Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Traumatic brain injury and the...
Neurogenic niche cells in...
Communication, crosstalk and...
The promising method for...
Conclusion and future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(1):368-378. doi:10.7150/ijbs.52169 This issue Cite
Review
Niche Cells Crosstalk In Neuroinflammation After Traumatic Brain Injury
Yibin Jia1#, Guanyi Wang1#, Yuqing Ye1,2#, Enming Kang1, Huijun Chen1, Zishuo Guo3, Xiaosheng He1 ![]()
1. Department of Neurosurgery, Xijing Hospital, Airforce Military Medical University (Fourth Military Medical University), Xi'an 710032, China
2. Department of Neurosurgery, PLA 163rd Hospital (Second Affiliated Hospital of Hunan Normal University), Changsha 410000, China
3. State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, AMMS, Beijing 100071, China
# These authors contributed equally to this work
Received 2020-8-27; Accepted 2020-11-23; Published 2021-1-1
Abstract

Traumatic brain injury (TBI) is recognized as the disease with high morbidity and disability around world in spite of the work ongoing in neural protection. Due to heterogeneity among the patients, it's still hard to acquire satisfying achievements in clinic. Neuroinflammation, which exists since primary injury occurs, with elusive duality, appear to be of significance from recovery of injury to neurogenesis. In recent years, studied have revealed that communication in neurogenic niche is more than “cell to cell” communication, and study on NSCs represent it as central role in the progress of neural regeneration. Hence, the neuroinflammation-affecting crosstalk after TBI, and clarifying definitive role of NSCs in the course of regeneration is a promising subject for researchers, for its great potential in overcoming the frustrating status quo in clinic, promoting welfare of TBI patient.
Keywords: Traumatic brain injury, Neuroinflammation, Nerve regeneration, Extracellular vesicles, Microglia, Astrocytes, Neural stem cell (NSC)
Introduction
For years, despite decreasing incidence of severe types, traumatic brain injury (TBI) is still the disease that causes the morbidity and mortality around world [1, 2]. Canonically, researchers mainly pour energy and captivity into exploring the neural recovery methods, such as medicine or cell treatment to enhance the neurogenesis [3], whereas little achievements were acquired [4]. In common, though no delimitation indeed exists, we customarily categorize TBI into “primary injury” and “secondary injury” [5]. Initiated with tissue damage and cell death, innate immune cells are activated, releasing “expanding-signal” to recruit other immune components to enlarge the reaction. Observation from the beds proves secondary injury as major reason contributing to the loss of neural cognition and function, with lasting and chronic damage to the brain, such as loss of glutamate homeostasis [6, 7], proteins and phospholipid membranes damaged by free radicals [8], and the inflammatory response comprised of both classic and neurogenic inflammation [9]. In addition, absence of qualitied TBI model exactly representing complex clinical situation [10, 11], and the lack of knowledge on elucidating the pattern of the secondary injury, account for poor understanding on treatment and rehabilitation [2].
The neuroinflammation occurs after the primary injury, is thought to be of great significance in either the recovery of injury, or proliferation and differentiation of the NSCs [12, 13]. It takes a lot for researchers to figures out the definitive role of inflammation in the neurogenesis or neural regeneration, which turns out to be beneficial in early stage but converts to causing damage and neurodegeneration in the long run. To further complicate matters, simply aims to inhibit the inflammatory response, including activation of immune cells or the release of some chemokine and cytokine, didn't work well in the outcome of the TBI patient [14]. Understanding the hiding mechanism of the neuroinflammation, which has been proved to have a double-edged function, and then wielding its helpful role in promoting the neurogenesis, is a promising approach to tackle with this clinical dilemma.
“Niche” was first proposed as the microenvironment sustaining the properties of proliferation and differentiation [15]. In TBI, the conception is thought as the region echo to the injury, and then gradually regulate this progress and promote recovery (through extracellular vesicles, microRNAs, et). SVZ (subventricular zone) and SGZ (subgranular zone) show as the most potential place to support the regeneration [16, 17]. The aim of treatment on injury comes to recovery of both function and structure, referred as neural regeneration in brain. The component in the neurogenic niche constitute of different cells (neuron, glial cells) and structures, ranging from simple to delicate. Each member has unique but cooperative function to maintain the hemostasis or be activated to make a response to the irritation [18-21]. NSCs and its progenitor cell, the cluster have the ability to promote regeneration and repair the damage caused by TBI, is considered as the central players in communication and crosstalk in the regeneration progress [18, 22]. Providing an panorama of neurogenic niche activity, may shed a light on modulating neuroinflammation toward to a regeneration-beneficial way, ensued with enhancing neural function and cognition in patients with TBI.
A number of the progress and literature emerging these years, makes it necessary to further summary the inflammation on neurogenesis after the TBI. In this review, we propose a sketch of post-traumatic neuroinflammatory events, and a constellation of resident cells in the niche reacting to the inflammation, promoting recovery and neurogenesis. Following the communication and interaction between the niche cells, involving factors are mentioned. In the ensuing sections, we aim to investigate NSCs as the leading role in the modulation of neurogenesis in the inflammation after TBI.
Traumatic brain injury and the inflammatory response
To struggle for clear understanding on TBI, even nowadays, there still remain controversaries that if any model could appropriately mimic the complex process in vivo [4, 23]. The primary injury of TBI is mainly the injuries about mechanical damage to the tissue, cell membrane and BBB [24]. As a heterogeneous entity, TBI comprise a series of mechanical injury patterns such as extrinsic compression from mass lesion, concussion, diffuse axonal injury [25], following a range of pathological mechanisms by which neuronal injury can be aroused, such as ischemia, apoptosis [26], mitochondrial autophagy [27], cortical spreading depression [28], edema [29] and microvascular spasm. The neural inflammation is thought to company with these courses all along, and the long-lasting inflammatory response seems to link the TBI with other neurodegenerative disease (AD, PD, CTE) [30].
The inflammatory response in neural system is a sophisticated, complex composition of processes including the ignition of the sensor, signals transducing, activation of the immune cells and at last, continuous inflammation. As a paradigm [2], at the time of injury happens, the alert signals (ATP, HSP, HGMB1, bradykinin) was released into neural system, which are generally considered as PAMP or DAMP, binding to the receptors on the sensor cells [31-33]. Pattern recognition receptors (PRRs), like Toll-like receptors (TLR), receptors of advanced glycosylation end-products (RAGE) or purinergic receptors that are commonly expressed on the innate immune cells to trigger the downstream reactivity facilitated by the immune active molecules [34]. It is established that some receptors and signals are associated with the outcome of TBI patient, on the evidence that blockade or knockout those can effectively reduce cerebral edema, lesion volume and the release of inflammatory factors like IL-1β, IL-6 [35, 36].
Normally, the inflammation in the neural system can be classified into “classical inflammation”, which immune response reacts just as other tissue injury (the secretion of the substance P) with plot similar with periphery, and “neural inflammation”, (involving the cells and molecules exclusive in the brain) [14, 37]. The damaged tissue or innate immune cells are the first to react to the injury, then by secreting chemokines and cytokines, the system can in turn recruit the glia cells, T cells to aggravate at the injured site. Consequently, the inflammation is further expanded and augmented, and the resident cells, like microglia (MG) or astrocyte (AG), neuron would make a difference in inflammation and neurogenesis [20, 38], in cooperation with other immune cells from periphery.
The inflammation response doesn't play a clarified role in neurogenesis, for existing evidence are unable to come to a definite outcome [39]. Inhibition or blockade of the releasing of the cytokines like IL-6, IL-1α and TNFα, seems to have beneficial results in the inactivation of the immune cells and the enlargement of immune response, while has little improvement in clinic outcome [40]. Similarly, depleting the neutrophil in mice effectively attenuates the immune injuries like edema or tissue loss, accompanied by reducing MG activation, but with a poor function recovery [41]. And using minocycline was found able to reduces chronic MG activation, but with an increasing neurodegeneration and aggravated cognitive deficits [42, 43]. It seems that in starting episode, there exists a mixture component of “pro” or “anti” inflammatory, and more “pro” in long term [44]. Anyhow, more convincing evidence and finding are urged to illustrate this dualism in the inflammation.
Neurogenic niche cells in neuroinflammation
Microglia
As the "macrophages" of the central nervous system, MG accounts for about 5-15% of all brain cells [45]. Unlike macrophages in non-nerve tissues, MG originates from the yolk sac in the embryonic stage, expressing the transcription factor RUNX1, and the tyrosine kinase receptor c-kit, developing from the red-myeloid precursor cells through the transcription factor Pu. 1- and Irf8-dependent pathway [46]. MG is highly branched, sensitive to the changes in neural environment, constantly detecting changes or external stimuli in surrounding environment, responding quickly, receiving, transducing and integrating corresponding signals to produce a series of biochemical activities to maintain the steady state of the internal environment [47]. At the same time, with changes in various conditions (aging, injury, disease, etc.), MG will also show new phenotypic characteristics [48]. It's believed that activation of microglial receptor involves not only changes in morphology or movement, but also changes in neurotransmitter release, modulation of neuronal-glial synaptic transmission, and cytokines secretion, generation of ROS. Especially, these changes are further involved in the process of secondary injury. The different types and severity of TBI, through the activation of the pertinent receptors, will also make the activation of MG follow different patterns, thereby showing the pro-inflammatory or anti-inflammatory state at different moments [49].
Traditionally, at the time inflammation fires, MG in resting state will be induced by different environments or factors, and "proinflammatory" or "anti-inflammatory" effects will appear (For example, stimulated by LPS, IFN-γ, the cell shows M1 type, and after IL-4 and IL-13 stimulation, it tends to represent as M2 type) [50]. Usually, M1 will participate in the secretion of pro-inflammatory factors (TNF-α, IL-6, et), aggravating inflammatory response of the environment, while M2 type may secrete protective, anti-inflammatory factors such as IL-10 to regulate the inactivation of pro-inflammatory cell phenotype, thereby maintaining the homeostasis of the environment [49]. Nevertheless, "polarization" in MG is not an issue on absolute conversion of phenotypes, but a relative status that overlaps in time and space. In recent years, with the endeavor in neuroinflammation research, the study on MG has become more comprehensive, and more scholars have advocated abandoning “M1/M2 typing” to represent the function state of MG in the study, but combining the technical knowledge of transcriptomics, proteomics, and single cell sequencing to dynamically observe or describe the changes of MG throughout the inflammatory response.
Astrocytes
With findings in these years, AG, just like MG, are thought to make a significant contribution to immune response after TBI [51, 52]. Apart from the functions in homoeostasis, such as secreting active factors, supporting the neuron and the uptake of Ca2+, AG also have unique role in neural inflammation [53]. Following TBI, the cells are activated by mechanical damage, for they are sensitive to the physical stretch or pressure, enriched with corresponding mechanosensitive ion channels. Then, partly like injury tissue, some alert signals are released from the cells. Particularly, along with the calcium dysfunction [54], ATP participates in the cascade amplification reaction, that is, response in activation and recruitment of reactive AG, as well as other immune members (MG, neutrophil, et). In the meantime, with reaction to the injury, other factors also be secreted, like MMP-9, endothelin-1, and isoprostanes, which as well take part in the repairing or maintain the structures and functions in the brain. It is well known that AG is of importance in the integrity of the BBB [55, 56], the excitotoxicity aroused by glutamate, and the formation of glial scar, while just like the dualism of the inflammation, the reactivity of the AG also presents a double-edged effect [53, 57].
Reactive astrogliosis is the consequence of the response to TBI, with the changing in the morphology and the ethology of the AG, which is found to be both protective and harmful in the regeneration of the neural system. AG not only produce alarmins or DAMP (HMGB1, HSP, S100 proteins), interestingly, similar with the MG, but also have the TLR4 and RAGE to amplify neuroinflammation, inducing NF-κB transducing and resulting in the release of inflammatory factors such as cyclooxygenase-2, TNFα, connective tissue growth factors (CTGF), and MMP-9 [58]. It is also found that NF-κB can trigger cell swelling and edema associated with TBI [59], probably influenced by the dysfunction of the AQP1 rather than AQP4 [60]. In addition, in Sox2-deletion mice, markedly ameliorated injury-induced tissue loss and behavioral deficits was observed [61], which indicates the correlation between neuroinflammation and other pathologic response, is far beyond illustrating. Notably, the secretion of these initial signals hides the silver lining of the inflammation in TBI, for their can simultaneously aggravate inflammatory impairment or restore neural microenvironment.
Neurons
Neurons are the fundamental constituents and basic functional unit in CNS. As fragile and delicate, even the mild trauma can lead to neuron loss and axonal injury [62]. As the main “victim” in TBI, neurons experience a series of pathological course which ultimately result in the enlargement of the neuroinflammation. The death of the neurons is accompanied with the degeneration of the associated axons, the two both act as powder-hose in the initiation of the neuroinflammation. In closed-head TBI model, the activation of the MG is localized at the region with injured neurons, which starts as the central of the inflammation [63].
TBI leads to series of profiles changes in neuron to present protective or harmful effects, initiating consequential inflammation and oxidative stress. For instance, induction of nociceptin/orphanin FQ [64], accounts for the pain, anxiety and even inflammation in rat model [65], and down-regulating IRF6 is proven to attenuate neuronal apoptosis [66]. The key to enhance the survival of new-born neurons from neurogenic niche, is lessening aberrant development and ectopic localization [67].
Oligodendrocytes
The study on the oligodendrocytes (OLs) in the inflammation after TBI is limited, in spite of the existed evidence that the cells are involved in the chronic demyelinating diseases. Different from the precursor cells (OPCs), those who have the ability in proliferation and differentiation, OLs is mature, non-differentiated, and sensitive to the excitotoxicity [68]. After traumatic axon injury, the OLs are prone to demyelination, along with the loss of action potential [69]. The loss of myelination, resulted from primary injury or secondary injury, may be accompanied with the loss of the axons, and induces OLs death end up with caspase 3-mediated apoptosis [70, 71]. Afterwards, axonal regeneration is inhibited by myelin debris and MG is activate as a further trigger of chronic neuroinflammation [44, 72, 73], resulting in neurodegeneration and cognitive impairment at last.
OPCs are characterized with PDGFRα and NG2, namely NG2+ cells, with various functions in CNS like morphological changes, axons protection and glia scar formation [74]. It is also found that, after TBI, OPCs express GFAP and plausibly have ability to differentiate into AG [75]. As reaction to damage, an increasing amount of OPCs gather in the injured region [70, 76], regulated by NG2, growth factors and glutamate [77, 78]. Myelination might be of importance in plasticity and recovery of function after TBI [79]. However, the role of OPCs in inflammation is ambiguous, for their interaction with vessel seems to disrupt the BBB and exacerbate neuroinflammation in MS [80] (Figure 1).
Niche cells in initiation and aggravation of neuroinflammation. Diagram succinctly shows the course of neuroinflammation after TBI. The alarmin signals are released from damage tissue and, via activating corresponding receptors on glia cells, initiating microglia, as well astrocytes activation, followed with the cascade in enlargement of immune response with inflammatory factors releasing. And reciprocally, inflammation becomes chronic, long-lasting condition in during the crosstalk (function of peripheral immune cells has not been illustrated in this figure).
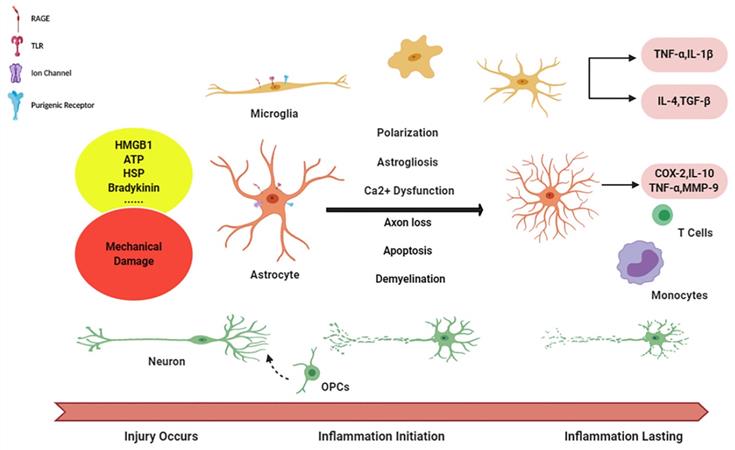
Communication, crosstalk and integration in neuroinflammation
Crosstalk among cells in inflammation and neural regeneration
TBI changes the reaction pattern in neurogenic niche, activating unique cell interaction in post-injury neurogenic response. The relationship between these niche cells tends to be more complex, though rather novel molecules emerge. It's likely that these findings in cells and molecule are weaving signal network to present the series on neuroinflammation after TBI.
For the controversy above, in recent years, researchers incline to categorize the AG into two groups, A1 and A2, with different phenotype, which resemble MG. A1 characterized as the upregulation of C3 and can be harmful for the neurogenesis. Especially, it is elicited by the reactive MG that secret lL-1α, TNF and C1q, leading to the death of neurons and OLs [81]. On the contrary, IL-10 originated from AG change the phenotype of MG, as well as lymphocyte density, to improve the neural survival [82]. The crosstalk between MG and AG after TBI is critical in the persistent neuroinflammation [51]. AG and OLs can produce IL-33 as the alarmins to promote the aggregation of the MG, followed by an altered cytokine/chemokine profile [83].
Using CSF1R antagonism (PLX5622) to eliminate the MG, which is activated by injured neuron, can attenuates rod microglial formation and astrogliosis [84]. And via downregulation of the purinergic receptor P2Y1, microglia could facilitate AG with scar formation, to behave as a neuroprotective phenotype [85]. After the depletion of the NG2, it comes with enhanced astrogliosis and up-regulated anti-inflammatory M2 biomarker Arg-1, followed with decreasing total number of microglia/macrophages, whereas worsening outcome was observed [86], then implicating the complicated mechanism in the neuroinflammation.
The crosstalk among the cells need the participation of specific molecule, like thyroid hormone (T3) might promote the neurogenesis through NSCs indirectly, probably with the neurons [87].
EVs, or specifically referred as exosomes, constituting diverse cargo like RNA, protein or lipids, capable of transmitting contents through BBB, appear to be the most effective courier in the neuroinflammation and nerve regeneration [21, 88, 89]. Neurons can generate exosomes enriched with miR-21-5p to regulate the MG towards to M1 type [90]. Correspondingly, glia EVs tend to modulate synaptic plasticity via miR-146a-5p [91], even with the sphingolipid on the membrane, leading to the excitatory transmission increase in neuron [92]. As the media in the crosstalk between MG and NSCs, exosomes are of key influence on repair of injury [93], and increasing microRNA are explored to regulate the polarization status of MG, further switching the direction of the neuroinflammation [94-97] (Table 1).
Effector molecules among niche cells in neuroinflammation crosstalk after TBI
| Cell Type | Molecules | Effect | Ref |
|---|---|---|---|
| Microglia | miR-146a-5p | Suppresses Syt1 and Nlg1 expression in receiving neurons; leading to dendritic spine loss as well as a decrease in the density and strength of excitatory synapses | [91] |
| IL-6, IL-1β,TNF-α | Enhance reactive astrogliosis and transformation the cells into neuroprotective type | [85] | |
| IL-1β, miR-155 | Activate additional microglia, progressive immune response in CNS | [98] | |
| sphingosine | Modulate synaptic plasticity | [92] | |
| Astrocytes | miR-873a-5p | Inhibit NF-κB signaling pathway and attenuate neuroinflammation mediated by microglia | [95] |
| GJA1-20 k | Downregulated the apoptosis rate and upregulated mitochondrial function to promote neuronal recovery | [99] | |
| IL-10 | Ameliorate microglial response and lymphocyte recruitment, promote neuronal survival. | [82] | |
| Oligodendrocytes | IL-33 | Aggravation of microglia/macrophages | [83] |
| Neuron | miR-21-5p | Promote polarization of M1 microglia | [90] |
| Mononuclear Phagocyte | succinate | Activate succinate receptor 1 (SUCNR1)/GPR91 and represent with anti-inflammatory effects | [100] |
| NSC/NPC | IFN-γ | Activate Stat1 Signaling in Target Cells | [101] |
| TGF-β2 | Reprogramming infiltrating monocyte-derived cells into anti-inflammatory type | [102] |
NSC might integrate the inflammatory signal in neural regenerative microenvironment
As the all-around player in CNS, NSCs and its progenitor (NPCs) play a pivotal role in the communication between different cells with high plasticity [18, 103], In the phase after TBI, different molecules, cytokines, chemokines, metabolites and neurotrophic factors from injured site or regulating cell, play roles in the differentiation and proliferation of the NSCs [22], requiring an exquisite cooperation. Observation on cytokine responses after TBI suggest that certain factors induced inflammation may function in an indirect way [104], that is to say, some intermediary exists. Just as transplantation of NSCs can generate OLs promote myelination [105], NSCs exhibit potential in regulating or remodeling microenvironment. Depletion of the transcriptional regulator Id3 leads to a decreasing number of AG generated from SVZ NSPCs and Id3(-/-) adult NSPCs are unable to differentiate into BMP-2-induced AG. While NSPCs deficient for transcription factor E47, which is found to be downregulated by Id3, shows performance of differentiation again, in the absence of BMP-2 [106], indicating a balance in the differentiation course of NSCs. Neonatal AG also promote the proliferation of NSCs with secreted protein [107]. Represented with similar function, NPCs secrete factors like TGF-β2 to transform MG into protective type [102], who in turn to support the maintenance and growth. The reparation on demyelination can also be initiated by M2 microglia, while through NSCs to bring about oligodendrogenesis and myelination [108].
Accumulating evidence has demonstrated that NSCs niches is the utmost region as the origin of the neural regenerative microenvironment, though its program of neurogenesis is a bit distinguished from the pattern after TBI. Apart from well accepted SGZ and SVZ, some new regions need more evidence to play a role in neurogenesis after TBI [109, 110]. And intriguingly, these regions seem to show response to the TBI rather than SCI [111], probably, on account of the different signals in blood.
BBB might connect the crosstalk between periphery and CNS, the collapse of BBB accounts for lasting neurodegeneration disease [112]. The degeneration of the ECM leads to the disruption of the BBB, and ECM-remodeling transcriptional changes can be induced by the serum protein albumin via TGFβ signaling in primary AG [113, 114].
NPCs may be of particular influence in secondary cytokine releasing [104], in response to the simultaneous inflammatory factors. Researchers firstly found that acute inflammation may induce neuronal regeneration more than damage in zebrafish CNS [115], coupled with unique role of Gata3 [116]. It is notable that proliferation of NSCs coincide with the expression of TLR4 [117], which can be conjectured the subtle connection between neurogenesis and inflammation, attracting the interest in inflammation regulation after TBI, like NSCs regulating NLRP3 and IL-1β, hence attenuating the neurotoxic cascade induced by MG [118]. Otherwise, after transplantation, NSC/NPCs tend to alter phenotype of MG and ameliorate inflammation [105, 119], that betokens EVs from NSCs also show anti-apoptotic and anti-inflammatory properties [120]. This might attribute to the role of inflammation on spatiotemporal reason. It's reasoned to speculate that immune response only assumes beneficial in the primary injured part (Figure 2).
NSCs might integrate the inflammatory signals crosstalk in neuroinflammation. Different cell may down/up-regulate specific molecule/receptors to enhance or weaken others. Microglia and astrocytes might mutually influence their phenotype as “pro” or “anti” effect on inflammation, and change the fate of neurons and OPCs. Finally, NSPC might act as intermediary among various crosstalk, modulating signals, restructuring microenvironment, and further promote recovery from the injury.
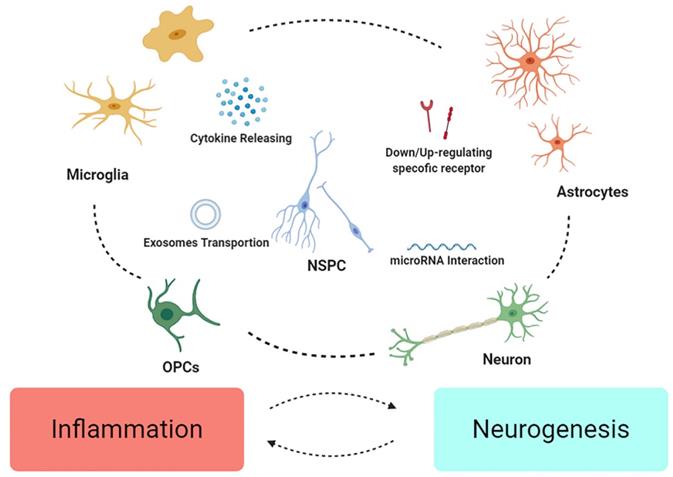
The promising method for regulation of neuroinflammation and enhance the neurogenesis
As promising treatments, those focused on the regulation of the inflammation, metabolism or the neurogenesis are emerging, it is worth believing TBI patients can acquire better outcome and welfare.
Existing evidence has proposed potential method to regulate the inflammation. knockdown of TLR4 can ameliorate neuroinflammation after TBI, in a way by inhibiting autophagy and AG activation [121], and salsalate, an unacetylated salicylate, is utilized to relieve inflammation and promote function recovery [122]. Downregulation of the inflammasomes is also a potential method to attenuate the damage caused by inflammation response [123].
New target for the treatment of TBI has been aroused, such as CCR5 [124]. And some dynamic biomarker, like Nrf2 [125], may be useful to evaluate the process TBI came through. Combined using minocycline and N-acetylcysteine, inducing remyelination and regulating neuroinflammation, seems superior to the monotherapy attempts before [126, 127].
Apart from the regulation of the inflammation, methods are exploited to improve the oxygenation or metabolism status, and weaken neuronal pyroptosis [128]. Hyperbaric oxygen therapy and Hydrogen gas both seem to be effective to attenuate the injury extent [129]. Lactate metabolism in brain is an essential feature after TBI [130], and hypertonic sodium lactate solution is proved to reverse brain oxygenation and metabolism dysfunction after traumatic brain injury through vasodilatory, mitochondrial, and anti-edema effects [131].
Stem cell therapy, for its multi-differentiated capability, is a promising method [132, 133]. However, though application of iPSC, is still limited for ethical issues or bio-safety. MSCs, which is thought to be the most optimum candidate to treat the neurodegeneration disease, no matter injecting cells directly or the extracellular vesicles (exosomes) [89, 134], striking improvement in both neurogenesis or recovery of the function have been acquired [135, 136]. Furthermore, a host of ncRNA are involved in the TBI diagnosis and treatment [137].
Conclusion and future perspectives
TBI is still the disease affecting long-term quality of life with protracted course of neuroinflammation [1]. The types of it, though, differ in pathology and outcome, the long-last and annoying course of inflammation seems to be a common situation. In spite of substantial evidence showing that reactivity in cell lineage is unable to imitate the exact procedures ongoing in vivo, coupled with sufficient microenvironment factors, it's still reasonable to make most use of the established model [10], for direct or indirect regulation in promising treatments. And it's worth celebrating that certain attempts seems able to balance the function of inflammation in neurogenesis.
To have an explicit vision of neuroinflammation in neurogenesis and nerve regeneration, detailed work and evidence are needed [2, 14, 39]. Evidence available is unable to clarify definitive role of inflammation after TBI, in view of complicated and mixing essence itself. Existing evidence shows feature as “protective” in acute inflammation and “harmful” in long term [13], while absence of particular point in time makes it harder to make utmost of advantages in neuroprotection and promotion in regeneration. Switching the polarization status of the MG may be potential, and increasing number of drugs or molecules have been found to be effective [138-140]. It shall bear in mind that the concept in polarization is probably appropriate in modulation on inflammatory outcome rather than an absolute condition. More study on multi-dimensional and comprehensive in these immune cells, with burgeoning progress in single-cell technology and integrated perception in CNS [18, 39, 141].
Besides MG, other niche cells represent peculiar functions in initiation, enlargement, or modulation of the neuroinflammation. AG and MG both serve as “mixed bag” [38]. Based on the double function on transforming immune response, the crosstalk, or glia-crosstalk, makes it complex to understand the mechanism underlying. Cytokine, chemokine and recently, exosomes are all participating in this interplay, depicting a more panoramic network in this long-studied subject.
Despite dwindling since the neonatal [142], NSCs and its progenitor cells are capable of repairing injury via differentiation, proliferation or replacement. A host of work on the stem cells transplantation in TBI or SCI have shown gratifying results, whereas the translation and clinical application is limited for frustrating ethical or bio-safety issues. Exosomes, by virtue of its various contents and excellent affinity with nervous system, may reveal a better therapeutic potential [19, 88]. Acquiring a more comprehensive awareness on the relation between neuroinflammation and neurogenic niche is essential, in the premise that glia, neuron and NSCs all engage in the communication and crosstalk.
Inflammation in neural regeneration is a commonplace, that being said, with more intriguing achievements in these years, it's requisite to balance rather than simply inhibiting it. It is important to reiterate that, among glias, MG are the most potential candidates at present, for its role in magnifying inflammatory response, and AG, based on its affluent secretion property, is a promising participant as well. We didn't refer to ECM, and vascular endothelial in this interplay, those both are of particular importance in pathophysiology. What's more, NSCs, shows fundamentality in the course all long. Clarifying its activity in integrating all the signals, as well as functional signals in promoting proliferation, survival, and migration, is of high-priority. It's believable enough that we are laying the key-stone for the avenue towards clinic translation and the welfare of TBI patients.
Acknowledgements
The present work was supported by the Natural Science Foundation of China (NSFC) grant to Xiaosheng He (Nos. 81971156 and 2020XC015).
Author Contributions
Yibin Jia had the idea for the article. Literature search was performed by Yibin jia, Guanyi Wang and Enming Kang. The manuscript was written by Yibin Jia and Yuqing Ye. Zishuo Guo and Huijun Chen critically revised the work. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khellaf A, Khan DZ, Helmy A. Recent advances in traumatic brain injury. J Neurol. 2019;266:2878-2889 DOI: 10.1007/s00415-019-09541-4
2. Jassam YN, Izzy S, Whalen M, McGavern DB, El KJ. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron. 2017;95:1246-1265 DOI: 10.1016/j.neuron.2017.07.010
3. Galgano M, Toshkezi G, Qiu X, Russell T, Chin L, Zhao LR. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017;26:1118-1130 DOI: 10.1177/0963689717714102
4. DeWitt DS, Hawkins BE, Dixon CE, Kochanek PM, Armstead W, Bass CR. et al. Pre-Clinical Testing of Therapies for Traumatic Brain Injury. J Neurotrauma. 2018;35:2737-2754 DOI: 10.1089/neu.2018.5778
5. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. Jama Neurol. 2015;72:355-362 DOI: 10.1001/jamaneurol.2014.3558
6. Dorsett CR, McGuire JL, DePasquale EA, Gardner AE, Floyd CL, McCullumsmith RE. Glutamate Neurotransmission in Rodent Models of Traumatic Brain Injury. J Neurotrauma. 2017;34:263-272 DOI: 10.1089/neu.2015.4373
7. Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798-800 DOI: 10.1126/science.2567056
8. Anthonymuthu TS, Kenny EM, Bayir H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016;1640:57-76 DOI: 10.1016/j.brainres.2016.02.006
9. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171-191 DOI: 10.1038/nrneurol.2017.13
10. Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019;56:5332-5345 DOI: 10.1007/s12035-018-1454-5
11. Eyolfson E, Khan A, Mychasiuk R, Lohman AW. Microglia dynamics in adolescent traumatic brain injury. J Neuroinflammation. 2020;17:326. DOI: 10.1186/s12974-020-01994-z
12. Skaper SD, Facci L, Zusso M, Giusti P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci. 2018;12:72. DOI: 10.3389/fncel.2018.00072
13. Xiong Y, Mahmood A, Chopp M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. 2018;21:137-151 DOI: 10.1016/j.cjtee.2018.02.003
14. Corrigan F, Mander KA, Leonard AV, Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation. 2016;13:264. DOI: 10.1186/s12974-016-0738-9
15. Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7-25
16. Chang EH, Adorjan I, Mundim MV, Sun B, Dizon ML, Szele FG. Traumatic Brain Injury Activation of the Adult Subventricular Zone Neurogenic Niche. Front Neurosci. 2016;10:332. DOI: 10.3389/fnins.2016.00332
17. Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB. et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219-1227 DOI: 10.1016/j.cell.2013.05.002
18. Vicidomini C, Guo N, Sahay A. Communication, Cross Talk, and Signal Integration in the Adult Hippocampal Neurogenic Niche. Neuron. 2020;105:220-235 DOI: 10.1016/j.neuron.2019.11.029
19. Hou BR, Jiang C, Wang ZN, Ren HJ. Exosome-mediated crosstalk between microglia and neural stem cells in the repair of brain injury. Neural Regen Res. 2020;15:1023-1024 DOI: 10.4103/1673-5374.270302
20. Clark D, Perreau VM, Shultz SR, Brady RD, Lei E, Dixit S. et al. Inflammation in Traumatic Brain Injury: Roles for Toxic A1 Astrocytes and Microglial-Astrocytic Crosstalk. Neurochem. Res. 2019;44:1410-1424 DOI: 10.1007/s11064-019-02721-8
21. Delpech JC, Herron S, Botros MB, Ikezu T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019;42:361-372 DOI: 10.1016/j.tins.2019.02.007
22. Christie KJ, Turnley AM. Regulation of endogenous neural stem/progenitor cells for neural repair-factors that promote neurogenesis and gliogenesis in the normal and damaged brain. Front Cell Neurosci. 2012;6:70. DOI: 10.3389/fncel.2012.00070
23. Najem D, Rennie K, Ribecco-Lutkiewicz M, Ly D, Haukenfrers J, Liu Q. et al. Traumatic brain injury: classification, models, and markers. Biochem Cell Biol. 2018;96:391-406 DOI: 10.1139/bcb-2016-0160
24. Cash A, Theus MH. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int J Mol Sci. 2020 21. DOI: 10.3390/ijms21093344
25. Wang ML, Li WB. Cognitive impairment after traumatic brain injury: The role of MRI and possible pathological basis. J. Neurol. Sci. 2016;370:244-250 DOI: 10.1016/j.jns.2016.09.049
26. Hutchison JS, Derrane RE, Johnston DL, Gendron N, Barnes D, Fliss H. et al. Neuronal apoptosis inhibitory protein expression after traumatic brain injury in the mouse. J Neurotrauma. 2001;18:1333-1347 DOI: 10.1089/08977150152725632
27. Zeng Z, Zhang Y, Jiang W, He L, Qu H. Modulation of autophagy in traumatic brain injury. J. Cell. Physiol. 2020;235:1973-1985 DOI: 10.1002/jcp.29173
28. Soldozy S, Sharifi KA, Desai B, Giraldo D, Yeghyayan M, Liu L. et al. Cortical Spreading Depression in the Setting of Traumatic Brain Injury. World Neurosurg. 2020;134:50-57 DOI: 10.1016/j.wneu.2019.10.048
29. Jayakumar AR, Taherian M, Panickar KS, Shamaladevi N, Rodriguez ME, Price BG. et al. Differential Response of Neural Cells to Trauma-Induced Swelling In Vitro. Neurochem. Res. 2018;43:397-406 DOI: 10.1007/s11064-017-2434-2
30. Impellizzeri D, Campolo M, Bruschetta G, Crupi R, Cordaro M, Paterniti I. et al. Traumatic Brain Injury Leads to Development of Parkinson's Disease Related Pathology in Mice. Front Neurosci. 2016;10:458. DOI: 10.3389/fnins.2016.00458
31. Chao H, Liu Y, Lin C, Xu X, Li Z, Bao Z. et al. Activation of bradykinin B2 receptor induced the inflammatory responses of cytosolic phospholipase A(2) after the early traumatic brain injury. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2957-2971 DOI: 10.1016/j.bbadis.2018.06.006
32. Braun M, Vaibhav K, Saad NM, Fatima S, Vender JR, Baban B. et al. White matter damage after traumatic brain injury: A role for damage associated molecular patterns. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2614-2626 DOI: 10.1016/j.bbadis.2017.05.020
33. Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano A, Aleksovska K. et al. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front Neurosci. 2018;12:628. DOI: 10.3389/fnins.2018.00628
34. Wofford KL, Loane DJ, Cullen DK. Acute drivers of neuroinflammation in traumatic brain injury. Neural Regen Res. 2019;14:1481-1489 DOI: 10.4103/1673-5374.255958
35. Wang KK, Yang Z, Zhu T, Shi Y, Rubenstein R, Tyndall JA. et al. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev. Mol. Diagn. 2018;18:165-180 DOI: 10.1080/14737159.2018.1428089
36. Kumar RG, Rubin JE, Berger RP, Kochanek PM, Wagner AK. Principal components derived from CSF inflammatory profiles predict outcome in survivors after severe traumatic brain injury. Brain Behav. Immun. 2016;53:183-193 DOI: 10.1016/j.bbi.2015.12.008
37. McDonald SJ, Sharkey JM, Sun M, Kaukas LM, Shultz SR, Turner RJ. et al. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J Neurotrauma. 2020;37:770-781 DOI: 10.1089/neu.2019.6885
38. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692-702 DOI: 10.1111/bph.13125
39. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171-191 DOI: 10.1038/nrneurol.2017.13
40. Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353:783-785 DOI: 10.1126/science.aaf6260
41. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223-228 DOI: 10.1038/nature12808
42. Scott G, Zetterberg H, Jolly A, Cole JH, De Simoni S, Jenkins PO. et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain. 2018;141:459-471 DOI: 10.1093/brain/awx339
43. Hanlon LA, Huh JW, Raghupathi R. Minocycline Transiently Reduces Microglia/Macrophage Activation but Exacerbates Cognitive Deficits Following Repetitive Traumatic Brain Injury in the Neonatal Rat. J Neuropathol Exp Neurol. 2016;75:214-226 DOI: 10.1093/jnen/nlv021
44. Taib T, Leconte C, Van Steenwinckel J, Cho AH, Palmier B, Torsello E. et al. Neuroinflammation, myelin and behavior: Temporal patterns following mild traumatic brain injury in mice. Plos One. 2017;12:e184811. DOI: 10.1371/journal.pone.0184811
45. Tay TL, Hagemeyer N, Prinz M. The force awakens: insights into the origin and formation of microglia. Curr. Opin. Neurobiol. 2016;39:30-37 DOI: 10.1016/j.conb.2016.04.003
46. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013;16:273-280 DOI: 10.1038/nn.3318
47. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat. Med. 2017;23:1018-1027 DOI: 10.1038/nm.4397
48. Anderson SR, Vetter ML. Developmental roles of microglia: A window into mechanisms of disease. Dev Dyn. 2019;248:98-117 DOI: 10.1002/dvdy.1
49. Younger D, Murugan M, Rama RK, Wu LJ, Chandra N. Microglia Receptors in Animal Models of Traumatic Brain Injury. Mol. Neurobiol. 2019;56:5202-5228 DOI: 10.1007/s12035-018-1428-7
50. Shields DC, Haque A, Banik NL. Neuroinflammatory responses of microglia in central nervous system trauma. J Cereb Blood Flow Metab. 2020: 271678X-20965786X. DOI: 10.1177/0271678X20965786.
51. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692-702 DOI: 10.1111/bph.13125
52. Hsu ET, Gangolli M, Su S, Holleran L, Stein TD, Alvarez VE. et al. Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol. 2018;136:955-972 DOI: 10.1007/s00401-018-1902-3
53. Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2016; 275 Pt 3: 305-315. DOI: 10.1016/j.expneurol. 2015 03.020
54. Sompol P, Norris CM. Ca(2+), Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front Aging Neurosci. 2018;10:199. DOI: 10.3389/fnagi.2018.00199
55. Rui Q, Ni H, Lin X, Zhu X, Li D, Liu H. et al. Astrocyte-derived fatty acid-binding protein 7 protects blood-brain barrier integrity through a caveolin-1/MMP signaling pathway following traumatic brain injury. Exp Neurol. 2019;322:113044. DOI: 10.1016/j.expneurol.2019.113044
56. Ikeshima-Kataoka H, Yasui M. Correlation between astrocyte activity and recovery from blood-brain barrier breakdown caused by brain injury. Neuroreport. 2016;27:894-900 DOI: 10.1097/WNR.0000000000000619
57. Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R. et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195-200 DOI: 10.1038/nature17623
58. Lu M, Yan XF, Si Y, Chen XZ. CTGF Triggers Rat Astrocyte Activation and Astrocyte-Mediated Inflammatory Response in Culture Conditions. Inflammation. 2019;42:1693-1704 DOI: 10.1007/s10753-019-01029-7
59. Jayakumar AR, Tong XY, Ruiz-Cordero R, Bregy A, Bethea JR, Bramlett HM. et al. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31:1249-1257 DOI: 10.1089/neu.2013.3169
60. Rauen K, Pop V, Trabold R, Badaut J, Plesnila N. Vasopressin V1a Receptors Regulate Cerebral Aquaporin 1 after Traumatic Brain Injury. J Neurotrauma. 2020;37:665-674 DOI: 10.1089/neu.2019.6653
61. Chen C, Zhong X, Smith DK, Tai W, Yang J, Zou Y. et al. Astrocyte-Specific Deletion of Sox2 Promotes Functional Recovery After Traumatic Brain Injury. Cereb. Cortex. 2019;29:54-69 DOI: 10.1093/cercor/bhx303
62. Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013;246:35-43 DOI: 10.1016/j.expneurol.2012.01.013
63. Wofford KL, Harris JP, Browne KD, Brown DP, Grovola MR, Mietus CJ. et al. Rapid neuroinflammatory response localized to injured neurons after diffuse traumatic brain injury in swine. Exp. Neurol. 2017;290:85-94 DOI: 10.1016/j.expneurol.2017.01.004
64. Witta J, Buzas B, Cox BM. Traumatic brain injury induces nociceptin/orphanin FQ expression in neurons of the rat cerebral cortex. J Neurotrauma. 2003;20:523-532 DOI: 10.1089/089771503767168456
65. Mallimo EM, Kusnecov AW. The role of orphanin FQ/nociceptin in neuroplasticity: relationship to stress, anxiety and neuroinflammation. Front Cell Neurosci. 2013;7:173. DOI: 10.3389/fncel.2013.00173
66. Hou MS, Cao J, Chen B, Liu XB. Down-Regulation of IRF6 Protects Cortical Neurons Against Traumatic Neuronal Injury Through Activating Akt-eNOS Pathway. Cell. Mol. Neurobiol. 2017;37:587-594 DOI: 10.1007/s10571-016-0394-8
67. Littlejohn EL, Scott D, Saatman KE. Insulin-like growth factor-1 overexpression increases long-term survival of posttrauma-born hippocampal neurons while inhibiting ectopic migration following traumatic brain injury. Acta Neuropathol Commun. 2020;8:46. DOI: 10.1186/s40478-020-00925-6
68. Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790-1815 DOI: 10.1002/glia.22658
69. Arancibia-Carcamo IL, Attwell D. The node of Ranvier in CNS pathology. Acta Neuropathol. 2014;128:161-175 DOI: 10.1007/s00401-014-1305-z
70. Flygt J, Djupsjo A, Lenne F, Marklund N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur. J. Neurosci. 2013;38:2153-2165 DOI: 10.1111/ejn.12179
71. Lotocki G, de Rivero VJ, Alonso O, Molano JS, Nixon R, Safavi P. et al. Oligodendrocyte vulnerability following traumatic brain injury in rats. Neurosci. Lett. 2011;499:143-148 DOI: 10.1016/j.neulet.2011.05.056
72. Clarner T, Diederichs F, Berger K, Denecke B, Gan L, van der Valk P. et al. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia. 2012;60:1468-1480 DOI: 10.1002/glia.22367
73. Geoffroy CG, Zheng B. Myelin-associated inhibitors in axonal growth after CNS injury. Curr. Opin. Neurobiol. 2014;27:31-38 DOI: 10.1016/j.conb.2014.02.012
74. Dent KA, Christie KJ, Bye N, Basrai HS, Turbic A, Habgood M. et al. Oligodendrocyte birth and death following traumatic brain injury in adult mice. Plos One. 2015;10:e121541. DOI: 10.1371/journal.pone.0121541
75. Komitova M, Serwanski DR, Lu QR, Nishiyama A. NG2 cells are not a major source of reactive astrocytes after neocortical stab wound injury. Glia. 2011;59:800-809 DOI: 10.1002/glia.21152
76. Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J. Neurosci. 1994;14:4716-4730
77. Scheller A, Bai X, Kirchhoff F. The Role of the Oligodendrocyte Lineage in Acute Brain Trauma. Neurochem. Res. 2017;42:2479-2489 DOI: 10.1007/s11064-017-2343-4
78. Gudz TI, Komuro H, Macklin WB. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J. Neurosci. 2006;26:2458-2466 DOI: 10.1523/JNEUROSCI.4054-05.2006
79. Armstrong RC, Mierzwa AJ, Sullivan GM, Sanchez MA. Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology. 2016;110:654-659 DOI: 10.1016/j.neuropharm.2015.04.029
80. Niu J, Tsai HH, Hoi KK, Huang N, Yu G, Kim K. et al. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat. Neurosci. 2019;22:709-718 DOI: 10.1038/s41593-019-0369-4
81. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481-487 DOI: 10.1038/nature21029
82. Villacampa N, Almolda B, Vilella A, Campbell IL, Gonzalez B, Castellano B. Astrocyte-targeted production of IL-10 induces changes in microglial reactivity and reduces motor neuron death after facial nerve axotomy. Glia. 2015;63:1166-1184 DOI: 10.1002/glia.22807
83. Wicher G, Wallenquist U, Lei Y, Enoksson M, Li X, Fuchs B. et al. Interleukin-33 Promotes Recruitment of Microglia/Macrophages in Response to Traumatic Brain Injury. J Neurotrauma. 2017;34:3173-3182 DOI: 10.1089/neu.2016.4900
84. Witcher KG, Bray CE, Dziabis JE, McKim DB, Benner BN, Rowe RK. et al. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia. 2018;66:2719-2736 DOI: 10.1002/glia.23523
85. Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K. et al. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y1 Receptor Downregulation. Cell Rep. 2017;19:1151-1164 DOI: 10.1016/j.celrep.2017.04.047
86. Huang C, Sakry D, Menzel L, Dangel L, Sebastiani A, Kramer T. et al. Lack of NG2 exacerbates neurological outcome and modulates glial responses after traumatic brain injury. Glia. 2016;64:507-523 DOI: 10.1002/glia.22944
87. Lin C, Li N, Chang H, Shen Y, Li Z, Wei W. et al. Dual effects of thyroid hormone on neurons and neurogenesis in traumatic brain injury. Cell Death Dis. 2020;11:671. DOI: 10.1038/s41419-020-02836-9
88. Vogel A, Upadhya R, Shetty AK. Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders. Ebiomedicine. 2018;38:273-282 DOI: 10.1016/j.ebiom.2018.11.026
89. Xiong Y, Mahmood A, Chopp M. Emerging potential of exosomes for treatment of traumatic brain injury. Neural Regen Res. 2017;12:19-22 DOI: 10.4103/1673-5374.198966
90. Yin Z, Han Z, Hu T, Zhang S, Ge X, Huang S. et al. Neuron-derived exosomes with high miR-21-5p expression promoted polarization of M1 microglia in culture. Brain Behav. Immun. 2020;83:270-282 DOI: 10.1016/j.bbi.2019.11.004
91. Prada I, Gabrielli M, Turola E, Iorio A, D'Arrigo G, Parolisi R. et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018;135:529-550 DOI: 10.1007/s00401-017-1803-x
92. Antonucci F, Turola E, Riganti L, Caleo M, Gabrielli M, Perrotta C. et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. Embo J. 2012;31:1231-1240 DOI: 10.1038/emboj.2011.489
93. Hou BR, Jiang C, Wang ZN, Ren HJ. Exosome-mediated crosstalk between microglia and neural stem cells in the repair of brain injury. Neural Regen Res. 2020;15:1023-1024 DOI: 10.4103/1673-5374.270302
94. Li G, Xiao L, Qin H, Zhuang Q, Zhang W, Liu L. et al. Exosomes-carried microRNA-26b-5p regulates microglia M1 polarization after cerebral ischemia/reperfusion. Cell Cycle. 2020;19:1022-1035 DOI: 10.1080/15384101.2020.1743912
95. Long X, Yao X, Jiang Q, Yang Y, He X, Tian W. et al. Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J Neuroinflammation. 2020;17:89. DOI: 10.1186/s12974-020-01761-0
96. Yang Y, Ye Y, Kong C, Su X, Zhang X, Bai W. et al. MiR-124 Enriched Exosomes Promoted the M2 Polarization of Microglia and Enhanced Hippocampus Neurogenesis After Traumatic Brain Injury by Inhibiting TLR4 Pathway. Neurochem. Res. 2019;44:811-828 DOI: 10.1007/s11064-018-02714-z
97. Jiang M, Wang H, Jin M, Yang X, Ji H, Jiang Y. et al. Exosomes from MiR-30d-5p-ADSCs Reverse Acute Ischemic Stroke-Induced, Autophagy-Mediated Brain Injury by Promoting M2 Microglial/Macrophage Polarization. Cell. Physiol. Biochem. 2018;47:864-878 DOI: 10.1159/000490078
98. Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N. et al. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation. 2017;14:47. DOI: 10.1186/s12974-017-0819-4
99. Chen W, Zheng P, Hong T, Wang Y, Liu N, He B. et al. Astrocytes-derived exosomes induce neuronal recovery after traumatic brain injury via delivering gap junction alpha 1-20 k. J Tissue Eng Regen Med. 2020;14:412-423 DOI: 10.1002/term.3002
100. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa A, Kwok CK, Leonardi T. et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell. 2018;22:355-368 DOI: 10.1016/j.stem.2018.01.020
101. Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, Drago D. et al. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol. Cell. 2014;56:193-204 DOI: 10.1016/j.molcel.2014.08.020
102. De Feo D, Merlini A, Brambilla E, Ottoboni L, Laterza C, Menon R. et al. Neural precursor cell-secreted TGF-beta2 redirects inflammatory monocyte-derived cells in CNS autoimmunity. J. Clin. Invest. 2017;127:3937-3953 DOI: 10.1172/JCI92387
103. Shear DA, Tate MC, Archer DR, Hoffman SW, Hulce VD, Laplaca MC. et al. Neural progenitor cell transplants promote long-term functional recovery after traumatic brain injury. Brain Res. 2004;1026:11-22 DOI: 10.1016/j.brainres.2004.07.087
104. Thelin EP, Hall CE, Gupta K, Carpenter K, Chandran S, Hutchinson PJ. et al. Elucidating Pro-Inflammatory Cytokine Responses after Traumatic Brain Injury in a Human Stem Cell Model. J Neurotrauma. 2018;35:341-352 DOI: 10.1089/neu.2017.5155
105. Koutsoudaki PN, Papastefanaki F, Stamatakis A, Kouroupi G, Xingi E, Stylianopoulou F. et al. Neural stem/progenitor cells differentiate into oligodendrocytes, reduce inflammation, and ameliorate learning deficits after transplantation in a mouse model of traumatic brain injury. Glia. 2016;64:763-779 DOI: 10.1002/glia.22959
106. Bohrer C, Pfurr S, Mammadzada K, Schildge S, Plappert L, Hils M. et al. The balance of Id3 and E47 determines neural stem/precursor cell differentiation into astrocytes. Embo J. 2015;34:2804-2819 DOI: 10.15252/embj.201591118
107. Dai Y, Sun F, Zhu H, Liu Q, Xu X, Gong P. et al. Effects and Mechanism of Action of Neonatal Versus Adult Astrocytes on Neural Stem Cell Proliferation After Traumatic Brain Injury. Stem Cells. 2019;37:1344-1356 DOI: 10.1002/stem.3060
108. Mecha M, Yanguas-Casas N, Feliu A, Mestre L, Carrillo-Salinas FJ, Riecken K. et al. Involvement of Wnt7a in the role of M2c microglia in neural stem cell oligodendrogenesis. J Neuroinflammation. 2020;17:88. DOI: 10.1186/s12974-020-01734-3
109. Bennett LB, Cai J, Enikolopov G, Iacovitti L. Heterotopically transplanted CVO neural stem cells generate neurons and migrate with SVZ cells in the adult mouse brain. Neurosci. Lett. 2010;475:1-6 DOI: 10.1016/j.neulet.2010.03.019
110. Bennett L, Yang M, Enikolopov G, Iacovitti L. Circumventricular organs: a novel site of neural stem cells in the adult brain. Mol. Cell. Neurosci. 2009;41:337-347 DOI: 10.1016/j.mcn.2009.04.007
111. Falnikar A, Stratton J, Lin R, Andrews CE, Tyburski A, Trovillion VA. et al. Differential Response in Novel Stem Cell Niches of the Brain after Cervical Spinal Cord Injury and Traumatic Brain Injury. J Neurotrauma. 2018;35:2195-2207 DOI: 10.1089/neu.2017.5497
112. Giannoni P, Claeysen S, Noe F, Marchi N. Peripheral Routes to Neurodegeneration: Passing Through the Blood-Brain Barrier. Front Aging Neurosci. 2020;12:3. DOI: 10.3389/fnagi.2020.00003
113. Kim SY, Senatorov VJ, Morrissey CS, Lippmann K, Vazquez O, Milikovsky DZ. et al. TGFbeta signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci Rep. 2017;7:7711. DOI: 10.1038/s41598-017-07394-3
114. Jiang R, Liu Q, Zhu H, Dai Y, Yao J, Liu Y. et al. The expression of TRIAD1 and DISC1 after traumatic brain injury and its influence on NSCs. Stem Cell Res Ther. 2018;9:297. DOI: 10.1186/s13287-018-1024-9
115. Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D. et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338:1353-1356 DOI: 10.1126/science.1228773
116. Kizil C, Kyritsis N, Dudczig S, Kroehne V, Freudenreich D, Kaslin J. et al. Regenerative neurogenesis from neural progenitor cells requires injury-induced expression of Gata3. Dev. Cell. 2012;23:1230-1237 DOI: 10.1016/j.devcel.2012.10.014
117. Ye Y, Xu H, Zhang X, Li Z, Jia Y, He X. et al. Association between toll-like receptor 4 expression and neural stem cell proliferation in the hippocampus following traumatic brain injury in mice. Int. J. Mol. Sci. 2014;15:12651-12664 DOI: 10.3390/ijms150712651
118. Chan E, Krishnansamy S, Wong C, Gan SY. The NLRP3 inflammasome is involved in the neuroprotective mechanism of neural stem cells against microglia-mediated toxicity in SH-SY5Y cells via the attenuation of tau hyperphosphorylation and amyloidogenesis. Neurotoxicology. 2019;70:91-98 DOI: 10.1016/j.neuro.2018.11.001
119. Gao J, Grill RJ, Dunn TJ, Bedi S, Labastida JA, Hetz RA. et al. Human Neural Stem Cell Transplantation-Mediated Alteration of Microglial/Macrophage Phenotypes after Traumatic Brain Injury. Cell Transplant. 2016;25:1863-1877 DOI: 10.3727/096368916X691150
120. Rong Y, Liu W, Lv C, Wang J, Luo Y, Jiang D. et al. Neural stem cell small extracellular vesicle-based delivery of 14-3-3t reduces apoptosis and neuroinflammation following traumatic spinal cord injury by enhancing autophagy by targeting Beclin-1. Aging (Albany NY). 2019;11:7723-7745 DOI: 10.18632/aging.102283
121. Jiang H, Wang Y, Liang X, Xing X, Xu X, Zhou C. Toll-Like Receptor 4 Knockdown Attenuates Brain Damage and Neuroinflammation After Traumatic Brain Injury via Inhibiting Neuronal Autophagy and Astrocyte Activation. Cell. Mol. Neurobiol. 2018;38:1009-1019 DOI: 10.1007/s10571-017-0570-5
122. Lagraoui M, Sukumar G, Latoche JR, Maynard SK, Dalgard CL, Schaefer BC. Salsalate treatment following traumatic brain injury reduces inflammation and promotes a neuroprotective and neurogenic transcriptional response with concomitant functional recovery. Brain Behav. Immun. 2017;61:96-109 DOI: 10.1016/j.bbi.2016.12.005
123. Yi HJ, Lee JE, Lee DH, Kim YI, Cho CB, Kim IS. et al. The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. J Neurosurg. 2019:1-9 DOI: 10.3171/2019.6.JNS1954
124. Joy MT, Ben AE, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas M. et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell. 2019;176:1143-1157 DOI: 10.1016/j.cell.2019.01.044
125. Dong W, Sun Y, Cheng H, Yang B, Wang L, Jiang Z. et al. Dynamic cell type-specific expression of Nrf2 after traumatic brain injury in mice. Eur. J. Neurosci. 2019;50:1981-1993 DOI: 10.1111/ejn.14399
126. Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J. et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J Cereb Blood Flow Metab. 2018;38:1312-1326 DOI: 10.1177/0271678X17718106
127. Haber M, Abdel BS, Grin'Kina NM, Irizarry R, Ershova A, Orsi S. et al. Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Exp. Neurol. 2013;249:169-177 DOI: 10.1016/j.expneurol.2013.09.002
128. Chen G, Gao C, Yan Y, Wang T, Luo C, Zhang M. et al. Inhibiting ER Stress Weakens Neuronal Pyroptosis in a Mouse Acute Hemorrhagic Stroke Model. Mol. Neurobiol. 2020;57:5324-5335 DOI: 10.1007/s12035-020-02097-9
129. Wang P, Zhao M, Chen Z, Wu G, Fujino M, Zhang C. et al. Hydrogen Gas Attenuates Hypoxic-Ischemic Brain Injury via Regulation of the MAPK/HO-1/PGC-1a Pathway in Neonatal Rats. Oxid Med Cell Longev. 2020;2020:6978784. DOI: 10.1155/2020/6978784
130. Patet C, Suys T, Carteron L, Oddo M. Cerebral Lactate Metabolism After Traumatic Brain Injury. Curr Neurol Neurosci Rep. 2016;16:31. DOI: 10.1007/s11910-016-0638-5
131. Millet A, Cuisinier A, Bouzat P, Batandier C, Lemasson B, Stupar V. et al. Hypertonic sodium lactate reverses brain oxygenation and metabolism dysfunction after traumatic brain injury. Br J Anaesth. 2018;120:1295-1303 DOI: 10.1016/j.bja.2018.01.025
132. Koliatsos VE, Xu L, Cummings BJ. Stem cell therapies for traumatic brain injury. Regen Med. 2015;10:917-920 DOI: 10.2217/rme.15.62
133. Blaya MO, Tsoulfas P, Bramlett HM, Dietrich WD. Neural progenitor cell transplantation promotes neuroprotection, enhances hippocampal neurogenesis, and improves cognitive outcomes after traumatic brain injury. Exp. Neurol. 2015;264:67-81 DOI: 10.1016/j.expneurol.2014.11.014
134. Ni H, Yang S, Siaw-Debrah F, Hu J, Wu K, He Z. et al. Exosomes Derived From Bone Mesenchymal Stem Cells Ameliorate Early Inflammatory Responses Following Traumatic Brain Injury. Front Neurosci. 2019;13:14. DOI: 10.3389/fnins.2019.00014
135. Kim C, Park JM, Kong T, Lee S, Seo KW, Choi Y. et al. Double-Injected Human Stem Cells Enhance Rehabilitation in TBI Mice Via Modulation of Survival and Inflammation. Mol. Neurobiol. 2018;55:4870-4884 DOI: 10.1007/s12035-017-0683-3
136. Zhang Y, Chopp M, Zhang ZG, Katakowski M, Xin H, Qu C. et al. Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem. Int. 2017;111:69-81 DOI: 10.1016/j.neuint.2016.08.003
137. Pinchi E, Frati P, Arcangeli M, Volonnino G, Tomassi R, Santoro P. et al. MicroRNAs: The New Challenge for Traumatic Brain Injury Diagnosis. Curr Neuropharmacol. 2020;18:319-331 DOI: 10.2174/1570159X17666191113100808
138. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649-665 DOI: 10.1111/bph.13139
139. Guo Y, Hong W, Wang X, Zhang P, Korner H, Tu J. et al. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma? Front Mol Neurosci. 2019;12:125. DOI: 10.3389/fnmol.2019.00125
140. Xu H, Wang Z, Li J, Wu H, Peng Y, Fan L. et al. The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention. Neural Plast. 2017;2017:5405104. DOI: 10.1155/2017/5405104
141. Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires-Afonso Y, Martina S. et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. Embo Rep. 2018 19. DOI: 10.15252/embr.201846171
142. Martin-Suarez S, Valero J, Muro-Garcia T, Encinas JM. Phenotypical and functional heterogeneity of neural stem cells in the aged hippocampus. Aging Cell. 2019;18:e12958. DOI: 10.1111/acel.12958
Author contact
![]() Corresponding author: Xiaosheng He, Department of Neurosurgery, Xijing Hospital, Airforce Military Medical University (Fourth Military Medical University), Xi'an 710032, China. E-mail: hexiaosedu.cn
Corresponding author: Xiaosheng He, Department of Neurosurgery, Xijing Hospital, Airforce Military Medical University (Fourth Military Medical University), Xi'an 710032, China. E-mail: hexiaosedu.cn