Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(15):3075-3084. doi:10.7150/ijbs.49735 This issue Cite
Research Paper
The NAD-dependent deacetylase SIRT2 regulates T cell differentiation involved in tumor immune response
Cui Jiang1,3, Jingwei Liu1, Min Guo1, Xiaoxin Gao4, Xuan Wu1, Ning Bai1, Wendong Guo1, Na Li1, Fei Yi1, Rong Cheng1, Hongde Xu1, Tingting Zhou1, Bo Jiang1, Tao Sun3, Shi Wei2 ![]() , Liu Cao1
, Liu Cao1 ![]()
1. Institute of Translational Medicine, Key Laboratory of Cell Biology of Ministry of Public Health, and Key Laboratory of Medical Cell Biology of Ministry of Education, Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis, Treatment and Prevention, China Medical University, No. 77, Puhe Road, Shenyang North New Area, Shenyang, 110042, Liaoning, China.
2. Department of Pathology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35249-7331, USA.
3. Department of Medical Oncology, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and & Institute, 44 Xiaoheyan Road, Dadong District, Shenyang 110042, Liaoning, China.
4. Central laboratory, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and & Institute, 44 Xiaoheyan Road, Dadong District, Shenyang 110042, Liaoning, China.
Received 2020-6-20; Accepted 2020-9-16; Published 2020-10-3
Abstract

Sirtuin 2 (SIRT2), an NAD+-dependent deacetylase, regulates multiple biologic and pathologic processes including mitosis, genomic integrity, cell homeostasis and tumorigenesis. However, the role of SIRT2 in the immune response to cancer remains largely elusive. In this study, we found significantly lower expression of SIRT2 in peripheral T lymphocytes from breast cancer patients when compared to normal individuals. Moreover, SIRT2 levels positively correlated with CD8+ effector memory T (TEM) cells in breast cancer patients. In keeping with these findings, altered T cells differentiation manifested as decreased TEM cells and increased naive T cells were observed in Sirt2 deficient mice. The upregulation of CD8+ TEM by SIRT2 might attribute to the activation of aerobic oxidation as well as the inhibition of GSK3β acetylation in CD8+ T cells. Taken together, these results suggest that SIRT2 participate in tumor immune response by regulating T cell differentiation, which may provide novel insight for tumor prevention and immune therapy.
Keywords: SIRT2, T cell, differentiation, breast cancer
Introduction
Sirtuins family members are evolutionarily conserved NAD+-dependent protein deacetylases with diverse subcellular localization and function [1]. Seven mammalian sirtuins (SIRT1-7) share a highly conserved core NAD+-binding sequence and a common catalytic domain [2]. SIRT2 is the only sirtuin protein mainly localized in the cytoplasm, but has also been reported to exist in the mitochondria and the nucleus [3]. Earlier studies have suggested that SIRT2 is a key regulator of multiple biological processes including mitosis, genomic integrity, autophagy, cell homeostasis and differentiation [4]. In addition, SIRT2 participates in the regulation of pathologic conditions including multiple nervous diseases, infection and inflammatory reaction [5].
Recent emerging evidence has suggested the potential regulatory role of SIRT2 in carcinogenesis, as Sirt2-/- mice develop cancers in multiple organ systems. SIRT2 expression was found to be downregulated in human breast cancers when compared to normal tissues [6]. The molecule inhibited proliferation and metastasis of colon cancer cells and demonstrated decreased expression in colon cancer tissues [7]. In addition, SIRT2 suppressed proliferation in lung cancer cells and enhances sensitivity to Cisplatin therapy [8]. To date, the role of SIRT2 in the immune response against tumor remains largely unknown.
CD8+ T cells act as critical regulator of various physiologic and pathologic processes such as infection, autoimmune diseases and cancer [9, 10]. In response to tumor, naïve CD4+ and CD8+ T cells are activated and then differentiate into effector and memory T (TEM) cells, of which memory CD8+ T cells respond more rapidly than naïve T (TN) cells with a more robust response upon repeat stimulation [11, 12]. Memory CD8+ T cells in circulating blood and lymphoid organs constitute an essential component of long-lived T cell immunity [13]. Following tumor infiltration, these resting memory CD8+ T cells rapidly exert effector functions and are induced to provide protective immunity against subsequent invasion. CD8+ T cell differentiation is modulated by costimulatory signals from antigen presenting cells (APCs) and metabolic pathways as well as epigenetic and transcriptional factors [14-16]. Understanding the specific regulatory mechanisms involved in the differentiation of CD8+ T cells may provide further insight into antitumor immune therapy.
In the present study, we sought to examine the expression of SIRT2 in T lymphocytes of breast cancer patients and normal individuals. We further investigated the potential molecular mechanisms using Sirt2 deficient mice. Our findings have suggested that SIRT2 may participate in tumor immune response by regulating T cell differentiation.
Materials and Methods
Mice
Sirt2-/-mice with a deletion of exon 5-8 were a kind gift from Deng CX [6]. C57BL/6N wild type mice were purchased from Vital River Laboratory Animal Technology Company as controls. All mice were kept in a specific pathogen-free facility in the Animal Resource Center at China Medical University and were enrolled at 6-8 weeks old. The animal protocols were approved by the Animal Care and Use Committee of China Medical University.
Patients
The study was approved by the Ethics Committee of Cancer Hospital of China Medical University. A total of 84 breast cancer patients and 24 healthy controls from Cancer Hospital of China Medical University were included in this study. All patients are treatment-naive women who underwent surgery. Patients were consecutively recruited from November 2017 to February 2018. The informed consents were obtained from the patients and healthy controls. The detailed information of all patients and controls were listed in Table 1. The diagnoses of all patients were pathologically confirmed. Whole blood samples (10 mL each) were collected in ethylenediaminetetraacetic acid (EDTA) tubes from healthy donors and patients before surgery.
Patient characteristics in the study
| N | % | variable | N | % | |||
|---|---|---|---|---|---|---|---|
| Normal controls | |||||||
| Age | ≤35y | 6 | 25 | Age | >35y | 18 | 75 |
| Breast cancer patients | |||||||
| Age | ≤35y | 4 | 4.8 | PR | - | 29 | 34.5 |
| >35y | 80 | 95.2 | + | 55 | 65.5 | ||
| Tumor size | T1:≤2cm | 55 | 65.5 | HER2 | - | 58 | 33.3 |
| T2:>2cm≤5cm | 26 | 31 | + | 26 | 66.7 | ||
| T3:>5cm | 3 | 3.5 | Ki-67 | ≤14% | 25 | 29.8 | |
| Lymphatic metastasis | N0:0 | 56 | 66.7 | >14% | 59 | 70.2 | |
| N1:1-3 | 17 | 20.2 | Luminal A | 17 | 20.2 | ||
| N2:4-9 | 11 | 13.1 | Molecular | Luminal B | 47 | 56 | |
| Stage | I | 36 | 42.8 | subtype | HER2+ | 12 | 14.3 |
| II | 32 | 38.1 | Basal-like | 8 | 9.5 | ||
| III | 16 | 19.1 | Dangerous degree classification | low-risk | 11 | 29.5 | |
| ER | - | 22 | 26.2 | intermediate-risk | 57 | 67.9 | |
| + | 62 | 73.8 | high-risk | 16 | 2.6 | ||
Abbreviation: TNBC: triple-negative breast cancer.
Flow cytometry
Human peripheral blood mononuclear cells (PBMCs) were isolated from blood samples through the Ficoll-Paque Plus (17144002, GE) density gradient separation method. The mice spleens were obtained from wild type and Sirt2-/-mice removing the red blood cells by red blood cell lysis buffer (R1010, Solarbio). Human PBMCs were assessed for live vs. dead status by Zombie NIR dye (423105, Biolegend), while mouse cells were evaluated with PI dye. To detect surface antigens of PBMC, 5×106/cells were washed twice by D-Hanks solution, then suspended in D-Hanks solution and incubated with allophycocyanin (APC), fluorescein isothiocyanate (FITC), phycoerythrin (PE), Brilliant Violet 421 (BV421), PerCP/Cy5.5, Alexa Fluor 647-conjugated monoclonal antibodies on ice for 30 min without light. The antibodies adopted were CD3, CD19, CD4, CD8, CD45RA, CD45RO, CCR7, CD25, CD44, CD62L (BD, Biolegend). The cells stained with surface markers CD4, CD25 were further fixed and permeabilized with IC fixation buffer and permeabilization buffer (00552300, Invitrogen) according to manufacturer instructions, and then incubated with foxp3 antibody or isotype control (Biolegend). CD8+ T cells were positively selected with BD FACS Aria II or Sony SH800s cell sorting/analysis instrument. Data were acquired by Sony SH800s, BD FACS Aria II or BD AccuriC6 flow cytometer and analyzed with FCS Express 6 Flow Cytometry RUO Professional software (De novo Software; USA) and FlowJo 10.4 (Tree Star; USA).
Cell culture
The isolated CD8+T cells (1×106) were sorted directly into TRIzol reagent (15596026, Invitrogen) and stored at -80 °C prior to RNA extraction. The other sorted cells were cultured at 37 °C in RPMI-1640 medium containing FBS (20%, CLARK, Australia, heat inactivated at 56 °C for 30 min), penicillin (100 U) and streptomycin (100 μg/ml) coated with anti-mouse CD3, clone 145-2C11 (2 ug/106 cells) (100314, Biolegend) and anti-CD28, clone 37.51 (5 ug/106 cells) (102116, Biolegend). SIRT2 specific inhibitor AGK2 (10 μM) with DMSO as control was incubated 24 h for further exploring SIRT2-induced experiments. HEK293T and Jurkat cells were obtained from cell bank of Cao's lab. Cells were cultured at 37 °C in Dulbecco's modified Eagle's medium (DMEM) or RPMI-1640 medium supplemented with 10% FBS.
Lentiviral production
In order to perform lentiviral production and infection, the control shRNA (shCtrl) lentivirus, shRNA against Sirt2 (shSirt2) and stably express Sirt2 lentivirus were purchased from Shanghai GeneChem Company. The Sirt2 sequence was 5'- CAACCATCTGTCACTACTT -3'; the stably overexpress Sirt2 sequence was 5'- GGAGCCATTTATTGAAACT-3'. Freshly sorted T cells were infected with the lentivirus for at least 60 hours, and the infected efficiency of the target cells was identified by western blot.
Antibodies and reagents
Antibodies used in this study included SIRT2 (1:1000, S8447, Sigma), GSK3a/β (1:1000, sc-7291, Santa Cruz), GSK3β (1:1000, 12456T, CST), α-tubulin (1:5000, AC012, Abclonal), GAPDH (1:1000, AC012, Abclonal), Flag (1:1000, SG4110-16, Shanghai Genomics Technology) and GFP (1:1000, YM3124, Immunoway). AGK2 (S7577) was purchased from Selleck. DMSO was from Sigma.
Plasmid constructions and transfection
Human SIRT2 was cloned into pcDNA3.1-flag/HA. Human GFP-GSK3β-isoform1 was purchased from Genechem, China (geneID: 2932, Bank ID: NM_002093). Flag-P300, Flag-CBP and Myc-GCN5 were kindly provided by Qunying Lei (Shanghai Medical College, Shanghai, China). Flag-PCAF was a gift from Weiguo Zhu (Shenzhen University, Shenzhen, China). The plasmids were verified by sequencing and then transfected into HEK293T and MCF-7 cells using lipofectamine 3000 regent (Thermo Fisher Scientific, USA) according to the manufacturer's instructions. Cells were collected 48h after transfection.
Western blot and Immunoprecipitation
Western blot was performed as previously described [17]. For immunoprecipitation, cell lysates were incubated with antibody and Protein A/G-Sepharose beads (sc-2003, Santa Cruz) overnight at 4 °C. The protein-antibody complexes were then washed three times at 4 °C with cold lysis buffer and eluted with SDS loading buffer by boiling for 10 min.
Quantitative reverse transcriptase polymerase chain reaction (QRT-PCR)
Total RNA was isolated using TRIzol regent, and complementary DNA (cDNA) was synthesized using PrimeScriptII 1st strand cDNA synthesis kit (6210A; TAKARA). QRT-PCR was performed with the Quanti-TectSYBR Green PCR kit (RR820A; TAKARA) using a Roche Light Cycler 480 II sequence detection system. We determined the expression level of Sirt2 in human CD3+T cells, and Sirt2, GSK3β and OPA1 in mice CD8+T cells. Analyses were performed using the cycle threshold (Ct) method, using the formula 2-△△Ct. The following primers were synthesized by Synbio Tech (Suzhou, China).
PCR primary pairs sequences:
- Human Sirt2: forward primer (FP), 5- CTGTCACTACTTCATGCGCCTG-3; and reverse primer (RP) 5- CCTCCACCAAGTCCTCCTGTT-3.
- Human GAPDH: FP, 5- TCAAGGCTGAGAACGGGAAG-3; and RP, 5-TCGCCCCACTTGATTTTGGA-3.
- Mouse Sirt2: FP, 5-CTTCCTTACCCAGAGGCCATC-3; and RP, 5- TCAGCAGGCGGATGAAGTAGT-3.
- Mouse GSK3β: FP, 5-AGAACTGGTTGCCATCAAGAAAG-3; and RP, 5- GAAATACCGCAGTCGGACTATGT-3.
- Mouse OPA1: FP, 5-TGATCTCACCAAGGAGGAAGATC-3; and RP, 5-CCCAGGGCCTTTGACATTT-3.
- Mouse GAPDH: FP, 5- GAGCTGAACGGGAAGCTCAC-3; and RP, 5- TCAGATGCCTGCTTCACCAC-3.
Measurement of OCR and ECAR
The oxygen consumption rate (OCR) as well as extracellular acidification rate (ECAR) were detected on the basis of Seahorse XFp analyzer (Seahorse Bioscience, 103020-100). 1×106 CD8+T cells/well were plated on Seahorse XFp plates for 24 h. The detailed procedure has been previously described [18].
Statistics
All the statistical analyses were performed using SPSS version 22.0 software (SPSS Inc, Chicago IL, USA) and p-values less than 0.05 were considered statistically significant.
Results
SIRT2 levels positively correlate with CD8+ TEM in breast cancer patients
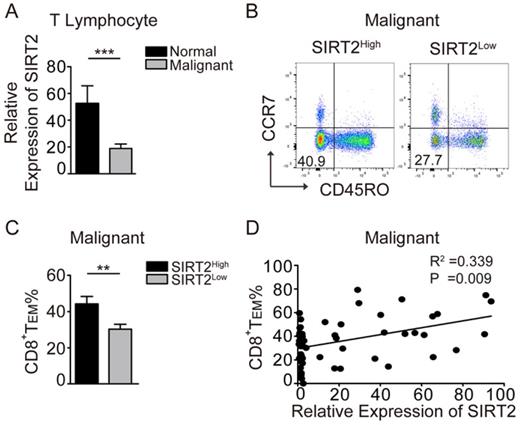
To explore the role of SIRT2 in antitumor immunity, we examined SIRT2 mRNA expression in the peripheral blood CD3+ T cells from breast cancer patients and normal individuals. To that end, significantly decreased SIRT2 expression in T lymphocytes was observed in breast cancer patients (Fig. 1A). Furthermore, the patients with SIRT2high T cells had a significantly higher percentage of CD45R0-CCR7-CD8+TEM than those with SIRT2low T cells (Fig. 1B, 1C). Moreover, SIRT2 levels and CD8+TEM showed a positive correlation in breast cancer patients (R2=0.339, P=0.009) (Fig. 1D). In contrast, no significant correlation was found between SIRT2 and CD4+ T cells (data not shown). The lower SIRT2 expression in T lymphocytes suggested insufficient antitumor immunity in breast cancer patients.
SIRT2 levels positively correlate with CD8+ TEM in breast cancer patients. A: The expression of SIRT2 was determined by real-time PCR within the T lymphocytes isolated from normal controls (n=19) and breast cancer patients (n=70). B and C: The comparison of CD8+TEM (CD45R0-CCR7-CD8+) was carried out in the SIRT2high and SIRT2low groups by flow cytometry. D: The correlation between relative expression of SIRT2 expression and CD8+TEM percentage within T lymphocytes was accessed by linear correlation analysis. The data were presented as mean±SD. *P<0.05; **P<0.01; ***P<0.005.
CD8+ TEM cells increase in the peripheral immune system during breast cancer progression
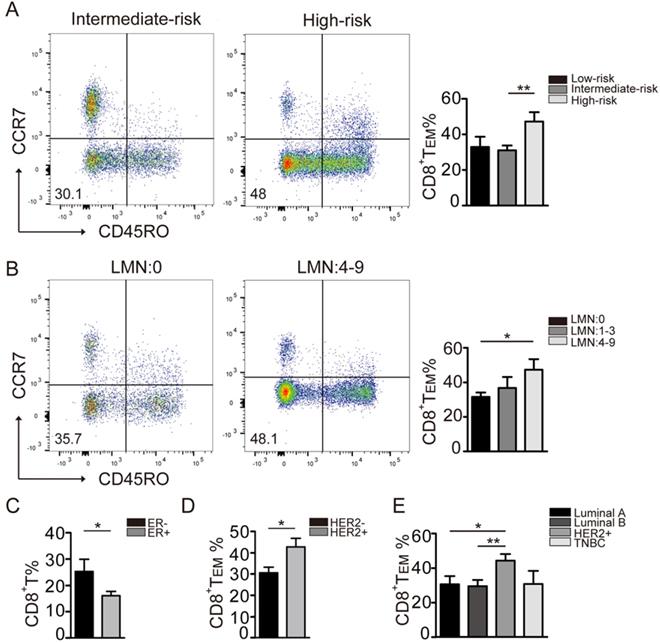
The distribution of immune cells varies in breast cancer with different molecular subtypes and risk levels [19]. Therefore, we further analyzed the immune status of breast cancer patients in this cohort. The patients' characteristics were summarized in Table 1. The patients were divided into four groups according to the hormone receptor (HR) ER, PR, HER2 and Ki-67 index: Luminal A (n=17), Luminal B (n=47), HER2-enriched (n=12), and triple-negative (n=8). According to the risk assessment for breast cancer recurrence by 2007 St. Gallen standard, these patients were divided into three groups: low-risk (n=11), intermediate-risk (n=57) and high-risk (n=16).
To that end, a significantly higher level of CD8+ TEM cells was found in the high-risk group when compared to that in the moderate-risk group (Fig. 2A). Furthermore, the levels of CD8+ TEM cells positively correlated with lymph node status. Significantly higher levels of CD8+ TEM were observed in patients with 4-9 positive lymph nodes when compared to those with no nodal metastasis (Fig. 2B). In addition, CD8+ TEM cells were more abundant in patients with ER negative tumors when compared to those with ER positive tumors (Fig. 2C). Moreover, the CD8+ TEM cells were significantly increased in the patients with a HER2 subtype tumor (Fig. 2D, 2E). Due to the small number of triple-negative patients enrolled, no obvious trend of CD8+ TEM cells was seen in this group. Taken together, these results suggested that CD8+ TEM cells were significantly increased in breast cancer subtypes with worse biologic behavior and unfavorable prognosis.
Distribution of CD8+ T cell phenotypes in different breast cancer subtypes. A: The comparison of CD8+TEM (CD45R0-CCR7-CD8+) was performed in different breast cancer recurrence risk groups: Low-risk (n=10), Intermediate-risk (n=47), High-risk (n=12). B: The CD8+TEM percentage in pathologic Lymph Nodes metastasis groups: LMN: 0 (n=50), LMN: 1-3 (n=12), LMN: 4-9 (n=8). C: The CD8+ T percentage in ER negative (n=17) and ER positive (n=53) groups. D: The CD8+TEM percentage in HER2-negative (n=41) and HER2-positive (n=21) groups. E: The CD8+TEM percentage in the biologic subtype: Luminal A (n=16), Luminal B (n=26), Her2+ (n=20), TNBC (n=6). The data were presented as mean±SD. *P<0.05; **P<0.01.
Sirt2 deficiency lead to abnormal T cells differentiation
In response to tumor cells, human naïve CD4 and CD8 T cells were activated and differentiate into effector T cells and memory T cells, and the latter react more rapidly than naïve T cells and provide a more robust response upon repeat stimulation [11]. In keeping with this early observation, the subsets of CD45R0+CCR7+CD8+ TN cells in the peripheral blood mononuclear cells (PBMCs) declined in breast cancer patients, with increased proportion of T cells differentiating into CD8+ TEM cells (Fig. 3A).
Distribution of CD4+ and CD8+ T cells in mice. A: The CD8+ T cell subsets in the normal controls (n=19) and breast cancers (n=70). B.C: The CD8+ and CD4+ T cell subsets in the homologous mouse: wildtype mice (n=11) and Sirt2-/- (n=9). The data were presented as mean±SD. *P<0.05; **P<0.01; ***P<0.005.
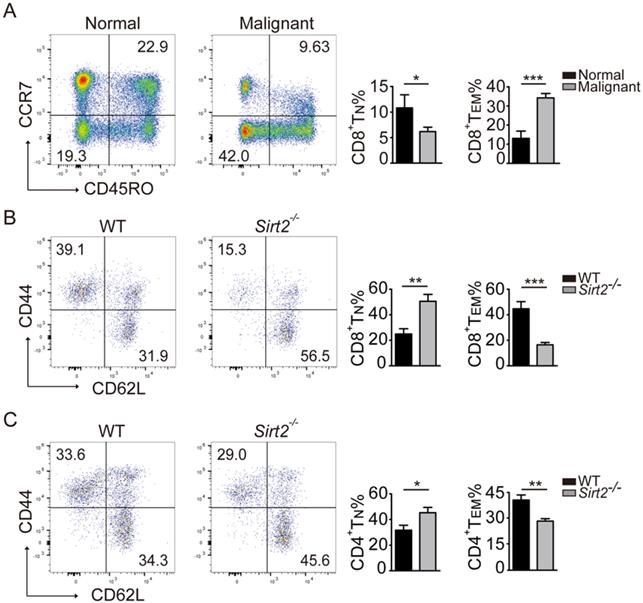
We next used the Sirt2 knockout mouse model to further investigate the impact of SIRT2 on the immune status. To that end, while there was no significant difference in the distribution of CD4+ and CD8+T cells (Fig. S2A), consistently increased CD44-CD62L+ naive T cells (TN) and decreased memory T cells (TM) in CD4+ or CD8+ T cells were observed in Sirt2-/- mice. Furthermore, this altered differentiation was mainly manifested as a decline in CD44+CD62L-effector memory T cells (TEM) (Fig. 3B, 3C). We did not observe the association between SIRT2 and CD4+ T cells in human samples. However, when the mouse Sirt2 gene was knocked out, SIRT2 also affected CD4+ T cells differentiation. Altogether, there results indicate that Sirt2 deficiency leads to abnormal T cells differentiation including the accumulation of TN cells and decline of TEM cells.
Sirt2 deletion restrains aerobic oxidation in CD8+ T cells
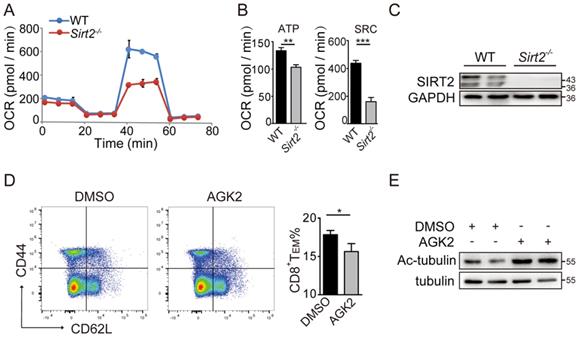
CD8+ T cells, especially the metabolically quiescent TE and TM cells, depend primarily on oxidative phosphorylation to provide sufficient energy for metabolism. Therefore, we sorted the freshly isolated CD8+ T cells of Sirt2-/- or wild type mice and assessed mitochondrial respiration as well as aerobic glycolysis via detecting the OCR and ECAR in basal conditions or drug-induced mitochondrial stress, to analysis T cells metabolic-flux. For EACR, a small decrease was found in basal glycolysis in Sirt2-/- CD8+ T cells when compared to the wild type CD8+ T cells (Fig. S2B & 2C). For OCR, there was no measurable difference in their basal respiration, leak or non-mitochondrial respiration. Nevertheless, a significant decline in spare respiratory capacity (SRC) in Sirt2-/-CD8+ T cells was detected when compared to wild type CD8+ T cells, and a small difference in ATP-coupled was found between the two groups (Fig. 4A-C). In addition, the inner mitochondrial membrane (IMM) fusion protein optic atrophy 1(OPA1), a molecule essential for mitochondrial aerobic oxidation reactions, demonstrated a significant decrease in Sirt2-/- CD8+ T cells (Fig. S2D & 2E ). Taken together, these finding suggest that SIRT2 regulates CD8+ T cell differentiation by altering its aerobic glycolysis.
SIRT2 affects the metabolism of CD8+ T cells. A. C: OCR of wild-type (n=6) and Sirt2-/- (n= 6) CD8+ T cells in real time after the addition of oligomycin,2,4-dinitrophenol (DNP) and retenone. B: ATP production and spare respiratory capacity of cells. D, E: wild-type CD8+ T cells were re-stimulated with anti-CD3 and anti-CD28 for 72 hours and treated with 10 uM/ml AGK2 or DMSO as the control for 24 hours (E). The harvested CD8+ T cells were analyzed by flow cytometry (D). The data were presented as mean±SD. *P<0.05; **P<0.01; ***P<0.005.
SIRT2-mediated CD8+ T differentiation relies on glycogen synthase kinase 3β (GSK3β) acetylation
Memory cells are enabled to respond more effectively to tumor invasion by multiple mechanisms, including alteration of cytokine receptor expression and epigenetic pathways. Here, we sought to test whether GSK3β was involved in the regulation of SIRT2 on CD8+ T cell differentiation. The wild type CD8+ T cells in the activation of CD3 and CD28 were cultured for 72 hours and treated with 10 uM/ml AGK2, a selective SIRT2 inhibitor, or DMSO as the control. In AGK2-treated cells, CD8+ TEM were slightly decreased when compared to the control group (Fig. 4D & 4E). Furthermore, infecting the cells with lentivirus overexpressing Sirt2 followed by 12h activation resulted in a higher level of CD8 TEM (Fig. 5A & 5B). Subsequent immunoprecipitation (IP) assay confirmed that GSK3β interacted with SIRT2 in Jurkat cells and HEK-293T cells (Fig. 5C & 5D).
SIRT2-mediated CD8+ T differentiation relies on GSK3β acetylation. A.B. wild-type CD8+ T cells were re-stimulated with anti-CD3 and anti-CD28 for 72 hours and infected with lentivirus overexpressing Sirt2 followed by 12 h activation (A). The harvested CD8+ T cells were analyzed by flow cytometry (B). C: SIRT2 interacts with GSK3β in vivo. Jurkat cell lysates were subjected to immunoprecipitation with control IgG and anti-GSK3β antibody. The immunoprecipitates were then blotted with the indicated antibodies. D: SIRT2 bind with GSK3β. Flag-tagged SIRT2 was individually transfected into HEK293T cells. The interaction was detected by IP and western blot. E: Catalytic activity of SIRT2 is required for GSK3β deacetylation. Flag-tagged SIRT2 was individually transfected into HEK293T cells. CHK2 acetylation was detected by immunoprecipitation using an anti-acetylated lysine antibody. F: Overexpression of p300, but not other histone acetyltransferases (HATs), could significantly increase GSK3β acetylation. HEK293 cells were transfected into HATs P300, CBP, GCN5 and PCAF. Acetylated GSK3β was purified from cells transfected with Flag-tagged P300, CBP, PCAF or Myc-tagged PCAF. Acetylated GSK3β was determined by immunoblotting. G: SIRT2 overexpression decreases GSK3β acetylation. Flag-tagged GSK3β and P300 was co-transfected with either Flag or Flag-tagged SIRT2 into HEK 293T cells. Acetylation of purified proteins was determined by immunoblotting and western blot. The data were presented as mean±SD. *P<0.05.
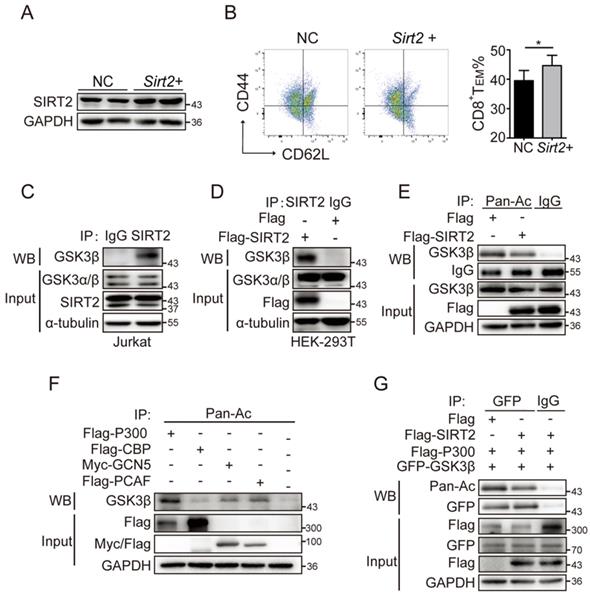
While SIRT2 is a well-known deacetylase, acetylation is also involved in the mechanism of regulating tumor immune responses [20]. To assess the acetylation status of GSK3β, HEK293T cells were transiently transfected with Flag-SIRT2, and then assayed for acetylation of GSK3β via IP assay with an anti-acetylated lysine antibody. A significantly decreased acetylated GSK3β was observed when SIRT2 was overexpressed (Fig. 5E). Thus, we next investigated the four classic acetyltransferases to determine which could affect the acetylation of GSK3B. To that end, the ectopic expression of P300, but not CBP, GCN5 and PCAF, significantly enhanced the acetylation of GSK3β, which was verified by Co-IP assay (Fig. 5F & 5G). Together, these results demonstrated that SIRT2 may promote CD8+ T cells differentiation by regulating GSK3β acetylation.
Discussion
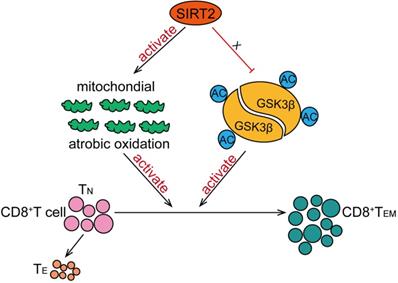
In this study, we have found that SIRT2 participate in T-cell-mediated immune response in breast cancer. SIRT2 levels positively correlated with CD8+ TEM cells in breast cancer patients. In addition, Sirt2 deficiency resulted in abnormal T cells differentiation manifested as declined TEM cells and increased TN cells in mice. Specifically, SIRT2 promoted aerobic oxidation, inhibited GSK3β acetylation in CD8+ T cells, thus mediating the differentiation of CD8+ T cells from TN into TEM (Fig. 6).
Schematic model proposed for the role of SIRT2 in T-cell-mediated immune response in breast cancer. SIRT2 promoted aerobic oxidation, inhibited GSK3β acetylation in CD8+ T cells, thus mediating the differentiation of CD8+ T cells from TN into TEM.
Sirt2-/- mice develop gender-specific cancers in multiple organs, which indicate that SIRT2 may function as a tumor suppressor. Indeed, previous studies have reported the disorder of SIRT2 in various types of cancers, suggesting that SIRT2 might suppress tumorigenesis via multiple mechanisms. Here, we discovered previously unrecognized function of SIRT2 in regulating T-cell-mediated immune response: T lymphocytes in breast cancer patients showed decreased SIRT2 expression compared to healthy controls. SIRT2high patients had a significantly higher CD45R0-CCR7-CD8+TEM percentage than the SIRT2low patients. Furthermore, SIRT2 and CD8+TEM showed a positive correlation in breast cancer patients. CD8+ T cells, including short-lived effector T cells and memory T cells, derive from naive CD8+ T cell precursor and supply long-term immunity against tumors [21]. The deficiency of T cells and destruction of cytotoxic mechanisms can prompt susceptibility to cancer in animal models [22]. In breast cancer, the primary effector immune cells eliminating cancer cells are CD8+ CTLs and NK cells [23]. Not surprisingly, elevated infiltration of CD8+T cells in breast cancer correlated with a favorable clinical outcome [24]. In addition, HER2-enrich and basal-like carcinomas are characterized by highest CD8 tumor infiltrating lymphocytes and highest frequencies of memory T cells [25, 26]. Researches in both human and mouse models have indicated that CD8+ TEM cells are enriched in the bone marrow of breast cancer patients/animals, but deficient in malignant effusions [27-29]. During the development of breast cancer, the human immune system is activated, and both CD4+ and CD8+ TE cells are mobilized to fight tumor cells (Fig. S1). CD8+ T cells perform more prominently in the immune response wherein CD8+ TN cells became activated, followed by differentiation into cytotoxic effector cells and TM cells, which are simultaneously changed during the progression of the breast cancer (Fig. S1). Altogether, SIRT2 promotes differentiation of CD8+ T cells into a memory phenotype. Bulk of CD8+ TEM cells are mobilized immediately in immune response and accumulate in more aggressive breast cancer.
We next explored the possible mechanisms by which SIRT2 regulates the immune status in cancer by altering CD8+ T cells differentiation. It has been suggested that memory CD8+ T cells, known as metabolically quiescent cells, rely primarily on oxidative phosphorylation as their energy source [30, 31]. One important feature of CD8+ T cell is the upregulation of mitochondrial respiratory capacity, a precondition for the growth and population expansion of CD8+ T cells [32, 33]. Under mitochondrial stress, CD8+ TEM cells demonstrate immediate increase of metabolic characteristics and sustain aerobic glycolysis [30]. Therefore, we examined whether differentiation disorder of Sirt2-/- CD8+ T cells was caused by imbalance of energy metabolism. Using extracted CD8+ T cells from SIRT2 knockout mice to detect their aerobic oxidation and glycolysis metabolism, we found a significant decline in SRC and slightly decreased ATP. Importantly, memory CD8+ T cells have been suggested to possess an increased SRC and mitochondrial content in CD8+ T cells [31]. It has been well known that SIRT2 is the only sirtuin mainly located in the cytoplasm. However, recent studies have shown that SIRT2 could also be localized to mitochondria and interact with mitochondrial candidate proteins to direct mitochondrial metabolism [34]. In the present study, we found that OPA1, a protein essential for the fusion events of the inner mitochondrial membrane during CD8+memory T cells metabolic reactions [10, 35], was also decreased in Sirt2-/- CD8+ T cells. Therefore, SIRT2 deficiency leads to an energy-deprivation as well as dysfunction in activating CD8+ T cells and thus a significant decrease of effective CD8 TEM cells. Our findings suggest that SIRT2 may functions as a critical energetic regulator of CD8+ T cells metabolism and differentiation, particularly through aerobic oxidation.
Another potential mechanism is that SIRT2 induces enrichment of CD8+ TEM in PBMCs by affecting key signaling pathways of tumor immune system. Among multiple and complex signaling pathways of T cell differentiation, several core molecules regulating memory T cell differentiation were verified and well-characterized [36-39]. It has been reported that AKT and its direct substrate GSK3β are critical regulators of CD8+memory T cells differentiation [40-42]. Several reports indicate that GSK3β is involved in various signaling pathways controlling T cells differentiation, metabolism, cell death and survival [43, 44]. Expression of constitutively active GSK3β decreases proliferation of CD8+ T cells [39, 45]. Additionally, AKT/GSK3β signal pathway activation occurs at mitochondria-endoplasmic reticulum contact sites, which is critical for early mitochondrial reprogramming of memory CD8+T cells [33]. Here, our findings suggest that SIRT2 induce the deacetylation of GSK3β. The interaction of GSK3β with SIRT2 has been confirmed by IP assay. Therefore, SIRT2 may mediate CD8+ T differentiation through the inhibition of GSK3β acetylation. However, more specific and detailed impact of SIRT2 protein on the functionality of antitumor immunity requires further investigation.
Conclusion
In summary, we have identified an association of SIRT2 with enhanced CD8+ TEM cell differentiation in breast cancer, an event mediated at least in part by activated aerobic oxidation as well as the inhibition of GSK3β acetylation in CD8+ T cells. These findings provide potential insight into the full biological repertoire of SIRT2 in the immune response against cancer thus warrant further investigation.
Abbreviations
SIRT2: Sirtuin 2; effector memory T: TEM; TN: naive T; APCs: antigen presenting cells; QRT-PCR: quantitative reverse transcriptase polymerase chain reaction; OCR: oxygen consumption rate; ECAR: extracellular acidification rate; HR: hormone receptor; SRC: spare respiratory capacity; IMM: inner mitochondrial membrane; OPA1: optic atrophy 1; GSK3β: glycogen synthase kinase 3β; IP: immunoprecipitation.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Funding
This work was supported by National Key R&D Program of China (2016YFC1302400), the Ministry of Education Innovation Team Development Plan (IRT_17R107&IRT13101), the Key Project of the National Natural Science Foundation (81130042, 81770001), the National Science Foundation of China (31300963, 81502400, 81502447, and 81502414), the Liaoning Education Department General Project (LK201627), and funded by the National Research Foundation of Korea (NRF-2014R1A1A3051320, NRF-2014M3A9D8034459, and NRF-2016R1D1A1B04934603).
Author contributions
LC and SW designed the experiments. CJ performed most of the experiments, JWL, XXG, XW, NB, NL RC and BJ performed part of the experiments. NB performed and analyzed the result of mouse model. WDG, FY and TTZ provided some antibodies and experimental material. HDX provided the data of mice. TS provided the data of patients. CJ, LC and WZ prepared and wrote the manuscript. JWL, SW and LC reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.
Ethics approval and consent to participate
Ethics approval was granted by the Ethics Committee of Cancer, Hospital of China Medical University. Animal experiments were carried out according to China Medical University Animal Care and Use Guidelines, and the experimental protocols were approved by the Animal Care and Use Committee of China Medical University. Written informed consents were obtained from all patients.
Availability of data and materials
The datasets used in current study are available from the corresponding author on reasonable request.
Consent for publication
All authors give consent for publication of the manuscript in Molecular Cancer.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kida Y, Goligorsky MS. Sirtuins, Cell Senescence, and Vascular Aging. The Canadian journal of cardiology. 2016;32:634-41
2. Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS. et al. Sirtuin functions and modulation: from chemistry to the clinic. Clinical epigenetics. 2016;8:61
3. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes & development. 2006;20:1256-61
4. Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55:100961
5. Gomes P, Fleming Outeiro T, Cavadas C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends in pharmacological sciences. 2015;36:756-68
6. Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X. et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487-99
7. Hu F, Sun X, Li G, Wu Q, Chen Y, Yang X. et al. Inhibition of SIRT2 limits tumour angiogenesis via inactivation of the STAT3/VEGFA signalling pathway. Cell Death Dis. 2018;10:9
8. Li Z, Xie QR, Chen Z, Lu S, Xia W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung cancer (Amsterdam, Netherlands). 2013;82:9-15
9. Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput Struct Biotechnol J. 2019;17:1-13
10. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599-611
11. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107-19
12. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A. et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. 2017;552:404-9
13. Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol. 2009;9:662-8
14. Delgoffe GM, Powell JD. Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Mol Immunol. 2015;68:492-6
15. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE. et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166:63-76
16. Luigia Pace CG, Elina Zueva, Paul Gueguen, Nina Burgdorf, Joshua J. Waterfall,Jean-Pierre Quivy, Geneviève Almouzni,Sebastian Amigorena. The epigenetic control of stemness in CD8+ T cell fate commitment. Science. 2018;359:177-86
17. Zhang W, Feng Y, Guo Q, Guo W, Xu H, Li X. et al. SIRT1 modulates cell cycle progression by regulating CHK2 acetylation-phosphorylation. Cell Death Differ. 2019 [Epub ahead of print]
18. Li M, Liu J, Li S, Feng Y, Yi F, Wang L. et al. Autophagy-related 7 modulates tumor progression in triple-negative breast cancer. Lab Invest. 2019;99:1266-74
19. Solinas C, Carbognin L, De Silva P, Criscitiello C, Lambertini M. Tumor-infiltrating lymphocytes in breast cancer according to tumor subtype: Current state of the art. Breast (Edinburgh, Scotland). 2017;35:142-50
20. Piccirillo AR, Cattley RT, D'Cruz LM, Hawse WF. Histone acetyltransferase CBP is critical for conventional effector and memory T-cell differentiation in mice. J Biol Chem. 2019;294:2397-406
21. Federica Sallusto DL, Reinhold Forster, Martin Lipp, Antonio Lanzavecchia. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. NATURE. 1999;401:708-12
22. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161-8
23. Jiang X, Shapiro DJ. The immune system and inflammation in breast cancer. Molecular and cellular endocrinology. 2014;382:673-82
24. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer. 2016;4:59
25. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ. et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13:228-41
26. Yi Huang CM, Qunyuan Zhang, Jian Ye, Fang Wang, Yanping Zhang, Pamela Hunborg, Mark A. Varvares, Daniel F. Hoft, Eddy C. Hsueh and Guangyong Peng. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget. 2015;19:17462-78
27. Markus FEUERER MR, Lianhua BAI, Victor UMANSKY, Erich-Franz SOLOMAYER, Gunther BASTERT, Ingo J. DIEL and, SCHIRRMACHER1 V. ENRICHMENT OF MEMORY T CELLS AND OTHER PROFOUND IMMUNOLOGICAL CHANGES IN THE BONE MARROW FROM UNTREATED BREAST CANCER PATIENTS. Int J Cancer. 2001;92:96-105
28. Arnaud Scherpereel BDG, Marc Noppen, Thomas Gey, Bachar Chahine, Simon Baldacci, Jacques Trauet, Marie-Christine Copin, Jean-Paul Dessaint, Henri Porte, Labalette aM. Defect in recruiting effector memory CD8+ T-cells in malignant pleural effusions compared to normal pleural fluid. BMC Cancer. 2013;13:324
29. Schirrmacher V. Cancer-reactive memory T cells from bone marrow: Spontaneous induction and therapeutic potential (Review). Int J Oncol. 2015;47:2005-16
30. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G. et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064-72
31. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. 2016;28:514-24
32. van der Windt Gerritje JW, Everts B, Chang C-H, Curtis Jonathan D, Freitas Tori C, Amiel E. et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity. 2012;36:68-78
33. Bantug GR, Fischer M, Grahlert J, Balmer ML, Unterstab G, Develioglu L. et al. Mitochondria-Endoplasmic Reticulum Contact Sites Function as Immunometabolic Hubs that Orchestrate the Rapid Recall Response of Memory CD8(+) T Cells. Immunity. 2018;48:542-55 e6
34. Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y. et al. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid Redox Signal. 2017;26:849-63
35. Yang F, Wu R, Jiang Z, Chen J, Nan J, Su S. et al. Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. 2018;9:556
36. Chi H. Regulation and function of mTOR signalling in T cell fate decision. Nat Rev Immunol. 2012;12:325-38
37. Shehata HM, Khan S, Chen E, Fields PE, Flavell RA, Sanjabi S. Lack of Sprouty 1 and 2 enhances survival of effector CD8(+) T cells and yields more protective memory cells. Proc Natl Acad Sci U S A. 2018;115:E8939-E47
38. Suresh EHKaM. Role of PI3K:Akt signaling in memory CD8 T cell differentiation. Front Immunol. 2013;4:20
39. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z. et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808-13
40. Koichi Araki APT, Virginia Oliva Shaffer, Shivaprakash Gangappa, Susanne A. Keller MFB, Christian P. Larsen, and Rafi Ahmed. mTOR regulates memory CD8 T cell differentiation. Nature. 2009;460:108-12
41. Sarikhani M, Mishra S, Maity S, Kotyada C, Wolfgeher D, Gupta MP. et al. SIRT2 deacetylase regulates the activity of GSK3 isoforms independent of inhibitory phosphorylation. Elife. 2018 7
42. Zahra Zahid Piracha HK, Umar Saeed, Jumi Kim, Jaesung Jung. et al. Sirtuin 2 Isoform 1 Enhances Hepatitis B Virus RNA Transcription and DNA Synthesis through the AKT/GSK-3 / - Catenin Signaling Pathway. Journal of Virology. 2018;92:e00955-18
43. Toshiaki Ohteki MP, Arsen Zakarian, Russell G. Jones, Linh T. Nguyen, James R. Woodgett, and Pamela S. Ohashi. Negative Regulation of T Cell Proliferation and Interleukin 2 Production by the Serine Threonine Kinase GSK-3. J Exp Med. 2000;192:99-104
44. Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. GSK-3 - at the crossroads of cell death and survival. Journal of Cell Science. 2014;127:1369-78
45. Prlic M, Bevan MJ. Cutting Edge: β-Catenin Is Dispensable for T Cell Effector Differentiation, Memory Formation, and Recall Responses. The Journal of Immunology. 2011;187:1542-6
Author contact
![]() Corresponding authors: E-mail: lcaoedu.cn; sweiedu.
Corresponding authors: E-mail: lcaoedu.cn; sweiedu.