Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Brief introduction of the...
Vaccine development for previous...
mRNA vaccine development for...
Delivery route and working...
Cellular fates of mRNA vaccines
Preclinical and clinical results...
Conclusion and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(6):1446-1460. doi:10.7150/ijbs.59233 This issue Cite
Review
mRNA vaccines for COVID-19: what, why and how
Jung Woo Park, Philip N.P. Lagniton, Yu Liu, Ren-He Xu ![]()
Institute of Translational Medicine, and Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macau, China.
Received 2021-2-9; Accepted 2021-3-15; Published 2021-4-10
Abstract

The Coronavirus disease-19 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus -2 (SARS-CoV-2), has impacted human lives in the most profound ways with millions of infections and deaths. Scientists and pharmaceutical companies have been in race to produce vaccines against SARS-CoV-2. Vaccine generation usually demands years of developing and testing for efficacy and safety. However, it only took less than one year to generate two mRNA vaccines from their development to deployment. The rapid production time, cost-effectiveness, versatility in vaccine design, and clinically proven ability to induce cellular and humoral immune response have crowned mRNA vaccines with spotlights as most promising vaccine candidates in the fight against the pandemic. In this review, we discuss the general principles of mRNA vaccine design and working mechanisms of the vaccines, and provide an up-to-date summary of pre-clinical and clinical trials on seven anti-COVID-19 mRNA candidate vaccines, with the focus on the two mRNA vaccines already licensed for vaccination. In addition, we highlight the key strategies in designing mRNA vaccines to maximize the expression of immunogens and avoid intrinsic innate immune response. We also provide some perspective for future vaccine development against COVID-19 and other pathogens.
Keywords: COVID-19, SARS-CoV-2, mRNA vaccine, efficacy and safety
Brief introduction of the coronavirus disease 2019 (COVID-19)
The COVID-19 pandemic has thus far caused infection of more than 100 million people and over 2 million deaths worldwide. These numbers also reflect an astonishing increase compared to more than 80,000 infections and over 3,000 deaths by March 2020 [1] when the World Health Organization declared COVID-19 a world pandemic. Moreover, the pandemic brought nearly the entire world to stop due to the consequent global crisis in health, economy, and psychology. COVID-19 is caused by severe acute respiratory syndrome coronavirus -2 (SARS-CoV-2), a name adapted from SARS-CoV that caused the infection of SARS in 2003 [2]. Since none of the explored therapies can directly kill the virus, vaccines have become the last hope to stop the pandemic. However, vaccine development is generally a time-consuming process, taking years to complete. As a great surprise, Moderna Biotechnology, Inc. delivered a vaccine named mRNA-1273 in only 42 days from the date when the spike protein-coding sequence of SARS-CoV-2 was published on January 10, 2020 [3]. Together, it took less than one year to complete the design, manufacture, efficacy and safety tests, and evaluation and approval for use.
Both mRNA vaccines have been found to be highly effective and safe in preventing COVID-19 according to clinical trials [4, 5]. Compared to other vaccine platforms, mRNA vaccines possess unique advantages including versatility, efficient delivery, use of the protein translational machinery of the host, and short developmental time. In this review, we introduce the general principles for the design, optimization, working mechanisms, and challenges of mRNA vaccine development. In addition, we will summarize up-to-date clinical trial data on mRNA vaccines. Hopefully, the review will help readers comprehend the history, current status, and prospective of mRNA vaccines for immunization against COVID-19 and also future infectious diseases.
Etiology and pathophysiology of COVID-19
COVID-19 is caused by SARS-CoV-2, which is an enveloped virus with a positive-strand and single-stranded RNA genome that belongs to the β-coronavirus subfamily [6]. The RNA genome of SARS-CoV-2, approximately 30 kb, encodes 14 open reading frames. The 5'-proximal end of the genome encodes two polypeptides, pp1a and pp1b, by a programmed -1 ribosomal frameshift [7]. Two polypeptides are cleaved into 16 non-structural proteins (nsp1-16), which mediate the delivery of the viral replication complexes to subcellular domains and viral replication, transcription, and post-transcriptional processes [8]. A surface envelope glycoprotein, Spike (S), encoded by the 3,822-bp S gene, mediates the viral entry into host cells via binding to its functional receptor, angiotensin-converting enzyme 2 (ACE2), expressed highly in lung alveolar epithelial cells and epithelial cells of many other organs including the heart, kidney, bladder, and ileum [9, 10]. Therefore, these organs are considered more vulnerable to SARS-CoV-2 [11]. The primary symptoms of COVID-19 include fever, dry cough, shortness of breath, muscle ache, dizziness, headache, sore throat, rhinorrhea, chest pain, diarrhea, nausea, and vomiting [1]. When the viral load is high or when the infection happens in patients with other severe diseases, the patients often develop acute respiratory syndrome and sepsis in a short time [1].
Mutation rate and transmissibility of SARS-CoV-2
RNA viruses such as influenza virus and HIV typically undergo mutation at a much higher rate than DNA viruses because RNA viruses usually lack the proof-reading activity. Although SARS-CoV-2 possesses the proof-reading activity, facilitated by nsp14 (Exonuclease N)-nsp10 complexes [12], scientists cataloged more than 12,000 different mutations in the SARS-CoV-2 genome [13]. Recently, several mutant variants including D614G mutation have replaced the original Wuhan strain and spread as dominant strains, possibly due to their increased infectivity to ACE2 [14, 15]. The South African strain, known as 501.V2 or B.1.351, underwent three amino acids substitutions, K417N, E484K, and N501Y in S protein of the original strain, among which N501Y is located in the receptor-binding domain (RBD).
Another variant recently identified in the UK, known as B1.1.7 or VOC202012/01, contains eight mutations in S protein, among which the N501Y mutation is overlapped with 501.V2 variant. Although the overall effect of the mutations is not yet known, the high transmission rate of UK iB1.1.7 in the UK may result from the increased affinity of the N501Y mutation to ACE2. In fact, a recent computational analysis predicts that the N501Y mutation likely results in an increased number of interactions with the Y41 and K353 amino acids of ACE2 hence elevated affinity [16]. There is a growing concern that the current vaccines may not protect people from the newly emerging variants of SARS-CoV-2. However, the faster development of mRNA vaccines than the other vaccine types may be a solution to prevent against the current and future variants as well as other outbreaks of viral diseases like COVID-19.
Vaccine development for previous coronavirus epidemics
During the outbreaks of Severe Acute Respiratory Syndrome (SARS) in 2002-2004 and Middle East Respiratory Syndrome (MERS) in 2012, vaccines were developed for these diseases but never licensed for humans. Two vaccine types, one inactivated form of SARS-CoV-1 virus developed by Sinovac Biotech Ltd. and one DNA-based vaccine developed by National Institute of Allergy and Infectious Disease (NIAID), were tested for phase I clinical trials but never licensed for human vaccination and commercial use [17, 18]. The majority of the vaccine development against SARS-CoV-1 was completely dropped since the virus never re-emerged after the first outbreak. As for MERS coronavirus, nine vaccines derived from various platforms were tested for phase I/II clinical trials [19, 20]. For example, ChAdOx developed by the University of Oxford was based on the replication-deficient adenovirus vector, expressing full-length S protein of MERS-CoV [21]. The coding sequence was optimized for protein translation. As demonstrated in animal studies, the vaccine was highly immunogenic and able to activate CD8+ T cells for exerting cytotoxicity and B cells for producing neutralizing antibodies [21]. It was also documented that two of 42 anti-MERS mRNA vaccine candidates were developed and tested at the pre-clinical stage, but neither proceeded to a large-scale clinical trial to be a licensed vaccine [22]. Currently, 63 anti-SARS-CoV-2 candidate vaccines have been being tested on clinical trials (Table 1). Although mRNA vaccines represent only 11% of all the vaccines developed on various platforms, two mRNA vaccines, mRNA-1273 (developed by Moderna) and BNT162b (developed by Pfizer and BioNTech Ltd.) were the first vaccines approved for emergency use in many countries.
Current vaccine platforms in clinical trials*
| Vaccine platform | Number of candidate vaccines | % of the total number |
|---|---|---|
| Protein subunit | 19 | 30% |
| Viral vector (non-replicating) | 10 | 16% |
| DNA | 8 | 13% |
| Inactivated virus | 9 | 14% |
| RNA | 7 | 11% |
| Viral vector (replicating) | 4 | 6% |
| Virus-like particle | 2 | 3% |
| VVr + antigen-presenting cell | 2 | 3% |
| Live attenuated virus | 1 | 2% |
| VVnr + antigen-presenting cell | 1 | 2% |
*The data were from the WHO Novel coronavirus Landscape as of January 8, 2021.
History of mRNA vaccines
mRNA vaccine is based on the principle that mRNA is an intermediate messenger to be translated to an antigen after the delivery into host cells via various routes. RNA molecules have been utilized as therapeutic and research tools for more than two decades, with the usage ranging from in vitro transcribed (IVT) mRNA, small interference RNA (siRNA), RNA aptamers, riboswitches, antisense RNA to the recent developed mRNA vaccines [23-26]. The idea that mRNA molecules can be directly delivered into cells for manipulating gene expression or producing proteins of interest was first tested in the late 1980s. Malone, et al., first demonstrated the efficient transfection of mRNA in NIH 3T3 fibroblasts using a cationic lipid N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride [27]. The idea of transfecting mRNA molecules into host cells for the expression of a gene of interest underwent several technical improvements over the next two decades or so [28].
In early 1990s, direct expression of external mRNA molecules in host animals for therapeutic effects was first tested by delivering RNA vectors encoding a reporter gene such as luciferase and β-galactosidase into murine muscle cells and transfecting vasopressin mRNA into rats to reverse Diabetes-Insipidus [29, 30]. In 1993 Martinson, et al., demonstrated that an in vitro synthesized mRNA vaccine encoding nucleoprotein of influenza virus triggered the activation of cytotoxic T lymphocytes in mice [31]. Later, it was found that in vivo application of mRNA induced both activation of cytotoxic T cells and humoral response of B cells to produce specific antibodies [32].
However, the possibility of using mRNA as a vaccine was not seriously taken due to the easy degradability of RNA, the ubiquitous presence of ribonucleases, and the lack of scalability. A series of advancements occurred recently when mRNA vaccines were used to prevent triple-negative breast cancer and lung carcinoma using mRNAs encoding MUC1 and herpes simplex virus I thymidine kinase, respectively [33, 34]. The application has been utilized in prevention of cancer and infectious diseases and treatment of allergy and other diseases that need protein replacement. Numerous mRNA vaccines are under clinical trials or already available today against infectious pathogens such as Zika virus, cytomegalovirus, influenza virus, metapneumovirus, and parainfluenza virus as well as cancer [35].
Advantages and disadvantages of mRNA vaccines over other vaccine platforms
The vaccine development can generally be classified into two categories: gene-based and protein-based. The protein-based approach has been the conventional method that relies on attenuated or recombinant proteins directly delivered as immunogens to activate the adaptive and humoral immune response. The gene-based vaccines are delivered via a DNA or RNA vector to host cells where they will be expressed to produce corresponding antigens to induce the immune response in the host. Both protein- and gene-based vaccines (including DNA and RNA) have been explored for COVID-19 and currently on clinical trials. There are several advantages of mRNA vaccines over the other platforms.
The first advantage of mRNA vaccines is the easiness and fast speed for their manufacturing. The core principle of mRNA vaccines is to deliver a transcript that encodes a target antigen or immunogen. The RNA synthesis can immediately be carried out on the same platform as soon as the sequence encoding the immunogen is available and the process can be easily scalable and cell-free, requiring minimal platform change during mRNA formulation and manufacturing [36]. Second, a mRNA vaccine expresses target protein (antigen) via translation from the mRNA rapidly after its transfection. mRNA vaccines possess much higher biosafety than DNA-based vaccines as the translation of the antigens takes place in the cytoplasm rather than the nucleus, thus much less possible for the mRNA to integrate into the genome than a DNA-based vaccine. Moreover, mRNA is a safer vector than DNA as mRNA carries a short sequence to be translated, is a transient molecule, and does not interact with the host genome. Third, protein-based vaccines are often produced from bacteria, whereas mRNA vaccines are translated by the host translation machinery thus likely to form an antigen that mimics the protein's structure expressed from the viral genome including the post-translational modifications.
However, the storage and transportation of mRNA vaccines require ultralow temperatures, whereas protein-based vaccines can be stored and transported in less stringent conditions. It has been tested that the leading COVID-19 mRNA vaccines remain stable up to 24 hours at room temperature [37]. Thus, it is a huge technical hurdle and economic burden to store and transfer millions of mRNA vaccines to and in warm countries and regions. Nevertheless, with the development of lipid nanoparticle technologies, the stability of mRNA vaccines can be sustained at less stringent conditions [38].
In addition to conventional mRNA vaccines, there is another type of RNA vaccines called self-amplifying RNA (saRNA) vaccines that have been tested and evaluated. saRNA vaccines can replicate after delivery, thus expressing more target antigens in a host at lower doses than conventional mRNA vaccines [39]. saRNA vaccines are genetically engineered replicons derived from self-replicating single-stranded RNA viruses and can be delivered as viral replicon particles or as a completely synthetic saRNA produced after IVT. They have been developed and tested in multiple animal models and humans against infectious diseases such as rabies, influenza, RSV, HIV, and Ebola [40].
mRNA vaccine development for COVID-19
Optimization of mRNA vaccine design
Typical vaccine development using live-attenuated or inactivated virus or a pseudovirus system involves tedious and time-consuming steps and has become a bottleneck for responding to an epidemic or pandemic caused by newly emerging viruses. As described above, mRNA vaccines possess distinctive advantages of rapid development and versatility as exemplified by the swift development of multiple COVID-19 mRNA vaccines. More importantly, recent preliminary data from clinical trials have shown that two licensed mRNA vaccines, mRNA-1273 and BNT162b, have higher protective efficacy than ChAdOx1 vaccine developed using a chimpanzee adenovirus (~95% vs. ~70%) [4, 41].
However, some intrinsic features of mRNA molecules demand special strategies to guarantee the stability, efficacy and safety of mRNA vaccines. First, mRNA are intrinsically unstable and prone to degradation due to the omnipresence of RNases in the serum and plasma [42]. Second, the cellular machinery recognizes exogenous RNA molecules as immunological mimic of viral infection, which results in an immediate immune response [43]. Thus, it is a prerequisite for the design of mRNA vaccines to maximize the stability of RNA and translation efficiency and avoid the innate immune response by host cells [44, 45]. Below we will discuss the major strategies used in designing mRNA vaccines, including 5'-capping, nucleoside modification, codon optimization, and efficient delivery of mRNA molecules with nanoparticles (Table 2).
Strategies for developing seven mRNA candidate vaccines
| Name of vaccine | IVT pol | 5'-cap | Codon optimization | Antigen design | Modified nucleotide | Purification method | Ref. |
|---|---|---|---|---|---|---|---|
| mRNA-1273 | T7 | m7GpppNmN | Yes | Full length | N1-methyl pseudouridine | Oligo-dT | [56, 121, 122] |
| S protein | |||||||
| K986P/V987P | |||||||
| BNT162b (3 LNP-mRNAs) | T7 | m7GpppNmN | Yes | S protein | N1-methyl pseudouridine | Magnetic purification | |
| RBD subunit | [57, 123] | ||||||
| K986P/V987P | |||||||
| CVnCoV | T7 | m7GpppNmN | Yes, GC rich | Full length | N1-methyl pseudouridine | LiCl | |
| S protein | precipitation | [124] | |||||
| K986P/V987P | |||||||
| LUNAR-COV19 | T7 | m7GpppNmN | Yes | VEEV-FL-S | N1-methyl pseudouridine | Silicon column | |
| protein | [54] | ||||||
| LNP-nCoVsaRNA | T7 | m7GpppNmN | Unknown | VEEV-FL-S | Unknown | LiCl | |
| Protein | precipitation | ||||||
| K986P/V987P | LiCl | [55] | |||||
| GGGGSGGGGS linker | |||||||
| ARCoV | T7 | m7GpppNmN | Yes | S protein | Unknown | Unknown | |
| RBD subunit | [86] | ||||||
| (AA319-541) | |||||||
| ChulaCov19 mRNA vaccine | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | NA |
Note: IVT, in vitro transcription; pol, polymerase; m7GpppN, 7-methylguanosine 5'-triphosphate; VEEV, Venezuelan equine encephalitis virus.
5'-capping of mRNA vaccines
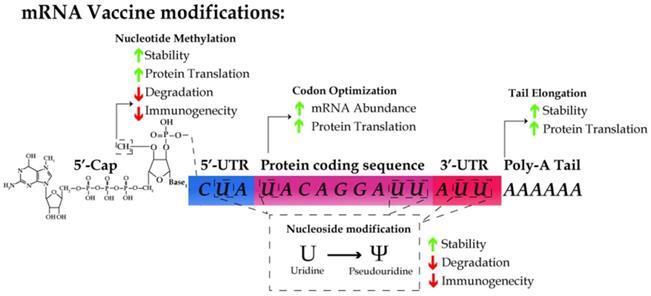
Endogenous mRNA molecules undergo post-transcriptional modifications, including 5'-capping and polyadenylation, for the stability of mRNA and efficient translation. Naturally, 7-methylguanosine cap (m7G) is added to the first nucleotide of a mRNA chain via 5' to 5' linkage. The 2'-OH of the ribose of the first nucleotide is further methylated to form m7GpppNm, also known as cap1. 5'-capping is critical for protecting mRNA from exonuclease activity, facilitating pre-mRNA splicing, and serving as the binding site for eIF4F, the heterodimeric translation initiation complex [46-49]. Recent studies have indicated the 5'-cap structure as a major determinant by which the host can discriminate between self vs. non-self mRNA molecules [50-53]. A m7GpppNm cap was added to the 5'-end of the majority of the mRNA vaccines reported thus far during their IVT [54-57].
Optimization of 5'- and 3'-untranslated regions and the length of polyadenylation tail
Regulatory elements in the 5′-untranslated region (UTR) [58] and the length of 3′-UTR [59] increase protein translation. In addition, the polyadenylation (polyA) tail stabilizes mRNA and increases protein translation. Several recent reports have shown that the length of polyA tail is closely associated with the translation efficiency [60]. However, the information on 5'- and 3'-UTRs and the nature of polyA signal sequence remains proprietary and undisclosed for the seven reported mRNA vaccines.
Nucleoside modification during IVT
Kariko, et al., demonstrated that RNA recognition by Toll-like receptors (TLRs) is suppressed via modification of the nucleosides in mRNA molecules [61, 62]. Incorporating m5C, m6A, m5U, s2U, or pseudouridine into mRNA molecules abrogates the immune response by evading the activation of TLR-3, -7, and -8 [61]. For all the seven reported vaccines, pseudouridine was incorporated into the mRNA vaccines in the place of uridine. In addition, the substitution with pseudouridine, m6A, and s2U in RNA molecules suppresses the degradation of RNA by RNase L [63]. Thus, the nucleoside modifications not only enhance the stability of RNA but also reduce the innate immune response.
The schematic for designing an mRNA vaccine. mRNA molecules are synthesized in vitro with cap1 structure (m7GpppNm), the substitution of uridine with pseudouridine, and the use of the preferred codon in humans, optimized UTR and polyA tail sequence. These modifications results in the increase of RNA stability and translation efficiency as well as the reduction of immunogenicity.
Purification of IVT
The contaminating impurities during IVT can massively affect the safety of mRNA vaccines once they are introduced to human cells. Even residual amounts of double-stranded RNA and DNA-RNA hybrid molecules can trigger the innate immune response as they can be recognized by the cellular sensors pattern recognition receptors. Various purification techniques have been used to remove residual impurities from IVT reactions for all the seven mRNA vaccines currently on clinical trials. A previous study indicates that the purification of mRNA reduces the expression of type I interferon and increases the protein translation [64]. As summarized in Table 2, various purification techniques such as Oligo dT column, LiCl precipitation, and silicone column have been employed to remove contaminants from in vitro synthesized mRNA [45].
Codon optimization
Several parameters have been considered for the codon optimization, which affects the translation efficiency, protein folding, and mRNA abundance. One example is that the GC content in the sequence. Although GC-rich sequences may be problematic for the secondary structure formation of mRNA, the translation efficiency of a GC-rich sequence can be 100-fold higher than that of a GC-poor sequence [65]. The translation elongation rate highly depends on the availability of the cognate tRNA species and the optimization of the codon usage to avoid sequences that match rare tRNA species and incorporate sequences that match more abundant tRNA species [66]. Moreover, the codon optimization is essential for the mRNA stability as the codon-dependent translation elongation rate has been implicated as a major determinant of the mRNA stability [67]. Mechanistically, reduced translational elongation of mRNA with suboptimal codons results in the recruitment of the DEAD-box RNA helicase, Dhh1p, which triggers mRNA decay [68]. Two additional codon optimization methods involve the use of the codons with human bias and the maximum adaptation index [69, 70]. Other bioinformatics approaches can be explored to further enhance the stability of mRNA, e.g., via design of the secondary structures and prediction of the expression level based on deep learning [71, 72].
Designing platform and target immunogen for the seven mRNA candidate vaccines
Each of the seven mRNA candidate vaccines was synthesized in vitro from a DNA template encoding either the full-length S protein or RBD of SARS-CoV-2 using bacteriophage T7 RNA polymerase. mRNA-1273, CVnCoV, LUNAR-CoV19, and LNP-nCoVsaRNA mRNA vaccines used the template encoding the full-length S protein with 2P substitutions at K986 and V987 positions to produce the stable pre-fusion form of S protein [73]. Pfizer/BioNTech have developed two immunogens, the RBD (BNT162b1) and the full-length S protein (BNT162b2). BNT162b2 has been shown to be safer than BNT162b1, especially in older adults in a preliminary clinical trial, and thus was chosen for a phase 3 clinical trial [74]. ARCoV vaccines are based on the RBD of SARS-CoV-2. Whereas the sequences of the 5'- and 3'-UTR of the mRNA templates were not revealed in the literature, the 3'-UTR of BNT162b mRNA vaccine derived empirically by screening naturally occurring 3'-UTRs for the highest RNA stability [75]. On the other hand, CVnCoV and LNP-nCoVsaRNA were built on the saRNA platform containing a self-replicating replicon of Trinidad donkey Venezuelan equine encephalitis virus (VEEV). The viral protein-encoding gene of the replicon is replaced with a modified S protein-encoding gene of SARS-CoV-2 with two proline mutations in the S2 subunit, K986P and V987P [54, 55]. Consistent with the notion that saRNA vaccines can self-amplify after delivery into host cells, the dose used for vaccination was one to two magnitude lower than conventional mRNA vaccines. As shown in Table 2, the dosage range for CVnCoV and LNP-nCoVsaRNA was 2-12 μg and 0.01-10 μg, respectively. In comparison, the typical dose range for the conventional mRNA vaccines was 30-100 μg.
Packaging mRNA vaccines with lipid nanoparticle (LNPs)
An early study has shown that the transfection efficiency of naked mRNAs is nearly two orders of magnitude lower than that of mRNA bound to lipofectin formulation [27]. The lipofectin-based carriers effectively help mRNA delivery into target cells and protect mRNA from RNase [36, 76]. The formulation of liposome-based transfection reagents containing cationic lipids has remarkably been improved in recent years [77]. In particular, LNPs, composed of proprietary components including positively charged lipids, cationic polypeptides, polymers, micelles or dendrimers, have been widely used for in vivo RNA delivery [78, 79]. LNPs encapsulate mRNA and assemble it into the stable lipid bilayers, which are then ingested by cells through a variety of endocytosis pathways. Below is the information for packaging of mRNA vaccines with various LNPs.
1. mRNA-1273: It was loaded into two proprietary cationic LNPs, WO2017070626 and WO2018115527. Although the exact formulation is not known, the composition of the LNPs was described as follows, SM-102, polyethylene glycol-2000-dimyristoyl glycerol (PEG2000-DMG), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) [80].
2. BNT162b mRNA: It was encapsulated by patented LNPs with improved efficiency of the mRNA delivery according to its clinical trial report (#NCT04368728) [81, 82]. The LNPs are composed of ionizable amino lipid, phospholipid, cholesterol and a PEGylated lipid prepared at a ratio of 50:10:38.5:1.5 mol/mol [82, 83]. It is interesting to note that BTN162b and mRNA-1273 vaccines are suggested to be shipped and stored at -80˚C and -20°C, respectively [80, 82].
3. CVnCoV: It was formulated with a proprietary LNP, referred to as RNActive® technology platform. The LNP consists of four lipid components: cholesterol, DSPC, PEGylated lipid, and a cationic lipid, however the detailed formulation information was not disclosed. CVnCoV remains stable for at least three months when it is stored at 5℃ as suggested by its manufacturer. Moreover, CVnCoV can be stored at room temperature as a ready-to-use the vaccine for up to 24 hours [84, 85].
4. ARCoV: It was encapsulated in LNPs of a proprietary composition using a preformed vesicle method and found thermostable at different temperatures, including 4°C, 25°C, and 37°C for up to one week [86].
5. ARCT-021: Currently undergoing phase 1/2 clinical trials, it combines two technologies, i.e., saRNA STARR™ and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [87]. In addition, LUNAR® lipids are pH-sensitive and biodegradable, causing minimal lipid accumulation in cells after multiple dosing [87]
6. LNP-nCoVsaRNA: Developed by Imperial College London using cationic liposome as the carrier, it has just entered phase 1 clinical trial [55].
4. ARCoV: It was encapsulated in LNPs of a proprietary composition using a preformed vesicle method and found thermostable at different temperatures, including 4°C, 25°C, and 37°C for up to one week [86].
5. ARCT-021: Currently undergoing phase 1/2 clinical trials, it combines two technologies, i.e., saRNA STARR™ and LUNAR® lipid-mediated delivery method. It was designed to enhance and extend antigen expression, enabling vaccination at lower doses [87]. In addition, LUNAR® lipids are pH-sensitive and biodegradable, causing minimal lipid accumulation in cells after multiple dosing [87]
6. LNP-nCoVsaRNA: Developed by Imperial College London using cationic liposome as the carrier, it has just entered phase 1 clinical trial [55].
RNA candidate vaccines on clinical trials
As of January 22, 2021, 173 candidate vaccines were on preclinical development and 63 on clinical trials. Seven mRNA candidate vaccines (11% of the 63) have completed the preclinical development or are now on clinical trials, of which mRNA-1273, BNT162b, and CVnCoV are undergoing or have completed phase 3 trials. LNP-encapsulated CoVsaRNA, ARCoV, ChulaCov19, and ARCT-021 mRNA vaccines are currently undergoing phase 1 or 2 trials. All vaccines except Lunar-COVID19 were administered at two doses at day 0 and day 21 or 28, respectively. Self-reported adverse effects including pain, swelling, redness in the local injection site, allergy, paralysis, chills, fever, and headache were observed in recipients of three mRNA vaccines mRNA-1273, BNT162b, and CVnCoV during phase 3 trials. Relevant information on all the seven mRNA vaccine candidates has been summarized in Table 3, including the developers, number of doses, dosage, vaccination method, side effects, and stage of clinical trials.
Seven RNA candidate Vaccines in clinical development as of January 25,221
| Vaccine name | # of doses | Dosing schedule | Dosage Tested | Route of administration | Developers | Clinically observevd side effects | Clinical trial phase | Clinical Trial ID | References |
|---|---|---|---|---|---|---|---|---|---|
| mRNA -1273 | 2 | Day 0 + 28 | 100 ug (Phase 3 result) | IM | Moderna + National Institute of Allergy and Infectious Diseases (NIAID) | Pain, Swelling Redness, Allergy, Paralysis, Chills, Tiredness, Headache | Phase 3 | NCT04470427 NCT04283461 NCT04405076 NCT04649151 | [4, 122, 125, 126] |
| BNT162b2 (3LNP-mRNAs) | 2 | Day 0 + 21 | 30 ug (Phase 3 result) | IM | Pfizer/BioNTech + Fosun Pharma | Pain, Swelling, Redness, Allergy, Paralysis, Chills, Tiredness, Headache, Anaphylactic shock | Phase 3 | NCT04368728 | [5, 57, 74, 127, 128] |
| CVnCoV Vaccine | 2 | Day 0 + 28 | 2 μg and 12 μg (Phase 1 result) | IM | CureVac/Bayer | Pain, Swelling, Redness, Chills, Tiredness, Headache | Phase 3 | NCT04674189 NCT04449276 NCT04515147 NCT04652102 EudraCT-2020-004066-19 | [124] |
| LUNAR- COV19 | 1 | Day 0 | 0.2 μg and 10 μg (Preclinical) | IM | Arcturus Therapeutics | N/A | Phase 2 | NCT04668339 NCT04480957 | [54] |
| LNP- nCoVsaRNA | 2 | ND | 0.01 μg to 10 μg (Preclinical) | IM | Imperial College London/ VacEquity Global Health | N/A | Phase 1 | ISRCTN170726-92 | [55] |
| SARS-CoV-2 mRNA vaccine (ARCoV) | 2 | Day 0 + 14 or Day 0 + 28 | 100 μg and 1000 μg (Preclinical) | IM | Academy of Military Science (AMS), Walvax Biotechnology and Suzhou | N/A | Phase 1 | ChiCTR2000034-112 ChiCTR2000039-212 | [86] |
| ChulaCov19 mRNA vaccine | 2 | Day 0 + 21 | N/A | IM | Chulalongkorn University | N/A | Phase 1 | NCT04566276 | N/A |
Note: IM, intramuscular; N/A, not apply; ND, not done.
Delivery route and working mechanisms of mRNA vaccines
According to published preclinical and clinical data, all the seven LNP-encapsulated mRNA vaccines were administered intramuscularly (IM). IM injection is one of the common methods, and the vaccines are injected into deeper tissues under the dermal and subcutaneous layers [88]. Shortly after the injection, the LNP-mRNA cargos enter muscle cells through endocytosis and then the mRNA is translated and the translates form metastable trimeric prefusion S protein. Later, a network of blood vessels adjacent to the muscles can recruit infiltrating antigen-presenting cells (APCs).
One superior advantage of mRNA vaccines is their use of the cellular translational machinery and other cytosolic components in producing a properly folded and fully functional protein from each injected mRNA. In the case of mRNA vaccines designed with the full-length S protein, the translated product contains a signal peptide from amino acids 1 to 15, enabling the S protein to be transported to the plasma membranes or secreted out of the cytoplasm. Meanwhile, the majority of the protein will be degraded in endosome-derived proteasome and subsequently incorporated as a part of the class I major histocompatibility complexes (MHCs), and presented to CD8+ and CD4+ T cells, respectively [53,59]. Dendritic cells transfected by an mRNA vaccine or its endocytosed immunogens process the assembly of the class II MHC complex and present it to immune cells (Figure 2).
Delivery and working mechanism of a mRNA vaccine. mRNA vaccine, containing the coding region of S protein flanked by the optimized 5'- and 3'-UTRs and polyA tail, is synthesized via IVT, followed by 5'-capping with a 5'-cap analogy and encapsulation with LNP for IM injection (step 1). The vaccine is delivered into muscle cells or antigen-presenting cells such as dendritic cells or macrophages via endocytosis (step 2). mRNA molecules are unloaded from LNPs and translated to S protein in the ribosome (step 3). Newly synthesized S protein is secreted to extracellular space, internalized via endocytosis into antigen-presenting cells and incorporated as a part of MHC class II antigen presentation complex (steps 5b, 6b, and 7) to present the antigen to immune cells including T and B cells [132]. Partially degraded S peptides by proteosomes are incorporated into MHC class I complexes, which are then transported to plasma membranes and also presented as antigens to immune cells (steps 4a, 4b, 5a, and 7).
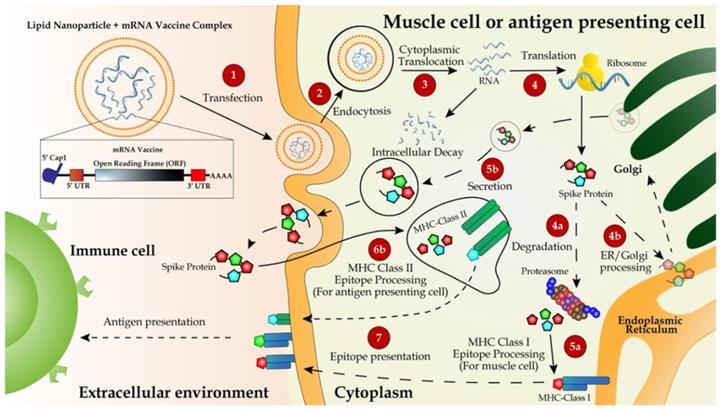
However, the major mechanism for immunization with an mRNA vaccine is humoral immune response via the activation of B cells. Once naïve B cells are activated by interacting with the cognate CD4+ T cells and the ligation of CD40, the activated B cells will proliferate and differentiate to either memory B cells or antibody-secreting plasma cells in lymphoid organs. The newly activated B cells with a high and low affinity will differentiate to short-lived plasma cells and quiescent memory B cells, respectively [89, 90]. Upon the secondary antigen exposure, the circulating antibodies produced from plasma cells will bind and neutralize the antigen, thus blocking the antigen-carrying virus from infecting it target cells. An insufficient amount of antibodies will activate memory B cells either to trigger secondary immune response [91].
Cellular fates of mRNA vaccines
RNA degradation
mRNA vaccines took the vaccine development stage by storm mainly due to their rapid development and versatility of design. However, as described above there are two significant intrinsic limitations of mRNA as a vaccine: 1) the instability of mRNA molecules and 2) the activation of the innate immune response. Although it is generally difficult to estimate the degradation rate of a particular messenger RNA in vivo, studies have estimated that the most endogenous mRNA transcripts are rapidly degraded, usually within 10-15 minutes [92, 93]. Two pathways degrade mRNA: 1) 5′ to 3′ exonuclease reaction mediated by Xrn1p after de-capping of 5'-methylguanosin; 2) 3' to 5' digestion mediated by a nuclear multi-protein complex called exosome after the removal of polyA tail, which does not require the removal of the 5'-cap [93, 94]. Following injection into muscles, synthetically made mRNA likely undergoes rapid RNA degradation by both extracellular and intracellular RNases. Since the half-life of mRNA in the cytoplasm is directly associated with protein expression, it is critical to maximizing the stability of mRNA. 5'-cap protects mRNA from the action of 5' to 3' Xrn1p-mediated exonuclease and enhances the binding to eukaryotic translation initiation factor 4E (eIF4E) [95]. Reciprocally, a recent finding shows that eIF4E promotes the 5'-capping of mRNAs, implicating an intimate crosstalk between 5'-cap and the translation initiation [96].
Chan, et al., demonstrates that the rate of mRNA decay is inversely proportional to the kinetics of translational initiation [93]. Thus, one way to increase mRNA stability is to promote the translation efficiency by optimizing the codon usage and UTR sequences, which is implemented for mRNA vaccines. It is worth noting that ARCoV, a recently reported mRNA vaccine by Zhang, et al., can be stored at least one week at room temperature without compromising the stability of the vaccine, mainly due to the proprietary protective nanoparticles against ribonucleases [86]. This is an important milestone for mRNA vaccines, given the unstable nature of mRNA molecules.
Potential activation of immune response by exogenous mRNA and impurities in vaccines
The innate immune system has evolved to defend against viral genomes and replicating intermediates via the potent pathogen-associated molecular patterns (PAMPs). PAMPs sense dsRNA through pattern recognition receptors (PRRs) in most cell types and subsequently activate the expression of pro-inflammatory cytokines and type I interferons (IFNs) [97]. Poly (I:C) has been long used as a TLR3 agonist to mimic the viral infection and as an immunostimulatory adjuvant for experimental vaccines [98]. Unlike other antigenic features of pathogens, such as flagellin or LPS, mRNAs are common to both host and pathogens and thus it requires the cellular machinery to discriminate the non-self mRNA from the self. Exogenously introduced mRNAs are inherently immunostimulatory [45]. Therefore, the principle of distinguishing the non-self mRNA from the self is based on the structural distinction, subcellular localization, and availability of the mRNA. For example, dsRNA, 3'-triphosphate RNA, partially degraded or damaged RNAs, and A to I editing level represent the structural signatures for the non-self mRNA and these features activate the innate immune response via PRRs [99].
RNA impurities during in vitro transcription (IVT) of mRNA vaccines potentially trigger the innate immune response, primarily by activating pro-inflammatory genes and type I IFNs (Figure 3). Mainly, dsRNA and DNA-RNA molecules, generated as the by-products of IVT reactions, differentially interact with specific members of PRRs and induce PRR-associated immune responses. Endosome-mediated sensing of long and short dsRNA and ssRNA by TLR family and cytosolic dsRNA sensing via retinoic acid-inducible gene I (RIG-I) are the two pathways most characterized thus far.
Mechanism of innate immune response to external mRNA. IVT-synthesized mRNA vaccines are recognized by PRRs including the endosomal TLR3, -7, and -8, and cytoplasmic innate immune receptors, RIG-I and MDA5. dsRNA, produced by inaccurate T7 polymerase activity, is recognized by TLR8 and RIG-I to induce the expression of proinflammatory cytokines and promote RNA degradation and translation inhibition mediated by 2′-5′-oligoadenylate synthase/RNase L and PKR-dependent phosphorylation of eIF2α.
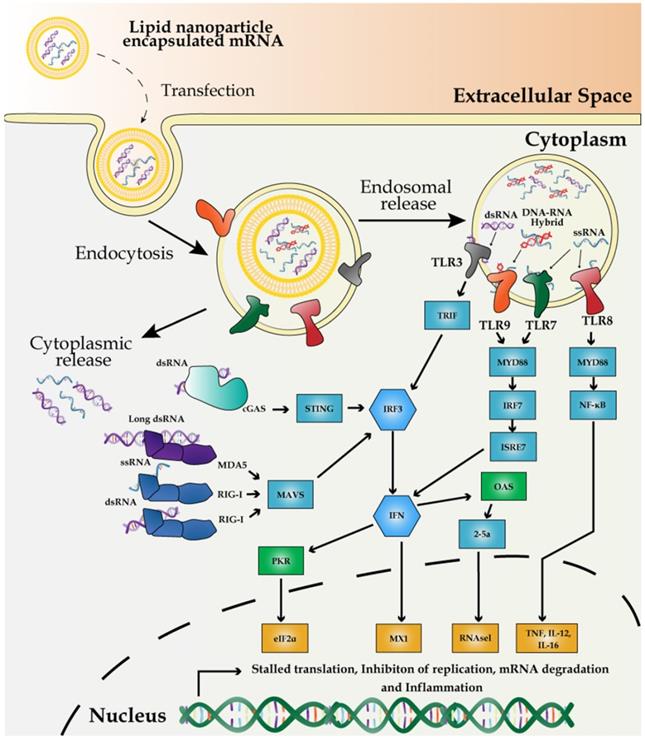
First, the primary source of the potentially immune-stimulatory molecule is dsRNA, derived from IVT reaction, in which T7 polymerase transcribes the antisense RNA from the promoter-less DNA template, forming dsRNA via base-pairing with the sense strand [100]. dsRNA is recognized by TLR3 in endosomes [101]. Alternatively, dsRNA induces antiviral response via another pathway of cGMP-AMP synthase-simulator of interferon genes (cGAS-STING) [102]. Second, DNA-RNA hybrid molecules generated during IVT trigger TLR9-mediated sensing of PAMPs and subsequent activation of pro-inflammatory cytokines and type I IFNs in dendritic cells [103]. Finally, viral or exogenously introduced single-stranded mRNA (ssRNA) molecules are themselves a PAMP after delivery to host cells, which can also trigger type I IFN production via the endosomal sensors TLR7 and -8 [104, 105].
Besides, partially degraded dsRNA in varying sizes can be differentially recognized as dsRNA PAMPs by two cytosolic sensors: melanoma differentiation-associated gene 5 (MDA5) and RIG-I [106]. For reducing the potential innate immune response, post-IVT purification has been widely implemented via high-performance liquid chromatography (HPLC) which prevents the activation of type I IFN production [107] and fast protein liquid chromatography (FPLC) which enhances the protein production up to 1,000-fold in primary human dendritic cells [107].
Independent of the innate cellular immune response against the viral and exogenous mRNA [108], dsRNA also triggers the activation of dsRNA-dependent protein kinase PKR, which, in turn, phosphorylates eIF2α, reducing the protein synthesis [109]. Thus, the contamination of dsRNA can both trigger type I IFN activation and shut down the protein synthesis. Furthermore, dsRNA activates IFN-induced expression of 2′-5′-oligoadenylate synthetase/RNase L, which promotes RNA degradation [24]. In addition, impurities in an mRNA vaccine can also trigger immune response via TLR3 [110].
Appropriate purification of IVT-synthesized mRNA is critical to avoid the cellular immune response against the exogenous mRNA and maximize the protein yield. Moreover, the incorporation of chemically modified nucleosides such as pseudouridine and 1-methylpseudouridine allows mRNA molecules to escape the recognition by TLR7 and -8 as well as other innate immune sensors [62, 111]. Surprisingly, pseudouridine in mRNA molecules enhances the translation efficiency from ssRNA by reducing the PKR activity [112]. Moreover, pseudouridine-modified mRNA can be translated in primary dendritic cells and even in mice by evading innate immune surveillance and increasing the protein yield [62].
Preclinical and clinical results of COVID-19 mRNA vaccines
Several experimental approaches are often considered to determine the efficacy of any vaccine. Induction of immune response, concentration of antigen-binding IgG, and antigen-neutralizing titres were determined in many preclinical studies for the seven mRNA vaccines. As summarized in Table 4, several mRNA vaccines with their experimental data publicized have demonstrated strong immunogenic activity by inducing both CD4+ and CD8+ T cells. Besides, the dose-dependent geometric mean of the antigen-neutralizing titre was observed on various animal models.
Preclinical studies on immune response to the seven mRNA candidate vaccines
| Vaccine name | Immune reaction | Immune cells | 50% inhibitory dilution (ID50) Geometric mean titer | Model | ||
|---|---|---|---|---|---|---|
| Reference | ||||||
| 1st | 2nd | |||||
| mRNA -1273 | Humoral response | Th1 CD4+ T- cells | 10 μg: 63 100 μg: 305 | 10 μg: 103100 μg: 1862 | Rhesus Macaque | [121] |
| BNT162 (3 LNP-mRNAs) | Humoral and cellular responses | CD4+ T-cells, | Mice:0.2 ug: 26 1ug: 176 5ug: 296 | Rhesus macaque:30μg: 962 100μg: 1689 | Mice, Rhesus macaque | [129] |
| CD8+ T-cells | Rhesus macaque:30μg: 65 100μg: 81 | |||||
| CVnCoV | Humoral and cellular responses | CD4+ T-cells, CD8+ T-cells | N/A | N/A | Rhesus macaque | [124] |
| LUNAR-COV19 | Humoral response | Th1 CD4+ T-cells, CD8+ T-cells | 0.2 μg : 57.72 2 μg: 217.9 10 μg: 320 | N/A | Mice | [54] |
| LNP-nCoVsaRNA | Cellular response | Th1 CD4+ T | N/A | N/A | Mice, Rat | [130] |
| ARCoV | N/A | CD4+ T cells, CD8+ T cells | NT50 2ug:278 30ug:559 | NT50 2ug: 2540 30ug: 7079 | Mice | [86] |
| ChulaCov19 mRNA | N/A | N/A | N/A | N/A | Mice, Monkey | [131] |
Preliminary results from phase 3 clinical trials have shown that the efficacy of the two mRNA vaccines, mRNA-1273 and BNT162b, reached 95% and 94.1%, respectively, comparably higher than that of another licensed vaccine, ChAdOx1 developed using a chimpanzee adenovirus (Oxford-AstraZeneca) which was 70% based on interim results of a phase 3 clinical trial [4, 5, 41]. Both mRNA vaccines were equally effective at all age groups tested for them (Table 5). Among 30,420 volunteers, 15,210 each were assigned in the placebo and vaccine groups in the observer-blinded clinical trial of mRNA-1273. 84.2% (vs. 19.8% in the placebo group) and 88.6% (vs. 18.8% in the placebo group) in the vaccine group reported adverse effects after the first and second doses, respectively. These adverse effects include pain, erythema, swelling, and lymphadenopathy on the injection sites [4]. 54.9% and 79.4% of all the participants after the first and second doses, respectively, also reported mild to moderate systemic adverse effects such as fever, headache, fatigue, myalgia, nausea, and chills [4].
Efficacy comparison of approved mRNA vaccines*
| mRNA-1273 Vaccine efficacy % (95% confidence interval) | BNT162b Vaccine efficacy % (95% confidence interval) | |
|---|---|---|
| Overall | 94.1 (89.3-96.8) | 95.0 (90.0-97.9) |
| Age group | ||
| 16 to 55 years | 95.6 (89.4-98.6) | |
| ≥18 to <65 years | 95.6 (90.6-97.9) | |
| > 55 years | 93.7 (80.6-98.8) | |
| ≥65 years | 86.4 (61.4-95.2) | 94.7 (66.7-99.9) |
| ≥75 years | 100.0 (-13.1-100.0) | |
| Sex | ||
| Male | 95.4 (87.4-98.3) | 96.4 (88.9-99.3) |
| Female | 93.1 (85.2-96.8) | 93.7 (84.7-98.0) |
As for BNT162b, 43,448 participants were recruited to a placebo-controlled phase 3 clinical trial with 21,728 and 21,720 assigned to the placebo and vaccine groups, respectively. 83% (vs. 14% in the placebo group) and 78% (vs. 12% in the placebo group) of the 16-55 years-old vaccine groups and 71% (vs. 9% in the placebo group) and 66% (vs. 8% in the placebo group) of the 55+ years-old vaccine groups reported mild to moderate local injection-site pain after the first and second doses, respectively. Common local adverse effects include redness and swelling and the systemic adverse effects were mild to moderate including fever, fatigue, headache, chills, vomiting, diarrhea, muscle pain, and join pain [5]. Between December 14 and 23, 2020, 1,893,360 doses of BTN162b were administered, of which 1,177,527 doses were for females, 648,327 doses for males, and 67,506 doses missing sex information). 4,393 (0.2%) people reported adverse effects after administration of the first dose of the vaccine, and the adverse effects developed within 30 minutes after the vaccination in 75% of the cases [113]. Among 21 people who reported anaphylaxis, 19 (90%) were female and 18 (86%) had allergy history.
Although continuous study of the long-term protection against SARS-CoV-2 and adverse effects remain to be carried out, the efficacy and safety of the two mRNA vaccines appear to be higher than the vaccines against other infectious diseases. According to a meta-analysis of 31 studies associated with the assessed efficacy of the licensed influenza virus, trivalent inactivated vaccine produced 59% efficacy in adults aged 18-65 years [114]. Overall, the efficacy of mRNA-1273 and BTN162b was considerably higher than the influenza vaccine against the seasonal influenza.
Conclusion and perspectives
The COVID-19 pandemic caused by the novel virus SARS-CoV-2 has mobilized a historically great number of scientists, clinicians, and government officials to work together in developing vaccines to cope with the health crisis all over the world. It is remarkable to witness that two mRNA vaccines, mRNA-1273 and BNT162b were developed and manufactured in less than one year. Without the technical advancements in RNA synthesis in a GMP-grade and large-scale manner, nanoparticle formulations, and the “smart” design of RNA vaccines, scientists would not have achieved the feat in producing highly effective and safe RNA vaccines. With the rapidly evolving SARS-CoV-2 and the ever-growing emergence of novel pathogens worldwide, the RNA vaccine platforms will be more widely applicable than ever. A recent screen of the monoclonal antibodies in the blood plasma isolated from mRNA-1273-vaccinated people showed slight to moderate decrease in neutralizing effects against the E484K, N501Y, and the K417N-E484K-N501Y combination variants. However, the rapid design and large-scale production features of mRNA vaccines can reduce the concern that an mRNA vaccine may lose the efficacy against novel SARS-CoV-2 variants.
New and improved methodologies will continue to be explored to optimize the stability and translation efficiency of mRNA and the delivery of LNP-mRNA complexes. Novel approaches, including deep learning and genome-wide screening method to identify the optimal codon usage and UTR design of mRNA are already being tested empirically [72, 115]. Recent studies have screened a library of the total mRNA containing 5'-UTR using computational and empirical analyses and determined the optimal 5'-UTR for the maximum RNA stability and translation efficiency in vitro and in vivo [116, 117]. Other considerations in codon design, including the GC content, repetitive sequences, secondary structure, and the incorporation of the immunologically less reactive nucleoside analogs, will improve the translation efficiency of mRNA vaccines. It is predictable that saRNA will serve as a more powerful platform than non-replicating mRNA for developing future mRNA vaccines as a smaller dose can produce a sufficient level of mRNA and protein via the self-replicating mechanism. Moreover, direct delivery of mRNA vaccines into antigen-presenting cells such as dendritic cells potentially improves the overall immune response [88].
Regardless of the platform type of the vaccines, there has been a concern about their antibody-dependent enhancement (ADEs). Vaccination often leads to the production of non-effective neutralizing antibodies in the host, which can exacerbate the pathological symptoms by triggering the harmful immunological cascades to facilitate the viral entry and produce excess amounts of cytokines and complements [118]. It is difficult to predict ADE of any vaccine based on in vitro antibody-dependent effects or pre-clinical animal studies, due to the incompatibility between human IgG and its counterpart animal receptors [119, 120]. Moreover, partially degraded mRNA could be transcribed to truncated proteins and proteins with conformational changes, which then induce the production of neutralizing antibodies that won't bind the native immunogen and can also lead to ADE. Thus, there is a continuous demand to identify and overcome ADE. Nevertheless, after more than a year of massive disturbance and destruction of human lives, mRNA vaccines, together with the other platforms of vaccines, have finally provided a great hope to save mankind from the unprecedented pandemic.
Abbreviations
COVID-19: Coronavirus disease 2019
SARS-CoV: Severe acute respiratory syndrome coronavirus
MERS: Middle east respiratory syndrome
SARS-CoV-2: Severe acute respiratory syndrome coronavirus 2
ACE2: Angiotensin-converting enzyme 2
IVT: In vitro transcription
siRNA: Small interference RNA
saRNA: Self-amplifying RNA
m7G: 7-methylguanosine
eIF4F: Elongation initiation factor 4F
UTR: Untranslated region
TLR: Toll-like receptors
PAMP: Pathogen-associated molecular patterns
RIG-I: Retinoic acid-inducible gene I
RBD: Receptor binding domain
VEEV: Venezuelan equine encephalitis virus
LNP: Lipid nanoparticle
IM: Intramuscular
cGAS-STING: cGMP-AMP synthase-simulator of interferon genes
IFN: Interferon
ADE: Antibody-dependent enhancement
VVr: Viral vector replicating
VVnr: Viral vector non-replicating
Acknowledgements
This work was supported by University of Macau Research Committee MYRG #2017‐00124‐FHS and Chair Professor Grant #CPG2021-00031-FHS, and Macau Science and Technology Development Fund grants #095/2017/A2 and 095/2017/A1 to R.X. and MYRG #2017‐00249‐FHS to J.P. The authors thank the council members of the Macau Society for Stem Cell Research for inspiring discussion.
Author Contributions
J.P. and R.X. conceived and designed the manuscript. J.P, P.L., Y.L., and R.X wrote the manuscript. R.X. gave the final approval of the manuscript.
Competing Interests
R.X. is a founder of ImStem Biotechnology, Inc., a stem cell company. The other authors declare no competing financial interests.
References
1. Yi Y, Lagniton PNP, Ye S, Li EQ, Xu RH. COVID-19: what has been learned and to be learned about the novel coronavirus disease. Int J Biol Sci. 2020;16:1753-66
2. Wu YT, Ho WZ, Huang YW, Jin DY, Li SY, Liu SL. et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet. 2020;395:949-50
3. Hodgson J. The pandemic pipeline. Nat Biotechnol. 2020;38:523-32
4. Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R. et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2020
5. Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020
6. Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2020
7. Bhatt PR, Scaiola A, Loughran G, Leibundgut M, Kratzel A, McMillan A. et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. bioRxiv. 2020. 2020 10.26.355099
8. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914-21 e10
9. Li WH, Moore MJ, Vasilieva N, Sui JH, Wong SK, Berne MA. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450-4
10. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. bioRxiv. 2020. 2020 01.26.919985
11. Roca-Ho H, Riera M, Palau V, Pascual J, Soler MJ. Characterization of ACE and ACE2 Expression within Different Organs of the NOD Mouse. Int J Mol Sci. 2017 18
12. Robson F, Khan KS, Le TK, Paris C, Demirbag S, Barfuss P. et al. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol Cell. 2020;80:1136-8
13. Callaway E. The coronavirus is mutating - does it matter? Nature. 2020;585:174-7
14. Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S. et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell. 2020;182:1284-94 e9
15. Ozono S, Zhang Y, Ode H, Sano K, Tan TS, Imai K. et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat Commun. 2021;12:848
16. Santos JC, Passos GA. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. bioRxiv. 2021: 2020.12.29.424708
17. Lin JT, Zhang JS, Su N, Xu JG, Wang N, Chen JT. et al. Safety and immunogenicity from a phase I trial of inactivated severe acute respiratory syndrome coronavirus vaccine. Antivir Ther. 2007;12:1107-13
18. Martin JE, Louder MK, Holman LA, Gordon IJ, Enama ME, Larkin BD. et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a Phase I clinical trial. Vaccine. 2008;26:6338-43
19. Folegatti PM, Bittaye M, Flaxman A. Safety and immunogenicity of a candidate Middle East respiratory syndrome coronavirus viral-vectored vaccine: a dose-escalation, open-label, non-randomised, uncontrolled, phase 1 trial (vol 20, pg 816, 2020). Lancet Infect Dis. 2020;20:148 -
20. Koch T, Dahlke C, Fathi A, Kupke A, Krahling V, Okba NMA. et al. Safety and immunogenicity of a modified vaccinia virus Ankara vector vaccine candidate for Middle East respiratory syndrome: an open -label, phase 1 trial. Lancet Infect Dis. 2020;20:827-38
21. Alharbi NK, Padron-Regalado E, Thompson CP, Kupke A, Wells D, Sloan MA. et al. ChAdOx1 and MVA based vaccine candidates against MERS-CoV elicit neutralising antibodies and cellular immune responses in mice. Vaccine. 2017;35:3780-8
22. List of candidate vaccines developed against MERS-CoV. Available from: https://www.who.int/blueprint/priority-diseases/key-action/list-of-candidate-vaccines-developed-against-mers.pdf
23. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nature Reviews Drug Discovery. 2010;9:537-50
24. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics-developing a new class of drugs. Nat Rev Drug Discov. 2014;13:759-80
25. Serganov A, Nudler E. A Decade of Riboswitches. Cell. 2013;152:17-24
26. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16:543-52
27. Malone RW, Felgner PL, Verma IM. Cationic Liposome-Mediated Rna Transfection. P Natl Acad Sci USA. 1989;86:6077-81
28. Oh S, Kessler JA. Design, Assembly, Production, and Transfection of Synthetic Modified mRNA. Methods. 2018;133:29-43
29. Jirikowski GF, Sanna PP, Maciejewskilenoir D, Bloom FE. Reversal of Diabetes-Insipidus in Brattleboro Rats - Intrahypothalamic Injection of Vasopressin Messenger-Rna. Science. 1992;255:996-8
30. Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A. et al. Direct Gene-Transfer into Mouse Muscle Invivo. Science. 1990;247:1465-8
31. Martinon F, Krishnan S, Lenzen G, Magne R, Gomard E, Guillet JG. et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993;23:1719-22
32. Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. European Journal of Immunology. 2000;30:1-7
33. Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J. et al. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol Ther. 2018;26:45-55
34. Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P. et al. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther. 2013;21:358-67
35. Wadhwa A, Aljabbari A, Lokras A, Foged C, Thakur A. Opportunities and Challenges in the Delivery of mRNA-based Vaccines. Pharmaceutics. 2020 12
36. Jackson NA, Kester KE, Casimiro D, Gurunathan S, DeRosa F. The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines. 2020;5:1-6
37. Crommelin DJA, Anchordoquy TJ, Volkin DB, Jiskoot W, Mastrobattista E. Addressing the Cold Reality of mRNA Vaccine Stability. J Pharm Sci. 2020
38. Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. 2016;7:319-34
39. Perri S, Greer CE, Thudium K, Doe B, Legg H, Liu H. et al. An alphavirus replicon particle chimera derived from Venezuelan equine encephalitis and Sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77:10394-403
40. Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2020
41. Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK. et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397:99-111
42. Houseley J, Tollervey D. The Many Pathways of RNA Degradation. Cell. 2009;136:763-76
43. Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. 2006;16:141-7
44. Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA Vaccines for Infectious Diseases. Front Immunol. 2019;10:594
45. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-79
46. Gao M, Fritz DT, Ford LP, Wilusz J. Interaction between a poly(A)-specific ribonuclease and the 5' cap influences mRNA deadenylation rates in vitro. Mol Cell. 2000;5:479-88
47. Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657-68
48. Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991;5:2108-16
49. Furuichi Y, LaFiandra A, Shatkin AJ. 5'-Terminal structure and mRNA stability. Nature. 1977;266:235-9
50. Schuberth-Wagner C, Ludwig J, Bruder AK, Herzner AM, Zillinger T, Goldeck M. et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N1-2'O-Methylated Self RNA. Immunity. 2015;43:41-51
51. Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG. et al. Structural basis for m7G recognition and 2'-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci U S A. 2016;113:596-601
52. Galloway A, Cowling VH. mRNA cap regulation in mammalian cell function and fate. Biochim Biophys Acta Gene Regul Mech. 2019;1862:270-9
53. Hyde JL, Diamond MS. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology. 2015;479-480:66-74
54. de Alwis R, Gan ES, Chen S, Leong YS, Tan HC, Zhang SL. et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv. 2020. 2020 09.03.280446
55. McKay PF, Hu K, Blakney AK, Samnuan K, Brown JC, Penn R. et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat Commun. 2020;11:3523
56. Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. 2020;586:567-71
57. Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature. 2020;586:594-9
58. Leppek K, Das R, Barna M. Functional 5 ' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Bio. 2018;19:158-74
59. Tanguay RL, Gallie DR. Translational efficiency is regulated by the length of the 3' untranslated region. Mol Cell Biol. 1996;16:146-56
60. Park JE, Yi H, Kim Y, Chang H, Kim VN. Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol Cell. 2016;62:462-71
61. Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165-75
62. Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S. et al. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Molecular Therapy. 2008;16:1833-40
63. Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Kariko K. Nucleoside modifications in RNA limit activation of 2'-5'-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011;39:9329-38
64. Sorgi FL, Bhattacharya S, Huang L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997;4:961-8
65. Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells. Plos Biol. 2006;4:e180
66. Hanson G, Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol. 2018;19:20-30
67. Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N. et al. Codon Optimality Is a Major Determinant of mRNA Stability. Cell. 2015;160:1111-24
68. Radhakrishnan A, Chen YH, Martin S, Alhusaini N, Green R, Coller J. The DEAD-Box Protein Dhh1p Couples mRNA Decay and Translation by Monitoring Codon Optimality. Cell. 2016;167:122-32 e9
69. Quax TEF, Claassens NJ, Soll D, van der Oost J. Codon Bias as a Means to Fine-Tune Gene Expression. Molecular Cell. 2015;59:149-61
70. Sharp PM, Li WH. The Codon Adaptation Index - a Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications. Nucleic Acids Research. 1987;15:1281-95
71. Wayment-Steele HK, Kim DS, Choe CA, Nicol JJ, Wellington-Oguri R, Sperberg RAP. et al. Theoretical basis for stabilizing messenger RNA through secondary structure design. bioRxiv. 2020. 2020 08.22.262931
72. Fu H, Liang Y, Zhong X, Pan Z, Huang L, Zhang H. et al. Codon optimization with deep learning to enhance protein expression. Sci Rep. 2020;10:17617
73. Hsieh CL, Goldsmith JA, Schaub JM, DiVenere AM, Kuo HC, Javanmardi K. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science. 2020;369:1501-5
74. Walsh EE, Frenck R, Falsey AR, Kitchin N, Absalon J, Gurtman A. et al. RNA-Based COVID-19 Vaccine BNT162b2 Selected for a Pivotal Efficacy Study. medRxiv. 2020
75. Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S. et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3' UTRs Identified by Cellular Library Screening. Mol Ther. 2019;27:824-36
76. Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA. et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nature nanotechnology. 2020;15:646-55
77. Felgner JH, Kumar R, Sridhar CN, Wheeler CJ, Tsai YJ, Border R. et al. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J Biol Chem. 1994;269:2550-61
78. Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther. 2017;25:1467-75
79. Lin Q, Chen J, Zhang Z, Zheng G. Lipid-based nanoparticles in the systemic delivery of siRNA. Nanomedicine (Lond). 2014;9:105-20
80. FACT SHEET FOR HEALTHCARE PROVIDERS ADMINISTERING VACCINE (VACCINATION PROVIDERS) EMERGENCY USE AUTHORIZATION (EUA) OF THE MODERNA COVID-19 VACCINE TO PREVENT CORONAVIRUS DISEASE 2019 (COVID-19). Available from: https://www.modernatx.com/covid19vaccine-eua/
81. Study to Describe the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals
82. Vogel AB, Kanevsky I, Che Y, Swanson KA, Muik A, Vormehr M. et al. Immunogenic BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature. 2021
83. Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W. et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 2013;21:1570-8
84. Activities regarding CVnCoV, CureVac's mRNA-based vaccine candidate against COVID-19
85. A Study to Determine the Safety and Efficacy of SARS-CoV-2 mRNA Vaccine CVnCoV in Adults for COVID-19. Available from: https://clinicaltrials.gov/ct2/show/NCT04652102
86. Zhang NN, Li XF, Deng YQ, Zhao H, Huang YJ, Yang G. et al. A Thermostable mRNA Vaccine against COVID-19. Cell. 2020;182:1271-83 e16
87. Arcturus Therapeutics Announces Positive Interim ARCT-021 (LUNAR-COV19) Phase 1/2 Study Results for Both Single Shot and Prime-boost Regimens, and Up to 0 Million in Additional Financial Commitments from Singapore
88. Zeng C, Zhang C, Walker PG, Dong Y. Formulation and Delivery Technologies for mRNA Vaccines. Curr Top Microbiol Immunol. 2020
89. Palm AE, Henry C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front Immunol. 2019;10:1787
90. Yoshida T, Mei H, Dorner T, Hiepe F, Radbruch A, Fillatreau S. et al. Memory B and memory plasma cells. Immunol Rev. 2010;237:117-39
91. Weisel F, Shlomchik M. Memory B Cells of Mice and Humans. Annu Rev Immunol. 2017;35:255-84
92. Neymotin B, Athanasiadou R, Gresham D. Determination of in vivo RNA kinetics using RATE-seq. Rna. 2014;20:1645-52
93. Chan LY, Mugler CF, Heinrich S, Vallotton P, Weis K. Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. Elife. 2018 7
94. Zinder JC, Lima CD. Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 2017;31:88-100
95. Fischer PM. Cap in hand: targeting eIF4E. Cell Cycle. 2009;8:2535-41
96. Culjkovic-Kraljacic B, Skrabanek L, Revuelta MV, Gasiorek J, Cowling VH, Cerchietti L. et al. The eukaryotic translation initiation factor eIF4E elevates steady-state m(7)G capping of coding and noncoding transcripts. Proc Natl Acad Sci U S A. 2020;117:26773-83
97. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86:2900-10
98. Ziegler A, Soldner C, Lienenklaus S, Spanier J, Trittel S, Riese P. et al. A New RNA-Based Adjuvant Enhances Virus-Specific Vaccine Responses by Locally Triggering TLR- and RLH-Dependent Effects. J Immunol. 2017;198:1595-605
99. Bartok E, Hartmann G. Immune Sensing Mechanisms that Discriminate Self from Altered Self and Foreign Nucleic Acids. Immunity. 2020;53:54-77
100. Mu X, Greenwald E, Ahmad S, Hur S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018;46:5239-49
101. Tatematsu M, Funami K, Seya T, Matsumoto M. Extracellular RNA Sensing by Pattern Recognition Receptors. J Innate Immun. 2018;10:398-406
102. Ni GX, Ma Z, Damania B. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. Plos Pathog. 2018 14
103. Rigby RE, Webb LM, Mackenzie KJ, Li Y, Leitch A, Reijns MA. et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 2014;33:542-58
104. Sioud M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J Mol Biol. 2005;348:1079-90
105. Gantier MP, Tong S, Behlke MA, Xu D, Phipps S, Foster PS. et al. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J Immunol. 2008;180:2117-24
106. Brisse M, Ly H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front Immunol. 2019;10:1586
107. Kariko K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011;39:e142
108. Linares-Fernandez S, Lacroix C, Exposito JY, Verrier B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol Med. 2020;26:311-23
109. Vattem KM, Staschke KA, Wek RC. Mechanism of activation of the double-stranded-RNA-dependent protein kinase, PKR: role of dimerization and cellular localization in the stimulation of PKR phosphorylation of eukaryotic initiation factor-2 (eIF2). Eur J Biochem. 2001;268:3674-84
110. Xu S, Yang K, Li R, Zhang L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int J Mol Sci. 2020 21
111. Kariko K, Buckstein M, Ni HP, Weissman D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165-75
112. Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D. et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Research. 2010;38:5884-92
113. Allergic Reactions Including Anaphylaxis After Receipt of the First Dose of Pfizer-BioNTech COVID-19 Vaccine — United States, December 14-23, 2020. Morbidity and Mortality weekly report; 2021
114. Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12:36-44
115. Cao J, Novoa EM, Zhang Z, Chen WCW, Liu D, Choi GCG. et al. High-Throughput 5' UTR Engineering for Enhanced Protein Production in Non-Viral Gene Therapies. bioRxiv. 2020. 2020 03.24.006486
116. Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR. et al. Human 5 ' UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol. 2019;37:803 -+
117. Suknuntha K, Tao LH, Brok-Volchanskaya V, D'Souza SS, Kumar A, Slukvin I. Optimization of Synthetic mRNA for Highly Efficient Translation and its Application in the Generation of Endothelial and Hematopoietic Cells from Human and Primate Pluripotent Stem Cells. Stem Cell Rev Rep. 2018;14:525-34
118. Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C. et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. 2020;584:353-63
119. Crowley AR, Ackerman ME. Mind the Gap: How Interspecies Variability in IgG and Its Receptors May Complicate Comparisons of Human and Non-human Primate Effector Function. Frontiers in Immunology. 2019 10
120. Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J. et al. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs. 2017;9:767-73
121. Corbett KS, Flynn B, Foulds KE, Francica JR, Boyoglu-Barnum S, Werner AP. et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N Engl J Med. 2020;383:1544-55
122. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN. et al. An mRNA Vaccine against SARS-CoV-2-Preliminary Report. New Engl J Med. 2020;383:1920-31
123. Vogel AB, Kanevsky I, Che Y, Swanson KA, Muik A, Vormehr M. et al. BNT162b vaccines are immunogenic and protect non-human primates against SARS-CoV-2. bioRxiv. 2020. 2020 12.11.421008
124. Rauch S, Gooch K, Hall Y, Salguero FJ, Dennis MJ, Gleeson FV. et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv. 2020. 2020 12.23.424138
125. Anderson EJ, Rouphael NG, Widge AT, Jackson LA, Roberts PC, Makhene M. et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N Engl J Med. 2020;383:2427-38
126. Widge AT, Rouphael NG, Jackson LA, Anderson EJ, Roberts PC, Makhene M. et al. Durability of Responses after SARS-CoV-2 mRNA-1273 Vaccination. N Engl J Med. 2021;384:80-2
127. Mulligan MJ, Lyke KE, Kitchin N, Absalon J, Gurtman A, Lockhart S. et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature. 2020;586:589-93
128. Walsh EE, Frenck RW Jr, Falsey AR, Kitchin N, Absalon J, Gurtman A. et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N Engl J Med. 2020;383:2439-50
129. Vogel AB, Kanevsky I, Che Y, Swanson KA, Muik A, Vormehr M. et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. bioRxiv. 2020. 2020 09.08.280818
130. Nishikawa T, Chang CY, Tai JA, Hayashi H, Sun J, Torii S. et al. Anti-CoVid19 plasmid DNA vaccine induces a potent immune response in rodents by Pyro-drive Jet Injector intradermal inoculation. bioRxiv. 2021. 2021 01.13.426436
131. ChulaCov19 mRNA vaccine. Available from: https://go.drugbank.com/drugs/DB16445
132. Jackson NAC, Kester KE, Casimiro D, Gurunathan S, DeRosa F. The promise of mRNA vaccines: a biotech and industrial perspective. NPJ Vaccines. 2020;5:11
Author contact
![]() Corresponding author: Ren-He Xu, Faculty of Health Sciences, University of Macau, Taipa, Macau, China. Tel: 853-8822-4993; Fax: 853-8822-8345; Email address: renhexuedu.mo
Corresponding author: Ren-He Xu, Faculty of Health Sciences, University of Macau, Taipa, Macau, China. Tel: 853-8822-4993; Fax: 853-8822-8345; Email address: renhexuedu.mo