Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Epidemiology of AKI in COVID-19...
Inflammation may be a mechanism...
Therapeutic potential for...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(6):1497-1506. doi:10.7150/ijbs.58791 This issue Cite
Review
Inflammatory stress in SARS-COV-2 associated Acute Kidney Injury
Junzhe Chen1,2, Wenbiao Wang1,3, Ying Tang2, Xiao-ru Huang1,4, Xueqing Yu4, Hui-Yao Lan1,5 ![]()
1. Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, and Lui Che Woo Institute of Innovative Medicine, The Chinese University of Hong Kong, Hong Kong, China.
2. Department of Nephrology, The Third Affiliated hospital, Southern Medical university, Guangzhou, China.
3. Guangdong Key Laboratory of Virology, Institute of Medical Microbiology, Jinan University, Guangzhou, China.
4. Guangdong-Hong Kong Joint Laboratory for Immunity and Genetics of Chronic Kidney Disease, Guangdong Academy of Medical Science, Guangdong Provincial People's Hospital, Guangzhou, China.
5. Guangdong-Hong Kong Joint Laboratory for Immunity and Genetics of Chronic Kidney Disease, The Chinese University of Hong Kong, Hong Kong, China.
Received 2021-1-28; Accepted 2021-3-1; Published 2021-4-10
Abstract

Increasing clinical evidence shows that acute kidney injury (AKI) is a common and severe complication in critically ill COVID-19 patients. The older age, the severity of COVID-19 infection, the ethnicity, and the history of smoking, diabetes, hypertension, and cardiovascular disease are the risk factor for AKI in COVID-19 patients. Of them, inflammation may be a key player in the pathogenesis of AKI in patients with COVID-19. It is highly possible that SARS-COV-2 infection may trigger the activation of multiple inflammatory pathways including angiotensin II, cytokine storm such as interleukin-6 (IL-6), C-reactive protein (CRP), TGF-β signaling, complement activation, and lung-kidney crosstalk to cause AKI. Thus, treatments by targeting these inflammatory molecules and pathways with a monoclonal antibody against IL-6 (Tocilizumab), C3 inhibitor AMY-101, anti-C5 antibody, anti-TGF-β OT-101, and the use of CRRT in critically ill patients may represent as novel and specific therapies for AKI in COVID-19 patients.
Keywords: COVID-19, AKI, cytokines, inflammation, mechanisms
Introduction
COVID-19 is a progressive viral pneumonia with a broad spectrum of clinical manifestations, ranging from asymptomatic to mild (80%), severe (10-15%) or critical and death (2-5%) [1, 2]. Among critically ill COVID-19 patients, acute respiratory distress syndrome (ARDS) and multiorgan failure including acute kidney injury (AKI) are the most common co-morbidities [3-5]. In this review article, we are focusing on SARS-CoV-2-associated AKI. The possible mechanisms and pathways related to SARS-CoV-2-associated AKI are discussed.
Epidemiology of AKI in COVID-19 patients
Increasing evidence shows that there is high prevalence of AKI in COVID-19 patients [6, 7]. The manifestations of AKI are diverse, from proteinuria, hematuria, elevated serum creatinine (Scr) or blood urea nitrogen (BUN) levels to acute renal failure. A meta-analysis shows that more than half (57%) of COVID-19 patients develop proteinuria, accompanied by elevated serum levels of Scr (9.6%-15.5%) and BUN (13.7-14.1%) [5, 6]. The CT scan also shows renal inflammation and edema [8]. Pathologically, diffuse proximal tubule injury with loss of brush border and frank necrosis is found in COVID-19 patients with AKI [9, 10].
Compared to patients with Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) in which the incidence of AKI is 6.7% and 42% respectively [11, 12]. The incidence of AKI in COVID-19 patients is highly variable. In the early reports from China, COVID-19 patients with AKI was rare [13, 14], but increased to 10% in a later study [15], and became more severe with the incident rate of 25%-29% in those admitted to ICU [16, 17]. The large cohort studies in the western countries revealed that the incidence of AKI was 27%-37% [18, 19] and became more severe (68%) in critically ill COVID-19 patients who were admitted to ICU in the New York city [20]. Nevertheless, it is now clear that the incidence of AKI in COVID-19 patients is associated with the age, smoking, the cytokine storm, the severity of disease, the ethnicity, and the history of diabetes, hypertension, and cardiovascular disease [7]. Thus, AKI is an independent risk factor for the poor long-term renal outcome and mortality in critically ill COVID-19 patients [21, 22]. During the follow-up study, AKI is a major cause of in-hospital mortality. In addition, the complete kidney recovery rate of AKI in COVID-19 infection is only about 30-45% based on the recent reports [15, 20, 23]. Thus, AKI is one of severe complications and mortality of in-hospital COVID-19 patients, however, mechanisms of COVID-19-associated AKI remain largely unclear and need further studies.
Inflammation may be a mechanism of AKI in COVID-19 patients
Multiple factors such as direct virus infection, cytokine storm, hypoxia, sepsis shock, hemodynamic instability and rhabdomyolysis, hypertension, and diabetes may be associated with AKI in COVID-19 patients. Of these factors, inflammation stress may be a mechanism of AKI in COVID-19 patients, which is discussed below.
Angiotensin II (Ang II) and hypertensive stress
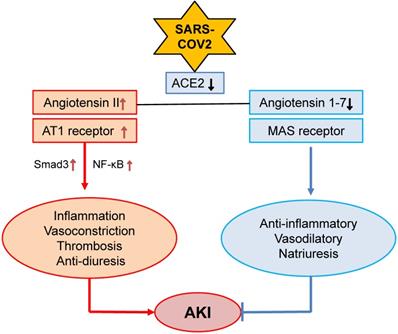
Kidney is a target organ of SARS-COV-2 virus infection due to the high expression levels of angiotensin-converting enzyme 2 (ACE2), a receptor for SARS-COV-2 virus [24], in the kidney tissues, particularly in renal tubular epithelial cells (TECs) [25-27]. Thus, SARS-COV-2 may be able to directly bind to ACE2 and infect kidney cells, which is supported by high levels of SARS-COV-2 spike (S) and nucleoprotein (N protein) in COVID-19 patients with AKI [9, 10, 28]. In the kidney, renin-angiotensin-aldosterone system (RAAS) maintains renal hemodynamic and regulates renal sodium transport in both normal physiological states and pathological conditions. Ang II and Ang 1-7 are the two major effectors of RAAS and are tightly controlled by two major enzymes of ACE and ACE2 [29]. Ang II acts via its receptor-1 (AT1) to mediate renal inflammation and fibrosis by activating NF-kB and Smad signaling crosstalk pathways, whereas Ang 1-7 binds receptor Mas to counter-regulate these pathological effects of Ang II [29]. The primary function of ACE2 is to covert Ang II to Ang 1-7 to exert its anti-inflammatory, vasodilatory and natriuretic properties [30] (Figure 1). After binding to ACE2, SARS-COV-2 significantly downregulates ACE2 expression [31, 32], resulting in a inhibition or loss of Ang 1-7 while enhancing Ang II-AT1-dependent renal inflammation, vasoconstriction, thrombosis and anti-diuresis effects [33] (Figure 1). It has been well documented that Ang II is a key mediator of AKI [34-36], whereas, the ACE2-Ang-1-7-Mas axis is renoprotective [37]. Thus, SARS-COV-2 viral infection to the kidney may downregulate ACE2-Ang1-7-Mas signaling while promoting the Ang II-AT1 signaling to mediate renal inflammation and AKI. A similar mechanism is also found in patients with ARDS [38]. Thus, the interaction between SARS-COV-2 virus and ACE2 may eventually impair the ACE2-Ang 1-7 while enhancing Ang II signaling, resulting in hypertension and inflammatory stress both systemically and locally in the kidney. This may well explain that hypertension is an independent risk factor in COVID-19 patients [39]. However, the role of Ang II signaling in COVID-19 patients with progressive renal injury remains yet to be determined.
Alterations of Ang II and Ang 1-7 signaling in COVID-19 associated AKI. SARS-COV-2 binds and downregulates ACE2, which may result in downregulation of Ang 1-7 while upregulating Ang II-AT signaling to promote AKI.
Diabetes and metabolic stress
Diabetes is also a risk factor for AKI [40]. Patients with diabetes are associated with the severity and death in pandemic influenza (H1N1) [41], SARS-COV [42] and MERS-COV [43]. Recent studies also reported that COVID-19 patients with diabetes have higher AKI and mortality rate than those with non-diabetes [14, 44]. This is also confirmed in a recent meta-analysis in 5497 COVID-19 patients [45].
It is now well accepted that metabolic stress including hyperglycemia, obesity, insulin resistance and high levels of glycosylation end products (AGEs) in patients with diabetes can trigger the production of pro-inflammatory cytokines and promotes the oxidative stress [46]. Hyperglycemia is a risk factor for AKI in patients with diabetes [21]. The IκB kinase-β (IKKβ)/NF-κB axis is a key inflammatory pathway in diabetes in response to hyperglycemia and insulin resistance [47]. AGEs can also induce activation of NF-κB, resulting in production of pro-inflammatory cytokines [48]. By comparing with non-diabetic COVID-19 patients, COVID-19 patients with diabetes have significantly higher levels of IL-6 and CRP [49]. In addition, patients with diabetes also develop hypertension, which is associated with activation of Ang II-AT1 while inhibiting ACE2-Ang 1-7 signaling [50]. In addition, since ACE2 is also expressed in pancreas, infection of SARS-COV-2 may also damage pancreatic islet β-cells and aggravate hyperglycemia [51]. Thus, enhanced metabolic inflammation in the diabetic kidney may be another mechanism contributing to the development of AKI in COVID-19 patients. However, mechanisms responsible for the metabolic stress in AKI after COVID-19 infection remain yet to be explored.
Cytokine storm
Inflammatory cytokines has been recognized as a critical factor in the progression of COVID-19 [52, 53]. The inflammatory response triggered by SARS-COV, MERS-COV or SARS-COV-2 can recruit and activate monocytes, macrophage and dendritic cells to produce inflammatory cytokines [54, 55], which may be essential in controlling the viral replication and cleaning the infected cells [56]. However, overactive immune responses may cause excessive and persistent cytokine production that leads to cytokine storm and results in multiple organ dysfunction as seen in patients with severe SARS [57], MERS [58] and COVID-19 infection [59]. In these patients, a number of proinflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-12, interferon γ (IFN-γ) and monocyte chemoattractant peptide (MCP-1) are associated with extensive lung damage in SARS patients [60]. The blood levels of IL-10, IL-15 and TGF-β1 are also positively correlated with the disease severity in patients with MERS [61]. In COVID-19 patients, levels of IL-1β, IL-1RA, IL-7, IL-8, IL-9, IL-10, granulocyte colony stimulating factor (G-CSF), IFN-γ, interferon γ inducible protein (IP)-10, tumor necrosis factor-alpha (TNF-α) and MCP-1 are also increased over the healthy controls and become worsen in those admitted to ICU with severe acute lung injury [62, 63].
Cytokine storm can also trigger AKI under various clinical conditions including secondary haemophagocytic lymphohistiocytosis (sHLH) [64-68]. sHLH is also found in patients with SARS and COVID-19 [69, 70]. In addition, cytokine storm may also result in the development of antiphospholipid syndrome in AKI patients with COVID-19 [71, 72]. Of these inflammatory cytokines, IL-6 has been recognized as a key mediator in COVID-19 patients, which is further described below.
IL-6
Many studies have demonstrated that among the inflammatory cytokines, IL-6 is a most strong and important mediator in COVID-19 patients [73, 74]. Meta-analysis involving 12681 COVID-19 patients confirms that IL-6 is significantly higher in those with severe disease conditions [75, 76]. Indeed, serum levels of IL-6 positively correlate with the severity of COVID-19 [75, 77, 78] and also predict the mechanical ventilation need for COVID-19 patients [79]. In COVID-19 patients with older age, IL-6 is an independent risk factor for in-hospital mortality [80].
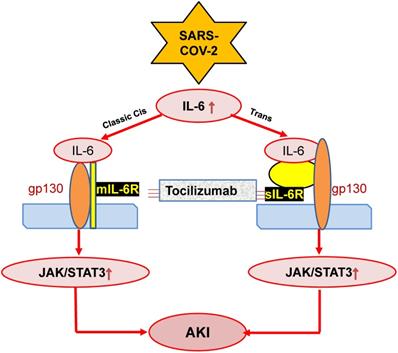
IL-6 is also a predictor for AKI in patients under various clinical conditions including cardiovascular disease, kidney diseases and liver transplantation [81-83]. This is also found in ischemic AKI animal model [84]. In response to injury, IL-6 is upregulated and released from renal TECs and plays an important role in the pathophysiology of AKI [85]. Increasing evidence shows that IL-6 is not only a biomarker but also a mediator for AKI as mice lacking IL-6 are resistant to HgCl2-induced AKI [85-88]. In patients with COVID-19, serum levels of IL-6 are elevated in those with AKI [89], and become further increased in those with critically ill [71, 90]. In addition, serum levels of IL-6 can also predict the clinical outcomes of AKI as it is significantly reduced in those when AKI is recovered after effective treatment [91]. Mechanistically, JAK-STAT3 is a downstream signal transduction of IL-6-membrane-bound-IL-6 receptor (mIL-6R)/soluble-bound-IL-6 receptor (sIL-6R). The IL-6-mIL-6R/sIL-6R-JAK-STAT3 signing pathways are activated during cytokine storm in severe COVID-19 patients [54], which is outlined in Figure 2. However, the functional role and molecular mechanisms of IL-6 in the pathogenesis of COVID-19 associated AKI remain largely unclear.
Possible mechanisms through which SARS-COV-2 may induce AKI by stimulating IL-6 signaling. SARS-COV-2 may activate IL-6-mIL-6/sIL6-JAK-STAT3 signaling, resulting in AKI, which can be blocked by Tocilizumab antibodies.
C-reactive protein (CRP)
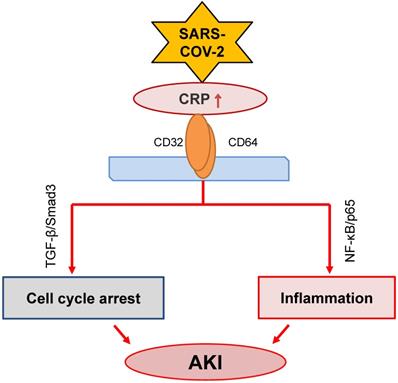
CRP, produced by liver and many inflammatory cells, is an acute phase protein. It has been widely used in clinical settings as an acute inflammation biomarker. CRP is proved as a predictor of postoperative AKI in patients undergoing Coronary Artery Bypass Graft (CABG) [92]. High sensitive CRP is associated with AKI in patients with acute myocardial infarction [93, 94]. Meanwhile, it is an independent predictor for AKI among ST elevation myocardial infarction patients undergoing primary percutaneous intervention [95]. Increasing evidence has suggested that CRP is also a pathogenic factor contributing to the development of inflammatory diseases including atherosclerosis [96] and AKI [97-101]. The mechanisms of CRP in the progression of AKI include stimulating macrophage activation [97], inducing cell death by causing G1 cell cycle arrest and autophagy [99], and promoting inflammation [101] (Figure 3). The activation of NF-κB/p65 and TGF-β/Smad3 signaling pathways are the major mechanisms through which CRP mediates AKI [98, 100, 101].
Possible role of CRP signaling in COVID-19-associated AKI. SARS-COV-2 infection may activate CRP signaling to cause AKI via TGF-β/Smad3-mediated G1 cell cycle arrest and NF-κB-dependent renal inflammation.
COVID-19 patients with AKI show higher levels of serum CRP over those without AKI [102]. Serum levels of CRP are also a risk factor of AKI in COVID-19 patients [103]. Tan et al. reported that serum levels of CRP are significantly elevated after SARS-CoV-2 infection, which becomes further increased when the disease is progressive but declines dramatically when COVID-19 is recovered [104]. Thus, levels of serum CRP may be a predictor for the clinical outcomes of COVID-19 patients. Meta-analysis confirms this notion that in contrast to mild and survival subgroup of COVID-19 patients, high levels of CRP are associated with severe and death subgroup of COVID-19 patients [75, 76, 105]. In addition, CRP is also an indicator for renal replacement therapy and the need for mechanical ventilation in COVID-19 patients [106]. Thus, elevated CRP is independently associated with poor clinical outcomes in COVID-19 patients [107, 108]. However, the pathogenic role and mechanisms of CRP in COVID-19-associated AKI remain largely unknown.
TGF-β
TGF-β is a pleiotropic cytokine and signals through its downstream canonical and non-canonical pathways to diversely regulate renal inflammation and fibrosis [109, 110]. It has been reported that SARS-COV nucleocapsid protein can interact with Smad3 to activate the canonical pathway [111]. Whereas, the non-canonical TGF-β signaling pathway is also activated by the papain-like protease of SARS-COV by inducing expression of TGF-β1 [112]. In COVID-19 patients, plasma levels of TGF-β are significantly elevated and associated with the disease severity and poor clinical outcomes [113, 114]. Elevated TGF-β2 mRNA is also found in the bronchoalveolar lavage (BAL) fluid of COVID-19 patients [115], which may contribute to lung inflammation and fibrosis because TGF-β1 is also a growth factor associated with fibrosis [109.110]. It is reported that SARS-COV-2 encoded microRNAs are able to target TGF-β signaling pathway to induce TGF-β-dominated adaptive immune response [116]. Upregulation of TGF-β1 in COVID-19 patients is responsible for the recruitment of neutrophils into the site of inflammation [117]. TGF-β can also induce MCP-1 to activate macrophage-dependent inflammation in the diabetic kidney via a Smad3-dependent LRNA9884 [118]. Induction of IL-6 production by TGF-β1 also leads to systemic inflammation and “cytokine storm” [119]. Furthermore, TGF-β can induce IgA class switching, which may contribute to vasculitis in critically ill patients with COVID-19 [120]. Thus, TGF-β may significantly contribute to the immediate and long-term effects of COVID-19.
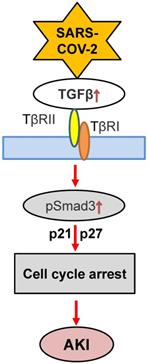
Although the role of TGF-β in COVID-19 associated AKI is still unclear, recent findings that conditional deletion of TGF-β receptor II from renal proximal tubules protects against mercuric chloride and cisplatin-induced AKI reveal a critical role of TGF-β signaling in AKI [121, 122]. Furthermore, mice specifically lacking bronchial epithelial TGF-β1 (epTGFβKO) are protected against influenza-induced weight loss, airway inflammation, and viral replication [123], suggesting the impact of TGF-β in viral infection. Mechanistically, TGF-β1 may act via Smad3 to cause AKI as genetic deletion or pharmacological inhibition of Smad3 can block AKI in ischemic mice with or without high human CRP conditions [124]. Indeed, Smad3 can be activated by both TGF-β-dependent and independent mechanisms including Ang II, advanced end products (AGE), and CRP under various disease conditions such as hypertension and diabetes [125-127]. Thus, activation of Smad3 signaling may account for the clinical notion that patients with diabetes and hypertension are high risk for COVID-19 [7]. Mechanistically, Smad3 promotes AKI by directly binding to p21/p27 to suppress CDKs/cyclin E to cause the G1 cell cycle arrest [128, 129] (Figure 4). Thus, it is highly possible that TGF-β/Smad3 signaling may contribute to COVID-19-associated AKI, which is warranted for further investigation.
Proposed TGF-β signaling in COVID-19-associated AKI. SARS-COV-2 may induce G1 cell cycle arrest and cell death via TGF-β/Smad3-p21/p27 mechanism.
Complement activation
The complement system is the first response of host immune system that recognizes and eliminates virus, such as SARS-COV or SARS-COV-2 [7, 130]. There are several pathways involving in systemic complement activation including the lectin pathway, classical pathway, and alternative pathway [130, 131]. The lectin pathway is triggered by the binding of mannose-binding lectin (MBL) with SARS-COV spike (S) protein [132], which leads to the activation of mannan-binding lectin-associated serine protease 2 (MASP-2). The N protein of coronavirus is also associated with the severity of lung injury according to MASP-2-mediated complement overactivation. Thus, alteration of MASP-2-binding motif or blocking the MASP-2-N protein interaction attenuates lung injury [133]. The classical pathway is activated by the binding of antibodies, which forms the immune complexes with viral antigens to complement C1 complex [130, 131]. The classical, lectin and alternative pathways resulting in the formation of C3 convertase to activate the complement system, which is observed in the lung after SARS-COV infection [130, 131, 134]. The role of C3 in SARS-COV-induced lung injury is confirmed in C3 deficient mice in which deletion of C3 protects against ARDS with lower levels of cytokine and inflammatory monocytes infiltration [135].
The complement system is activated during the progression of COVID-19 [136]. The activation of complement system is related to the disease severity and the respiratory failure in COVID-19 patients [134, 136, 137]. It has been shown that the C5a-C5aR1 axis plays an important role in the development of ARDS in COVID-19 [138], whereas, C4d and C5-9 are colocalized with the SARS-COV-2 S protein in the lung and skin vasculature [139].
The activation of alternative pathway of complement also participates in the pathogenesis of AKI [140, 141]. It has been reported that local synthesis or deposition and activation of complement by renal epithelium is an important cause of AKI [142, 143]. In addition, strong C5b-9 staining has been demonstrated on the apical brush border of TECs of the kidney with SARS-COV-2 infection [10]. Activation of the classical complement pathway is also observed in the AKI kidney in critically ill children with COVID-19 [90]. However, the functional role and mechanisms of complement activation in COVID-19-assicated AKI remain largely unclear.
The lung-kidney crosstalk pathway
The crosstalk between the lung and kidney has been observed in patients with AKI and ARDS [144]. Clinically, acute lung injury and AKI are complications often encountered in patients with critical illness [145]. Mechanical ventilation can improve lung function but is a risk factor for AKI in critically ill patients [146, 147]. It has been reported that positive pressure of mechanical ventilation can increase the risk of AKI by almost 8-fold [106], which is associated with systemic hemodynamic and neurohormonal changes and biotrauma [65, 148, 149]. ARDS can trigger AKI via mechanisms associated with systemic hypoxia, hypercapnia, systemic inflammatory response syndrome (SIRS), and mechanical ventilation. Severe hypoxemia in ICU patients is associated with AKI required for renal replacement therapy [145]. Kidney is susceptible to hypoxic injury due to the high rate of oxygen consumption [65, 150]. Thus, hypoxia can induce AKI and tubular necrosis or apoptosis [149]. Hypercapnia in COVID-19 patients can also affect the renal blood flow by stimulating renal vasoconstriction [151]. More importantly, the lung-kidney cross-talk is also associated with the cytokine storm [152]. The inflammatory reaction caused by the lung injury can damage the kidney to release abundant inflammatory cytokines, which, in turn, promotes the damage in the lung [8]. Thus, the approaches to limit ventilator-induced lung injury and decrease the duration of mechanical ventilatory support to protect against AKI in critically ill patients are proposed for treatment of critically ill COVID-19 with acute lung and kidney injury.
Therapeutic potential for COVID-19-associated AKI
Continuous renal replacement therapy (CRRT)
CRRT is an advanced approach to treat patients with AKI by improving overload water status and removing inflammatory factors [153], particularly in those with septic AKI [154]. CRRT has been utilized in AKI patients with severe MERS and in critically ill COVID-19 [155]. However, whether the early initiation or high intensity of CRRT can improve the progression of AKI COVID-19 patients remains to be determined.
Tocilizumab (TCZ)
TCZ, a recombinant humanized monoclonal antibody against the human membrane and soluble IL-6 receptors, is widely used for treatment of immunoinflammatory rheumatic diseases [156, 157]. TCZ has been shown to block the IL-6/NF-κB/JNK pathway to have a protective effect against sepsis-induced acute lung injury and AKI [158]. As IL-6 is important in COVID-19, TCZ has been used to treat COVID-19 patients clinically (Figure 2). The early treatment with TCZ has been shown to effectively improve the oxygen status in COVID-19 patients [156]. The meta-analysis of TCZ studies in a total of 1675 and 6279 COVID-19 patients with critically ill shows that TCZ treatment can significantly reduce the in-hospital mortality rate, although patients remain the need for hemodialysis and ventilation [159]. Thus, the therapeutic efficacy of TCZ on COVID-19-associated AKI needs to be further studied.
Complement inhibitor
A C3 inhibitor AMY-101
AMY-101 is a highly selective and potent C3 inhibitor and is currently tested in Phase II clinical trials in patients with sepsis, hemodialysis-induced inflammation or malarial anemia [160-163]. It is reported that treatment with AMY-101 is safe and can significantly improve the clinical manifestations in severe COVID-19 patients [164]. Further Phase II and III clinical trials are still going.
An anti-C5 antibody
An anti-C5 antibody has been clinically used in patients with C3 glomerulopathy and several types of AKI including atypical hemolytic uremic syndrome and paroxysmal nocturnal hemoglobinuria [165]. Treatment with the anti-C5 antibody has shown to improve the kidney function and ameliorate the intra-renal complement activation and systemic inflammation in ischemia reperfusion-induced AKI mouse model [165]. The first result of anti-C5 treatment has also revealed a rapid and promising effect on COVID-19 patients [166]. However, more clinical trials are needed for the conclusive results of the anti-C5 antibody treatment on COVID-19 patients with AKI.
Anti-TGF-β treatment
As SARS coronavirus can upregulate TGF-β and patients with coronavirus infection have elevated levels of TGF-β [113, 114], it has been proposed that TGF-β could be a valid target for the treatment of COVID-19 [167, 168]. In a recent Phase II clinical trial, inhibition of TGF-β expression by OT-101, an anti-sense to TGF-β1, has been shown to suppress SARS-COV and SARS-COV-2 replication and allow patients to recover without going into respiratory crisis [169], suggesting that COVID-19 can be treated with TGF-β inhibition. However, it should be noted that TGF-β has diverse roles in renal inflammation and fibrosis [109, 110] and targeting the upstream of TGF-β may also cause adverse effects. Our recent studies showed that TGF-β can trigger AKI via the Smad3-dependent mechanism and treatment with Smad3 inhibitors such as SIS3 or a natural product of Traditional Chinese Medicine Quercetin can effectively suppress AKI even under high human CRP conditions [100, 170]. These findings suggest that targeting Smad3 specifically, rather than the entire TGF-β signaling, may represent as a novel and effective therapy for AKI in COVID-19 patients clinically.
Conclusion
AKI is a common complication in critically ill COVID-19 patients. Inflammation may be a key mechanism triggering this process. Many inflammatory stress molecules and pathways including Ang II-associated hypertensive stress, diabetes-related metabolic stress, cytokine storm, high CRP, overreactive TGF-β signaling, complement activation, and lung-kidney crosstalk may promote AKI in COVID-19 patients. Thus, treatments by targeting these molecules and pathways may represent as a novel and specific therapy for AKI in COVID-19 patients.
Abbreviations
ACE2: angiotensin-converting enzyme 2; Ang I: Angiotensin II; AKI: Acute kidney injury; ARDS: acute respiratory distress syndrome; BUN: Blood urea nitrogen; CRP: C-reactive protein; CABG: Coronary Artery Bypass Graft; CRRT: Continuous renal replacement therapy; G-CSF: granulocyte colony stimulating factor; IFN-γ: interferon γ; IKKβ: IκB kinase-β; IL-6: Interleukin-6; IP: inducible protein; MASP-2: mannan-binding lectin-associated serine protease 2; MCP-1: monocyte chemoattractant peptide; mIL-6R: membrane-bound-IL-6 receptor; RAAS: renin-angiotensin-aldosterone system; Scr: serum creatinine; sIL-6R: soluble-bound-IL-6 receptor; TCZ: Tocilizumab; TNF-α: tumor necrosis factor-alpha.
Acknowledgements
This study was supported by Lui Che Woo Institute of Innovative Medicine (CARE program), Research Grants Council of Hong Kong (GRF 14163317, 14106518, 14111019, C7018‐16G, R4012-18), Health and Medical Research Fund (03140486, 05161326, 14152321), and the Guangdong-Hong Kong-Macao-Joint Labs Program from Guangdong Science and Technology Department (2019B121205005).
Author Contributions
J.C. and W.W. wrote the manuscript and Y.T and X.R.H collected and analyzed data. X.Y. and H.Y.L. designed and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239-42
2. Gagliardi I, Patella G, Michael A. et al. COVID-19 and the Kidney: From Epidemiology to Clinical Practice. J Clin Med. 2020;9:2506
3. Soleimani M. Acute Kidney Injury in SARS-CoV-2 Infection: Direct Effect of Virus on Kidney Proximal Tubule Cells. Int J Mol Sci. 2020;21:3257
4. Xu Z, Shi L, Wang Y. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420-2
5. Cheng YC, Luo R, Wang K. et al. Kidney disease is associated with in -hospital death of patients with COVID-19. Kidney International. 2020;97:829-38
6. Yang X, Jin Y, Li R. et al. Prevalence and impact of acute renal impairment on COVID-19: a systematic review and meta-analysis. Crit Care. 2020;24:356
7. Stasi A, Castellano G, Ranieri E. et al. SARS-CoV-2 and Viral Sepsis: Immune Dysfunction and Implications in Kidney Failure. J Clin Med. 2020;9:4057
8. Li Z WM, Yao JW. et al. Caution on Kidney Dysfunctions of COVID-19 Patients. medRxiv. 2020
9. Su H, Yang M, Wan C. et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98:219-27
10. Diao B WC, Wang RH. et al. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. medRxiv. 2020
11. Chu KH, Tsang WK, Tang CS. et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005;67:698-705
12. Zhou S, Xu J, Xue C. et al. Coronavirus-associated kidney outcomes in COVID-19, SARS, and MERS: a meta-analysis and systematic review. Ren Fail. 2020;43:1-15
13. Wang LW, Li X, Chen H. et al. Coronavirus Disease 19 Infection Does Not Result in Acute Kidney Injury: An Analysis of 116 Hospitalized Patients from Wuhan, China. American Journal of Nephrology. 2020;51:343-8
14. Guan WJ, Ni ZY, Hu Y. et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382:1708-20
15. Pei G, Zhang Z, Peng J. et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J Am Soc Nephrol. 2020;31:1157-65
16. Yang X, Yu Y, Xu J. et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475-81
17. Yu Y, Xu D, Fu S. et al. Patients with COVID-19 in 19 ICUs in Wuhan, China: a cross-sectional study. Crit Care. 2020;24:219
18. Hirsch JS, Ng JH, Ross DW. et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020;98:209-18
19. Richardson S, Hirsch JS, Narasimhan M. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA. 2020;323:2052-9
20. Chan L, Chaudhary K, Saha A. et al. Acute Kidney Injury in Hospitalized Patients with COVID-19. medRxiv. 2020
21. Chen J, Zeng H, Ouyang X. et al. The incidence, risk factors, and long-term outcomes of acute kidney injury in hospitalized diabetic ketoacidosis patients. BMC Nephrol. 2020;21:48
22. Tang Y, Chen J, Huang K. et al. The incidence, risk factors and in-hospital mortality of acute kidney injury in patients after abdominal aortic aneurysm repair surgery. BMC Nephrol. 2017;18:184
23. Ng JH, Hirsch JS, Hazzan A. et al. Outcomes Among Patients Hospitalized With COVID-19 and Acute Kidney Injury. Am J Kidney Dis. 2021;77:204-15
24. Staico MF, Zaffanello M, Di Pietro G. et al. The kidney in COVID-19: protagonist or figurant? Panminerva Med. in press
25. Wan Y, Shang J, Graham R. et al. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94:e00127-20
26. Wrapp D, Wang N, Corbett KS. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260-3
27. Wu H, Uchimura K, Donnelly EL. et al. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell. 2018;23:869-81
28. Braun F, Lutgehetmann M, Pfefferle S. et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet. 2020;396:597-8
29. Ni J, Yang F, Huang XR. et al. Dual deficiency of angiotensin-converting enzyme-2 and Mas receptor enhances angiotensin II-induced hypertension and hypertensive nephropathy. J Cell Mol Med. 2020;24:13093-103
30. Santos RAS, Sampaio WO, Alzamora AC. et al. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol Rev. 2018;98:505-53
31. Malha L, Mueller FB, Pecker MS. et al. COVID-19 and the Renin-Angiotensin System. Kidney Int Rep. 2020;5:563-5
32. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. European Journal of Internal Medicine. 2020;76:14-20
33. Mizuiri S, Ohashi Y. ACE and ACE2 in kidney disease. World J Nephrol. 2015;4:74-82
34. Kontogiannis J, Burns KD. Role of AT1 angiotensin II receptors in renal ischemic injury. Am J Physiol. 1998;274:79-90
35. Jerkic M, Miloradovic Z, Jovovic D. et al. Relative roles of endothelin-1 and angiotensin II in experimental post-ischaemic acute renal failure. Nephrol Dial Transplant. 2004;19:83-94
36. Mackie FE, Campbell DJ, Meyer TW. Intrarenal angiotensin and bradykinin peptide levels in the remnant kidney model of renal insufficiency. Kidney Int. 2001;59:1458-65
37. da Silveira KD, Pompermayer Bosco KS, Diniz LR. et al. ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin Sci (Lond). 2010;119:385-94
38. Reddy R, Asante I, Liu S. et al. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS One. 2019;14:e0213096
39. Lippi G, Wong J, Henry BM. Hypertension in patients with coronavirus disease 2019 (COVID-19): a pooled analysis. Pol Arch Intern Med. 2020;130:304-9
40. Muroya Y, He X, Fan L. et al. Enhanced renal ischemia-reperfusion injury in aging and diabetes. Am J Physiol Renal Physiol. 2018;315:1843-54
41. Schoen K, Horvat N, Guerreiro NFC. et al. Spectrum of clinical and radiographic findings in patients with diagnosis of H1N1 and correlation with clinical severity. BMC Infect Dis. 2019;19:964
42. Yang JK, Feng Y, Yuan MY. et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet Med. 2006;23:623-8
43. Banik GR, Alqahtani AS, Booy R, Rashid H. Risk factors for severity and mortality in patients with MERS-CoV: Analysis of publicly available data from Saudi Arabia. Virol Sin. 2016;31:81-4
44. Onder G, Rezza G, Brusaferro S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA. 2020;323:1775-6
45. Hansrivijit P, Qian C, Boonpheng B. et al. Incidence of acute kidney injury and its association with mortality in patients with COVID-19: a meta-analysis. J Investig Med. 2020;68:1261-70
46. Hussain A, Bhowmik B, do Vale Moreira NC. COVID-19 and diabetes: Knowledge in progress. Diabetes Res Clin Pract. 2020;162:108142
47. Shoelson SE, Lee J, Yuan M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. International Journal of Obesity. 2003;27:S49-S52
48. Ott C, Jacobs K, Haucke E. et al. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411-29
49. Guo WN, Li MY, Dong YL. et al. Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes-Metabolism Research and Reviews. in press
50. Tikellis C, Pickering R, Tsorotes D. et al. Interaction of diabetes and ACE2 in the pathogenesis of cardiovascular disease in experimental diabetes. Clin Sci (Lond). 2012;123:519-29
51. Yang JK, Lin SS, Ji XJ, Guo LM. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetologica. 2010;47:193-9
52. Mehta P, McAuley DF, Brown M. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033-4
53. Stebbing J, Phelan A, Griffin I. et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20:400-2
54. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368:473-4
55. Ye Q, Wang BL, Mao JH. The pathogenesis and treatment of the 'Cytokine Storm' in COVID-19. Journal of Infection. 2020;80:607-13
56. Tufan A, Avanoglu Guler A, Matucci-Cerinic M. COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk J Med Sci. 2020;50:620-32
57. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529-39
58. Fehr AR, Channappanavar R, Perlman S. Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus. Annu Rev Med. 2017;68:387-99
59. Lipworth B, Chan R, Lipworth S, RuiWen Kuo C. Weathering the Cytokine Storm in Susceptible Patients with Severe SARS-CoV-2 Infection. J Allergy Clin Immunol Pract. 2020;8:1798-801
60. Wong CK, Lam CW, Wu AK. et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95-103
61. Min CK, Cheon S, Ha NY. et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep. 2016;6:25359
62. Huang C, Wang Y, Li X. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506
63. Yang Y SC, LI JX. et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. medRxiv. 2020
64. Ahmed AR, Ebad CA, Stoneman S. et al. Kidney injury in COVID-19. World J Nephrol. 2020;9:18-32
65. Chong WH, Saha BK. Relationship Between Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Etiology of Acute Kidney Injury (AKI). Am J Med Sci. in press
66. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A. et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-16
67. Aulagnon F, Lapidus N, Canet E. et al. Acute Kidney Injury in Adults With Hemophagocytic Lymphohistiocytosis. American Journal of Kidney Diseases. 2015;65:851-9
68. Karras A. What nephrologists need to know about hemophagocytic syndrome. Nature Reviews Nephrology. 2009;5:329-36
69. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev. 2020;19:102537
70. Dimopoulos G, de Mast Q, Markou N. et al. Favorable Anakinra Responses in Severe Covid-19 Patients with Secondary Hemophagocytic Lymphohistiocytosis. Cell Host Microbe. 2020;28:117-23
71. Xia P, Wen Y, Duan Y. et al. Clinicopathological Features and Outcomes of Acute Kidney Injury in Critically Ill COVID-19 with Prolonged Disease Course: A Retrospective Cohort. J Am Soc Nephrol. 2020;31:2205-21
72. Zhang Y, Xiao M, Zhang S. et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med. 2020;382:e38
73. Chen XH, Zhao BH, Qu YM. et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated With Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clinical Infectious Diseases. 2020;71:1937-42
74. Fara A, Mitrev Z, Rosalia RA, Assas BM. Cytokine storm and COVID-19: a chronicle of pro-inflammatory cytokines. Open Biol. 2020;10:200160
75. Ji P, Zhu J, Zhong Z. et al. Association of elevated inflammatory markers and severe COVID-19: A meta-analysis. Medicine (Baltimore). 2020;99:e23315
76. Zeng F, Huang Y, Guo Y. et al. Association of inflammatory markers with the severity of COVID-19: A meta-analysis. Int J Infect Dis. 2020;96:467-74
77. Chen L, Liu HG, Liu W. et al. [Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:203-8
78. Luo X, Zhou W, Yan X. et al. Prognostic Value of C-Reactive Protein in Patients With Coronavirus 2019. Clin Infect Dis. 2020;71:2174-9
79. Herold T, Jurinovic V, Arnreich C. et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol. 2020;146:128-36
80. Yan Q, Zuo P, Cheng L. et al. Acute Kidney Injury Is Associated With In-hospital Mortality in Older Patients With COVID-19. J Gerontol A Biol Sci Med Sci. in press
81. Chae MS, Kim Y, Chung HS. et al. Predictive Role of Serum Cytokine Profiles in Acute Kidney Injury after Living Donor Liver Transplantation. Mediators Inflamm. 2018;2018:8256193
82. Zhang WR, Garg AX, Coca SG. et al. Plasma IL-6 and IL-10 Concentrations Predict AKI and Long-Term Mortality in Adults after Cardiac Surgery. J Am Soc Nephrol. 2015;26:3123-32
83. Greenberg JH, Whitlock R, Zhang WR. et al. Interleukin-6 and interleukin-10 as acute kidney injury biomarkers in pediatric cardiac surgery. Pediatric Nephrology. 2015;30:1519-27
84. Grigoryev DN, Liu M, Hassoun HT. et al. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. 2008;19:547-58
85. Nechemia-Arbely Y, Barkan D, Pizov G. et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol. 2008;19:1106-15
86. Kielar ML, John R, Bennett M. et al. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol. 2005;16:3315-25
87. Patel NS, Chatterjee PK, Di Paola R. et al. Endogenous interleukin-6 enhances the renal injury, dysfunction, and inflammation caused by ischemia/reperfusion. J Pharmacol Exp Ther. 2005;312:1170-8
88. Lemay S, Rabb H, Postler G, Singh AK. Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation. 2000;69:959-63
89. Joseph A, Zafrani L, Mabrouki A. et al. Acute kidney injury in patients with SARS-CoV-2 infection. Ann Intensive Care. 2020;10:117
90. Wang XW, Chen XF, Tang F. et al. Be aware of acute kidney injury in critically ill children with COVID-19. Pediatric Nephrology. 2021;36:163-9
91. Villa G, Romagnoli S, De Rosa S. et al. Blood purification therapy with a hemodiafilter featuring enhanced adsorptive properties for cytokine removal in patients presenting COVID-19: a pilot study. Crit Care. 2020;24:605
92. Han SS, Kim DK, Kim S. et al. C-Reactive Protein Predicts Acute Kidney Injury and Death After Coronary Artery Bypass Grafting. Ann Thorac Surg. 2017;104:804-10
93. Cosentino N, Genovese S, Campodonico J. et al. High-Sensitivity C-Reactive Protein and Acute Kidney Injury in Patients with Acute Myocardial Infarction: A Prospective Observational Study. J Clin Med. 2019;8:2192
94. Fu EL, Franko MA, Obergfell A. et al. High-sensitivity C-reactive protein and the risk of chronic kidney disease progression or acute kidney injury in post-myocardial infarction patients. Am Heart J. 2019;216:20-9
95. Shacham Y, Leshem-Rubinow E, Steinvil A. et al. High sensitive C-reactive protein and the risk of acute kidney injury among ST elevation myocardial infarction patients undergoing primary percutaneous intervention. Clinical and Experimental Nephrology. 2015;19:838-43
96. Verma S. C-reactive protein incites atherosclerosis. Can J Cardiol. 2004;20:S29-S31
97. Pegues MA, McCrory MA, Zarjou A, Szalai AJ. C-reactive protein exacerbates renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2013;304:1358-65
98. Tang Y, Huang XR, Lv J. et al. C-reactive protein promotes acute kidney injury by impairing G1/S-dependent tubular epithelium cell regeneration. Clin Sci (Lond). 2014;126:645-59
99. Bian A, Shi MJ, Flores B. et al. Downregulation of autophagy is associated with severe ischemia-reperfusion-induced acute kidney injury in overexpressing C-reactive protein mice. Plos One. 2017;12:e0181848
100. Lai W, Tang Y, Huang XR. et al. C-reactive protein promotes acute kidney injury via Smad3-dependent inhibition of CDK2/cyclin E. Kidney Int. 2016;90:610-26
101. Li ZI, Chung AC, Zhou L. et al. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Lab Invest. 2011;91:837-51
102. Taha M, Sano D, Hanoudi S. et al. Platelets and renal failure in the SARS-CoV-2 syndrome. Platelets. 2021;32:130-7
103. Hamer M, Kivimaki M, Gale CR, Batty GD. Lifestyle risk factors, inflammatory mechanisms, and COVID-19 hospitalization: A community-based cohort study of 387,109 adults in UK. Brain Behav Immun. 2020;87:184-7
104. Tan C, Huang Y, Shi F. et al. C-reactive protein correlates with computed tomographic findings and predicts severe COVID-19 early. J Med Virol. 2020;92:856-62
105. Smilowitz NR, Kunichoff D, Garshick M. et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J. in press
106. Doher MP, Torres de Carvalho FR, Scherer PF. et al. Acute Kidney Injury and Renal Replacement Therapy in Critically Ill COVID-19 Patients: Risk Factors and Outcomes: A Single-Center Experience in Brazil. Blood Purif. 2020:1-11
107. Malik P, Patel U, Mehta D. et al. Biomarkers and outcomes of COVID-19 hospitalisations: systematic review and meta-analysis. BMJ Evid Based Med. in press
108. Vena A, Giacobbe DR, Di Biagio A. et al. Clinical characteristics, management and in-hospital mortality of patients with coronavirus disease 2019 in Genoa, Italy. Clin Microbiol Infect. 2020;26:1537-44
109. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38
110. Gu YY, Liu XS, Huang XR. et al. TGF-beta in renal fibrosis: triumphs and challenges. Future Med Chem. 2020;12:853-66
111. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem. 2008;283:3272-80
112. Li SW, Wang CY, Jou YJ. et al. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep. 2016;6:25754
113. Ghazavi A, Ganji A, Keshavarzian N. et al. Cytokine profile and disease severity in patients with COVID-19. Cytokine. 2021;137:155323
114. Agrati C, Sacchi A, Bordoni V. et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 2020;27:3196-207
115. Xiong Y, Liu Y, Cao L. et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerging Microbes & Infections. 2020;9:761-70
116. Ferreira-Gomes M. KA, Durek P. et al. In severe COVID-19, SARS-CoV-2 induces a chronic, TGF-β-dominated adaptive immune response. medRxiv. 2020
117. Barnes BJ, Adrover JM, Baxter-Stoltzfus A. et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med. 2020;217:e20200652
118. Zhang YY, Tang PMK, Tang PCT. et al. LRNA9884, a Novel Smad3-Dependent Long Noncoding RNA, Promotes Diabetic Kidney Injury in db/db Mice via Enhancing MCP-1-Dependent Renal Inflammation. Diabetes. 2019;68:1485-98
119. Turner M, Chantry D, Feldmann M. Transforming growth factor beta induces the production of interleukin 6 by human peripheral blood mononuclear cells. Cytokine. 1990;2:211-6
120. Yu HQ, Sun BQ, Fang ZF. et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur Respir J. 2020;56:2001526
121. Gewin L, Vadivelu S, Neelisetty S. et al. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J Am Soc Nephrol. 2012;23:2001-11
122. Yang Q, Ren GL, Wei B. et al. Conditional knockout of TGF-betaRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics. 2019;9:8277-93
123. Denney L, Branchett W, Gregory LG. et al. Epithelial-derived TGF-beta 1 acts as a pro-viral factor in the lung during influenza A infection. Mucosal Immunology. 2018;11:523-35
124. Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301:436-42
125. Wang W, Huang XR, Canlas E. et al. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98:1032-9
126. Li JH, Huang XR, Zhu HJ. et al. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176-8
127. You YK, Huang XR, Chen HY. et al. C-Reactive Protein Promotes Diabetic Kidney Disease in db/db Mice via the CD32b-Smad3-mTOR signaling Pathway. Sci Rep. 2016;6:26740
128. Matsuura I, Denissova NG, Wang G. et al. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226-31
129. Fu S, Tang Y, Huang XR. et al. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the G1 cell cycle arrest. Clin Sci (Lond). 2017;131:1955-69
130. Java A, Apicelli AJ, Liszewski MK. et al. The complement system in COVID-19: friend and foe? JCI Insight. 2020;5:e140711
131. Noris M, Benigni A, Remuzzi G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020;98:314-22
132. Ip WK, Chan KH, Law HK. et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;191:1697-704
133. Gao T HM, Zhang XP. et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. 2020
134. de Nooijer AH, Grondman I, Janssen NAF. et al. Complement activation in the disease course of COVID-19 and its effects on clinical outcomes. J Infect Dis. 2020
135. Gralinski LE, Sheahan TP, Morrison TE. et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio. 2018;9:e01753-18
136. Cugno M, Meroni PL, Gualtierotti R. et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J Autoimmun. 2021;116:102560
137. Holter JC, Pischke SE, de Boer E. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:25018-25
138. Carvelli J, Demaria O, Vely F. et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588:146-50
139. Magro C, Mulvey JJ, Berlin D. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1-13
140. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305-10
141. David S, Biancone L, Caserta C. et al. Alternative pathway complement activation induces proinflammatory activity in human proximal tubular epithelial cells. Nephrol Dial Transplant. 1997;12:51-6
142. Farrar CA, Zhou WD, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. Faseb Journal. 2006;20:217-26
143. Thurman JM, Lucia MS, Ljubanovic D, Holers VM. Acute tubular necrosis is characterized by activation of the alternative pathway of complement. Kidney International. 2005;67:524-30
144. Panitchote A, Mehkri O, Hastings A. et al. Factors associated with acute kidney injury in acute respiratory distress syndrome. Ann Intensive Care. 2019;9:74
145. Darmon M, Clec'h C, Adrie C. et al. Acute Respiratory Distress Syndrome and Risk of AKI among Critically Ill Patients. Clinical Journal of the American Society of Nephrology. 2014;9:1347-53
146. Doi K, Ishizu T, Fujita T, Noiri E. Lung injury following acute kidney injury: kidney-lung crosstalk. Clin Exp Nephrol. 2011;15:464-70
147. Husain-Syed F, Birk HW, Seeger W, Ronco C. A Protective Kidney-Lung Approach to Improve Outcomes in Mechanically Ventilated Patients. Blood Purif. 2016;42:214-8
148. Basu RK, Wheeler DS. Kidney-lung cross-talk and acute kidney injury. Pediatr Nephrol. 2013;28:2239-48
149. Domenech P, Perez T, Saldarini A. et al. Kidney-lung pathophysiological crosstalk: its characteristics and importance. Int Urol Nephrol. 2017;49:1211-5
150. Ricksten SE, Bragadottir G, Redfors B. Renal oxygenation in clinical acute kidney injury. Crit Care. 2013;17:221
151. Husain-Syed F, Slutsky AS, Ronco C. Lung-Kidney Cross-Talk in the Critically Ill Patient. Am J Respir Crit Care Med. 2016;194:402-14
152. Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S. et al. Covid-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev Med Virol. 2020:e2176.
153. Silvester W. Mediator removal with CRRT: Complement and cytokines. American Journal of Kidney Diseases. 1997;30:S38-S43
154. Tian H, Sun T, Hao D. et al. The optimal timing of continuous renal replacement therapy for patients with sepsis-induced acute kidney injury. Int Urol Nephrol. 2014;46:2009-14
155. Cha RH, Joh JS, Jeong I. et al. Renal Complications and Their Prognosis in Korean Patients with Middle East Respiratory Syndrome-Coronavirus from the Central MERS-CoV Designated Hospital. Journal of Korean Medical Science. 2015;30:1807-14
156. Okoh AK, Bishburg E, Grinberg S, Nagarakanti S. Tocilizumab use in COVID-19-associated pneumonia. Journal of Medical Virology. 2021;93:1023-8
157. Choy EH, De Benedetti F, Takeuchi T. et al. Translating IL-6 biology into effective treatments. Nature Reviews Rheumatology. 2020;16:335-45
158. Ibrahim YF, Moussa RA, Bayoumi AMA, Ahmed AF. Tocilizumab attenuates acute lung and kidney injuries and improves survival in a rat model of sepsis via down-regulation of NF-kappa B/JNK: a possible role of P-glycoprotein. Inflammopharmacology. 2020;28:215-30
159. Aziz M, Haghbin H, Abu Sitta E. et al. Efficacy of tocilizumab in COVID-19: A systematic review and meta-analysis. J Med Virol. in press
160. Lindorfer MA, Cook EM, Reis ES. et al. Compstatin Cp40 blocks hematin-mediated deposition of C3b fragments on erythrocytes: Implications for treatment of malarial anemia. Clin Immunol. 2016;171:32-5
161. van Griensven M, Ricklin D, Denk S. et al. Protective Effects of the Complement Inhibitor Compstatin CP40 in Hemorrhagic Shock. Shock. 2019;51:78-87
162. Mastellos DC, Reis ES, Biglarnia AR. et al. Taming hemodialysis-induced inflammation: Are complement C3 inhibitors a viable option? Clin Immunol. 2019;198:102-5
163. Reis ES, Berger N, Wang X. et al. Safety profile after prolonged C3 inhibition. Clin Immunol. 2018;197:96-106
164. Mastaglio S, Ruggeri A, Risitano AM. et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450
165. Zilberman-Itskovich S, Abu-Hamad R, Stark M, Efrati S. Effect of anti-C5 antibody on recuperation from ischemia/reperfusion-induced acute kidney injury. Ren Fail. 2019;41:967-75
166. Laurence J, Mulvey JJ, Seshadri M. et al. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin Immunol. 2020;219:108555
167. Al-helfawi MA. Potential Approach for Fighting Against Corona Virus Disease. ASRJETS. 2020;66:127-44
168. Chen W. A potential treatment of COVID-19 with TGF-beta blockade. Int J Biol Sci. 2020;16:1954-5
169. Uckun FM TV. Medical-Scientific Rationale for a Randomized, Placebo-Controlled, Phase 2 Study of Trabedersen/OT-101 in COVID-19 Patients with Hypoxemic Respiratory Failure. Anna pul and Cri Car Med. 2020;3:01-9
170. Gu YY, Zhang M, Cen H. et al. Quercetin as a potential treatment for COVID-19-induced acute kidney injury: Based on network pharmacology and molecular docking study. PLoS One. 2021;16:e0245209
Author contact
![]() Corresponding author: Hui-Yao Lan, Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China. E-mail: hylanedu.hk.
Corresponding author: Hui-Yao Lan, Departments of Medicine & Therapeutics, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China. E-mail: hylanedu.hk.