Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pathophysiology of COVID-19
Generation of ROS at sites of...
Myeloperoxidases could play a...
Hypochlorous acid alters...
Toxicity of free iron
MPO and other members of the...
COVID-19 affects virtually every...
Summary
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(1):62-72. doi:10.7150/ijbs.51811 This issue Cite
Review
A Multiple-Hit Hypothesis Involving Reactive Oxygen Species and Myeloperoxidase Explains Clinical Deterioration and Fatality in COVID-19
Pravin T Goud1,2, David Bai3, Husam M Abu-Soud3,4,5, ![]()
1. Division of Reproductive Endocrinology and Infertility & California IVF Fertility Center, Department of Obstetrics and Gynecology, University of California Davis, Sacramento, CA, 95833, USA.
2. California Northstate University Medical College, Elk Grove, CA, 95757, USA.
3. Department of Obstetrics and Gynecology, The C.S. Mott Center for Human Growth and Development, Wayne State University School of Medicine, Detroit, MI, 48201, USA.
4. Department of Physiology, Wayne State University School of Medicine, Detroit, MI, 48201, USA.
5. Department of Microbiology, Immunology and Biochemistry, Wayne State University School of Medicine, Detroit, MI, 48201, USA.
Received 2020-8-11; Accepted 2020-10-7; Published 2021-1-1
Abstract

Multi-system involvement and rapid clinical deterioration are hallmarks of coronavirus disease 2019 (COVID-19) related mortality. The unique clinical phenomena in severe COVID-19 can be perplexing, and they include disproportionately severe hypoxemia relative to lung alveolar-parenchymal pathology and rapid clinical deterioration, with poor response to O2 supplementation, despite preserved lung mechanics. Factors such as microvascular injury, thromboembolism, pulmonary hypertension, and alteration in hemoglobin structure and function could play important roles. Overwhelming immune response associated with “cytokine storms” could activate reactive oxygen species (ROS), which may result in consumption of nitric oxide (NO), a critical vasodilation regulator. In other inflammatory infections, activated neutrophils are known to release myeloperoxidase (MPO) in a natural immune response, which contributes to production of hypochlorous acid (HOCl). However, during overwhelming inflammation, HOCl competes with O2 at heme binding sites, decreasing O2 saturation. Moreover, HOCl contributes to several oxidative reactions, including hemoglobin-heme iron oxidation, heme destruction, and subsequent release of free iron, which mediates toxic tissue injury through additional generation of ROS and NO consumption. Connecting these reactions in a multi-hit model can explain generalized tissue damage, vasoconstriction, severe hypoxia, and precipitous clinical deterioration in critically ill COVID-19 patients. Understanding these mechanisms is critical to develop therapeutic strategies to combat COVID-19.
Keywords: Coronavirus. COVID-19, reactive oxygen species, free iron, myeloperoxidase, HOCl
Introduction
Coronavirus disease 2019 (COVID-19), a disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has shaken the world with its rapid and continual global spread in pandemic proportions [1]. Moreover, the economic and social impact of the pandemic is enormous and unprecedented. Currently, over 33 million individuals around the world have been affected, and over 1 million have succumbed to the severe respiratory distress associated with COVID-19 or related complications [2]. In particular, the United States continues to lead the world in cases, with over 7 million infected and at least 200,000 dead as of the time of writing [3]. Clinical management of COVID-19 is uncertain, especially due to the novelty of the virus and poorly understood pathophysiology, combined with the urgency of the situation limiting time available for clinical trials and further testing [4-9]. As a result, as consensus guidelines for COVID-19 treatment emerge, many clinicians are presently focusing on supportive treatments such as ventilator support [10]. However, mortality among the hospitalized cases continues to be high. Administration of oxygen and support via mechanical ventilation sometimes fail to prevent the profound hypoxia associated with COVID-19 from causing significant clinical decline [10, 11]. Thus, the pathophysiology of COVID-19 may be unique compared to known respiratory conditions such as typical acute respiratory infections, pneumonia, or acute respiratory distress syndrome (ARDS) [11, 12].
Published reports indicate that approximately 20% of COVID-19 patients tend to have severe or critical disease, with a mortality rate of 50% or more in critical cases [13-17]. According to various reports, COVID-19 mortality has been predicted by factors such as decreased hemoglobin, elevated cytokines, D-dimer, cardiac and/or renal injury, leukocytosis, and elevation of neutrophil : lymphocyte ratio (NLR) [16, 17]. One possible explanatory aspect for these factors contributing to severe COVID-19 cases is an inappropriately exaggerated immune response previously described as a “cytokine storm,” which results in the formation of reactive oxygen species (ROS) [18-20]. Such an acute severe immunological response in similar viral infections is known to release a cascade of inflammatory mediators, including various interleukins, tumor necrosis factor (TNF α) and other chemokines. Interleukin-6 (IL-6), which is capable of signaling through both membrane-bound receptors (IL-6R) and soluble receptors, is one of the most important [21]. The IL-6 pathway involves increased vascular permeability and immune cell recruitment through modulation of endothelial activation and dysfunction [21]. Furthermore, infiltrating neutrophils, a hallmark of COVID-19, can release myeloperoxidase (MPO), which can activate several pathways that lead to elevated cytokines and production of ROS such as hypochlorous acid (HOCl), superoxide (O2•-), and hydrogen peroxide (H2O2) [22-24]. Notably, HOCl can both compete with O2 at hemoglobin heme binding sites and also cause heme degradation and subsequent release of free iron (Fe2+). Free iron can then undergo the Fenton reaction to produce an array of ROS, including the highly reactive hydroxyl radical (•OH) [23-27]. Another possible facet of the observed pathophysiology in critical cases of COVID-19 is a decline in nitric oxide (NO), a key mediator of vasodilation [28, 29]. Vasodilation mediated by NO deficiency combined with the effect of excessive ROS on the structure and function of hemoglobin (Hb) could impact pulmonary and peripheral circulation, possibly eventually leading to critical or fatal hypoxia. Therefore, connection of these pathways establishes a multi-hit model that relates simultaneous activation of ROS-mediated mechanisms to key features of critically ill COVID-19 patients. Understanding these pathways will provide multiple clues to develop therapeutic strategies that will prevent severe morbidity and mortality related to COVID 19.
Pathophysiology of COVID-19
SARS-CoV-2 is a positive sense singled-stranded RNA virus with a nucleocapsid [30]. Human-to-human transmission occurs via respiratory droplets imparted by close contact, contaminated surfaces, or aerosol in closed spaces [22]. Animal-to-human transmission has been speculated on, but the evidence remains insubstantial. SARS-CoV-2 is thought to enter the host cells via angiotensin converting enzyme-2 (ACE-2) receptors ubiquitously expressed on the cells of the lungs, gastrointestinal tract, blood vessels, heart, and kidney [31]. The primary target of SARS CoV-2 appears to be the lower respiratory tract, rather than the ciliated epithelial cells of the conducting airways; cells in the lower respiratory tract are indeed known to express ACE-2 receptors [32]. Once inside host cells, the virus hijacks cellular machinery for viral replication, ultimately leading to cellular destruction and further proliferation of virions [32, 33].
Current speculations on the immediate host immune response to SARS-CoV-2 is based mostly on previous research on SARS-CoV, Middle East respiratory syndrome related coronavirus (MERS-CoV), and other related viral infections. SARS-CoV is the most similar virus, showing 79% and 69% similarity in terms of genome and amino acid sequence, respectively, to SARS-CoV-2 [34]. Direct infection of macrophages and T cells may also be possible [32]. As postulated based on SARS-CoV, the initial innate immunological response subsequent to viral entry into monocytes/macrophages begins with pathogen associated molecular patterns (PAMP) being recognized by the endosomal RNA receptors TLE3 and TLR7 and cytosolic RNA sensors RIG-1/MDA5, activating a downstream signaling cascade involving nuclear translocation of NF-κB and TRF3 causing transcription of type I interferon (IFN). This in turn activates IFNAR, phosphorylating STAT1 and STAT2 via the JAK-STAT pathway and has the final result of viral destruction through IFN [35]. Based on information gathered from studies on SARS-CoV and MERS-CoV, a putative delayed or dysfunctional IFN-1 host response may result in neutrophil and monocyte-macrophage hyper response, especially in lethal cases of SARS CoV and MERS. Continued viral propagation can accordingly continually activate neutrophil and therefore cause release of proinflammatory cytokines in the aforementioned cytokine storm [20, 36]. Other immunological responses include an adaptive immune response, where the cytokine milieu created by antigen presenting cells directs T cell responses in the form of helper T cell actions that orchestrate the overall adaptive immune response and cytotoxic T cell actions kill virus infected cells. Additionally, the humoral immune response results in seroconversion, with secretion of anti-IgM antibody as early as 4-14 days after onset of symptoms in patients with SARS-CoV 2 [37, 38]. The clinical course of COVID-19 begins with an asymptomatic or mildly symptomatic phase, when the disease could be locally progressive, and there may be some innate immune response. Currently, it is estimated that about 80% of patients suffer nothing more than mild to moderate upper respiratory symptoms [39]. Meanwhile, progressive viral replication continues in individuals unable to clear the virus; these 20% of patients progress to the severe stage of COVID-19. This stage is marked by lower respiratory tract and lung symptoms, particularly infection of type II alveolar pneumocytes in the peripheral and subpleural lungs [40]. Poor prognosis is generally marked by relative lymphopenia, elevated inflammatory markers characteristic of the cytotoxic storm, and markers of cardiac, renal or coagulation system anomaly [41]. Tissue destruction through viral destruction and induced apoptosis then results in alveolar damage. Recovery is dependent on the specificity and degree of both the innate and adaptive immune response against the virus. However, the repair process itself could be aberrant, especially in case of a hyperimmune response and may result in fibrosis and lung damage, especially that associated with excessive ROS. Proper activation of cellular antioxidant machinery is important to regulate the high degree of stimulation of ROS pathways in the immune or hyperimmune response and prevent cellular damage.
Generation of ROS at sites of inflammation
Typically, in COVID-19 and similar viral infections, the inflammatory immune response begins with chemokines released from inflamed respiratory tissue, followed by infiltration of leukocytes, especially neutrophils and macrophages [42, 43]. In the earliest stages of the immune response, neutrophils in particular play a crucial role by neutralizing and destroying viral proteins with the aid of ROS such as O2•-, H2O2, •OH and HOCl, essential components of the immune response [44]. In addition to their direct roles in the immune response, ROS can also act in cellular messaging by modifying the activity of immune and other cells; however, they can exert significant cytotoxic effects when released in the large amounts typical in some immune responses [45, 46]. Consequently, understanding the activity of ROS in infection such as COVID-19 is critical to grasping the pathology.
A model showing the link between neutrophil MPO activity generated during the “cytokine storm” provoked by COVID-19, ROS, and its role in NO consumption and heme destruction as well as subsequent iron release is shown in Figure 1. In this model, neutrophils, eosinophils, monocytes, macrophages, mitochondrial damage, and NADPH oxidase are the major sources of generation of O2•- at sites of inflammation [47-50]. Another ROS-generating enzyme is xanthine oxidoreductase (XOR), which metabolites hypoxanthine and xanthine to uric acid to instantaneously generate O2•- [51]. In COVID-19, like other inflammatory diseases, a major damaging pathway mediated by overproduction of O2•- and subsequent oxidative stress is caspase-3 activation, which is closely associated apoptosis and therefore DNA fragmentation [52]. However, O2•- is a short-lived molecule and is quickly consumed either through nonenzymatic pathways or a superoxide dismutase-catalyzed reaction to produce H2O2 [53, 54]. Meanwhile, H2O2 is considerably more stable than O2•-, diffuses freely through biological membranes, and is equally capable of inducing cytotoxicity when overproduced, as in COVID-19 [55, 56]. In addition to the dismutation of O2•-, enhancement of H2O2 during infection is also related to the activity of a variety of oxidases such as glucose/glucose, monoamine, and amino acid oxidase [57]. In addition to its direct effects, H2O2 can also react with O2•- or Fe2+, through the Fenton reaction, to generate the highly reactive and toxic hydroxyl radical. These can contribute to tissue damage, further worsening the condition of infected individuals [58]. Catalase, a key regulator of H2O2, scavenges H2O2 and catalyzes its decomposition, thereby protecting cells from H2O2 toxicity [59].
Neutrophil MPO -generated oxidants and their potential role in COVID-19. Activated neutrophils, eosinophils, and mitochondrial damage competently generate reduced O2 species such O2•- and H2O2 by a process known as oxidative burst. MPO is secreted from activated neutrophil which is abundant in human blood. MPO utilizes H2O2 as an oxidative substrate. In the presence of plasma levels of chloride, the enzyme generates the oxidant HOCl. MPO, under normal conditions, releases ROS to eliminate invading microorganisms. Under abnormal conditions, the generated ROS is excessive and mediates DNA oxidative damage, tissue injury and apoptosis. It also mediates hemoprotein (e.g. hemoglobin, NOS, and catalase) heme destruction and release of heme degradation products, protein aggregation, and free iron release. Free iron (Fe2+/Fe3+) reacts through the Fenton reaction to generate the highly reactive and toxic hydroxyl radical. By generating ROS, consuming NO, mediating hemoprotein heme destruction, promoting free iron release and •OH generation, neutrophil MPO activity may contribute to COVID-19 pathology by promoting oxidative stress, vasoconstriction by consuming NO, hypoxia through hemoglobin heme destruction, and blood clotting by generating free iron, and DNA damage through generation of hydroxyl radical.
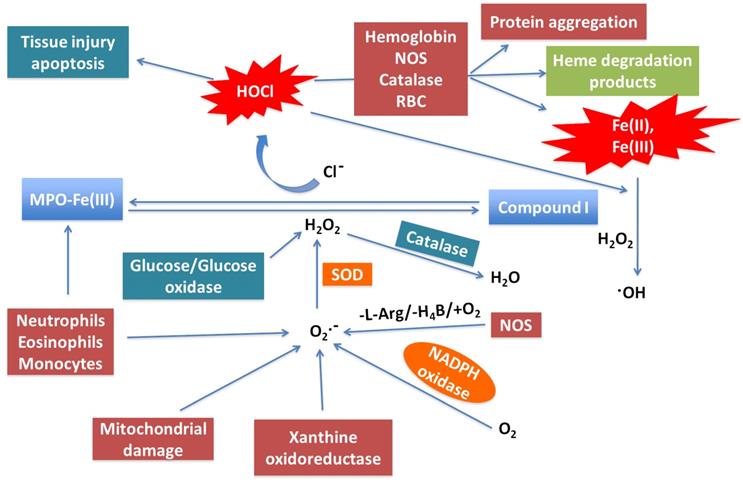
Myeloperoxidases could play a central role in COVID-19
Secondary to initial viral entry of SARS-CoV-2 into alveolar cells, a substantial number of neutrophils infiltrate [60]. They then release MPO, a major scavenger of H2O2 in the respiratory system, from azurophil granules. In the presence of Cl-, MPO catalyzes generation of HOCl from H2O2, contributing this important antimicrobial oxidant to the immune response [61, 62]. However, HOCl is also a long-lived oxidative species that can be the source of hydroxyl radical, e.g. upon reaction with O2•-, and there is evidence documenting its destructive interactions with hemoproteins [63, 64]. Moreover, MPO can utilize NO as a 1e- substrate for MPO Compound I and Compound II, generating nitrosonium cation (NO+) and nitrite (NO2-) as final products [65]) (Figure 2). Consumption of NO, a vasodilator, through this pathway contributes to vasoconstriction of the pulmonary vasculature. Therefore, the MPO-HOCl system and other ROS associated with MPO may take part in accelerating lung damage in infected patients. Accumulation of these species in the blood of COVID-19 patients can cause destruction of hemoproteins and generate free iron, which further promotes oxidative stress, also hindering normal physiological function, while damaging vital lipids, proteins, and nucleic acids [25, 64, 66]. A particularly significant hemoprotein affected by HOCl proliferation is Hb, which may be related to the hypoxia observed in COVID-19.
Major pathways of NO scavenging under inflammatory conditions. Three major pathways have been proposed to participate in consumption of NO in COVID-19. (1) The reaction of NO with oxy-hemoglobin to yield methemoglobin and NO3- is likely a major pathway of NO consumption in vivo. (2) The near diffusion-controlled rate reaction of NO with O2•- to yield ONOO-, which then decays to NO3-. This reaction is of specific importance wherever enhanced rates of both NO and O2•- production take place. (3) Overproduction of MPO in COVID-19 efficiently consumes NO as a 1e- substrate for both MPO Compounds I and II during steady-state catalysis, forming the very reactive NO+, which rapidly decays to NO2-. This pathway is particularly relevant at sites of inflammation where leukocyte peroxidases, NO, and H2O2 are present.
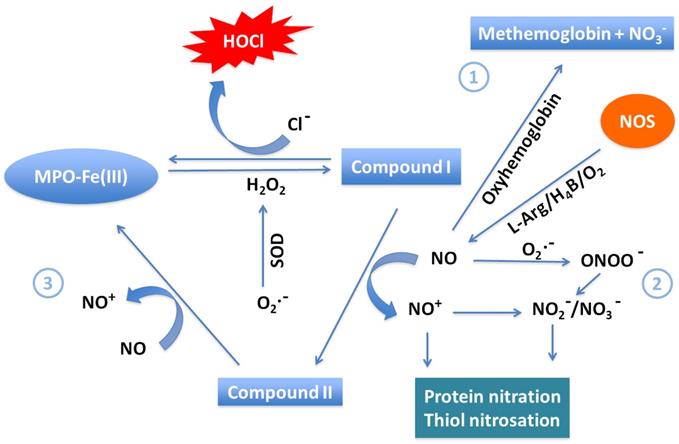
Hypochlorous acid alters hemoglobin function
Recently, it has been reported that many COVID-19 patients had low peripheral O2 saturation (50 -70%) but absence of common symptoms of O2 deficiency such as shortness of breath, troubled breathing, or dizziness [67, 68]. In COVID-19, a primary site of damage is the alveolar membrane, the site of O2 transfer from inspired air to blood. This is a possible explanation of the decreased O2 saturation in blood. However, when lung mechanics are preserved or the decrease of O2 saturation is disproportionate to the extent of lung damage, alternate possibilities must be considered. Here, neutrophil MPO activity may also contribute to O2 deprivation in these patients, rationalizing the phenomenon of patients with relatively low oxygen saturation without corresponding symptoms [67, 68].
Since CO2 levels are also low in COVID-19 patients, a defect in hemoglobin, the O2/CO2 carrier, is also possible [22]. Hemoglobin is a tetramer of four heme groups attached to four globin subunits [69]. These heme prosthetic groups transport O2 from the lung to peripheral tissue for cellular metabolism while ferrying CO2 back from peripheral tissue to the lung. To fulfill its function, Hb works against the concentration gradient by binding O2 in the oxygen-rich lungs to transport and release it in peripheral tissue, which are relatively O2-deficient [69]. Human Hb exists mainly in two reduced forms: oxygenated (Hb-Fe(II)-O2, oxy-Hb) and deoxygenated (Hb-Fe(II)-CO2). HOCl can disrupt Hb function by oxidizing oxyHb to methemoglobin and binding to the ferric heme iron to form HB-Fe(III)-OCl complex that can then decay to Hb-Fe(IV)=O (Compound II) through the formation of Hb-Fe(IV)=O+Π● (Compound I) and eventually lead to Hb heme destruction [64]. Heme destruction releases a variety of degradation products, particular the toxic free iron, and also encourages aggregation of globins [64]. Furthermore, methemoglobin and Hb ferryl intermediates are forms of Hb that cannot bind O2 [70]. Therefore, accumulation of these species in blood during COVID-19 could result in tissue hypoxia through the lack of normal Hb function. Furthermore, it is notable that the affinity of Hb toward OCl- is higher than the affinity of Hb toward O2, which further inhibits normal Hb function [64]. Therefore, during early stages of the disease, O2 supplementation could be beneficial in reoxygenation of Hb and correcting O2 saturation. However, at later stages of disease, there would be accordingly higher OCl- generation, which results in widespread, irreversible heme destruction of Hb. In this situation, supplementation with O2 could conversely be deadly due to additional generation of toxic ROS such as O2•-and H2O2.
HOCl-mediated cleavage reaction of the Hb heme and oxidation occurs randomly at any of the heme double bonds, independent of HOCl concentration. The degree of heme degradation depends mainly on the ratio of HOCl to heme, suggesting that multiple molecules of HOCl are required per molecule of heme. Additionally, excess ROS, especially O2•- and H2O2, have also been reported to destroy the heme of Hb and other hemoproteins such as cytochrome c, catalase and NOS with a similar mechanism [71]. Thus, inhibiting the chlorinating activity of MPO and/or direct scavenging of HOCl in the early stages of infection could be helpful to the patients with COVID-19. Recently, it has been shown that melatonin, lycopene, taurine, glutathione, and methionine not only serve as potent HOCl scavengers but also inhibit the chlorinating activity of MPO, thereby preventing HOCl-mediated heme destruction and subsequent free iron release [72, 73].
It is important to note that the mechanism of HOCl-mediated heme destruction completely differs from controlled heme oxygenase activity, which catalyzes the rate-limiting enzymatic step of heme degradation into carbon monoxide, free iron, and biliverdin [74]. Heme oxygenase-1 (HO-1) is an antioxidant enzyme, and its expression and activity are significantly up-regulated at sites of viral inflammation. Deficiencies in HO-1 expression have been reported to modulate chronic inflammation in both mice and humans [75].
Toxicity of free iron
The toxicity of extraneously generated free iron deserves special mention due to its ability to mediate the production of a variety of toxic ROS, such as the O2•-, H2O2, and •OH, that then cause cellular mitochondria poisoning, lipid peroxidation, damage to oxidative phosphorylation pathways [76, 77]. Furthermore, free iron can both damage blood vessels and promote cardiovascular complications such as hypertension and metabolic acidosis through vasodilation and increased vascular permeability [78-80]. Another cardiovascular condition free iron is associated with is increased coagulation through enhancement of fibrinogen activity, which is especially dangerous given the relationship between this phenomenon and thrombotic disease, i.e. stroke [81, 82]. Finally, increased free iron accumulation has been reported in a variety of other pathologies such as atherosclerosis, endometriosis, and cancer, which have also been correlated with increased MPO activity [78, 83, 84]. The enhanced MPO associated with these conditions further predisposes affected individuals towards both severe complications of COVID-19, other pathological conditions, and biochemical imbalances such as depletion of critical species, e.g., NO.
MPO and other members of the mammalian peroxidase superfamily serve as catalytic sinks for NO at sites of infection
As NO is produced in lungs, it may diffuse into the lumen of blood vessels, where it will mostly be scavenged by Hb at near diffusion rates in a controlled interaction with erythrocyte oxyhemoglobin, yielding ferric (met)hemoglobin and nitrate (NO3-) [85]. However, during advanced stages of COVID-19, hemoglobin levels and saturation are significantly decremented, and NO scarcity, vasoconstriction, and hypertension are present [86]. In particular, these observations imply either decreased production or increased consumption of NO in the subendothelial space of lung tissue and other inflamed tissues [1]. The reason for this has not yet been explicated, but a possible explanation is hypoxic suppression of NOS activity, in which low O2 ventilation (O2 concentration close to or lower than the Km(O2) values of NOSs) depresses cellular O2 below the level required for NOS activity. Importantly, it has been shown that in some cases of COVID-19, Hb O2 can reach as low as 50%, which is indeed close to or lower than the Km(O2) of both NOS-II and NOS-I [87]. Under these conditions, NOS generates O2•- instead of NO, which causes a cascade of other harmful effects.
Meanwhile, long term changes in NO levels through action of NO scavengers has been previously described. One major pathway for removal of NO in tissues is through its rapid reaction with O2•, producing peroxynitrite (ONOO-) [87]. This reaction may be of particular importance at sites of inflammation and phagocyte activation where both NO and O2•- production are elevated. However, NO removal only through this pathway does not explain the complete loss of NO-dependent signaling in vascular smooth muscle cells, suggesting alternative pathways of NO depletion [88].
Alternatively, the MPO-H2O2 system may also influence NO levels at sites of inflammation through two distinct mechanisms, namely, (1) consuming NO as a one e- substrate or (2) through HOCl-mediated NOS heme destruction. Previously, it has been shown that NO can serve as a physiological substrate of MPO compounds I and II, and other members of mammalian peroxidase superfamily, both in the presence and absence of plasma levels of Cl- (100 mM) or with a superoxide generating system, forming nitrosonium cation (NO+) [65]. The so generated NO+ is highly reactive (half-life < 0.3 ns) and rapidly decays, forming NO2- as a final product [65]. These interactions also suggest a complex and interdependent relationship between NO levels and modulation of peroxidase steady-state catalysis in vivo. For instance, tracheal ring investigations have studied the potential functional consequences of peroxidase-NO interactions in asthma. NO-dependent bronchodilation of preconstructed tracheal rings was reversibly inhibited when exposed to pathological levels of peroxidase and H2O2 [89]. It is possible that the interaction of NO and mammalian peroxidase serves as a regulatory pathway for the catalytic activities of those species and therefore inflammatory events. Furthermore, it has been observed that progression of COVID-19 is accompanied by impaired guanylate cyclase activetion and vascular response to endothelium-derived relaxing factor or NO [90]. It is therefore tempting to speculate that peroxidases like MPO might play a role in altering guanylate cyclase activation or other NO-dependent signaling events during development of vascular disease. For instance, there has been some evidence that deficiency of NO intensifies arterial thrombosis, while NO supplementation decreases it [91].
HOCl overproduction can also mediate NOS heme destruction through a mechanism similar to its destruction of Hb heme, although this pathway's role in modulating NO bioavailability in vivo remains to be determined. NO may also react with hemoprotein model compounds, generating the corresponding Fe-NO complexes [92]. NO is also a potent scavenger of a variety of radical species, such as lipid peroxyl and alkoxyl radicals [53]. Thus, it follows that these systems' contribution to increased net NO consumption likely limits NO bioavailability in COVID-19 patients, subsequently causing characteristic symptoms of vasoconstriction and hypertension. Therefore, NO supplementations could be helpful in combating COVID-19 symptoms and progression, especially before severe changes have occurred in the vasculature.
COVID-19 affects virtually every organ system
Multisystem involvement is a hallmark of COVID-19. Although direct viral spread to distant organs is a possibility, release of inflammatory mediators and downstream messengers are also likely important effectors of physiological systems. Major physiological systems affected in COVID-19 include, primarily, the respiratory and cardiovascular system, as well as the renal, hepatic, and nervous systems (Figure 3).
Involvement of multiple systems in COVID-19. Activation of the immune system following the entry into the respiratory system involves activation of neutrophils and monocytes with release of the MPO and ROS, which cause damage to both, local (alveoli and lung parenchyma) as well as distant tissues including gastrointestinal tract and liver, kidneys, brain, peripheral circulation and the heart. Damage to these tissues manifest as organ dysfunction and multi-system failure. Release of heme degradation products and iron along with ROS are particularly damaging to the kidneys, liver, heart, and brain, while inappropriate activation of the coagulation system and insufficiency of NO can result in pulmonary and peripheral vasoconstriction. Simultaneous occurrence of these events can result in severe, persistent, and potentially fatal hypoxemia.
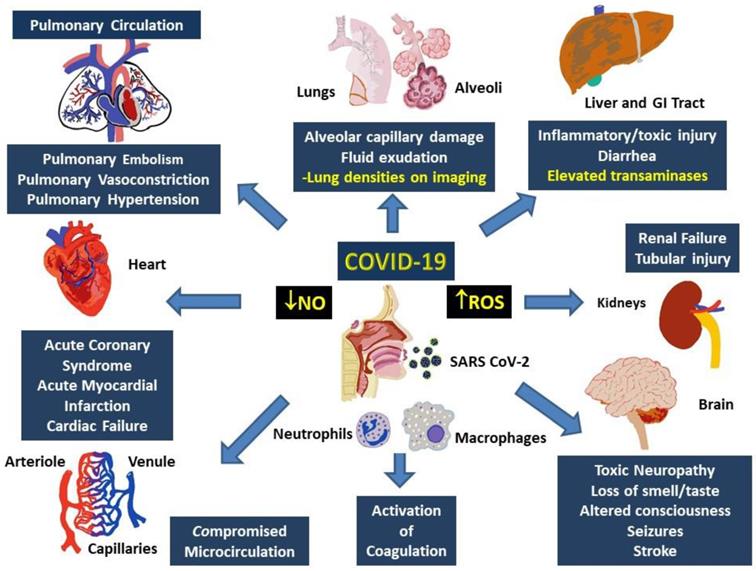
In the respiratory system, histopathology indicates diffuse alveolar damage with denuded alveolar cells, interstitial mononuclear infiltrates and type II pneumocyte hyperplasia, edema, intra-alveolar proteinaceous exudate, patchy inflammation and multinucleated giant cells as well as interstitial fibrosis and intra-alveolar loose fibrous plugs [93]. While some of these features represent ARDS, presence of respiratory distress with severe hypoxia and well-preserved lung mechanics indicate involvement of the pulmonary vasculature. Furthermore, absence of classical ARDS symptoms such as hyaline membrane or type II pneumocyte hyperplasia do not fully support a presentation of ARDS [94]. Varying degrees of elevations in D-dimer, with normal or slightly abnormal INR, PTT, and platelet counts, as well as activation of the complementary cascades combine with elevated ROS and decreased NO paves a path for explanation of the involvement of other organ systems in COVID 19.
Cardiac involvement is noted in 20-30% of hospitalized COVID-19 patients, contributing to ~40% mortality, and prognosticates the general outlook of pathological progression [95-97]. It is likely secondary to pulmonary damage, its accompanying hypoxia, and the systemic inflammatory response, as noted by elevation in the markers of inflammation including IL-6, IL-2, IL-7, TNF-α, interferon-γ inducible protein (IP)-10, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP) 1-α, granulocyte-colony stimulating factor (G-CSF), C-reactive protein (CRP), procalcitonin, and ferritin [40, 98-100]. Commonly occurring cardiovascular abnormalities in COVID-19 include hypertension, arrhythmias, acute coronary syndrome and acute myocardial infarction [101]. Typical markers of cardiac injury, such as troponin I and brain natriuretic peptide (BNP), accompany these pathologies and indicate particularly increased risk of mortality [101]. Other cardiovascular dysfunctions include inflammatory changes, formation of microthrombi, and/or alteration of blood viscosity, which have also been reported in patients with other acute respiratory disease, such as influenza, acute pneumonia, and H1N1, and lead to severe organ damage and eventual mortality [102, 103]. In particular, altered coagulation parameters, characterized by increased d-dimer, fibrin degradation products, PTT and thrombocytopenia, predict poor prognosis and are related to elevated risk of stroke and pulmonary embolism [104].
Abnormalities of renal function are known to occur in about 75% of hospitalized patients with COVID-19, and the degree of deficiency often predicts overall disease outcome [105]. COVID-19 associated renal injuries include diffuse proximal tubule injury, non-isometric vacuolar degeneration, and frank necrosis as well as hemosiderin pigmentation and pigmented casts, which are associated with heme degradation and iron toxicity [106-108]. Erythrocyte aggregates obstructing capillary lumens in the absence of vasculitis, platelet aggregates, or fibrinoid aggregates were also observed [106].
Hepatic injury in COVID-19 has been reported in form of elevated liver enzymes, especially in severe cases, and in diarrheas with detection of virus in the blood [109]. Other likely mechanisms could be related to hypoxia and systemic immune mediated inflammation, particularly in association with ROS elevation and/or iron toxicity [108, 110].
Nervous system damage in COVID-19 is likely similar to that found in its coronavirus cousins. In SARS-CoV infection, previous reports have indicated polyneuropathy, encephalitis, acute ischemic stroke, and demyelination [111, 112]. Similarly, MERS-CoV has been linked to mental disturbances, seizures, altered consciousness, ischemic stroke, or Guillain-Barre syndrome [113, 114]. About 35% of hospitalized patients with COVID-19 display neurological symptoms, including headache, altered consciousness, seizures, and sudden loss of taste and/or smell [115, 116]. Autopsies have revealed cerebral edema, degeneration, and encephalitis with viral RNA present in the CSF [117, 118]. Possible causative factors for neurological damage include direct infection after passage through the blood-brain barrier, immune-related hypoxic injury, infectious toxic encephalopathy, acute cerebrovascular disease, and iron toxicity resulting from heme degradation [119].
Summary
One of the characteristic changes in the blood parameters among the patients with COVID-19 includes leukocytosis with relative neutrophilia. Activation of neutrophils can trigger various cellular mechanisms, including the release of prostanoids, lysosomal enzymes, as well as highly reactive oxygen radicals and their intermediates. Myeloperoxidases released from the azurophil granules of neutrophils play a particularly critical role by participating in the synthesis of HOCl through a reaction involving H2O2 and chloride (Cl-). HOCl is highly reactive and not only causes lipid peroxidation of membranes, affecting their permeability; but also contributes to oxidative modification of free functional groups, inducing changes in the functionality of proteins. HOCl degrades heme with release of Fe2+, which in turn participates in generation of additional ROS. Furthermore, involvement of MPO and its related mechanisms result in a decrease in nitric oxide (NO), consequently leading to vasoconstriction. Taken together, these phenomena snugly fit into the clinical pathophysiology of severe/critical COVID-19 illness, which consist of alveolar capillary damage (secondary to the production of superoxide, H2O2 and HOCl), pulmonary vasoconstriction and pulmonary hypertension (secondary to NO depletion), elevated ferritin (following release of free iron secondary to heme-degradation), and deterioration of oxygen carrying capacity (secondary to heme degradation). Production of Fe3+ results in formation of methemoglobin, which further decreases the affinity of Hb to oxygen, shifting the O2 dissociation curve further to the right and deteriorating blood O2 saturation while preserving the tissue levels of O2. This causes possible delay in appearance of the classical symptoms of hypoxia and may not reflect the severity of depletion of O2 saturation. Another cardiovascular feature affected by these changes is thrombosis, which is exacerbated by both accumulation of free iron and depletion of NO. Thus, a multi-hit model of MPO and its multifaceted reactions that lead to production of ROS such as HOCl, O2•-, H2O2 and •OH, which then mediate decrease O2 diffusion, carriage, and delivery through interaction with heme proteins and other players could explain the rapid evolution of respiratory failure and subsequent mortality in patients in critical stages of COVID-19. Understanding these mechanisms can provide clues to therapies to prevent severe morbidity and mortality among patients affected with COVID-19.
Abbreviations
COVID-19: Coronavirus disease-19; ROS: Reactive oxygen species; MPO: myeloperoxidase; H2O2: hydrogen peroxide; HOCl: hypochlorous acid; O2•-: superoxide; •OH: hydroxyl radical.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470-3
2. Coronavirus disease (COVID-19) Situation Report - 149. World Health Organization; 2020
3. Coronavirus Disease 2019 (COVID-19) Cases and Deaths in the U.S.: United States Centers for Disease Control and Prevention; 2020
4. Thanh Le T, Andreadakis Z, Kumar A, Gomez Roman R, Tollefsen S, Saville M. et al. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19:305-6
5. Zhang L, Liu Y. Potential interventions for novel coronavirus in China: A systematic review. J Med Virol. 2020;92:479-90
6. Phadke M, Saunik S. COVID-19 treatment by repurposing drugs until the vaccine is in sight. Drug Dev Res. 2020
7. Ahn DG, Shin HJ, Kim MH, Lee S, Kim HS, Myoung J. et al. Current Status of Epidemiology, Diagnosis, Therapeutics, and Vaccines for Novel Coronavirus Disease 2019 (COVID-19). J Microbiol Biotechnol. 2020;30:313-24
8. Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G. et al. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol. 2020;38:337-42
9. Md Insiat Islam R. Current Drugs with Potential for Treatment of COVID-19: A Literature Review. J Pharm Pharm Sci. 2020;23:58-64
10. Alhazzani W, Moller MH, Arabi YM, Loeb M, Gong MN, Fan E. et al. Surviving Sepsis Campaign: guidelines on the management of critically ill adults with Coronavirus Disease 2019 (COVID-19). Intensive Care Med. 2020;46:854-87
11. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H. et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475-81
12. Li T, Lu H, Zhang W. Clinical observation and management of COVID-19 patients. Emerg Microbes Infect. 2020;9:687-90
13. Kakodkar P, Kaka N, Baig MN. A Comprehensive Literature Review on the Clinical Presentation, and Management of the Pandemic Coronavirus Disease 2019 (COVID-19). Cureus. 2020;12:e7560
14. Lythgoe MP, Middleton P. Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol Sci. 2020;41:363-82
15. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW. et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. Jama. 2020
16. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G. et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. Bmj. 2020;368:m1091
17. Zhang J, Wang X, Jia X, Li J, Hu K, Chen G. et al. Risk factors for disease severity, unimprovement, and mortality in COVID-19 patients in Wuhan, China. Clin Microbiol Infect. 2020;26:767-72
18. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363-74
19. Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox Biology of Respiratory Viral Infections. Viruses. 2018 10
20. Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016;13:3-10
21. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020
22. Tang D, Comish P, Kang R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020;16:e1008536
23. Abu-Soud HM, Maitra D, Shaeib F, Khan SN, Byun J, Abdulhamid I. et al. Disruption of heme-peptide covalent cross-linking in mammalian peroxidases by hypochlorous acid. J Inorg Biochem. 2014;140:245-54
24. Galijasevic S, Abdulhamid I, Abu-Soud HM. Potential role of tryptophan and chloride in the inhibition of human myeloperoxidase. Free Radic Biol Med. 2008;44:1570-7
25. Maitra D, Shaeib F, Abdulhamid I, Abdulridha RM, Saed GM, Diamond MP. et al. Myeloperoxidase acts as a source of free iron during steady-state catalysis by a feedback inhibitory pathway. Free Radic Biol Med. 2013;63:90-8
26. Galijasevic S, Maitra D, Lu T, Sliskovic I, Abdulhamid I, Abu-Soud HM. Myeloperoxidase interaction with peroxynitrite: chloride deficiency and heme depletion. Free Radic Biol Med. 2009;47:431-9
27. Maitra D, Byun J, Andreana PR, Abdulhamid I, Saed GM, Diamond MP. et al. Mechanism of hypochlorous acid-mediated heme destruction and free iron release. Free Radic Biol Med. 2011;51:364-73
28. Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci U S A. 2003;100:14766-71
29. Hazen SL, Zhang R, Shen Z, Wu W, Podrez EA, MacPherson JC. et al. Formation of nitric oxide-derived oxidants by myeloperoxidase in monocytes: pathways for monocyte-mediated protein nitration and lipid peroxidation In vivo. Circ Res. 1999;85:950-8
30. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. 2020;55:105924
31. Li Y, Zhou W, Yang L, You R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol Res. 2020;157:104833
32. Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 pathophysiology: A review. Clin Immunol. 2020;215:108427
33. Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W. et al. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses. 2020 12
34. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565-74
35. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523-34
36. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J Infect. 2020;80:607-13
37. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1-9
38. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX. et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382:1708-20
39. Team TNCPERE. Vital Surveillances: The Epidemiological Characteristics of an Outbreak of 2019 Novel Coronavirus Diseases (COVID-19) — China, 2020. China CDC Weekly. 2020;2:113-22
40. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054-62
41. Ponti G, Maccaferri M, Ruini C, Tomasi A, Ozben T. Biomarkers associated with COVID-19 disease progression. Crit Rev Clin Lab Sci. 2020:1-11
42. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J. et al. Inflammatory mechanisms in the lung. J Inflamm Res. 2009;2:1-11
43. Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol. 2016;38:471-82
44. Vatansever F, de Melo WC, Avci P, Vecchio D, Sadasivam M, Gupta A. et al. Antimicrobial strategies centered around reactive oxygen species- bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol Rev. 2013;37:955-89
45. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453-62
46. Kim HJ, Kim CH, Ryu JH, Kim MJ, Park CY, Lee JM. et al. Reactive oxygen species induce antiviral innate immune response through IFN-lambda regulation in human nasal epithelial cells. Am J Respir Cell Mol Biol. 2013;49:855-65
47. Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707-16
48. Robb CT, Regan KH, Dorward DA, Rossi AG. Key mechanisms governing resolution of lung inflammation. Semin Immunopathol. 2016;38:425-48
49. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol. 2017;7:373
50. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5-23
51. Guerin P, El Mouatassim S, Menezo Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum Reprod Update. 2001;7:175-89
52. Ivanova D, Zhelev Z, Aoki I, Bakalova R, Higashi T. Overproduction of reactive oxygen species - obligatory or not for induction of apoptosis by anticancer drugs. Chin J Cancer Res. 2016;28:383-96
53. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11-26
54. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244:6049-55
55. Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994-1003
56. Bartz RR, Piantadosi CA. Clinical review: oxygen as a signaling molecule. Crit Care. 2010;14:234
57. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527-605
58. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82-83:969-74
59. Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell Mol Life Sci. 2004;61:192-208
60. Astuti I, Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab Syndr. 2020;14:407-12
61. Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med. 2000;28:1717-25
62. Denys GA, Devoe NC, Gudis P, May M, Allen RC, Stephens JT Jr. Mechanism of Microbicidal Action of E-101 Solution, a Myeloperoxidase- Mediated Antimicrobial, and Its Oxidative Products. Infect Immun. 2019 87
63. Candeias LP, Patel KB, Stratford MR, Wardman P. Free hydroxyl radicals are formed on reaction between the neutrophil- derived species superoxide anion and hypochlorous acid. FEBS Lett. 1993;333:151-3
64. Maitra D, Byun J, Andreana PR, Abdulhamid I, Diamond MP, Saed GM. et al. Reaction of hemoglobin with HOCl: mechanism of heme destruction and free iron release. Free Radic Biol Med. 2011;51:374-86
65. Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524-32
66. Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood). 2002;227:671-82
67. Ottestad W, Seim M, Maehlen JO. COVID-19 with silent hypoxemia. Tidsskr Nor Laegeforen. 2020 140
68. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: A harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020
69. Gell DA. Structure and function of haemoglobins. Blood Cells Mol Dis. 2018;70:13-42
70. Jaffe ER. Methemoglobin pathophysiology. Prog Clin Biol Res. 1981;51:133-51
71. Nagababu E, Rifkind JM. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem Biophys Res Commun. 1998;247:592-6
72. Maitra D, Abdulhamid I, Diamond MP, Saed GM, Abu-Soud HM. Melatonin attenuates hypochlorous acid-mediated heme destruction, free iron release, and protein aggregation in hemoglobin. J Pineal Res. 2012;53:198-205
73. Pennathur S, Maitra D, Byun J, Sliskovic I, Abdulhamid I, Saed GM. et al. Potent antioxidative activity of lycopene: A potential role in scavenging hypochlorous acid. Free Radic Biol Med. 2010;49:205-13
74. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323-54
75. Vijayan V, Mueller S, Baumgart-Vogt E, Immenschuh S. Heme oxygenase-1 as a therapeutic target in inflammatory disorders of the gastrointestinal tract. World J Gastroenterol. 2010;16:3112-9
76. Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett. 2005;157:175-88
77. Crichton RR, Wilmet S, Legssyer R, Ward RJ. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J Inorg Biochem. 2002;91:9-18
78. Ong WY, Halliwell B. Iron, atherosclerosis, and neurodegeneration: a key role for cholesterol in promoting iron-dependent oxidative damage? Ann N Y Acad Sci. 2004;1012:51-64
79. Trinder D, Fox C, Vautier G, Olynyk JK. Molecular pathogenesis of iron overload. Gut. 2002;51:290-5
80. Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008;88:7-15
81. Nielsen VG, Pretorius E. Iron-enhanced coagulation is attenuated by chelation: thrombelastographic and ultrastructural analysis. Blood Coagul Fibrinolysis. 2014;25:845-50
82. Lipinski B, Pretorius E, Oberholzer HM, Van Der Spuy WJ. Iron enhances generation of fibrin fibers in human blood: implications for pathogenesis of stroke. Microsc Res Tech. 2012;75:1185-90
83. Fletcher NM, Jiang Z, Ali-Fehmi R, Levin NK, Belotte J, Tainsky MA. et al. Myeloperoxidase and free iron levels: potential biomarkers for early detection and prognosis of ovarian cancer. Cancer Biomark. 2011;10:267-75
84. Defrere S, Lousse JC, Gonzalez-Ramos R, Colette S, Donnez J, Van Langendonckt A. Potential involvement of iron in the pathogenesis of peritoneal endometriosis. Mol Hum Reprod. 2008;14:377-85
85. Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free Radic Biol Med. 2013;61:464-72
86. Geier MR, Geier DA. Respiratory conditions in coronavirus disease 2019 (COVID-19): Important considerations regarding novel treatment strategies to reduce mortality. Med Hypotheses. 2020;140:109760
87. Dweik RA, Comhair SA, Gaston B, Thunnissen FB, Farver C, Thomassen MJ. et al. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc Natl Acad Sci U S A. 2001;98:2622-7
88. White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA. et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93:8745-9
89. Abu-Soud HM, Khassawneh MY, Sohn JT, Murray P, Haxhiu MA, Hazen SL. Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: development of a model describing NO-peroxidase interactions. Biochemistry. 2001;40:11866-75
90. Kobayashi J, Murata I. Nitric oxide inhalation as an interventional rescue therapy for COVID-19-induced acute respiratory distress syndrome. Ann Intensive Care. 2020;10:61
91. Gresele P, Momi S, Guglielmini G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem Pharmacol. 2019;166:300-12
92. Henry Y, Guissani A. Interactions of nitric oxide with hemoproteins: roles of nitric oxide in mitochondria. Cell Mol Life Sci. 1999;55:1003-14
93. Zhang H, Zhou P, Wei Y, Yue H, Wang Y, Hu M. et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann Intern Med. 2020;172:629-32
94. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1-13
95. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F. et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020
96. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846-8
97. Akhmerov A, Marban E. COVID-19 and the Heart. Circ Res. 2020;126:1443-55
98. Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC. et al. [A pathological report of three COVID-19 cases by minimal invasive autopsies]. Zhonghua Bing Li Xue Za Zhi. 2020;49:411-7
99. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. Jama. 2020
100. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506
101. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259-60
102. Bogomolov BP, Deviatkin AV. [Microcirculation and hemostasis in influenza and acute viral respiratory infections complicated with pneumonia]. Ter Arkh. 2002;74:44-8
103. Salgado DR, Ortiz JA, Favory R, Creteur J, Vincent JL, De Backer D. Microcirculatory abnormalities in patients with severe influenza A (H1N1) infection. Can J Anaesth. 2010;57:940-6
104. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844-7
105. Pei G, Zhang Z, Peng J, Liu L, Zhang C, Yu C. et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J Am Soc Nephrol. 2020;31:1157-65
106. Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY. et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98:219-27
107. Gupta A. Iron overload in human disease. N Engl J Med. 2012;366:1549 author reply -50
108. Wang L, Vijayan V, Jang MS, Thorenz A, Greite R, Rong S. et al. Labile Heme Aggravates Renal Inflammation and Complement Activation After Ischemia Reperfusion Injury. Front Immunol. 2019;10:2975
109. Zhang C, Shi L, Wang FS. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5:428-30
110. Yan M, Huo Y, Yin S, Hu H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018;17:274-83
111. Hugkulstone CE. Use of a bandage contact lens in perforating injuries of the cornea. J R Soc Med. 1992;85:322-3
112. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y. et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415-24
113. Saad M, Omrani AS, Baig K, Bahloul A, Elzein F, Matin MA. et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: a single-center experience in Saudi Arabia. Int J Infect Dis. 2014;29:301-6
114. Kim JE, Heo JH, Kim HO, Song SH, Park SS, Park TH. et al. Neurological Complications during Treatment of Middle East Respiratory Syndrome. J Clin Neurol. 2017;13:227-33
115. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q. et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020
116. Giacomelli A, Pezzati L, Conti F, Bernacchia D, Siano M, Oreni L. et al. Self-reported olfactory and taste disorders in SARS-CoV-2 patients: a cross-sectional study. Clin Infect Dis. 2020
117. Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J. et al. A first case of meningitis/encephalitis associated with SARS- Coronavirus-2. Int J Infect Dis. 2020;94:55-8
118. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-2
119. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L. et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun. 2020;87:18-22
Author contact
![]() Corresponding author: Husam M Abu-Soud, PhD, Department of and Gynecology, Wayne State University School of Medicine, The C.S. Mott Center for Human Growth and Development, 275 E. Hancock Detroit, MI 48201, Tel. 313 577-6178, Fax. 313 577-8554, E-Mail: habusoudwayne.edu
Corresponding author: Husam M Abu-Soud, PhD, Department of and Gynecology, Wayne State University School of Medicine, The C.S. Mott Center for Human Growth and Development, 275 E. Hancock Detroit, MI 48201, Tel. 313 577-6178, Fax. 313 577-8554, E-Mail: habusoudwayne.edu