Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The role of miRNAs in regulating...
MiRNAs orchestrate the...
The role of miRNAs in modulation...
MiRNAs participate in the...
The role of miRNAs in NAFLD...
MiRNAs influence NAFLD by...
Extracellular vesicles (EVs) in...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(7):1851-1863. doi:10.7150/ijbs.59588 This issue Cite
Review
MicroRNAs in the Pathogenesis of Nonalcoholic Fatty Liver Disease
Zhiqiang Fang1, Guorui Dou2 ![]() , Lin Wang1
, Lin Wang1 ![]()
1. Department of Hepatobiliary Surgery, Xi-Jing Hospital, Fourth Military Medical University, Xi'an 710032, China.
2. Department of Ophthalmology, Xi-Jing Hospital, Fourth Military Medical University, Xi'an 710032, China.
Received 2021-2-23; Accepted 2021-4-14; Published 2021-4-29
Abstract

Nonalcoholic fatty liver disease (NAFLD), or, more accurately, metabolic associated fatty liver disease, accounts for a large proportion of chronic liver disorders worldwide and is closely associated with other conditions such as cardiovascular disease, obesity, and type 2 diabetes mellitus. NAFLD ranges from simple steatosis to nonalcoholic steatohepatitis (NASH) and can progress to cirrhosis and, eventually, also hepatocellular carcinoma. The morbidity and mortality associated with NAFLD are increasing rapidly year on year. Consequently, there is an urgent need to understand the etiology and pathogenesis of NAFLD and identify effective therapeutic targets. MicroRNAs (miRNAs), important epigenetic factors, have recently been proposed to participate in NAFLD pathogenesis. Here, we review the roles of miRNAs in lipid metabolism, inflammation, apoptosis, fibrosis, hepatic stellate cell activation, insulin resistance, and oxidative stress, key factors that contribute to the occurrence and progression of NAFLD. Additionally, we summarize the role of miRNA-enriched extracellular vesicles in NAFLD. These miRNAs may comprise suitable therapeutic targets for the treatment of this condition.
Keywords: miRNAs, Nonalcoholic fatty liver disease, NAFLD, Pathogenesis
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a clinicopathological syndrome caused by excessive fat deposition in hepatocytes of people who consume little or no alcohol. Due to dramatic lifestyle changes over recent decades, the number of NAFLD patients has increased significantly, accounting for a growing proportion of end-stage liver diseases [1]. It is estimated that approximately a quarter of the world's population has NAFLD, but its global distribution is not uniform [2]. NAFLD includes a collection of histopathological changes varying from simple steatosis to nonalcoholic steatohepatitis that can progress to cirrhosis and, eventually, hepatocellular carcinoma (HCC) [3].
Although the pathogenesis of NAFLD is very complex, existing evidence indicates that it is associated with the gut microbiome, bile acids, immunity, adipokines, oxidative stress, and genetic and epigenetic factors. Among the latter, miRNAs are suggested to play a key role in the occurrence and development of NAFLD. MiRNAs comprise at most 22 nucleotides and exert their activity by interfering with target mRNAs [4]. They are indispensable for numerous biological processes, such as proliferation, apoptosis, development, differentiation, metabolism, and carcinogenesis [5]. Evidence accumulated over recent years has indicated that miRNA dysregulation is involved in the occurrence and development of NAFLD, mainly through its effects on lipid metabolism, inflammation, apoptosis, fibrosis, insulin resistance, and oxidative stress. Recently, miRNA-enriched extracellular vesicles (EVs) have also been implicated in the pathogenesis of this condition. Accordingly, in this review, we focus on the role of miRNAs in the above-mentioned mechanisms, aiming to increase our understanding of the complex role of specific miRNAs in NAFLD and provide promising perspectives for therapeutic strategies to treat this disease.
The role of miRNAs in regulating lipid metabolism in NAFLD
The liver is an important organ for lipid metabolism. Once lipid metabolic homeostasis is disrupted, excessive fat will accumulate in the liver, mainly in hepatocytes, which may eventually lead to the development of NAFLD. Lipid metabolism is influenced by several biological processes, but mainly including de novo lipogenesis (DNL), fatty acid intake, fatty acid oxidation (FAO), and very-low-density lipoprotein (VLDL) export. Once some of the above processes become dysregulated, hepatic lipid metabolism will be disrupted, manifesting as excessive hepatic triglyceride (TG) accumulation [6]. Excessive TG deposition leads to the generation of hepatic steatosis, rendering the liver vulnerable to attack by factors such as proinflammatory cytokines, mitochondrial dysfunction, oxidative or endoplasmic reticulum (ER) stress, and the gut microbiome, further leading to the occurrence of inflammation, cellular apoptosis, or necrosis and fibrosis [7].
MiR-21, widely expressed in many types of tissues in humans [8], is dysregulated in cancer, inflammation, fibrosis, and NAFLD [9]. The level of circulating miR-21 and its expression in the liver is elevated in NAFLD patients and mouse models [9-11]. Calo et al. found that liver-specific miR-21/miR-21 knockout (LImiR21KO) mice fed a high-fat diet exhibited decreased hepatic steatosis through the regulation of several key transcription factors such as FOXA2, FOXO1, HNF4α, STAT3, and INSIG2 [12]. Moreover, miR-21 was also shown to inactivate the WNT/β-catenin signaling pathway by targeting LRP6, thereby aggravating lipid accumulation and inflammation [13], while Huang et al. reported that long noncoding RNA (lncRNA) MEG3 competitively bound to miR-21 with LRP6 and promoted lipid accumulation through inhibiting the mTOR pathway [14]. MiR-21 also promotes hepatosteatosis and cancer progression through the Hbp1-p53-Srebp1c pathway [15]. Several studies have found that inhibiting miR-21 could alleviate steatosis through activating PPARα [16, 17]; however, the role of miR-21 in targeting PPARα requires further investigation [18]. In addition, miR-21 also promotes the progression of NAFLD-related HCC (NAHCC) through PI3K/AKT, TGF-β, and STAT3 signaling [19], suggesting that miR-21 may be involved in various stages of NAFLD, from steatosis to NAHCC.
MiR-122, the most abundant miRNA in the liver [20], is expressed specifically in hepatocytes [8] and is involved in NAFLD development mainly through regulating lipid metabolism [21]. In NASH patients, the level of circulating miR-122 is significantly upregulated [22] and the hepatic expression of miR-122 is decreased [23], indicating that the miR-122 present in serum is released by hepatocytes [24]. MiR-122 can regulate lipid metabolism via multiple signaling pathways. Chai et al. found that free fatty acids (FFAs) could induce the expression of miR-122 and promote its secretion to circulating blood, and that miR-122 could suppress TG synthesis by targeting Agpat1 and Dgat1 as well as increasing beta-oxidation. The authors also found that inhibiting miR-122 led to the downregulation of FoxO1 and the upregulation of PPARγ, indicating that miR-122 is involved in multiple lipid metabolism-related pathways [25]. Intriguingly, adipose can secrete miR-122-containing exosomes that are absorbed by the liver, thereby alleviating NAFLD progression, which partly accounts for the paradoxically high levels of circulating miR-122 and its low expression in the liver of NAFLD patients [26]. Additionally, miR-122 can indirectly downregulate FASN and ACC and decrease cholesterol synthesis to ameliorate steatosis [27]. In addition to their function as upstream regulators of target mRNAs, microRNAs can themselves serve as downstream targets. For example, circRNA_002581 can suppress miR-122 expression, leading to the upregulation of CPEB1 and phosphorylated-mTOR and the concomitant suppression of autophagy, finally resulting in aggravated steatosis, inflammation, hepatocyte apoptosis, and oxidative stress [28]. In contrast, however, Long et al. reported that miR-122 could directly bind to the 3'-UTR of Sirt1, thereby inhibiting its expression and promoting lipogenesis [29]. These contradictory results suggest that the function of miR-122 in NAFLD pathogenesis is complex and warrants further investigation.
Although the expression of miR-34a in hepatocytes is not high, miR-34a may nonetheless be closely correlated with lipid metabolism. It is well known that Sirt1 is a direct target of miR-34a. Ding et al. demonstrated that inhibiting miR-34a could alleviate hepatosteatosis by targeting PPARα and Sirt1 [30]. Cholesterol metabolism also influences NAFLD severity. By inhibiting Sirt1 expression, miR-34a can promote that of HMGCR, which leads to increased free cholesterol levels and the aggravation of NAFLD [31]. Moreover, miR-34a decreases VLDL secretion by downregulating the expression of HNF4α, leading to increased lipid accumulation [32]. In addition to functioning as an upstream regulator, miR-34a is itself targeted and upregulated by FoxO3, which promotes palmitate (PA)-induced cholangiocyte lipoapoptosis [33]. Additionally, circRNA_0046367 releases the inhibitory effect of miR-34a on PPARα through disrupting the miRNA/mRNA interaction, thus alleviating lipid peroxidation [34]. Kim et al. identified the inhibitory role of LXRα in hepatocyte autophagy by upregulating the expression of miR-34a [35], while the farnesoid X receptor/miR-34a/sirtuin 1 pathway was proposed as a potential entry point for treating NAFLD [36]. MiR-33 also plays a pivotal role in lipid metabolism in NAFLD as excellently illustrated by Rottiers and Näär [37]. Other miRNAs with lipid metabolism regulatory roles in NAFLD are summarized in Table 1.
MiRNAs that regulate lipid metabolism in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets |
|---|---|---|---|---|
| miR-21 | ↑10,11 | ↑9,15 | ↑ | FOXA2, FOXO1, HNF4α, STAT3, INSIG2,12 LRP6,13 Hbp1,15 PPARα16,17 |
| miR-122 | ↑22,38 | ↑22↓23 | ↓25,28↑27 | Agpat1, Dgat1,25 CPEB1,28 Sirt129 |
| miR-34a | ↑10,11 | ↑32,30 | ↑ | Sirt1,30,31,36 HNF4α,32 PPARα30 |
| miR-27a | ↓39 | ↑40 | ↓40↑41 | FAS, SCD1,40 Nrf241 |
| miR-29a | ↓42 | ↓43in fibrosis | ↓ | HMGCR,44 DNMT3b,45 CD36,43 GSK3β46 |
| miR-132 | unknown | ↑47 | ↑ | FoxO3, PTEN, Sirt1, CYP2E1, P30047 |
| miR-125b | ↑22 | ↑48 | ↓ | FAS49 |
| miR-155 | unknown | ↑50,51 | ↓ | Ces3/TGH,50 LXα52 |
| miR-192 | ↑11,22 | ↓53 | ↓ | SREBF1,54 SCD153 |
| miR-378 | unknown | ↑55,56 | ↑ | Nrf156 |
* ↑: aggravating hepatic steatosis; ↓: alleviating hepatic steatosis.
In summary, this part mainly focused on the regulatory mechanisms underlying the roles of several miRNAs in lipid metabolism in NAFLD/NASH and briefly introduced their expression levels in peripheral blood and liver. Given that serum levels of some miRNAs, such as miR-21, miR-122, and miR-192, differ significantly between NAFLD/NASH patients and healthy controls, they have the potential to serve as biomarkers for noninvasive diagnosis. MiRNAs participate in every stage of lipid metabolism, including DNL, fatty acid oxidation, lipid transportation, and cholesterol metabolism. Lipid accumulation is regarded as the “first hit” in NAFLD. Consequently, alleviating steatosis represents an efficient strategy for blocking NAFLD progression and the above-mentioned miRNAs may provide a theoretical basis for the miRNA-based treatment of this disease.
MiRNAs orchestrate the progression of NAFLD via insulin signaling
Insulin resistance (IR) refers to a decrease in the efficiency of insulin in promoting glucose uptake and utilization. To compensate for increased blood glucose levels, the body produces and secretes excessive amounts of insulin, known as hyperinsulinemia [57]. Numerous studies have suggested that both genetic and environmental factors are related to the occurrence of IR, with obesity and hyperglycemia playing a key role [58]. Insulin resistance plays a critical role in the pathogenesis of NAFLD, increasing DNL and accelerating adipose tissue lipolysis, thereby leading to excessive fatty acid accumulation in the liver [57]. IR also disrupts the function of adipose tissue, consequently disrupting the normal regulation of inflammatory cytokines and adipokines [59]. The above consequences result in inflammation, oxidative stress, and apoptosis, which in turn maintain and promote the IR condition [58].
Numerous miRNAs have been shown to participate in insulin signal transduction and may be therapeutic targets in NAFLD. For instance, the PI3K/PDK1/AKT pathway is a key player in insulin signaling, and PI3K activity is regulated by phosphatase and tensin homolog (PTEN) and Src homology 2 domain-containing inositol 5'-phosphatase 2 (SHIP2). The hepatic expression of miR-152 was markedly downregulated in db/db mice and mice fed a high-fat diet, which leads to impaired hepatic glycogenesis—a hallmark of IR in hepatocytes—via PTEN upregulation [60]; meanwhile, miR-499-5p exerts a similar effect via the same target [61]. This results in dysregulated insulin signaling and excessive fatty acid accumulation in the liver. Several other miRNAs are known to target insulin signaling pathways related to the pathogenesis of NAFLD. For instance, Fu et al. showed that the downregulation of miR-26a contributed to the development of IR via multiple pathways [62], while Wang and colleagues reported that high miR-497 levels could inhibit insulin receptor expression and inactivate the IRS-1/PI3K/Akt/GSK-3b/GS pathway, thereby inducing hepatic IR [63]. MiR-15b exerts the same effect via InsR [64]. MiR-30b was upregulated in the liver of rats fed a high-fat diet and its overexpression can promote IR through suppressing sarco(endo)plasmic reticulum Ca2+-ATPase 2b (SERCA2b) translation [65]. Moreover, miR-206 can simultaneously facilitate insulin signaling and decrease hepatic lipogenesis through inhibiting protein tyrosine phosphatase non-receptor type 1 (PTPN1) [66]. In contrast, Xu et al. reported that miR-190b had the opposite effect on lipid metabolism and insulin sensitivity in NAFLD [67]. In addition, the serum level of miR-103 is higher in NAFLD patients than in healthy controls and is closely related to IR, and may serve as a biomarker for this condition in NAFLD [68]. The brief information of the above-mentioned miRNAs has been summarized in Table 2.
MiRNAs that regulate insulin resistance in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets |
|---|---|---|---|---|
| miR-152 | unknown | ↓60 | ↓ | PTEN60 |
| miR-499-5p | unknown | ↓61 | ↓ | PTEN61 |
| miR-26a | unknown | ↓62 | ↓ | GSK3β, PKCδ, PKCθ, ACSL3, ACSL4, PCK1, TCF7L262 |
| miR-497 | unknown | ↑63 | ↑ | InsR63 |
| miR-15b | ↑69 | ↑64,69 | ↑ | InsR64 |
| miR-30b | ↑65 | ↑65 | ↑ | SERCA2b65 |
| miR-206 | unknown | ↓66 | ↓ | PTPN166 |
| miR-190b | unknown | ↑67 | ↑ | IGF-1, ADAMTS967 |
* ↑: aggravating insulin resistance; ↓: alleviating insulin resistance.
In summary, we briefly introduced several miRNAs involved in the regulation of insulin signaling and, consequently, lipid metabolism. It is well known that IR contributes to excessive lipid accumulation in the liver, which can lead to steatosis and the progression from NAFLD to NASH. Moreover, therapy focusing on insulin signaling may also be beneficial for other metabolic disorders such as T2DM and obesity, as well as for NAFLD patients with these conditions.
The role of miRNAs in modulation of oxidative stress and ER stress in NAFLD
Redox homeostasis is vital for the maintenance of normal cellular functions and cell fate commitment [70]. Oxidative stress, which refers to disturbances in the regulatory role of reactive oxygen species (ROS), can interfere with various physiological processes and participate in the pathogenesis of various diseases [71]. ROS are widely defined as reactive oxidizing substances generated mainly in mitochondria, peroxisomes, and ER as a result of both enzymatic and nonenzymatic reactions [70]. Excessive ROS production disrupts normal biological processes by damaging molecules such as DNA, proteins, and lipids [70]. The ER is associated with protein maturation. When excessive FFAs flow into the liver, the ER in hepatocytes must process increased amounts of protein, leading to greater numbers of misfolded proteins, defined as ER stress, and the activation of a protective program called the unfolded protein response (UPR). Sustained activation of the UPR promotes NAFLD progression by affecting lipid accumulation, mitochondrial activities, and insulin signaling [72]. During NAFLD progression, an excess of FFAs leads to ROS overproduction, which can damage mitochondria and lead to lipotoxicity [73]. In turn, lipotoxicity leads to ER stress, increased inflammation, hepatocyte damage, and, finally, hepatocyte death [74]. Interestingly, ER stress also promotes ROS production [74]. Therefore, oxidative stress and ER stress can interact with each other and jointly promote NAFLD development.
In addition to IR, miR-26a also plays a role in ER stress. Xu et al. found that miR-26a was induced in liver cell lines treated with an ER stress inducer while hepatic expression of miR-34a is reduced in NAFLD patients, suggesting that there is an ER stress/miR-26a feedback circuit in hepatocytes. In other words, ER stress in NAFLD stimulates the upregulation of miR-26a, which, in turn, alleviates ER stress and lipid accumulation [75]. Cheng and colleagues proposed that miR-421 inhibited SIRT3, thereby interfering with normal mitochondrial function in NAFLD. In a mouse model of NAFLD, miR-421 expression was markedly increased, which aggravated oxidative stress and increased lipid accumulation through SIRT3/FOXO3 signaling. The authors concluded that suppressing hepatic miR-421 might alleviate oxidative stress-induced cellular damage in NAFLD [76]. Nuclear factor-erythroid 2-related factor 2 (Nrf2), a key modulator of the cellular antioxidant system, can also affect the pathogenesis of NAFLD. The expression of Nrf2 can be inhibited when miR-27a is overexpressed, leading to increased ROS production and lipid accumulation [41]. Furthermore, miR-136 was reportedly downregulated by high-content hydrogen water, resulting in increased levels of Nrf2 and MEG3 [77]. Cytochrome C participates in ROS production and mitochondrial apoptosis. Zhang et al. found that the upregulation of miR-96-5p ameliorated NAFLD through inhibition of the p66shc/cytochrome C cascade [78]. The brief information of the above-mentioned miRNAs has been summarized in Table 3.
MiRNAs that regulate oxidative stress and endoplasmic reticulum (ER) stress in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets(references) |
|---|---|---|---|---|
| miR-26a | unknown | ↓75 | ↓ | EIF2α75 |
| miR-421 | unknown | ↑76 | ↑ | Sirt376 |
| miR-27a | ↓39 | ↑41 | ↑ | Nrf241 |
| miR-136 | unknown | ↑77 | ↑ | MEG377 |
| miR-96-5p | unknown | ↓78 | ↓ | p66shc78 |
* ↑: promoting oxidative stress/ER stress; ↓: improving oxidative stress/ER stress.
In summary, we briefly described the roles of several miRNAs in the regulation of oxidative stress and ER stress. Oxidative stress and ER stress contribute to lipid accumulation and hepatocyte apoptosis, thus aggravating the progression of NAFLD. Many miRNAs, such as miR-26a, miR-27a, and miR-136, can improve the metabolic state of liver cells by regulating key redox pathways and molecules, thereby affecting NAFLD progression. These findings may provide clues for aiding the monitoring of disease progression and the development of miRNA-based targeted therapy for the treatment of NAFLD.
MiRNAs participate in the regulation of NADLD related hepatocyte apoptosis
Apoptosis is common in acute and chronic liver injury. Different from necrosis, apoptosis is executed through a precisely designed program that usually occurs in only a single cell, having little effect on surrounding cells such that tissue homeostasis or organ development is not disrupted [79]. Under physiological conditions, cellular apoptosis in the liver is relatively rare and well-organized. However, inflammation, lipotoxicity, mitochondrial dysfunction, or other injuries can result in excessive apoptosis in the liver. Several key molecular pathways, such as the Bcl-2, caspase, and c-Jun N-terminal kinase (JNK) signaling pathways, are involved in the regulation of hepatocyte apoptosis, a key factor in the pathogenesis of NAFLD/NASH. Apoptotic hepatocytes will, in turn, stimulate surrounding HSCs and immune cells, leading to aggravated hepatic inflammation and the occurrence and progression of fibrosis [80].
In addition to playing a role in hepatic inflammation and fibrosis, miR-223 is also involved in hepatocyte apoptosis. For instance, Qadir et al. found that knockdown of miR-223 could alleviate Fas-induced hepatocyte apoptosis via targeting IGF-1R [81], suggesting that miR-223 may also interfere with the progression of NAFLD through regulation of hepatocyte apoptosis. This possibility merits further investigation. Additionally, Feng et al. found that miR-24 could suppress hepatocyte apoptosis via the proapoptotic Bcl-2 homolog, BIM, a contributor to TNF-α-induced apoptosis, in a lipopolysaccharide (LPS)-induced mouse model of acute liver failure [82]; however, whether miR-24 exerts the same anti-apoptotic function in NAFLD remains unknown. Deoxycholic acid (DCA) is a free bile acid reported to induce apoptosis via multiple pathways [83]. Rodrigues et al. showed that DCA inhibited the NF-κB pathway, decreased the expression of miR-21, increased the expression of the miR-21 proapoptotic target programmed cell death 4 (PDCD4), and promoted apoptosis in primary rat hepatocytes. In contrast, the overexpression of miR-21 decreased the levels of hepatocellular apoptosis [83]. The same authors found that DCA activated miR-34a to induce apoptosis via targeting SIRT1 [84] and other groups have also reported on the proapoptotic role of miR-34a [85,86]. The BH3-only protein PUMA is upregulated during lipoapoptosis. Cazanave et al. showed that the liver expression of miR-296 was decreased in NASH patients, while the overexpression of miR-296 in Huh7 cells ameliorated lipoapoptosis via targeting PUMA [87]. The brief information of the above-mentioned miRNAs has been summarized in Table 4.
MiRNAs that regulate hepatocyte apoptosis in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets(references) |
|---|---|---|---|---|
| miR-223 | ↑88 | ↑89 | ↑? | IGF-1R81 |
| miR-24 | unknown | ↑90 | ↓? | BIM82 |
| miR-21 | ↑10,11 | ↑9,15 | ↓? | PDCD483 |
| miR-34a | ↑10,11 | ↑32,30 | ↑ | Sirt184-86 |
| miR-296 | unknown | ↓87 | ↓ | PUMA87 |
* ↑: promoting apoptosis; ↓: suppressing apoptosis.
In summary, we introduced several miRNAs involved in the regulation of hepatocyte apoptosis, mainly focusing on the associated mechanisms. Apoptotic hepatocytes can contribute to the progression of inflammation and fibrosis, indicating that hepatocyte apoptosis plays a central role in the pathogenesis of NAFLD and NASH. Alleviating hepatocyte apoptosis will likely limit inflammation and fibrosis, thereby inhibiting the progression of NAFLD. This observation highlights that miRNA-based targeted therapy for inhibiting hepatocyte apoptosis holds great promise for the treatment of NAFLD/NASH. Nevertheless, most studies investigating hepatocellular apoptosis-related miRNAs have focused on HCC; other miRNAs associated with hepatocellular apoptosis in NAFLD/NASH need to be identified and explored.
The role of miRNAs in NAFLD related hepatic inflammation
Inflammation participates in the pathogenesis of many liver diseases, including NAFLD [91]. It is a required pathological feature for defining NASH and differentiating NASH from simple steatosis [92]. Inflammation is also a key factor in the progression of NASH to cirrhosis and, finally, HCC [93]. Inflammation plays both injurious and beneficial roles in the pathogenesis of NAFLD, depending on the types of inflammatory cells and inflammatory mediators involved, as well as on the stage of the disease [94]. Inflammation can help heal the liver by promoting liver regeneration and the clearing of dead or unhealthy cells. In contrast, continuous inflammation can negatively affect normal physiological activities and destroy normal tissue structure, and can, in the end, lead to the occurrence of fibrosis. Hepatic inflammation in NAFLD likely originates outside the liver and inside the liver, a topic that has been reviewed elsewhere [95]. MiRNAs have been shown to participate in hepatic inflammatory responses and influence the progression of NAFLD/NASH [96, 97].
Until recently, miR-223 expression was thought to be neutrophil/myeloid-specific [8], however, it is now known that miR-223 is also expressed in hepatocytes [98]. MiR-223, an miRNA with anti-inflammatory functions, was reported to ameliorate hepatic inflammation in NASH via targeting Cxcl10 and transcriptional coactivator with PDZ-binding motif (TAZ) in hepatocytes [89]. Moreover, miR-223 plays a vital role in the crosstalk between inflammatory cells and hepatocytes via EVs. For example, IL-6 reportedly stimulates myeloid cells to release exosomes containing miR-223 that are absorbed by hepatocytes. Subsequently, miR-223 alleviates liver fibrosis by suppressing the expression of its profibrotic target genes, including Nlrp3, Igf1r, Cxcl10, and TAZ [88]. Additionally, hepatocytes can absorb neutrophil-derived miR-223-enriched EVs in a LDLR- and apolipoprotein E (APOE)-dependent manner [99]. The specific role of these two molecules in the selective control of miRNA-223 transfer has potential as a therapeutic target for the treatment of NASH.
MiR-194 is also involved in NAFLD-related inflammation, but not in HCC progression and metastasis [8]. Nie et al. demonstrated that miR-194 could suppress FXR/Nr1h4 expression, thereby promoting inflammatory responses and metabolic dysfunction [100], while Tian et al. showed that the overexpression of miR-194 could reduce the expression levels of TRAF6, a downstream effector of the TLR4 pathway, thus alleviating inflammatory responses [101]. These contradictory results illustrate the complexity associated with miRNA regulatory networks and that of the function of a single miRNA. Additionally, several miRNAs involved in the regulation of lipid metabolism also play a role in inflammation. For instance, exosomal miR-192-5p derived from NAFLD hepatocytes could promote macrophage activation through the Rictor/Akt/FoxO1pathway [102]. This suggests that hepatocyte-derived exosomes can also have an important effect on surrounding inflammatory cells. Besides increasing lipid accumulation, miR-378 can also promote hepatic inflammation through activating the NF-κB/TNF-α pathway [103]. Similarly, miR-125b can also enhance NF-κB signaling by targeting TNFAIP3, thereby aggravating inflammatory responses [48]. Further, miR-451 activates the AMPK/AKT pathway, thus decreasing hepatic inflammation by suppressing the activation of NF-κB via targeting Cab39 [104]. The brief information of the above-mentioned miRNAs has been summarized in Table 5.
MiRNAs that regulate hepatic inflammation in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets(references) |
|---|---|---|---|---|
| miR-194 | unknown | ↑100 | ↑100↓101 | FXR/Nr1h4,100 TRAF6101 |
| miR-223 | ↑88 | ↑89 | ↓ | Cxcl10, TAZ,89,88 NLRP3,88,105 Igf1r88 |
| miR-192-5p | ↑102 | ↑102 | ↑ | Rictor102 |
| miR-378 | unknown | ↑103 | ↑ | Prkag2103 |
| miR-125b | ↑22 | ↑48 | ↑ | TNFAIP348 |
| miR-451 | unknown | ↓104 | ↓ | Cab39104 |
* ↑: promoting inflammation; ↓: suppressing inflammation.
In summary, we introduced the specific mechanisms underlying the roles of several miRNAs involved in the regulation of hepatic inflammation in NASH. Several canonical inflammation-related pathways, such as the NF-κB/TNF-α and TLR4 pathways, are regulated by miRNAs, including miR-194, miR-378, miR-125b, and miR-451. MiR-223 can alleviate hepatic inflammation and fibrosis via multiple targets. The crosstalk between hepatocytes and surrounding cells, such as macrophages and neutrophils, plays a pivotal role in hepatic inflammation. It is increasingly recognized that EVs are involved in the occurrence and development of hepatic inflammation in NASH. The function of miR-223-enriched EVs in inflammation has been excellently illustrated by Wang et al. [88] and He et al. [99].
MiRNAs influence NAFLD by regulating the progression of liver fibrosis
Liver fibrosis is a pathological process in which extracellular matrix accumulates and damage repair persists [106]. Different stages of fibrosis indicate different outcomes in NAFLD and fibrosis is an important predictor of mortality [107]. Hepatocyte lipoapoptosis is the main driving force for the progression of fibrosis [108]. Pathological conditions promote the activation of a large number of HSCs, which are the main source of fibrogenic myofibroblasts (MFs; primary cells that produce extracellular matrix). An excessive number of MFs leads to massive EM deposition, the main characteristic of liver fibrosis [107]. Multiple signal transduction pathways can regulate HSC activation, including the TGF-β/Smad, Notch, Wnt, Hedgehog, and integrin pathways. MiRNAs are currently considered to be a turning point for the noninvasive diagnosis of NAFLD [109]. Given that many miRNAs are involved in the development of fibrosis and HSC activation, they may provide references for the treatment of NAFLD/NASH and the prevention of disease progression.
Using high-throughput sequencing, Leti et al. found that the levels of many miRNAs were altered in NAFLD-related fibrosis, thereby providing new insights into the mechanisms underlying the pathogenesis of this condition [110]. Dattaroy et al. showed that the leptin-NADPH oxidase-mediated induction of miR-21, via the TGF-β signaling pathway, was a key regulatory step in NASH-related fibrogenesis [111]. In addition to promoting hepatosteatosis, miR-21 also contributes to liver fibrosis by inhibiting PPARα signaling [16]. High miR-21 expression is mainly observed in bile duct cells and inflammatory cells in NASH patients and mouse models [16], suggesting that suppressing hepatic miR-21 expression may reduce fibrosis by suppressing inflammation. It has been proposed that miR-21 may serve as a plasma biomarker for fibrotic liver disease [112]. Intriguingly, miR-21 was reported to be unnecessary for HSC activation and the development of liver fibrosis [113]. Consequently, the role of miR-21 in liver fibrosis and HSC activation remains unclear and requires further investigation. MiR-122 is highly expressed in hepatocytes and is correlated with cholesterol metabolism [114]. Csak et al. reported that the overexpression of miR-122 alleviated liver fibrosis by targeting HIF-1α, vimentin, and MAP3K3 [114]. Moreover, Du et al. showed that the proliferation and activation of HSCs could be suppressed by miR-146a-5p in NASH via Wnt1 and Wnt5a [115].
The Wnt signaling pathway is involved in various biological processes, including apoptosis, and the canonical Wnt/β-catenin signaling pathway is one of the main inducers of HSC apoptosis [116,117]. MiR-214 is markedly upregulated during HSC activation, which results in the inhibition of the expression of suppressor-of-fused homolog (Sufu), a negative regulator of the Hedgehog pathway, and the consequent promotion of HSC activation and liver fibrosis [118]. Moreover, miR-214 also enhances the activity of the EGFR and TGF-β signaling pathways by targeting Mig6, a negative regulator of EGFR signaling, thereby promoting HSC activation and liver fibrosis [119]. However, the role of miR-214 in NASH-related fibrosis remains unclear. Wang et al. demonstrated that miR-130a-3p targeted TGFBR1 and TGFBR2, which inactivated HSCs, promoted their apoptosis, and ameliorated fibrosis in NASH [120]. The TGF-β/Smad pathway can block the progression of liver fibrosis by inhibiting HSC collagen secretion and cell activation. Using a CCL4-induced liver fibrosis mouse model, Tsay et al. showed that downregulating miR-221-3p in hepatocytes could mitigate the symptoms of liver fibrosis by suppressing HSC activation via G protein alpha inhibiting activity polypeptide 2 (GNAI2) [121]. MiR-29 can also decrease inflammation and fibrosis in NASH by suppressing CD36 [43]. The brief information of the above-mentioned miRNAs has been summarized in Table 6.
MiRNAs that regulate liver fibrosis in nonalcoholic fatty liver disease (NAFLD).
| MiRNA | Circulation level | Liver expression | Function* | Identified targets(references) |
|---|---|---|---|---|
| miR-21 | ↑10,11 | ↑9,15 | ↑ | SMAD7,111 PPARα16 |
| miR-122 | ↑22,38 | ↓114 | ↓ | HIF-1α, vimentin, MAP3K3114 |
| miR-146a-5p | unknown | ↓115 | ↓ | Wnt1, Wnt5a115 |
| miR-214 | unknown | unknown | ↑? | Sufu,118 Mig-6119 |
| miR-130a-3p | unknown | ↓120 | ↓ | TGFBR1, TGFBR2120 |
| miR-221-3p | unknown | ↑121 | ↑? | GNAI2121 |
| miR-223 | ↑88 | ↑89 | ↓ | Nlrp3, Cxcl10, TAZ, Igf1r88 |
| miR-29a | ↓42 | ↓43 | ↓ | CD3643 |
In this section, we summarized the function of some miRNAs involved in the regulation of liver fibrosis. Many miRNAs participate in the regulation of classical fibrosis-related signaling pathways such as the TGF-β/Smad, Wnt, and Hedgehog pathways. A few miRNAs, including miR-214 and miR-221-3p, play a role in drug-induced fibrosis instead of NASH, and the role of these miRNAs in NASH-induced fibrosis needs to be investigated. Many miRNAs affect the progression of NAFLD through multiple processes, including steatosis, apoptosis, inflammation, and fibrosis. Consequently, whether these miRNAs directly interfere with the process of fibrosis or indirectly promote/aggravate fibrosis through affecting steatosis or inflammation needs to be determined. This will help clarify the underlying mechanisms and provide a solid theoretical basis for miRNA-based targeted therapy.
Extracellular vesicles (EVs) in NAFLD: potential biomarkers and therapeutic targets
EVs refer to vesicles encapsulated by phospholipid bilayers that are released by various types of cells. They can be classified into three groups, namely, exosomes (40-150 nm in diameter), microvesicles (50-1,000 nm in diameter), and apoptotic bodies (500 nm in diameter), according to their cellular origins [122]. EVs can carry numerous particles, including enzymes, growth factors, proteins, lipids, and noncoding RNAs, thereby mediating cell-to-cell communication [123]. Over recent years, EVs have been found to contribute to various liver diseases, such as hepatitis B and C, alcoholic liver disease (ALD), NAFLD, and HCC. Given that EVs carry specific proteins and lipids, they can recognize specific target cells through receptor-mediated or membrane-mediated processes, which also allow EVs to serve as biomarkers and therapeutic targets [122].
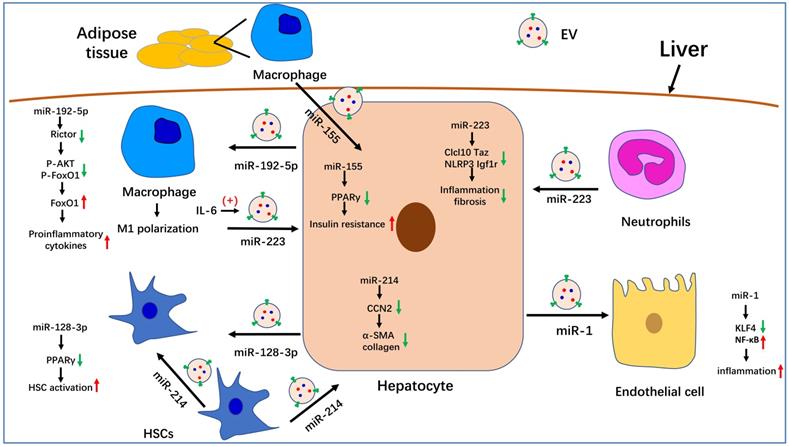
Several studies have suggested that circulating EV levels are increased in both experimental and human NASH [124]. Studies investigating the functions of EVs secreted by hepatocytes have mostly focused on their effects on liver cells, and EVs have been found to be mainly correlated with the regulation of inflammation and fibrosis. For example, in a NASH mouse model, hepatocytes released integrin β1-enriched EVs, which was absorbed by monocytes and increased their adhesion to liver sinusoidal endothelial cells, thus aggravating inflammation and fibrosis [125]. Jiang et al. showed that lipotoxic hepatocyte-derived and miR-1-enriched EVs promoted endothelial inflammation and atherogenesis, which was proposed to provide a theoretical basis for the treatment of atherosclerosis patients with NAFLD [126]. Lipotoxic hepatocyte-derived EVs can also be internalized by HSCs and induce their activation, likely through the downregulation of PPARγ induced by miR-128-3P contained in the EVs [127]. Hepatocyte-derived EVs can also be internalized by macrophages. For instance, sphingosine 1-phosphate (S1P)-enriched EVs released by lipotoxic hepatocytes can induce macrophage chemotaxis [128]. Similarly, mixed lineage kinase 3 (MLK3) is induced in lipotoxic hepatocytes, leading to the release of EVs containing C-X-C motif ligand 10 (CXCL10) and the induction of macrophage chemotaxis [129]. Endoplasmic reticulum to nucleus signaling 1 (ERN1) is also induced in hepatocytes of NASH livers and promotes EV release and macrophage chemotaxis [130]. Additionally, lipid stimulates death receptor 5 (DR5) and leads to increased release of hepatocyte-derived EVs containing TNF-related apoptosis-inducing ligand (TRAIL) and the upregulation of IL-1β and IL-6 expression in macrophages [131]. Furthermore, lipotoxic hepatocyte-derived exosomal miR-192-5p activates macrophages and increases the production of proinflammatory cytokines [102]. Inflammatory cells also release EVs to interact with hepatocytes. For example, IL-6 induces macrophages to release miR-223-enriched EVs, which are internalized by hepatocytes and lead to a reduction in the expression of profibrotic TAZ [88]. Neutrophils can also release miR-223-enriched EVs and ameliorate hepatic inflammation and fibrosis [99]. Interestingly, EVs can also be transferred among cells of the same type. For instance, miR-214-enriched exosomes can be transferred between HSCs, thereby reducing fibrogenesis [132]. Recent studies have demonstrated that adipose tissue-derived EVs can be transferred into the liver, such as that seen for adipose tissue macrophage-derived exosomal miR-155, which impairs the insulin sensitivity of hepatocytes via targeting PPARγ [133].
Given that circulating miRNAs are transported via EVs and those specific proteins and EVs can be absorbed by specific cells, miRNA-enriched EVs have great potential to serve as effective biomarkers and therapeutic targets. Considering that miRNAs in EVs are more stable than those exposed to blood, the diagnostic value of detecting miRNAs in EVs may be greater than that of detecting miRNAs extracted from serum. However, due to the lack of a uniform standard for separating EVs from serum, the diagnostic potential of EVs is currently limited. Recently, a few laboratories have tried to use EVs containing specific miRNAs to affect NAFLD progression. For example, Li et al. delivered miR-199a-5p- or anti-miR-199a-5p-containing exosomes to respectively aggravate or ameliorate lipid accumulation in a NAFLD mouse model [134]. However, the clinical translation of treatment based on miRNA-containing EVs is in its infancy and requires substantial investigation and confirmation.
In summary, we summarized the known roles of EVs in the pathogenesis of NAFLD. Serum EV levels are significantly increased in NASH mouse models and patients. Many molecules that are involved in NAFLD progression have been detected in EVs, especially miRNAs. The role of partial miRNA-enriched EVs in NAFLD has been summarized in Figure 1. Because EVs can be internalized by specific cells through specific membrane receptors, they function as a means of intercellular communication. These characteristics endow EVs with the potential to serve as biomarkers and therapeutic targets for the treatment of NAFLD/NASH.
The role of miRNA-enriched extracellular vesicles (EVs) in intercellular communication in nonalcoholic fatty liver disease (NAFLD). Adipose tissue-derived macrophages secrete miR-155-enriched EVs, which are internalized by hepatocytes and impair insulin sensitivity. MiR-223-enriched EVs released by neutrophils and macrophages are absorbed by hepatocytes and improve hepatic inflammation and fibrosis. MiR-214-containing EVs encapsulated by hepatic stellate cells (HSCs) can simultaneously be absorbed by HSCs and hepatocytes and decrease the expression of fibrogenesis-related genes by suppressing CCN2. Hepatocytes also secrete miR-192-5p-enriched EVs, which are internalized by macrophages, where they promote inflammation and induce M1 polarization. Hepatocytes also release miR-1-enriched EVs, which are transferred to endothelial cells to promote inflammation by targeting KLF4. Moreover, miR-128-3p-containing EVs released by hepatocytes can be taken up by HSCs and increase HSC activation by inhibiting PPARγ.
Conclusion
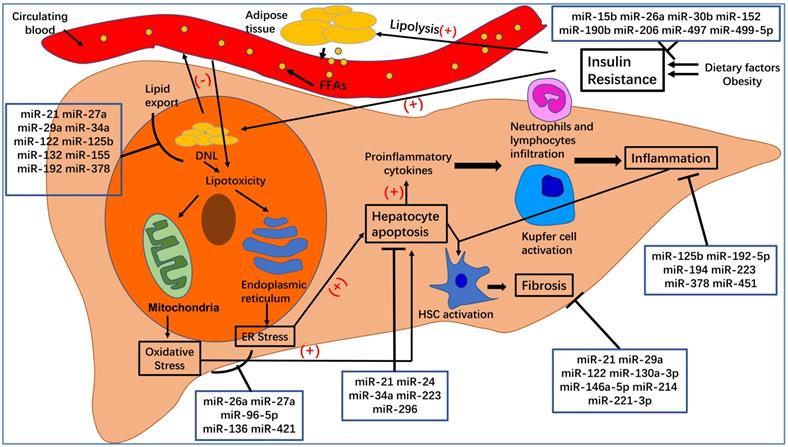
Here, we have summarized the roles of specific miRNAs in the pathogenesis of NAFLD (see Figure 2). We expected that this review may be of benefit to clinical research and disease treatment. MiRNAs undoubtedly play a critical role in the post-transcriptional regulation of target genes involved in the pathogenesis of NAFLD/NASH. Elaborating on the function of each miRNA in NAFLD is beyond the scope of this review. Instead, we have focused on several widely studied miRNAs, such as miR-21, miR-26a, miR-34a, miR-122, miR-194, and miR-223. Additionally, because exosomal miRNAs have attracted significant attention over recent years, we also summarized the role of EVs in NAFLD and the function of miRNA-enriched EVs in the pathogenesis of NAFLD. The relationship between miRNAs and their targets is complex. For example, miR-21 regulates NAFLD progression via multiple targets, including FoxA2, HNF4α, STAT3, [12] LRP6, [13] Hbp1, [15] PPARα, [16] SMAD7, [111] and PDCD4 [83]. One target gene can be regulated by several miRNAs. For instance, Sirt1 can be regulated by miR-34a, [30, 31, 36, 84-86] miR-122, [29] and miR-132 [47]. Moreover, multiple-to-multiple relationships between miRNAs and their target genes have been observed in HCC [135]. This phenomenon indicates that the miRNA regulatory network in NAFLD is complex and warrants further investigation.
The pathogenesis of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Dietary factors and obesity, important for the development of insulin resistance, increase the lipolysis of adipose tissue and de novo lipogenesis (DNL) in hepatocytes. An excess of free fatty acids from circulating blood enters the liver, which, together with increased DNL in hepatocytes, leads to hepatocyte steatosis and lipotoxicity. The latter can affect the normal functioning of mitochondria and endoplasmic reticulum, leading to oxidative stress and endoplasmic reticulum stress, which can increase hepatocyte apoptosis. Apoptotic hepatocytes increase the production of proinflammatory cytokines, recruit inflammatory cells to the liver, activate Kupfer cells, and lead to inflammation, which is a hallmark of NASH. At the same time, inflammation and hepatocyte apoptosis promote the activation of hepatic stellate cells (HSCs), which will transform into myofibroblasts and produce extracellular matrix. Excessive deposition of extracellular matrix leads to the occurrence of liver fibrosis.
Because of their stability, detectability, and evident changes in circulating levels in health and disease, miRNAs can serve as efficient, noninvasive biomarkers. For example, serum levels of miR-21 [10, 11], miR-122, [22] and miR-192 [102] are markedly upregulated in NAFLD and could serve as serum biomarkers for the early diagnosis of this disease. Moreover, miR-34a, miR-122, and miR-192 may be suitable for use as biomarkers to distinguish NAFLD and NASH severity [8]. However, it is difficult to diagnose NAFLD via a single miRNA because determining specificity remains problematic. For instance, the serum levels of miR-21 also increase in other liver diseases such as ALD, HCC, and viral hepatitis [136]. It is therefore more effective to use a combination of miRNAs or a combination of miRNAs and traditional biomarkers for the noninvasive diagnosis of NAFLD. Accordingly, Liu et al. proposed a composite biomarker that included miR-21, miR-192, miR-505, and alanine aminotransferase (ALT) to improve noninvasive NASH diagnosis [11], while Thietart et al. showed that a cluster of 12 miRNAs in serum EVs could distinguish patients with NASH from those with chronic hepatitis B and C or healthy controls [137]. Recently, an increasing number of studies have demonstrated that numerous miRNAs are transferred via EVs and that miRNAs in EVs are more stable than those in circulating blood. Therefore, isolating EVs from serum and detecting their miRNA concentrations may represent a novel and efficient strategy for noninvasive diagnosis. For instance, as mentioned above, exosomal miR-128-3p [127], miR-192-5p [102], miR-223, [88,99] and miR-155 [133] we have discussed before may serve as effective biomarkers in NAFLD. Moreover, our group also isolated plasma exosomes from mice with MCD-induced NASH and found that the expression levels of miR-144-3p, miR-23b-3p, and miR-126a-5p were notably upregulated; however, the roles of these miRNAs in NASH remain unknown. The lack of a unified standard for the isolation of EVs and limited sequencing technology capabilities constrain the development of EVs as biomarkers in NAFLD. Additionally, the validity and usefulness of utilizing circulating exosomal miRNAs as biomarkers in NAFLD require further investigation.
Given that miRNAs can regulate the progression of NAFLD via multiple target genes, treatment based on the relevant miRNAs has attracted increasing research interest. Many groups have already demonstrated the validity of miRNA-based treatment in animal models of NAFLD. For instance, Lin et al. reported that a miR-192 mimic delivered by lentivirus alleviated hepatic steatosis in mice [54]. Most studies exploring the function of specific miRNAs have used miRNA mimics or anti-miRNA delivered by viruses or transgenic mice; however, these methods are mostly not appropriate for use in a clinical setting. This highlights the need to find an appropriate miRNA delivery system. EVs are stable in circulating blood and can encapsulate many molecules, and packaging miRNAs (siRNAs or drugs) into EVs represents a promising strategy for the treatment of NAFLD. Li et al. packaged miR-199a-5p/anti-miR-199a-5p into exosomes and injected them into mice, resulting in aggravated/attenuated hepatic steatosis, respectively [134]. Additionally, He et al. reported the delivery of a miR-146b mimic to hepatocytes through lactosylated PDMAEMA nanoparticles [138]. However, the efficiency and safety of these targeted delivery systems remain to be determined. Meanwhile, because they have multiple targets and also target multiple cell types, the off-target effects of miRNA-based therapy must also be considered.
Due to the limitations of research techniques, our understanding of the miRNAs involved in the pathogenesis of NAFLD remains incomplete. Moreover, we could not elaborate on each miRNA that participates in NAFLD. However, with the development of bioinformatics and the progress of animal and clinical research, an increasing number of miRNAs involved in the occurrence and development of NAFLD will be identified, and these miRNAs might offer guidance for the noninvasive diagnosis and treatment of NAFLD.
Abbreviations
NAFLD: nonalcoholic fatty liver disease; T2DM: type 2 diabetes mellitus; NASH: nonalcoholic steatohepatitis; HCC: hepatocellular carcinoma; HSC: hepatic stellate cell; miRNAs: microRNAs; EVs: extracellular vesicles; DNL: de novo lipogenesis; FAO: fatty acid oxidation; VLDL: very-low-density lipoprotein; TG: triglyceride; ER: endoplasmic reticulum; NAHCC: NAFLD-related HCC; FFAs: free fatty acids; IR: insulin resistance; ROS: reactive oxygen species; UPR: unfolded protein response; JNK: c-Jun N-terminal kinase; DCA: deoxycholic acid; TAZ: transcriptional coactivator with PDZ-binding motif; MFs: fibrogenic myofibroblasts; ALD: alcoholic liver disease.
Acknowledgements
This study was supported by MOST (2016YFA0102100 [The National Key Research and Development Program of China: Stem Cell and Translational Research]), and NSFC 81422009, 81401940, 81670863, 81770560, 81800533, 31730041, 31671523).
Author Contributions
ZF wrote the manuscript. LW, GD gave constructive guidance and made critical revisions. All authors read and approved the submitted version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11-20
2. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E. et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology. 2019;69(6):2672-2682
3. Petroni ML, Brodosi L, Bugianesi E, Marchesini G. Management of non-alcoholic fatty liver disease. BMJ. 2021;372:m4747
4. Baffy G. MicroRNAs in Nonalcoholic Fatty Liver Disease. J Clin Med. 2015;4(12):1977-1988
5. Giordano S, Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57(2):840-847
6. Liu W, Cao H, Yan J, Huang R, Ying H. 'Micro-managers' of hepatic lipid metabolism and NAFLD. Wiley Interdiscip Rev RNA. 2015;6(5):581-593
7. Chao H-W, Chao S-W, Lin H, Ku H-C, Cheng C-F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2019;20(2):298
8. Wang X, He Y, Mackowiak B, Gao B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut. 2021;70(4):784-795
9. Benhamouche-Trouillet S, Postic C. Emerging role of miR-21 in non-alcoholic fatty liver disease. Gut. 2016;65(11):1781-1783
10. Pillai SS, Lakhani HV, Zehra M, Wang J, Dilip A, Puri N. et al. Predicting Nonalcoholic Fatty Liver Disease through a Panel of Plasma Biomarkers and MicroRNAs in Female West Virginia Population. Int J Mol Sci. 2020;21(18):6698
11. Liu J, Xiao Y, Wu X, Jiang L, Yang S, Ding Z. et al. A circulating microRNA signature as noninvasive diagnostic and prognostic biomarkers for nonalcoholic steatohepatitis. BMC Genomics. 2018;19(1):188
12. Calo N, Ramadori P, Sobolewski C, Romero Y, Maeder C, Fournier M. et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut. 2016;65(11):1871-1881
13. Wang X-M, Wang X-Y, Huang Y-M, Chen X, Lü M-H, Shi L. et al. Role and mechanisms of action of microRNA-21 as regards the regulation of the WNT/β-catenin signaling pathway in the pathogenesis of non-alcoholic fatty liver disease. Int J Mol Med. 2019;44(6):2201-2212
14. Huang P, Huang F-Z, Liu H-Z, Zhang T-Y, Yang M-S, Sun C-Z. LncRNA MEG3 functions as a ceRNA in regulating hepatic lipogenesis by competitively binding to miR-21 with LRP6. Metab Clin Exp. 2019;94:1-8
15. Wu H, Ng R, Chen X, Steer CJ, Song G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut. 2016;65(11):1850-1860
16. Loyer X, Paradis V, Hénique C, Vion A-C, Colnot N, Guerin CL. et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARα expression. Gut. 2016;65(11):1882-1894
17. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62(3):720-733
18. Mazzini GS, Khoraki J, Browning MG, Campos GM. Concurrent miR-21 suppression and FXR activation as a mechanism of improvement in nonalcoholic fatty liver disease. Cell Death Dis. 2018;9(3):354
19. Lai C-Y, Yeh K-Y, Lin C-Y, Hsieh Y-W, Lai H-H, Chen J-R. et al. MicroRNA-21 Plays Multiple Oncometabolic Roles in the Process of NAFLD-Related Hepatocellular Carcinoma via PI3K/AKT, TGF-β, and STAT3 Signaling. Cancers (Basel). 2021;13(5):940
20. Jopling C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2017;9(2):137-142
21. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M. et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87-98
22. Pirola CJ, Fernández Gianotti T, Castaño GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M. et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64(5):800-812
23. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW. et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48(6):1810-1820
24. Miyaaki H, Ichikawa T, Kamo Y, Taura N, Honda T, Shibata H. et al. Significance of serum and hepatic microRNA-122 levels in patients with non-alcoholic fatty liver disease. Liver Int. 2014;34(7):e302-e307
25. Chai C, Rivkin M, Berkovits L, Simerzin A, Zorde-Khvalevsky E, Rosenberg N. et al. Metabolic Circuit Involving Free Fatty Acids, microRNA 122, and Triglyceride Synthesis in Liver and Muscle Tissues. Gastroenterology. 2017;153(5):1404-1415
26. Baranova A, Maltseva D, Tonevitsky A. Adipose may actively delay progression of NAFLD by releasing tumor-suppressing, anti-fibrotic miR-122 into circulation. Obes Rev. 2019;20(1):108-118
27. Gjorgjieva M, Sobolewski C, Dolicka D, Correia de Sousa M, Foti M. miRNAs and NAFLD: from pathophysiology to therapy. Gut. 2019;68(11):2065-2079
28. Jin X, Gao J, Zheng R, Yu M, Ren Y, Yan T. et al. Antagonizing circRNA_002581-miR-122-CPEB1 axis alleviates NASH through restoring PTEN-AMPK-mTOR pathway regulated autophagy. Cell Death Dis. 2020;11(2):123
29. Long J-K, Dai W, Zheng Y-W, Zhao S-P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. 2019;25(1):26
30. Ding J, Li M, Wan X, Jin X, Chen S, Yu C. et al. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci Rep. 2015;5:13729
31. Min H-K, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J. et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665-674
32. Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat Commun. 2015;6:7466
33. Natarajan SK, Stringham BA, Mohr AM, Wehrkamp CJ, Lu S, Phillippi MA. et al. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J Lipid Res. 2017;58(5):866-875
34. Guo X-Y, Chen J-N, Sun F, Wang Y-Q, Pan Q, Fan J-G. circRNA_0046367 Prevents Hepatoxicity of Lipid Peroxidation: An Inhibitory Role against Hepatic Steatosis. Oxid Med Cell Longev. 2017;2017:3960197
35. Kim YS, Nam HJ, Han CY, Joo MS, Jang K, Jun DW. et al. LXRα activation inhibits autophagy and lipophagy in hepatocytes by dysregulating ATG4B and Rab-8B, reducing mitochondrial fuel oxidation. Hepatology. 2020 [Epub ahead of print]
36. Lim JY, Liu C, Hu K-Q, Smith DE, Wang X-D. Ablation of carotenoid cleavage enzymes (BCO1 and BCO2) induced hepatic steatosis by altering the farnesoid X receptor/miR-34a/sirtuin 1 pathway. Arch Biochem Biophys. 2018;654:1-9
37. Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13(5):239-250
38. Ye D, Zhang T, Lou G, Xu W, Dong F, Chen G. et al. Plasma miR-17, miR-20a, miR-20b and miR-122 as potential biomarkers for diagnosis of NAFLD in type 2 diabetes mellitus patients. Life Sci. 2018;208:201-207
39. Ando Y, Yamazaki M, Yamada H, Munetsuna E, Fujii R, Mizuno G. et al. Association of circulating miR-20a, miR-27a, and miR-126 with non-alcoholic fatty liver disease in general population. Sci Rep. 2019;9:18856
40. Zhang M, Sun W, Zhou M, Tang Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci Rep. 2017;7:14493
41. Teimouri M, Hosseini H, Shabani M, Koushki M, Noorbakhsh F, Meshkani R. Inhibiting miR-27a and miR-142-5p attenuate nonalcoholic fatty liver disease by regulating Nrf2 signaling pathway. IUBMB Life. 2020;72(3):361-372
42. Lin H-Y, Yang Y-L, Wang P-W, Wang F-S, Huang Y-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells. 2020;9(4):1041
43. Lin H-Y, Wang F-S, Yang Y-L, Huang Y-H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells. 2019;8(10):1298
44. Liu M-X, Gao M, Li C-Z, Yu C-Z, Yan H, Peng C. et al. Dicer1/miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta Pharmacol Sin. 2017;38(5):660-671
45. Yang Y-L, Kuo H-C, Wang F-S, Huang Y-H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int J Mol Sci. 2019;20(6):1499
46. Yang Y-L, Wang P-W, Wang F-S, Lin H-Y, Huang Y-H. miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. Int J Mol Sci. 2020;21(18):6884
47. Hanin G, Yayon N, Tzur Y, Haviv R, Bennett ER, Udi S. et al. miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut. 2018;67(6):1124-1134
48. Zhang Q, Yu K, Cao Y, Luo Y, Liu Y, Zhao C. miR-125b promotes the NF-κB-mediated inflammatory response in NAFLD via directly targeting TNFAIP3. Life Sci. 2021;270:119071
49. Zhang Z-C, Liu Y, Xiao L-L, Li S-F, Jiang J-H, Zhao Y. et al. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J Hepatol. 2015;63(6):1466-1475
50. Lin X, Jia J, Du T, Li W, Wang X, Wei J. et al. Overexpression of miR-155 in the liver of transgenic mice alters the expression profiling of hepatic genes associated with lipid metabolism. PLoS ONE. 2015;10(3):e0118417
51. Hartmann P, Tacke F. Tiny RNA with great effects: miR-155 in alcoholic liver disease. J Hepatol. 2016;64(6):1214-1216
52. Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA. et al. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS ONE. 2013;8(8):e72324
53. Liu X-L, Cao H-X, Wang B-C, Xin F-Z, Zhang R-N, Zhou D. et al. miR-192-5p regulates lipid synthesis in non-alcoholic fatty liver disease through SCD-1. World J Gastroenterol. 2017;23(46):8140-8151
54. Lin Y, Ding D, Huang Q, Liu Q, Lu H, Lu Y. et al. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(9):869-882
55. Zhang T, Duan J, Zhang L, Li Z, Steer CJ, Yan G. et al. LXRα Promotes Hepatosteatosis in Part Through Activation of MicroRNA-378 Transcription and Inhibition of Ppargc1β Expression. Hepatology. 2019;69(4):1488-1503
56. Zhang T, Zhao X, Steer CJ, Yan G, Song G. A negative feedback loop between microRNA-378 and Nrf1 promotes the development of hepatosteatosis in mice treated with a high fat diet. Metab Clin Exp. 2018;85:183-191
57. Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16(17):1941-1951
58. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab Clin Exp. 2016;65(8):1038-1048
59. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367-377
60. Wang S, Wang L, Dou L, Guo J, Fang W, Li M. et al. MicroRNA 152 regulates hepatic glycogenesis by targeting PTEN. FEBS J. 2016;283(10):1935-1946
61. Wang L, Zhang N, Pan H-P, Wang Z, Cao Z-Y. MiR-499-5p Contributes to Hepatic Insulin Resistance by Suppressing PTEN. Cell Physiol Biochem. 2015;36(6):2357-2365
62. Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X. et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest. 2015;125(6):2497-2509
63. Wang X, Wang M, Li H, Lan X, Liu L, Li J. et al. Upregulation of miR-497 induces hepatic insulin resistance in E3 rats with HFD-MetS by targeting insulin receptor. Mol Cell Endocrinol. 2015;416:57-69
64. Yang W-M, Jeong H-J, Park S-W, Lee W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol Nutr Food Res. 2015;59(11):2303-2314
65. Dai L-L, Li S-D, Ma Y-C, Tang J-R, Lv J-Y, Zhang Y-Q. et al. MicroRNA-30b regulates insulin sensitivity by targeting SERCA2b in non-alcoholic fatty liver disease. Liver Int. 2019;39(8):1504-1513
66. Wu H, Zhang T, Pan F, Steer CJ, Li Z, Chen X. et al. MicroRNA-206 prevents hepatosteatosis and hyperglycemia by facilitating insulin signaling and impairing lipogenesis. J Hepatol. 2017;66(4):816-824
67. Xu M, Zheng X-M, Jiang F, Qiu W-Q. MicroRNA-190b regulates lipid metabolism and insulin sensitivity by targeting IGF-1 and ADAMTS9 in non-alcoholic fatty liver disease. J Cell Biochem. 2018;119(7):5864-5874
68. Xu Q, Li Y, Shang Y-F, Wang H-L, Yao M-X. miRNA-103: molecular link between insulin resistance and nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21(2):511-516
69. Zhang Y, Cheng X, Lu Z, Wang J, Chen H, Fan W. et al. Upregulation of miR-15b in NAFLD models and in the serum of patients with fatty liver disease. Diabetes Res Clin Pract. 2013;99(3):327-334
70. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931-947
71. Engedal N, Žerovnik E, Rudov A, Galli F, Olivieri F, Procopio AD. et al. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid Med Cell Longev. 2018;2018:4968321
72. Liu C, Zhou B, Meng M, Zhao W, Wang D, Yuan Y. et al. FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J Hepatol. 2021 [Epub ahead of print]
73. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F. et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21(5):739-746
74. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res. 2015;49(12):1405-1418
75. Xu H, Tian Y, Tang D, Zou S, Liu G, Song J. et al. An Endoplasmic Reticulum Stress-MicroRNA-26a Feedback Circuit in NAFLD. Hepatology. 2020 [Epub ahead of print]
76. Cheng Y, Mai J, Hou T, Ping J. MicroRNA-421 induces hepatic mitochondrial dysfunction in non-alcoholic fatty liver disease mice by inhibiting sirtuin 3. Biochem Biophys Res Commun. 2016;474(1):57-63
77. Wang X, Wang J. High-content hydrogen water-induced downregulation of miR-136 alleviates non-alcoholic fatty liver disease by regulating Nrf2 via targeting MEG3. Biol Chem. 2018;399(4):397-406
78. Zhang Y, Wang C, Lu J, Jin Y, Xu C, Meng Q. et al. Targeting of miR-96-5p by catalpol ameliorates oxidative stress and hepatic steatosis in LDLr-/- mice via p66shc/cytochrome C cascade. Aging (Albany NY). 2020;12(3):2049-2069
79. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15(12):738-752
80. Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H. et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. 2018;24(25):2661-2672
81. Qadir XV, Chen W, Han C, Song K, Zhang J, Wu T. miR-223 Deficiency Protects against Fas-Induced Hepatocyte Apoptosis and Liver Injury through Targeting Insulin-Like Growth Factor 1 Receptor. Am J Pathol. 2015;185(12):3141-3151
82. Feng Z, Li Z, Zhu D, Ling W, Zheng L, Pu L. et al. Mir-24 regulates hepatocyte apoptosis via BIM during acute liver failure. Am J Transl Res. 2017;9(11):4925-4935
83. Rodrigues PM, Afonso MB, Simão AL, Borralho PM, Rodrigues CMP, Castro RE. Inhibition of NF-κB by deoxycholic acid induces miR-21/PDCD4-dependent hepatocellular apoptosis. Sci Rep. 2015;5:17528
84. Ferreira DMS, Afonso MB, Rodrigues PM, Simão AL, Pereira DM, Borralho PM. et al. c-Jun N-terminal kinase 1/c-Jun activation of the p53/microRNA 34a/sirtuin 1 pathway contributes to apoptosis induced by deoxycholic acid in rat liver. Mol Cell Biol. 2014;34(6):1100-1120
85. Castro RE, Ferreira DMS, Afonso MB, Borralho PM, Machado MV, Cortez-Pinto H. et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol. 2013;58(1):119-125
86. Shan W, Gao L, Zeng W, Hu Y, Wang G, Li M. et al. Activation of the SIRT1/p66shc antiapoptosis pathway via carnosic acid-induced inhibition of miR-34a protects rats against nonalcoholic fatty liver disease. Cell Death Dis. 2015;6(7):e1833
87. Cazanave SC, Mott JL, Elmi NA, Bronk SF, Masuoka HC, Charlton MR. et al. A role for miR-296 in the regulation of lipoapoptosis by targeting PUMA. J Lipid Res. 2011;52(8):1517-1525
88. Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y. et al. Myeloid cell-specific IL-6 signaling promotes miR-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology. 2020 [Epub ahead of print]
89. He Y, Hwang S, Cai Y, Kim S-J, Xu M, Yang D. et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology. 2019;70(4):1150-1167
90. Ng R, Wu H, Xiao H, Chen X, Willenbring H, Steer CJ. et al. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology. 2014;60(2):554-564
91. Feng D, Mukhopadhyay P, Qiu J, Wang H. Inflammation in Liver Diseases. Mediators Inflamm. 2018;2018:3927134
92. Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci. 2016;73(10):1969-1987
93. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation. 2019;103(1):e1-e13
94. Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology. 2016;150(8):1704-1709
95. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15(6):349-364
96. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76(1):99-128
97. Arner P, Kulyté A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat Rev Endocrinol. 2015;11(5):276-288
98. Ye D, Zhang T, Lou G, Liu Y. Role of miR-223 in the pathophysiology of liver diseases. Exp Mol Med. 2018;50(9):1-12
99. He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S. et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Invest. 2021;131(3):e141513
100. Nie H, Song C, Wang D, Cui S, Ren T, Cao Z. et al. MicroRNA-194 inhibition improves dietary-induced non-alcoholic fatty liver disease in mice through targeting on FXR. Biochim Biophys Acta Mol Basis Dis. 2017;1863(12):3087-3094
101. Tian H, Liu C, Zou X, Wu W, Zhang C, Yuan D. MiRNA-194 Regulates Palmitic Acid-Induced Toll-Like Receptor 4 Inflammatory Responses in THP-1 Cells. Nutrients. 2015;7(5):3483-3496
102. Liu X-L, Pan Q, Cao H-X, Xin F-Z, Zhao Z-H, Yang R-X. et al. Lipotoxic Hepatocyte-Derived Exosomal miR-192-5p Activates Macrophages via Rictor/Akt/FoxO1 Signaling in NAFLD. Hepatology. 2020;72(2):454-469
103. Zhang T, Hu J, Wang X, Zhao X, Li Z, Niu J. et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-κB-TNFα pathway. J Hepatol. 2019;70(1):87-96
104. Hur W, Lee JH, Kim SW, Kim J-H, Bae SH, Kim M. et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int J Biochem Cell Biol. 2015;64:265-276
105. Jimenez Calvente C, Del Pilar H, Tameda M, Johnson CD, Feldstein AE. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol Ther. 2020;28(2):653-663
106. Friedman SL. Liver fibrosis - from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38-S53
107. Angulo P, Machado MV, Diehl AM. Fibrosis in nonalcoholic Fatty liver disease: mechanisms and clinical implications. Semin Liver Dis. 2015;35(2):132-145
108. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol. 2018;68(2):238-250
109. Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int J Mol Sci. 2018;19(12):3966
110. Leti F, Malenica I, Doshi M, Courtright A, van Keuren-Jensen K, Legendre C. et al. High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Transl Res. 2015;166(3):304-314
111. Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M. et al. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2015;308(4):G298-312
112. Takeuchi-Yorimoto A, Yamaura Y, Kanki M, Ide T, Nakata A, Noto T. et al. MicroRNA-21 is associated with fibrosis in a rat model of nonalcoholic steatohepatitis and serves as a plasma biomarker for fibrotic liver disease. Toxicol Lett. 2016;258:159-167
113. Caviglia JM, Yan J, Jang M-K, Gwak G-Y, Affo S, Yu L. et al. MicroRNA-21 and Dicer are dispensable for hepatic stellate cell activation and the development of liver fibrosis. Hepatology. 2018;67(6):2414-2429
114. Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K. et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015;35(2):532-541
115. Du J, Niu X, Wang Y, Kong L, Wang R, Zhang Y. et al. MiR-146a-5p suppresses activation and proliferation of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis through directly targeting Wnt1 and Wnt5a. Sci Rep. 2015;5:16163
116. Lin X, Kong L-N, Huang C, Ma T-T, Meng X-M, He Y. et al. Hesperetin derivative-7 inhibits PDGF-BB-induced hepatic stellate cell activation and proliferation by targeting Wnt/β-catenin pathway. Int Immunopharmacol. 2015;25(2):311-320
117. Cheng JH, She H, Han Y-P, Wang J, Xiong S, Asahina K. et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G39-49
118. Ma L, Yang X, Wei R, Ye T, Zhou J-K, Wen M. et al. MicroRNA-214 promotes hepatic stellate cell activation and liver fibrosis by suppressing Sufu expression. Cell Death Dis. 2018;9(7):718
119. Okada H, Honda M, Campbell JS, Takegoshi K, Sakai Y, Yamashita T. et al. Inhibition of microRNA-214 ameliorates hepatic fibrosis and tumor incidence in platelet-derived growth factor C transgenic mice. Cancer Sci. 2015;106(9):1143-1152
120. Wang Y, Du J, Niu X, Fu N, Wang R, Zhang Y. et al. MiR-130a-3p attenuates activation and induces apoptosis of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis by directly targeting TGFBR1 and TGFBR2. Cell Death Dis. 2017;8(5):e2792
121. Tsay H-C, Yuan Q, Balakrishnan A, Kaiser M, Möbus S, Kozdrowska E. et al. Hepatocyte-specific suppression of microRNA-221-3p mitigates liver fibrosis. J Hepatol. 2019;70(4):722-734
122. Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF. et al. Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology. 2016;64(6):2219-2233
123. Hernández A, Arab JP, Reyes D, Lapitz A, Moshage H, Bañales JM. et al. Extracellular Vesicles in NAFLD/ALD: From Pathobiology to Therapy. Cells. 2020;9(4):817
124. Newman LA, Sorich MJ, Rowland A. Role of Extracellular Vesicles in the Pathophysiology, Diagnosis and Tracking of Non-Alcoholic Fatty Liver Disease. J Clin Med. 2020;9(7):2032
125. Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A. et al. Integrin β1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71(6):1193-1205
126. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72(1):156-166
127. Povero D, Panera N, Eguchi A, Johnson CD, Papouchado BG, Araujo Horcel L de. et al. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-γ. Cell Mol Gastroenterol Hepatol. 2015;1(6):646-663.e4
128. Liao C-Y, Song MJ, Gao Y, Mauer AS, Revzin A, Malhi H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front Immunol. 2018;9:2980
129. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA. et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016;63(3):731-744
130. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao C-Y. et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology. 2020;159(4):1487-1503.e17
131. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW. et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology. 2016;150(4):956-967
132. Chen L, Charrier A, Zhou Y, Chen R, Yu B, Agarwal K. et al. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology. 2014;59(3):1118-1129
133. Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB. et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell. 2017;171(2):372-384.e12
134. Li Y, Luan Y, Li J, Song H, Li Y, Qi H. et al. Exosomal miR-199a-5p promotes hepatic lipid accumulation by modulating MST1 expression and fatty acid metabolism. Hepatol Int. 2020;14(6):1057-1074
135. Borel F, Han R, Visser A, Petry H, van Deventer SJH, Jansen PLM. et al. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology. 2012;55(3):821-832
136. Zhang T, Yang Z, Kusumanchi P, Han S, Liangpunsakul S. Critical Role of microRNA-21 in the Pathogenesis of Liver Diseases. Front Med (Lausanne). 2020;7:7
137. Thietart S, Rautou P-E. Extracellular vesicles as biomarkers in liver diseases: A clinician's point of view. J Hepatol. 2020;73(6):1507-1525
138. He S, Guo W, Deng F, Chen K, Jiang Y, Dong M. et al. Targeted delivery of microRNA 146b mimic to hepatocytes by lactosylated PDMAEMA nanoparticles for the treatment of NAFLD. Artif Cells Nanomed Biotechnol. 2018;46(sup2):217-228
Author contact
![]() Corresponding authors: Guorui Dou, Email: douguoruicom; Lin Wang, Email: fierywangcom or wangdouedu.cn
Corresponding authors: Guorui Dou, Email: douguoruicom; Lin Wang, Email: fierywangcom or wangdouedu.cn