. YangMH WangH, HanSN et al. Circular RNA expression in isoproterenol hydrochloride-induced cardiac hypertrophy. Aging (Albany NY). 2020;12:2530-44
91. Hirsh BJ, Copeland-Halperin RS, Halperin JL. Fibrotic Atrial Cardiomy opathy, Atrial Fibrillation, and Thromboembolism: Mechanistic Links and Clinical Inferences. J Am Coll Cardiol. 2015;65:2239-51
92. Barrett TW, Vermeulen MJ, Self WH. et al. Emergency department management of atrial fibrillation in the United States versus Ontario, Canada. J Am Coll Cardiol. 2015;65:2258-60
93. Smaill BH. Fibrosis, myofibroblasts, and atrial fibrillation. Circ Arrhythm Electrophysiol. 2015;8:256-7
94. Zheng LY, Zhang MH, Xue JH. et al. Effect of angiotensin II on STAT3 mediated atrial structural remodeling. Eur Rev Med Pharmacol Sci. 2014;18:2365-77
95. Zuo K, Li M, Zhang X. et al. MiR-21 suppresses endothelial progenitor cell proliferation by activating the TGF beta signaling pathway via downregulation of WWP1. Int J Clin Exp Pathol. 2015;8:414-22
96. Moren A, Imamura T, Miyazono K. et al. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem. 2005;280:22115-23
97. Zhi X, Chen C. WWP1: a versatile ubiquitin E3 ligase in signaling and diseases. Cell Mol Life Sci. 2012;69:1425-34
98. Tao H, Zhang M, Yang JJ. et al. MicroRNA-21 via Dysregulation of WW 3 Domain-Containing Protein 1 Regulate 4 Atrial Fibrosis in Atrial Fibrillation. Heart Lung Circ. 2018;27:104-13
99. Martin-Serrano J, Eastman SW, Chung W. et al. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol. 2005;168:89-101
100. Mund T, Lewis MJ, Maslen S. et al. Peptide and Small Molecule Inhibitors of HECT-type Ubiquitin Ligases. Proc Natl Acad Sci USA. 2014;111:16736-41
101. Bernassola F, Karin M, Ciechanover A. et al. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer Development. Cancer Cell. 2008;14:10-21
102. Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol. 2016;17:626-42
103. Pirozzi G, McConnell SJ, Uveges AJ. et al. Identification of novel human WW domain-containing proteins by 441 cloning of ligand targets. J Biol Chem. 1997;272:14611-16
104. Lu PJ, Zhou XZ, Shen M. et al. Function of WW Domains as Phosphoserine- Or Phosphothreonine-Binding Modules. Science. 1999;283:1325-8
105. Wiesner S, Ogunjimi AA, Wang HR. et al. Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell. 2007;130:651-62
106. Liao B, Jin Y. Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell Res. 2010;20:332-44
107. Garfinkel AC, Seidman JG, Seidman CE. Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail Clin. 2018;14:139-46
108. Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355-65
109. Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207-12
110. Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383-408
111. Chen H, Moreno-Moral A, Pesce F. et al. WWP2 regulates pathological cardiac fibrosis by modulating SMAD2 signaling. Nat Commun. 2019;10:3616
112. Tang LY, Yamashita M, Coussens NP. et al. Ablation of Smurf2 reveals an inhibition in TGF-beta signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011;30:4777-89
113. Inman GJ, Nicolas FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2,3, and 4 permits sensing of TGF-beta receptor activity. Mol Cell. 2002;10:283-94
114. Edgley AJ, Krum H, Kelly DJ. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-beta. Cardiovasc Ther. 2012;30:e30-e40
115. Brooks CL, Li M, Gu W. Mechanistic studies of MDM2-mediated ubiquitination in p53 regulation. J Biol Chem. 2017;282:22804-15
116. Henning RJ, Bourgeois M, Harbison RD. Poly (ADP-ribose) polymerase (PARP) and PARP inhibitors: mechanisms of action and role in cardiovascular disorders. Cardiovasc Toxicol. 2018;18:493-506
117. Wang C, Xu W, Zhang Y. et al. PARP1 promote autophagy in cardiomyocytes via modulating FoxO3a transcription. Cell Death Dis. 2018;9:1047
118. Lu J, Zhang R, Hong H. et al. The poly (ADP-ribosyl) ation of FoxO3 mediated by PARP1 participates in isoproterenol-induced cardiac hypertrophy. Biochim Biophys Acta. 2016;1863:3027-39
119. Naijin Zhang, Ying Zhang, Hao Qian. et al. Selective targeting of ubiquitination and degradation of PARP1 by E3 ubiquitin ligase WWP2 regulates isoproterenol-induced cardiac remodeling. Cell Death Differ. 2020 [Epub ahead of print]
120. Karsan A, Harlan JM. Modulation of endothelial cell apoptosis: mechanisms and pathophysiological roles. J Atheroscler Thromb. 1996;3:75-80
121. Szymanski MK, Buikema JH, van Veldhuisen DJ. et al. Increased cardiovascular risk in rats with primary renal dysfunction; mediating role for vascular endothelial function. Basic Res Cardiol. 2012;107:242
122. Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014-30
123. N. Zhang, Y. Zhang, S. Zhao, et al. Septin4 as a novel binding partner of PARP1 contributes to oxidative stress induced human umbilical vein endothelial cells injure. Biochem Biophys Res Commun. 2018;496:621-7
124. Zhang N, Zhang Y, Wu B. et al. Role of WW domain E3 ubiquitin protein ligase 2 in modulating ubiquitination and Degradation of Septin4 in oxidative stress endothelial injury. Redox Biol. 2020;30:101419
125. Galiè N, Humbert M, Vachiery JL. et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed By: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67-119
126. Baptista R, Meireles J, Agapito A. et al. Pulmonary hypertension in Portugal: first data from a nationwide registry. Biomed Res Int. 2013;2013:489574
127. Morrell NW, Bloch DB, Dijke PT. et al. Targeting BMP signalling in cardiovascular disease and anaemia. Nat Rev Cardiol. 2016;13:106-20
128. Zhu H, Kavsak P, Abdollah S. et al. A SMAD ubiquitin ligase targetsthe BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687-93
129. Atkinson C, Stewart S, Upton PD. et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672-8
130. Baptista R, Marques C, Catarino S. et al. MicroRNA-424(322) as a newmarker of disease progression in pulmonary arterial hypertension and its role in right ventricular hypertrophy by targeting SMURF1. Cardiovasc Res. 2018;114:53-64
131. Rothman AMK, Arnold ND, Pickworth JA. et al. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest. 2016;126:2495-508
132. Landré V, Rotblat B, Melino S. et al. Screening for E3-Ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget. 2014;5:7988-8013
133. Jin Y, Kaluza D, Jakobsson L. VEGF, Notch and TGFbeta/ BMPs in regulation of sprouting angiogenesis and vascular patterning. Biochemical Society transactions. 2014;42:1576-83
134. Nam EH, Park SR, Kim PH. TGF-β1 induces mouse dendritic cells to express VEGF and its receptor (Flt-1) under hypoxic conditions. Exp Mol Med. 2010;42:606-13
135. de Vinuesa AG, Bocci M, Pietras K. et al. Targeting tumour vasculature by inhibiting activin receptor-like kinase (ALK)1 function. Biochemical Society transactions. 2016;44:1142-9
136. Li W, Salmon RM, Jiang H. et al. Regulation of the ALK1 ligands, BMP9 and BMP10. Biochemical Society transactions. 2016;44:1135-41
137. Ying Y, Takashi U, Jiang S. et al. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget. 2017;8:32794-806
138. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30-41
139. Oshima Y, Ouchi N, Shimano M. et al. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation. 2009;120:1606-15
140. Roh JD, Hobson R, Chaudhari V. et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med. 2019;11:eaau8680
141. Liu W, Zhang H, Mai J. et al. Distinct Anti-Fibrotic Effects of Exosomes Derived from Endothelial Colony-Forming Cells Cultured Under Normoxia and Hypoxia. Med Sci Monit. 2018;24:6187-99
142. Brown CB, Boyer AS, Runyan RB. et al. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 1999;283:2080-2
143. Townsend TA, Robinson JY, Deig CR. et al. BMP-2 and TGFβ2 Shared Pathways Regulate Endocardial Cell Transformation. Cells Tissues Organs. 2011;194:1-12
144. Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223-67
145. Duenas A, Exposito A, Munoz MDM. et al. MiR-195 enhances cardiomyogenic differentiation of the proepicardium/septum transversum by Smurf1 and Foxp1 modulation. Sci Rep. 2020;10:9334
146. Narimatsu M. et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137:295-307
147. Koefoed K, Rørdam JS, Andersen P. et al. The E3 ubiquitin ligase SMURF1 regulates cell-fate specification and outflow tract septation during mammalian heart development. Sci Rep. 2018;8:9542
148. Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012;366:34-54
149. Murakami K, Mathew R, Huang J. et al. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia. Exp Biol Med (Maywood). 2010;235:805-13
150. Izzi L, Attisano L. Regulation of the TGFbeta signaling pathway by ubiquitin-mediated degradation. Oncogene. 2004;23:2071-8
151. Imamura T, Oshima Y, Hikita A. Regulation of TGF-beta family signalling by ubiquitination and deubiquitination. J Biochem. 2013;154:481-9
152. Kavsak P, Rasmussen RK, Causing CG. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
153. Bonni S, Wang HR, Causing CG. et al. TGF-b induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587-95
154. Watson KE, Boström K, Ravindranath R. et al. TGF-beta 1 and 25-hydroxycholesterol Stimulate Osteoblast-Like Vascular Cells to Calcify. J Clin Invest. 1994;93:2106-13
155. Du Y, Wang Y, Wang L. et al. Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ Res. 2011;108:917-28
156. Song R, Fullerton DA, Ao L. et al. An epigenetic regulatory loop controls pro-osteogenic activation by TGF-β1 or bone morphogenetic protein 2 in human aortic valve interstitial cells. J Biol Chem. 2017;292:8657-66
157. Freed LA, Levy D, Levine RA. et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. 1999;341:1-7
158. Nakao A, Imamura T, Souchelnytskyi S. et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353-62
159. Bondi CD, Manickam N, Lee DY. et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93-102
160. Cucoranu I, Clempus R, Dikalova A. et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900-7
161. Pan D, Yang J, Lu F. et al. Platelet-derived growth factor BB modulates PCNA protein synthesis partially through the transforming growth factor beta signalling pathway in vascular smooth muscle cells. Biochem Cell Biol. 2007;85:606-15
162. Kietzmann T, Petry A, Shvetsova A. et al. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. 2017;174:1533-54
163. Münzel T, Camici GG, Maack C. et al. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J Am Coll Cardiol. 2017;70:212-29
164. Wang Y, Wang GZ, Rabinovitch PS. et al. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappab-mediated inflammation in macrophages. Circ Res. 2014;114:421-33
165. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1417-27
166. Kurian GA, Rajagopal R, Vedantham S. et al. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid Med Cell Longev. 2016;2016:1656450
167. Sinha N, Dabla PK. Oxidative stress and antioxidants in hypertension- a current review. Curr Hypertens Rev. 2015;11:132-142
168. Shang F, Zhang J, Li Z. et al. Cardiovascular protective effect of metformin and telmisartan: reduction of parp1 activity via the AMPK-PARP1 Cascade. PLoS One. 2016;11:e0151845
169. Xu S, Bai P, Little PJ. et al. Poly (adp-ribose) polymerase 1 (parp1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev. 2014;34:644-75
170. Qian H, Zhang N, Wu B. et al. The E3 ubiquitin ligase Smurf2 regulates PARP1 stability to alleviate oxidative stress-induced injury in human umbilical vein endothelial cells. J Cell Mol Med. 2020;24:4600-11
171. Dong W, Xie F, Chen XY. et al. Inhibition of Smurf2 translation by miR-322/503 protects from ischemia reperfusion injury by modulating EZH2/Akt/GSK3β signaling. Am J Physiol Cell Physiol. 2019;317:C253-61
172. Holmes JW, Yamashita H, Waldman LK. et al. Scar remodeling and transmural deformation after infarction in the pig. Circulation. 1994;90:411-20
173. Hao J, Ju H, Zhao S. et al. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol. 1999;31:667-78
174. Lijnen PJ, Petrov VV, Fagard RH. 2000. Induction of cardiac fibrosis by transforming growth factor-beta (1). Mol Genet Metab. 2000;71:418-35
175. Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. 2000;275:36818-22
176. Kavsak P, Rasmussen RK, Causing CG. et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
177. Cunnington RH, Nazari M, Dixon IM. c-Ski, Smurf2, and Arkadia as regulators of TGF-β signaling: new targets for managing myofibroblast function and cardiac fibrosis. Can J Physiol Pharmacol. 2009;87:764-72
Author contact
![]() Corresponding authors: 155 Nanjing North Street, Heping District, Shenyang, 110001, Liaoning Province, People's Republic of China. 77 Puhe Road, Shenbei New District, Shenyang, 110001, Liaoning Province, People's Republic of China. Telephone Number: +86 15040171605; +86 13804068889; +86 18900911888, E-mail: yxsunedu.cn (Y.S); njzhangedu.cn (N.Z); lcaoedu.cn (L.C).
Corresponding authors: 155 Nanjing North Street, Heping District, Shenyang, 110001, Liaoning Province, People's Republic of China. 77 Puhe Road, Shenbei New District, Shenyang, 110001, Liaoning Province, People's Republic of China. Telephone Number: +86 15040171605; +86 13804068889; +86 18900911888, E-mail: yxsunedu.cn (Y.S); njzhangedu.cn (N.Z); lcaoedu.cn (L.C).