Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
SMCs contribute to smooth muscle...
Smooth muscle cell plasticity
Transcriptional and epigenetic...
Role of SMCs in cardiovascular...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(14):2741-2751. doi:10.7150/ijbs.49871 This issue Cite
Review
Role of smooth muscle cells in Cardiovascular Disease
Yingzhi Zhuge1,2#, Jian Zhang1,2#, Fanyu Qian1,2, Zhengwang Wen2, Chao Niu1,2, Ke Xu3, Hao Ji3, Xing Rong2, Maoping Chu1,2 ![]() , Chang Jia1,2
, Chang Jia1,2 ![]()
1. Pediatric Research Institute, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, China.
2. Children's Heart Center, Institute of Cardiovascular Development and Translational Medicine, The Second Affiliated Hospital and Yuying children's Hospital of Wenzhou Medical University, Wenzhou 325027, China.
3. The Institute of Life Sciences, Wenzhou University, Wenzhou, Zhejiang, China.
#These authors contributed equally to this work.
Received 2020-6-24; Accepted 2020-8-6; Published 2020-8-21
Abstract

Normally, smooth muscle cells (SMCs) are localized in the tunica media of the vasculature, where they take responsibility for vascular contraction and extracellular matrix (ECM) generation. SMCs also play a significant role in obedience and elastic rebound of the artery in response to the haemodynamic condition. However, under pathological or stressed conditions, phenotype switching from contractile to synthetic state or other cell types will occur in SMCs to positively or negatively contribute to disease progression. Various studies demonstrated that functional changes of SMCs are implicated in several cardiovascular diseases. In this review, we present the function of vascular SMCs (VSMCs) and the involved molecular mechanisms about phenotype switching, and summarize the roles of SMCs in atherosclerosis, hypertension, arterial aneurysms and myocardial infarction, hoping to obtain potential therapeutic targets against cardiovascular disease in the clinical practices.
Keywords: smooth muscle cells, cardiovascular disease, phenotype switching, arterial aneurysms
Introduction
Muscle cells are active, contractile cells in animals, which are slender in shape, also known as muscle fibers. Unlike all other tissues, muscle cells contain a large number of muscle filaments, which have the characteristics of contractile movement and are the power source for the movement of body organs. Generally speaking, according to the different structure and contraction characteristics, the muscle tissues in human body are divided into three different types as follows: cardiac muscle, skeletal muscle and smooth muscle, among which cardiac muscle and skeletal muscle have alternating horizontal lines under the light microscope, so collectively referred to as striated muscle. Cardiac muscle is composed of cardiomyocytes, which can be divided into working cells and autonomic cells according to their histological and electrophysiological characteristics, and functional differences to maintain the systolic and autonomic activities of the heart. Skeletal muscle, mainly distributed in the limbs, is superior to smooth muscle and forms rapid and powerful contractions. Smooth muscle is composed of smooth muscle cells (SMCs), which are widely distributed in blood vessel walls and many internal organs, and maintain relatively slow and prolonged contraction. In recent years, more and more studies have shown that SMCs are involved in the occurrence and development of various cardiovascular diseases.
SMCs contribute to smooth muscle contraction
It is well known that smooth muscle is short of transverse striations found in skeletal and cardiac muscle cells, and is often found in the muscular layers of gastrointestinal, respiratory, urogenital, vascular, and lymphatic vessels, as well as in the tunica of some parenchymal organs. The tension contraction of smooth muscle not only resists the gravity or the applied load, but also maintains the normal shape of the organs, and achieves their motor function.
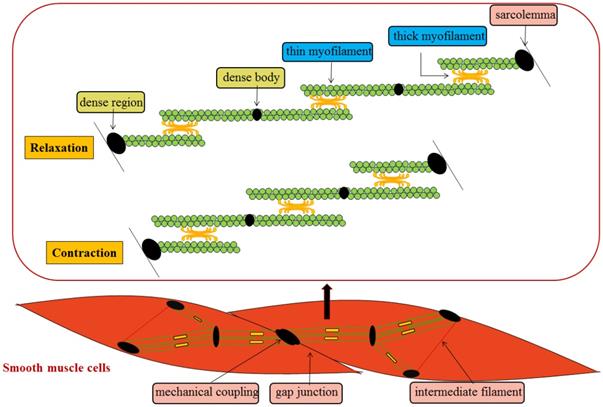
In general, SMCs are slender and spindle-shaped, with a diameter of 1~5 μm and great length variability. It is characterized by the existence of a cytoskeleton inside the cell, which contains some ovoid dense body structures. The spacers appear on the inner side of the cell membrane and become dense regions. The two membranes are closely connected to each other, forming a mechanical coupling to complete the transfer of inter-cell tension. At the same time, gap junctions exist between cells to realize the electric and chemical coupling between cells. Similar protein components to those in the z-band of skeletal muscle are found in the dense body and region, and there is also a filamentous polymer called intermediate filament between the dense body and region, from which the filamentous binding of the dense body and the dense region inside the membrane forms a complete intracellular framework. The thick myofilament of SMCs extend the cross-bridge in different directions in the opposite direction, so that the thin myofilament of different directions slide towards each other, and the sliding range between the thick myofilament and the thin myofilament can extend to the whole length of the thin myofilament, so that the contraction range is larger [1, 2] (Figure 1). Compared with skeleton muscle cells, SMCs contract slowly mainly due to that the formation of some longitudinal shape of the pouch concave on cell membrane and absence of embedded T tube result in the increased surface area of the cell membrane, and slower rate to deliver the action potential on the cell membrane.
Mechanisms of SMCs contraction and relaxation.
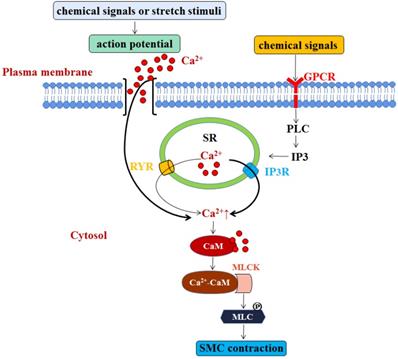
We know that Ca2+ is the main factor that triggers SMC contraction. Although the sarcoplasmic reticulum (SR) of SMCs is underdeveloped, both Ca2+-sensitive ryanodine receptor (RYR) and Inositol 1,4,5-trisphosphate (IP3)-sensitive receptors (IP3R) in the SR membrane act as calcium releasing channels [3]. When the cell is activated, Ca2+ enters the cell through electric-mechanical coupling and pharmaco-mechanical coupling. In the former manner, action potential is generated by chemical signals or stretch stimuli, which promotes extracellular Ca2+ influx into intracellular cytosol. Most cytosolic Ca2+ comes from extracellular sources, but a small part is released by the RYR. In the latter manner, chemical signals can activate the G-protein coupled receptor-PLC-IP3 pathway, which then activates IP3R in the SR membrane to release Ca2+ into the cytosol [4, 5]. SMCs do not contain troponin, but have a calmodulin (CaM). In the majority of SMCs, cytosolic Ca2+ concentration is increased, and increased Ca2+ combines with CaM to form Ca2+-CaM compounds. The compounds then combine with and activate cytoplasmic myosin light chain kinase (MLCK) to promote the phosphorylation of myosin light chain (MLC) for SMC contraction [6] (Figure 2). Otherwise, when the smooth muscle cells are relaxed, cytosolic Ca2+ will be returned back to SR through the SR membrane calcium pump, or be transported out of the cells through the muscle membrane Na+-Ca2+ exchanger and calcium pump, making the drop of the cytosolic Ca2+ concentration, MLCK deactivation, and the dephosphorylation of phosphorylated MLC by the action of MLC phosphatase (MLCP) in the cytoplasm, eventually resulting in relatively slow relaxation of SMCs [7]. Besides, endodermal nitric oxide can also increase the activity of MLCP by regulating cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP), and subsequent protein kinase G (PKG) and protein kinase A (PKA), and ultimately achieve SMCs relaxation [8, 9]. In addition to calcium-dependent mechanism, vascular contraction is also regulated by calcium-independent mechanisms which related to the alteration of calcium sensitization, the remodeling of actin cytoskeleton, and reactive oxygen species (ROS) [10-12].
SMC contraction is modulated by two manners that are both involve Ca2+. One is through action potential induced by chemical signals or stretch stimuli, which promotes extracellular Ca2+ influx into intracellular cytosol. Most cytosolic Ca2+ comes from extracellular sources, but a small part is released by the RYR. Another is G-protein coupled receptor-PLC-IP3 pathway triggered by chemical signals, which then activates IP3R in the SR membrane to release Ca2+ into the cytosol. The elevated level of Ca2+ combines with CaM to form Ca2+-CaM compounds, which then combine with and activate cytoplasmic myosin light chain kinase (MLCK). The activation of MLCK promotes the phosphorylation of myosin light chain (MLC) for SMC contraction.
Smooth muscle cell plasticity
SMCs play a key role in controlling blood pressure and blood distribution, and maintaining vascular structural integrity. SMCs are involved in physiological and pathological vascular remodeling by dynamically regulating its phenotypes. Nowadays, there are increasing researches on the phenotype plasticity of SMCs. The contractile and synthetic phenotypes of SMCs are considered as the most extreme ones, while the rest fall in between [13]. Under normal condition, SMCs exist as a highly differentiated contractile phenotype. Yet, SMCs turned to de-differentiated proliferative or migratory phenotype under stressed conditions. Therefore, SMCs become difficult to recognize if the expression of their marker proteins is remarkably reduced or completely lost, or the SMCs express marker proteins of macrophages, mesenchymal stem cells, osteochondrogenic cells and myofibroblast-like cells etc. [13, 14].
Several factors can modulate SMC phenotype transition, such as integrins and miRNAs. Integrins are transmembrane receptors that link the cell cytoskeleton to the ECM, which involves in the changes of SMC phenotypes. For example, the highly expressed integrins α1β1 and α7β1 in SMCs can bind to collagen IV and laminin, respectively. Their absence transforms the SMCs to a synthetic epithelioid state from a constrictive and spindle phenotype, and promotes the proliferation and accumulation of SMCs during the thickening of the experimental intima. In contrast, serum α2β1, α5β1 and αvβ3 are highly expressed in synthetic SMCs, which promote collagen deposition, participate in fibronectin deposition, and prevent oxygen-low density lipoprotein (Ox-LDL)-mediated SMC apoptosis to promote plaque stability [15-18]. Besides, miRNAs are also particularly involved in regulating phenotype switching of SMCs. Mature miRNA derives from Dicer-cleaved precursor miRNA. The generated miRNA is important for SMC function and development by modulating proliferation and contractile differentiation [19], and also function in the regulation of SMC phenotype. For example, miR-143/145 clusters strongly promote the contractility of the SMC phenotype, while miR-21, miR-221, and miR-222 contribute to the proliferation of SMCs [20, 21].
In recent years, researches on SMC phenotype conversion have been conducted in the following three cases [22]: (1) In the process of vascular injury, SMC contraction gene expression is lost. (2) The expression of some classical marker genes used for SMC identification is not limited to SMC lineage, but can also be expressed by other cell types. (3) Cell types that do not express the SMC marker gene under physiological conditions can change to the SMC-like state in diseased or damaged blood vessels.
Transcriptional and epigenetic modulation of differentiated markers of SMCs
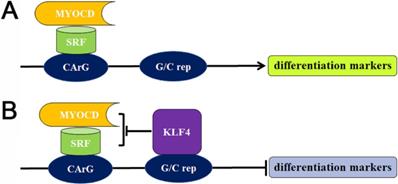
The different regulatory elements in SMC-specific cytoskeleton gene modulate the expression of these proteins. Myocardin (MYOCD) is a main protein that drives the formation of SMC contractile phenotype [23, 24], which forms a complex through combining with the ubiquitous serum response factor (SRF), followed by binding to the specific CArG sequence motif to promote the expression of differentiation markers. On the contrary, the pluripotent transcription factor Krüppel-like factor 4 (KLF4) is absent in contractile SMCs, which can promote the SMC dedifferentiation [25]. KLF can not only bind to G/C repressor elements nearby CArG boxes, but also interact with SRF [26]. KLF involves in the SMC dedifferentiation by inhibiting MYOCD expression, disrupting MYOCD/SRF complex, dissociating MYOCD/SRF complex, and modifying chromatin structure (Figure 3). In addition, other transcription factors, including phosphatase and tensin homolog (PTEN), phosphorylated ETS domain-containing protein-1 (Elk-1), Forkhead box protein O4 (FOXO4), Sp-1 and dedicator of Cytokinesis 2 (DOCK2), can also participate in the differentiation or de-differentiation of SMC via acting on the MYOD/SRF complex dynamics or interacting with KLF4 [27].
Transcriptional regulation of the differentiation or de-differentiation of SMC markers. (A) The MYOCD/SRF complex attaches to the CArG box for the expression of differentiation markers in contractile SMCs. (B) The absence of KLF4 in contractile SMCs, while increased level of KLF4 in synthetic SMCs. The binding of elevated KLF4 with G/C repressor suppresses the MYOCD/SRF complex, resulting in the transcriptional suppression of SMC differentiation markers. G/C rep: G/C repressor.
In addition to gene modulation, the SMC phenotypes are also subject to programmed chromatin remodeling. Chromatin is constituted of nucleosomes, which is composed of 146 bp DNA wrapped around an octamer of histones. The histones are formed by two copies of histones H2A, H3B, H3 and H4. Chromatin exhibits in two different conformations including heterochromatin and euchromatin. The heterochromatin is a condensed DNA form, which can cause silencing of gene transcription, whereas the euchromatin is a non-condensed DNA form that can lead to the activation of gene transcription. Chromatin conformation is modulated by the acetylation of histone and methylation of lysine residues [28]. In contractile SMCs, H3 and H4 relevant to CArG containing regulatory elements of α-SMA, SM22α, and SM-MHCs can be acetylated, leading to CArG elements bound by SRF. The increased activity of histone acetyltransferase (HAT) upregulates the expression of SM22α, while the elevated activity of histone deacetylase (HDACs) inhibits its expression [29]. In addition, DNA demethylation also contributes to SMC differentiation. For instance, the DNA demethylase Ten-eleven translocation-2 (TET2) can oxidize 5-methylcytosis (5mC) to 5-hydroxymethycytosis, and therefore increase the DNA accessibility to transcription factors. In de-differentiated SMCs, the downregulation of TET2 is evidenced by both in vitro and in human coronary artery atherosclerotic plaque and experimentally induced intimal thickening. In cultured SMCs, TET2 knockdown reduced the accessibility of chromatin at the promoters of SMC contractile proteins, including α-SMA, MYOCD, and SMMHCs [29, 30]. In a nutshell, SMC differentiation/dedifferentiation is regulated at both transcriptional and epigenetic levels.
Role of SMCs in cardiovascular diseases
SMCs in atherosclerosis
Atherosclerosis, a type of chronic progressive inflammatory disease, is a major cause of mortality worldwide. It is also a well-known process that lipid deposition occurred between the inner and middle layers of the blood vessel. This allows the macrophage infiltration and SMCs differentiation, and eventually leading to plaque formation [31]. The major clinical consequences of this disease such as myocardial infraction or stroke are mainly caused by thrombotic events related to acute rupture or erosion of an unstable plaque, rather than gradual narrowing of vascular lumen [14]. Historical observation in atherosclerosis showed that the migration and proliferation of SMCs within the intima allows the formation of initial atherosclerotic plaque, while they form fibrous caps to stabilize vulnerable plaques at the advanced stages. However, several studies on the genetic lineage tracing have suggested that the switching of SMC phenotype can not only lead to less differentiated forms in SMC 'markers' including macrophage-like cells, but also directly promote the development of atherosclerosis. Moreover, the proliferation of SMC may be beneficial for atherogenesis, however, the apoptosis of SMC, senescence, and SMC-derived macrophage-like cells may encourage atherosclerosis through inflammation.
Observation of atherosclerotic plaque composition in both animal models and human autopsies reveals that SMCs participate in the progression of atherosclerotic plaque at all stages, which include pre-atherosclerosis, early atherosclerosis, and late atherosclerosis [13]. Since diffuse intimal thickenings (DITs) exist from birth, they are regarded pre-atherosclerotic plaques [32]. Human DITs is composed of SMCs, proteoglycans and elastin, but lack of macrophages and thrombus. Most of SMCs that exhibit a synthetic phenotype in DITs are heterogeneous, as evidenced by a decreased genes encoding expression of contractile proteins and an increased ECM components generation. SMCs are considered as the major source of ECM in DITs, accounting for the increased intimal thickness, but, importantly for the development of atherosclerosis. DITs are full of proteoglycans, which are essential for the retaining apolipoproteins and subsequent inflammatory response.
ECM plays a crucial role in initiating atherosclerosis by the interaction between negative-charged proteoglycans and positive-charged apolipoproteins, which causes the retention of plasma-derived lipoproteins [33]. The modifications of lipoproteins including LDL oxidation trigger the recruitment of macrophages and the initiation of the inflammatory response in atherosclerosis [34]. During this process, the expression of contractile marker proteins, such as α-SMA, are attributed to both phenotypic switching and loss of SMCs through cell death including apoptosis, necrosis, and pyroptosis [34-36]. For instance, the uptake of LDL oxidation and the formation of SMC-derived foam cells are associated with the induction of cell apoptosis [37], while the free cholesterol might be triggered by the dead SMCs [38]. Therefore, SMCs play an essential role in initiation and early development of atherosclerosis.
Late atherosclerosis is featured by fibrous cap generation of SMCs and the formation of necrotic core, which are the consequences of efferocytosis defects of dead SMCs and macrophages [39]. It is previously believed that intimal SMCs in the later stages of atherosclerosis are conducive to the formation of fibrous caps and protect plaques from rupture. Yet, the SMCs lipid loading and altered interactions with ECM cause the phenotype changes of SMC and increase macrophage markers expression in SMCs. Switching of SMCs to macrophage-maker-positive cells leads to a significant decrease in the thickness of intima and the cell content of α-SMA+ in the fibrous cap [40, 41]. SMCs also play a critical role in the inflammatory milieu of the atherosclerotic plaque by recruiting macrophages. Further reports have strongly indicated that SMC-derived macrophage-like cells have direct impact on the progression of plaque. SMCs not only exhibit ECM-producing cells and macrophages of the fibrous cap, but also exhibit foam cells, mesenchymal stem cell (MSC)-like cells and osteochondrogenic cells in the plaque through phenotype switching [42, 43]. Moreover, SMCs play a critical role in calcification of atherosclerotic plaques through apoptosis and osteochondrogenic conversion [44, 45].
During the course of atherosclerosis, the aberrant interplay between endothelial cells (ECs) and vascular SMCs (VSMCs) also play essential roles [46]. For instance, the excessive turnover of VSMCs can be promoted by manipulating ECs in the formation of atherosclerotic plaque, while the EC injury-induced PDGF signaling is related to the proliferation of VSMC and the synthesis of ECM [47]. In addition, the DNA demethylation of PDGF in ECs can activate VSMCs under homocysteine treatment [48]. Moreover, ECs can suppress SMC proliferation and subsequent vascular injury through mediating vascular re-endothelialization through respectively inhibiting cell migration [49] and restenosis [50], enhancing the activity of peroxiredoxin [51], and inducing apoptosis of VSMCs [52].
According to these studies, an important target in the future is to identify and explore the potential factors and mechanisms that can result in the beneficial changes in atherosclerosis through mediating SMC phenotypes and functions, replacing these more conventional anti-atherosclerotic therapies.
SMCs in hypertension
Hypertension, a leading risk factor of various cardiovascular diseases including stroke, coronary artery disease, peripheral vascular disease and heart failure, is related to vascular changes featured by endothelial dysfunction, increased vasoconstriction, and vascular remodeling [53]. In hypertension, vascular remodeling involves changes to SMCs in the vessel wall, as well as endothelial cells, elastin and collagen contents. Among them, SMCs are critically involved in this process due to their plastic and dynamic features, as well as their ability of phenotypic switching. Pro-hypertensive stimuli, including oxidative stress, mechanical forces, renin-angiotensin-aldosterone system (RAAS), the activation of sympathetic nervous system and hemodynamic changes, can lead to vascular structural, functional and mechanical changes through stimulating SMC signaling [54-57].
As is known, the contraction and relaxation of SMCs quickly affects the diameter of blood vessels, which in turn affects blood flow velocity and changes the pressure on the blood vessel wall. The alteration of vascular diameter is an acute and rapid adaptive process, which is largely dependent on the activation and in-activation of the constrictor proteins in SMCs, namely the phosphorylation and dephosphorylation states. The contractile mechanisms of SMCs include actin and myosin as well as highly organized cytoskeleton [53]. The cross-bridge formation of actin and myosin, actin filaments re-organization, intermediate filaments, and microtubules contribute to the vascular contraction. In SMCs, increased actin polymerization, activation of small GTP binding proteins, tyrosine phosphorylation of paxillin, and conformational changes in focal adhesion sites allow the stiffening and re-organization of the cytoskeleton. This dynamic rearrangement of the actin cytoskeleton is very important in maintaining vascular tone and plasticity, and regulating vascular diameter in hypertension.
In addition, SMC contraction in hypertension is also modulated by Ca2+-dependent and independent mechanisms. Many calcium-dependent mechanisms involved in regulating intracellular calcium homeostasis, including Ca2+ channels, contraction-related and unrelated signaling pathways, and immune and inflammatory systems. The perturbance of this homeostasis can cause abnormal vascular Ca2+ handling and high intracellular free calcium concentration ([Ca2+]i). In contrast, the calcium-independent processes can modify MLC sensitivity to Ca2+, and then regulate SMC contraction. Two major signaling pathways that associated with the calcium sensitization are DAG-PLC-PKC pathway and RhoA-Rho pathway kinase (ROCK) [58-61]. Besides, other kinases such as p21-activated protein kinase, integrin-linked kinase (ILK), and zipper-interacting protein kinase (ZIPK) [62, 63], are also involved in this process. The calcium sensitization mechanism can regulate MLC20 phosphorylation independently of calcium-calmodulin-MLCK signaling.
Similar to calcium, reactive oxygen species (ROS) are also regarded as important second messenger molecules in SMCs. This is because the increased generation of vascular Nox-derived ROS in hypertension can result in increased concentration of intracellular free calcium and subsequently the rearrangement of cytoskeleton [64, 65], further causing the alteration of vascular reactivity and enhancement of contraction.
It is known that vascular contraction or vascular relaxation is modulated by calcium-dependent and calcium-independent manners. Emerging evidence indicated that the immune and inflammatory systems and the noncoding genome also contribute to the vascular dysfunction. Therefore, elucidating the complicated interactions between canonical pro-contractile calcium-regulated mechanisms and noncanonical pro-contractile mechanisms can provide better insights for the SMC phenotype switching in hypertension.
SMCs in arterial aneurysm
Arterial aneurysm (AA) is a bulging in the wall of an artery, whose rupture is a catastrophe, including abdominal aortic aneurysm, thoracic aortic aneurysm, coronary artery aneurysm, and so on. In terms of morphology, arterial aneurysms are defined as local dilatation of the artery wall with partial loss of wall parallelism, while on the grounds of functions, arterial aneurysms are regarded as the gradual loss of the ability of the artery wall to withstand the wall tension caused by intracavity pressure, finally leading to acute rupture. Arterial aneurysm is correlated with arterial wall deformities and injury, as a result of infiltration of inflammatory cells, ECM degradation, and dysfunction of SMCs. Studies reported that infiltration of inflammatory cells, and synthesis and modelling of extracellular matrix are associated with SMCs [66, 67], indicating that SMCs play a very important role in the formation and progression of arterial aneurysms.
Previous studies elucidated that the excessive production of the proteases by SMCs, the phenotype switching of SMCs, and the loss of SMCs contribute to aneurysms of larger vessels, such as abdominal aortic aneurysm and thoracic aortic aneurysm. For example, Zhang et al. reported that Smad4-dependent TGF-β signaling in SMCs restrains the excessive production of the proteases essential for elastin degradation and protects against the development of aortic aneurysms. In addition, they demonstrated that Smad4 deficiency in SMCs directly triggers an inflammation-mediated progression of aortic aneurysms. These results indicated that Smad4 deficiency in SMCs promotes the development of aortic aneurysms through inducing the production of proteases and inflammation [68]. As is known, a synthetic phenotype switched from a contractile phenotype in SMCs is considered as an important event in the development of abdominal aortic aneurysms. Peng et al. have demonstrated that VPO1 modulates the phenotypic switch of VSMC from contractile to synthetic via multiple signalling pathways including H2O2/VPO1/HOCl/ERK1/2 pathway. All of the above signaling pathways play a critical role in the development of abdominal aortic aneurysms [69]. Li et al. substantiated that lncRNA H19 is capable of mediating the expression levels, cellular localization, and functionality of hypoxia-induced factor 1α (HIF1α), which exerts pro-apoptotic and anti-proliferation effects in aortic SMCs, resulting in subsequent abdominal aortic aneurysm-like dilation [70].
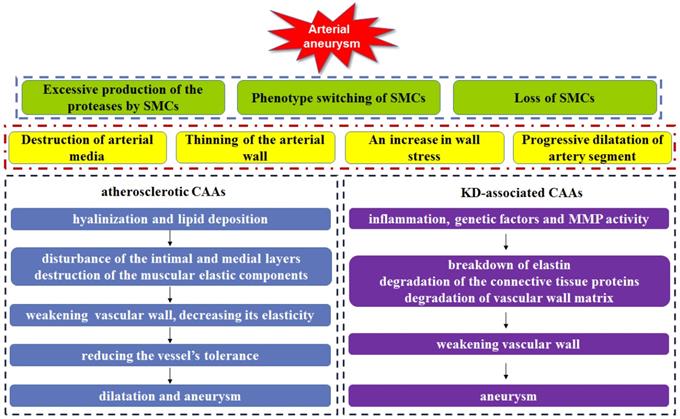
In addition to these aneurysms of larger vessels, coronary artery aneurysm is also a fatal cardiovascular disease, whose rupture will produce catastrophic effect in the patients. Coronary artery aneurysm (CAA) can cause a localized and irreversible dilatation of the blood vessel lumen, which increases the diameter of the adjacent normal segment by 1.5-fold [71]. At present, although the pathophysiology is still not fully understood, CAA is similar to the aneurysm in larger vessels, characteristics by arterial media destruction, arterial wall thinning, wall stress increasing, and the dilatation of the coronary artery segment. Atherosclerosis is responsible for > 90% of CAAs in adults, on the other hand, Kawasaki disease is accountable for most cases in children [72] (Figure 4). Currently, the treatment options of coronary artery aneurysms remain unresolved.
Arterial aneurysm can be induced by excessive production of the proteases by SMCs, phenotype switching of SMCs, and loss of SMCs, which lead to the arterial media destruction, the arterial wall thinning, an increase in wall stress and artery segment dilatation. Both hyalinization and lipid deposition in atherosclerotic CAAs result in the disturbance of the inner and medial layers in vessel wall, the muscular elastic components destruction, the vascular wall weakening and elasticity decreasing, allowing the reduction of vessel's tolerance, as well as vascular dilatation and aneurysm. In KD-associated CAAs, inflammation, genetic factors and MMPs activity lead to breakdown of elastin, degradation of the connective tissue proteins and vessel wall matrix, weakening vascular wall, finally inducing CAAs.
Histological observation results of atherosclerotic CAAs showed that the hyalinization and lipid deposition can not only disturb the inner and medial layers of the vessel wall, but also the destruct muscular elastic components, which was associated with SMC phenotype switching [71, 73]. In addition, several reports have also demonstrated the presence of calcification, fibrosis and large cholesterol crystals triggered the weakening of vessel wall and decreasing in elasticity. This caused further reduction of vessel tolerance to intraluminal pressures of blood flow, and thereby allowing the dilatation of vessels and the formation of aneurysms. Long-term transmural inflammation also contributes to the weakening of vessel walls. The over-stimulation of the vasodilator nitric oxide and the local mechanical stresses caused by stenosis may also induce the medial wall weakening of the coronary artery [74-76].
Kawasaki disease (KD), an acute inflammatory syndrome, is the most common causes of CAAs in children and the second in adults. KD may lead to acute vasculitis of the coronary arteries, which allows the subsequent dilatation of coronary artery and the formation of aneurysms. Accumulating studies have demonstrated that inflammation, genetic factors and matrix metalloproteinase activity contribute to the KD-associated CAAs. For instance, the elevated level of TNF-α is associated with the breakdown of elastin in KD patients [77], suggesting the vital role of inflammation in the progression of CAAs. Lin et al have clarified the close relationship between the genetic variant rs2833195 in the intron of the TIAM1 gene and the progression of CAAs in KD patients. TIAM1 protein may act a key role in chemokine-induced T cell migration, which allows the infiltration of lymphocytes into the vascular wall during the acute vasculitis stage of KD [78]. Matrix metalloproteinase (MMP) is an enzyme responsible for degradation of the connective tissue proteins, and therefore weakening vascular wall. Aneurysmal vessels have shown elevated MMPs levels, whereas declined level of tissue-specific inhibitors of MMPs (TIMPs). This proteolytic imbalance drives the degradation of vessel wall matrix, subsequently leading to CAAs [79].
What's more, changes of SMC functions can also lead to CAAs. For example, Zhang et al reported that without miR-223 VSMC dedifferentiation proceeds undamped resulting in medial damage, apoptosis, and MMP9 release. The elevated MMP9 levels drive the degradation of arterial wall matrix, and subsequently promoting CAA formation [80-82]. It is a well-known fact that SMCs senescence is also proven to enhance the formation and rupture of aneurysm as well [83, 84]. Therefore, many different vascular SMC functions and intracellular signaling pathways are associated with the occurrence and progression of AA, which a defect in vascular SMC may be pathogenic. In addition, SMCs are unique to induce repair in the damaged vessel. To sum up, SMCs play a significant role in AA and become a potential target for further study.
SMCs in MI
Acute myocardial infarction (MI) is myocardial necrosis induced by acute and persistent ischemia and hypoxia in the coronary arteries. Most of them occur on the basis of coronary atherosclerotic stenosis. Thrombus blocking coronary artery lumen can lead to myocardial ischemia and necrosis [85], which can also be induced by sharply increased myocardial oxygen consumption or coronary artery spasm. MI is a common cause that leads to elevated morbidity and mortality in patients with cardiovascular complications.
For the first time, Thidathip et al. analyzed transcripts of aortic vascular SMCs extracted by LCM from patients with MI and non-MI. Gene expression profiling revealed a new 21 gene SMCs-related classifier that distinguished the response of SMCs to MI at the transcriptome level. Vascular SMCs-related genes such as ATP1A2, MYOCD and SOD1 were found to be up-regulated in patients with acute MI [86]. TATP1A2 encodes Na+/K+-ATPase that regulates the exchange of Na+ and K+ ions in response to the condition of metabolic stress, triggering increased vascular contraction of the thoracic aorta in mice [86]. Myocardin (MYOCD) is a myogenic co-activator expressed in vascular SMCs. Previous study demonstrated that MYOCD interacts with SRF to induce gene expression in the CArG boxes, causing the differentiation of SMCs in mice [24]. Superoxide dismutase 1 (SOD1) can regulate the level of H2O2 in the vasculature, thereby modulating the cellular oxidative stress of SMCs in mice [87]. IPA analysis showed that in vascular SMCs of patients with MI, hypoxia signal of the cardiovascular system was the most important activation pathway, so the researchers also found the up-regulated HIF-1A, UBE2 and CREB1 genes that are associated with the hypoxia signaling pathway, while SOD1 of the aortic wall in MI patients was activated [71]. The degradation pathway of superoxide radicals in protein analysis is abundant, in which the antioxidant enzymes SOD1 and CAT play a significant role. Meanwhile, the results of proteomics and transcriptome suggested that SMCs in the aortic wall activate the degradation pathway of superoxide free radical in patients with myocardial infarction. SOD1, an antioxidant that can protect cells from oxidative stress, can catalyze the conversion of superoxide anions into hydrogen peroxide and oxygen to reduce oxidative stress [88-89]. Literature has shown that ROS production may be associated with the hypoxia and the level of electron transport chain cells in mitochondria [90]. Hypoxia can cause an increase in the expression of SOD1. The production of ROS induces the remodeling of vascular wall by enhancing the MYOCD expression [91]. In addition, the ROS can also regulate the expression level of cyclase-1- soluble A-3 (GUCY1A3), which is a risk gene of myocardial infarction [92].
Moreover, Guohong Ye et al. found that VSMCs secrete bFGF to the medium, and then activate the PI3K/AKT signaling pathway by binding to cardiomyocyte FGFR1 in the hypoxia state, thereby weakening the apoptosis and autophagy of myocardial I/R [93]. STIM1 plays an important role in regulating calcium signaling pathways in different cell types, including SMCs [94]. STIM1 detects changes in Ca2+ levels in the endoplasmic reticulum and interacts directly with Orai channels localized in the plasma membrane, coordinating to open Ca2+ channels (CRAC) activated by Ca2+ release [95]. Specific absence of STIM1 in SMCs can significantly reduce the myocardial infarction area by decreasing ER stress, oxidative stress, inflammation and apoptosis. Furthermore, blocking STIM1 in SMCs protects the heart from chronic ischemia [96]. Inactivation of the mineralocorticoid receptor (MR) in VSMCs has also been shown to improve left ventricular dysfunction and remodeling after myocardial infarction by maintaining coronary reserve and reducing oxidative stress-mediated coronary endothelial dysfunction [97]. Harada et al reported that SMC sheet transplantation can significantly shorten fractional area, suppress left ventricular dilation, preserve wall thickness of the area at risk and the posterior wall, decrease fibrosis, and preserve myocardium in the scar area, eventually protecting cardiac function and minimizing cardiac remodeling in a rat MI model [98].
In brief, the above studies indicate that SMCs play a critical role in the process of MI. Through regulating SMC function or relevant SMC proteins, we can preserve the cardiac function under the background of MI.
Conclusion
Cardiovascular disease is a major threat to human, especially for over 50 year-old patients. It is characterized by high morbidity, disability and mortality, so it is urgent to find the most advanced and perfect treatment. The research emphasized in this review contributes to our understanding that SMCs act as an essential role in the occurrence and development of cardiovascular diseases, such as atherosclerosis, hypertension, arterial aneurysms and myocardial infarction. Moreover, exploring its phenotypic changes, including contractile or synthetic phenotypes, and other cell types, such as macrophages, osteochondrogenic cells, mesenchymal stem cells and myofibroblast-like cells, provide a number of important implications for exploiting new targets and directions for clinical treatment. Although many successfully advances have been made in the study of SMCs, there is still much to be elucidated in this field. Therefore, the multifaceted roles of SMCs in cardiovascular disease should be further delineated so as to ensure to have a better understand of its mechanism, and obtain better therapeutic effects in clinical practices.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 81970435, 81770502, 81802251), the Zhejiang Provincial Natural Science Funding (LQ18C010003, LQ18H150003, LY18H020012), Zhejiang Provincial Science and Technology Project of Traditional Chinese Medicine (No. 2018ZZ019), and Wenzhou Science & Technology Bureau Foundation (Y20190064).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Herrera AM, Mcparland BE, Bienkowska A, Tait R, Pare PD, Seow CY. `Sarcomeres' of smooth muscle: functional characteristics and ultrastructural evidence. Journal of Cell Science. 2005;118:2381-92
2. Herzog W, Aithaddou R. Considerations on muscle contraction. Journal of Electromyography and Kinesiology. 2002;12:425-33
3. Taylor CW, Tovey SC. IP3 Receptors: toward understanding their activation. Cold Spring Harbor Perspectives in Biology. 2010 2
4. Zamponi GW, Currie KPM. Regulation of CaV2 calcium channels by G protein coupled receptors. Biochimica et Biophysica Acta. 2013;1828:1629-43
5. Billups D, Billups B, Challiss RAJ, Nahorski SR. Modulation of Gq-Protein-Coupled inositol trisphosphate and Ca2+ signaling by the membrane potential. The Journal of Neuroscience. 2006;26:9983-95
6. He W, Peng YJ, Zhang WC, Lv N, Tang J, Chen C. et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. 2008;135:610-20
7. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel J. The Vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194-204
8. Vanhoutte P, Shimokawa H, Feletou M, Tang EHC. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol. 2017;219:22-96
9. Bolz S, Vogel L, Sollinger D, Derwand R, De Wit C, Loirand G. et al. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation. 2003;107:3081-7
10. Matchkov VV, Kudryavtseva O, Aalkjaer C. Intracellular Ca2+ signalling and phenotype of vascular smooth muscle cells. Basic Clin Pharmacol. 2012;110:42-8
11. Tang DD. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respiratory Research. 2015;16:134
12. Cheng J, Cheng H, Tsai I, Jiang MJ. ROS-mediated downregulation of MYPT1 in smooth muscle cells: a potential mechanism for the aberrant contractility in atherosclerosis. Laboratory Investigation. 2013;93:422-33
13. Basatemur G, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nature Reviews Cardiology. 2019;16:727-44
14. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circulation Research. 2016;118:692-702
15. Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vascular Medicine. 2009;14:73-89
16. Finney AC, Stokes KY, Pattillo CB, Orr AW. Integrin signaling in atherosclerosis. Cellular and Molecular Life Sciences. 2017;74:2263-82
17. Pickering JG, Chow LH, Li S, Rogers KA, Rocnik EF, Zhong R. et al. α5β1 Integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am J Pathol. 2000;156:453-65
18. Jizhong C. Mechanical stretch inhibits oxidized low density lipoprotein-induced apoptosis in vascular smooth muscle cells by up-regulating integrin alphavbeta3 and stablization of PINCH-1. The Journal of biological chemistry. 2007 47
19. Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:1118-26
20. Maegdefessel L, Rayner KJ, Leeper NJ. MicroRNA regulation of vascular smooth muscle function and phenotype: early career committee contribution. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35:2-6
21. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705-10
22. Liu M, Gomez D. Smooth muscle cell phenotypic Diversity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39:1715-23
23. Miano JM. Myocardin in biology and disease. Journal of Biomedical Research. 2015;29:3-19
24. Wang Z, Wang D, Pipes GCT, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. P Natl Acad Sci USA. 2003;100:7129-34
25. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annual Review of Physiology. 2012;74:13-40
26. Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, Mcdonald OG, Owens GK. A G/C element mediates repression of the SM22α promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circulation Research. 2004;95:981-8
27. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochatonpiallat M. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114:540-50
28. Mcdonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circulation Research. 2007;100:1428-41
29. Gomez D, Swiatlowska P, Owens GK. Epigenetic control of smooth muscle cell identity and lineage memory. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35:2508-16
30. Liu R, Leslie KL, Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochimica et Biophysica Acta. 2015;1849:448-53
31. Chen Z, Ichetovkin M, Kurtz MM, Zycband E, Kawka DW, Woods J. et al. Cholesterol in human atherosclerotic plaque is a marker for underlying disease state and plaque vulnerability. Lipids in Health and Disease. 2010;9:61
32. Ikari Y, Mcmanus BM, Kenyon J, Schwartz SM. Neonatal intima formation in the human coronary artery. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19:2036-40
33. Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL. et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750-4
34. Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis update and therapeutic implications. Circulation. 2007;116:1832-44
35. Wu G, Cai J, Han Y, Chen J, Huang Z, Chen C. et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452-65
36. Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C. et al. Role of pyroptosis in cardiovascular diseases. International Immunopharmacology. 2019;67:311-8
37. Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A, Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation. 2000;102:2680-6
38. Tulenko TN, Chen M, Mason PE, Mason RP. Physical effects of cholesterol on arterial smooth muscle membranes: evidence of immiscible cholesterol domains and alterations in bilayer width during atherogenesis. Journal of Lipid Research. 1998;39:947-56
39. Clarke MCH, Talib S, Figg N, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circulation Research. 2010;106:363-72
40. Jacobsen K, Lund MB, Shim J, Gunnersen S, Fuchtbauer E, Kjolby M. et al. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI insight. 2017 2
41. Yu H, Stoneman V, Clarke MCH, Figg N, Xin H, Kotlikoff MI. et al. Bone marrow-derived smooth muscle-like cells are infrequent in advanced primary atherosclerotic plaques but promote atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:1291-9
42. Evans JF, Ragolia L. Systemic and local ACTH produced during inflammatory states promotes osteochondrogenic mesenchymal cell differentiation contributing to the pathologic progression of calcified atherosclerosis. Medical Hypotheses. 2012;79:823-6
43. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156-64
44. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D. et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circulation Research. 2015;116:1312-23
45. Lerouxberger M, Queguiner I, Maciel TT, Ho ATV, Relaix F, Kempf H. Pathologic calcification of adult vascular smooth muscle cells differs on their crest or mesodermal embryonic origin. Journal of Bone and Mineral Research. 2011;26:1543-53
46. Li M, Qian M, Kyler K, Xu J. Endothelial-Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front Cardiovasc Med. 2018;23:151
47. Qi YX, Jiang J, Jiang XH, Wang XD, Ji SY, Han Y. et al. PDGF-BB and TGF-{beta}1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc Natl Acad Sci USA. 2011;108:1908-13
48. Zhang DH, Chen YQ, Xie XN, Liu JL, Wang QJ, Kong W. et al. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol. 2012;53:487-96
49. Breen DM, Chan KK, Dhaliwall JK, Ward MR, Koudsi NA, Lam L. et al. Insulin increases reendothelialization and inhibits cell migration and neointimal growth after arterial injury. Arterioscler Thromb Vasc Biol. 2009;29:1060-6
50. Fuchs AT, Kuehnl A, Pelisek J, Rolland PH, Mekkaoui C, Netz H. et al. Inhibition of restenosis formation without compromising reendothelialization as a potential solution to thrombosis following angioplasty? Endothelium. 2008;15:85-92
51. Kang DH, Lee DJ, Kim J, Lee JY, Kim HW, Kwon K. et al. Vascular injury involves the overoxidation of peroxiredoxin type II and is recovered by the peroxiredoxin activity mimetic that induces reendothelialization. Circulation. 2013;128:834-44
52. Kim HJ, Kim JY, Lee SJ, Kim HY, Oh CJ, Choi YK. et al. α-Lipoic acid prevents neointimal hyperplasia via induction of p38 mitogen-activated protein kinase/Nur77-mediated apoptosis of vascular smooth muscle cells and accelerates postinjury reendothelialization. Arterioscler Thromb Vasc Biol. 2010;30:2164-72
53. Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A. et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529-39
54. Riet LT, Van Esch JHM, Roks AJM, Den Meiracker AHV, Danser AHJ. Hypertension renin-angiotensin-aldosterone system alterations. Circulation Research. 2015;116:960-75
55. Dikalov S, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. American Journal of Physiology-heart and Circulatory Physiology. 2013 305
56. Esler MD, Lambert E, Schlaich MP. Point: chronic activation of the sympathetic nervous system is the dominant contributor to systemic hypertension. Journal of Applied Physiology. 2010;109:1996-8
57. Kim M, Seo J, Cho K, Yoon S, Choi JH, Shin M. Echocardiographic Assessment of Structural and Hemodynamic Changes in Hypertension-Related Pregnancy. Journal of Cardiovascular Ultrasound. 2016;24:28-34
58. Calò LA, Davis PA, Pagnin E, Maso LD, Maiolino G, Teresa M. et al. Increased level of p63RhoGEF and RhoA/Rho kinase activity in hypertensive patients. Journal of hypertension. 2014;32:331-8
59. Crestani S, Webb RC, Silva-Santos JE. High-salt intake augments the activity of the RhoA/ROCK pathway and reduces intracellular calcium in arteries from rats. American journal of hypertension. 2017;30:389-99
60. Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017;113:1009-23
61. Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nature reviews Cardiology. 2010;7:637-47
62. Endo A, Surks HK, Mochizuki S, Mochizuki N, Mendelsohn ME. Identification and characterization of zipper-interacting protein kinase as the unique vascular smooth muscle myosin phosphatase-associated kinase. Journal of Biological Chemistry. 2004;279:42055-61
63. Deng JT, Van Lierop JE, Sutherland C, Walsh MP. Ca2+-independent smooth muscle contraction. a novel function for integrin-linked kinase. The Journal of biological chemistry. 2001;276:16365-73
64. Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. The Canadian journal of cardiology. 2015;31:631-41
65. Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM. Downregulation of nuclear factor erythroid 2-related factor and associated antioxidant genes contributes to redox-sensitive vascular dysfunction in hypertension. Hypertension. 2015;66:1240-50
66. Kim HW, Weintraub NL. Aortic Aneurysm: In defense of the vascular smooth muscle cell. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36:2138-40
67. Chen PY, Qin L, Li G, Malagon-Lopez J, Wang Z, Bergaya S. et al. Smooth muscle cell reprogramming in aortic aneurysms. Cell stem cell. 2020;26:542-57.e11
68. Zhang P, Hou S, Chen J, Zhang J, Lin F, Ju R. et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circulation Research. 2016;118:388-99
69. Peng H, Zhang K, Liu Z, Xu Q, You B, Li C. et al. VPO1 modulates vascular smooth muscle cell phenotypic switch by activating extracellular signal-regulated kinase 1/2 (ERK 1/2) in abdominal aortic aneurysms. Journal of the American Heart Association. 2018 7
70. Li DY, Busch A, Jin H, Chernogubova E, Pelisek J, Karlsson J. et al. H19 induces abdominal aortic aneurysm development and progression. Circulation. 2018;138:1551-68
71. Russo A, Favero L, Sacca S, Caico SI, Cernetti C. Coronary artery aneurysms. Giornale italiano di cardiologia. 2015;16:409-17
72. Sheikh A, Hailan A, Kinnaird T, Choudhury A, Smith D. Coronary artery aneurysm: evaluation, prognosis, and proposed treatment strategies. Heart Views. 2019;20:101
73. Dubland JA, Francis GA. So Much Cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol. 2016;27:155-61
74. Sherif SA, Tok OO, Taskoylu O, Goktekin O, Kilic ID. Coronary artery aneurysms: a review of the epidemiology, pathophysiology, diagnosis, and treatment. Front Cardiovasc Med. 2017;4:24
75. Zeb M, Mckenzie DB, Scott PA, Talwar S. Treatment of coronary aneurysms with covered stents: a review with illustrated case. Journal of Invasive Cardiology. 2012;24:465
76. Keyser A, Hilker M, Husser O, Diez C, Schmid C. Giant coronary aneurysms exceeding 5 cm in size. Interactive Cardiovascular and Thoracic Surgery. 2012;15:33-6
77. Huiyuen JS, Duong TT, Yeung RSM. TNF-α is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. Journal of Immunology. 2006;176:6294-301
78. Kim J, Park Y, Yoon D, Lee K, Song MS, Lee HD. et al. Identification of KCNN2 as a susceptibility locus for coronary artery aneurysms in Kawasaki disease using genome-wide association analysis. Journal of Human Genetics. 2013;58:521-5
79. Pahlavan SMDP, Niroomand F. Coronary artery aneurysm: a review. Clinical Cardiology. 2006;29:439-43
80. Zhang Y, Wang Y, Zhang L, Xia L, Zheng MH, Zeng Z. et al. Reduced platelet miR-223 induction in Kawasaki disease leads to severe coronary artery pathology through a miR-223/PDGFRβ vascular smooth muscle cell axis. Circulation Research. 2020 doi: 10.1161/CIRCRESAHA.120.316951
81. Mata KM, Prudente PS, Rocha FS, Prado CM, Floriano EM, Elias J. et al. Combining two potential causes, of metalloproteinase secretion causes abdominal aortic aneurysms in rats: a new experimental model. Int J Exp Pathol. 2011;92:26-39
82. Gavin PJ, Crawford SE, Shulman ST, Garcia FL, Rowley AH. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute Kawasaki disease. Arterioscler Thromb Vasc Biol. 2003;23:576-81
83. Guo W, Gao R, Zhang W, Ge W, Ren M, Li B. et al. IgE aggravates the senescence of smooth muscle cells in abdominal aortic aneurysm by upregulating lincRNA-p21. Aging and Disease. 2019;10:699-710
84. Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. Journal of Surgical Research. 2000;92:85-95
85. Lim SY. No-Reflow Phoenomenon by intracoronary thrombus in acute myocardial infarction. Chonnam Medical Journal. 2016;52:38-44
86. Wongsurawat T, Woo CC, Giannakakis A, Lin XY, Cheow ESH, Lee CN. et al. Distinctive molecular signature and activated signaling pathways in aortic smooth muscle cells of patients with myocardial infarction. Atherosclerosis. 2018;271:237-44
87. Madamanchi NR, Moon SK, Hakim ZS, Clark S, Mehrizi A, Patterson C. et al. Differential activation of mitogenic signaling pathways in aortic smooth muscle cells deficient in Superoxide Dismutase Isoforms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:950-6
88. Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Medical Science Monitor. 2009 15
89. Fukai T, Ushiofukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants & Redox Signaling. 2011;15:1583-606
90. Wang QS, Zheng YM, Dong L, Ho Y, Guo Z, Wang YX. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radical Bio Med. 2007;42:642-53
91. Chettimada S, Ata H, Rawat DK, Gulati S, Kahn AG, Edwards JG. et al. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. American Journal of Physiology-heart and Circulatory Physiology. 2014 306
92. Cote GJ, Zhu W, Thomas A, Martin E, Murad F, Sharina IG. Hydrogen Peroxide alters splicing of soluble Guanylyl cyclase and selectively modulates expression of splicing regulators in human cancer cells. PLOS ONE. 2012 7
93. Ye G, Fu Q, Jiang L, Li Z. Vascular smooth muscle cells activate PI3K/Akt pathway to attenuate myocardial ischemia/reperfusion-induced apoptosis and autophagy by secreting bFGF. Biomedicine & Pharmacotherapy. 2018;107:1779-85
94. Trebak M. STIM/Orai signalling complexes in vascular smooth muscle. The Journal of Physiology. 2012;590:4201-8
95. Amin AH, Choi S, Galan M, Kassan M, Partyka M, Kadowitz PJ. et al. Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. The Journal of Pathology. 2012;227:165-74
96. Mali VR, Haddox S, Belmadani S, Matrougui K. Essential role for smooth muscle cell stromal interaction molecule-1 in myocardial infarction. Journal of Hypertension. 2018;36:377-86
97. Gueret A, Harouki N, Favre J, Galmiche G, Nicol L, Henry J. et al. Vascular smooth muscle mineralocorticoid receptor contributes to coronary and left ventricular dysfunction after myocardial infarction. Hypertension. 2016;67:717-23
98. Harada S, Nakamura Y, Shiraya S, Fujiwara Y, Kishimoto Y, Onohara T. et al. Smooth muscle cell sheet transplantation preserve cardiac function and minimize cardiac remodeling in a rat myocardial infarction model. Journal of Cardiothoracic Surgery. 2016;11:131
Author contact
![]() Corresponding authors: Maoping Chu (E-mail: chmpingcom); Chang Jia (E-mail: jiashang0802com).
Corresponding authors: Maoping Chu (E-mail: chmpingcom); Chang Jia (E-mail: jiashang0802com).