Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(9):2135-2146. doi:10.7150/ijbs.60056 This issue Cite
Research Paper
Disruption of myofibroblastic Notch signaling attenuates liver fibrosis by modulating fibrosis progression and regression
Zhensheng Yue1,2*, Zijian Jiang1*, Bai Ruan1,3, Juanli Duan1, Ping Song1, Jingjing Liu1, Hua Han4,5 ![]() , Lin Wang1
, Lin Wang1 ![]()
1. Department of Hepatobiliary Surgery, Xi-Jing Hospital, Fourth Military Medical University, Xi'an 710032, China.
2. Department of Ophthalmology, Xi-Jing Hospital, Fourth Military Medical University, Xi'an 710032, China.
3. Aerospace Clinical Medical Center, School of Aerospace Medicine, Fourth Military Medical University, Xi'an 710032, China.
4. State Key Laboratory of Cancer Biology, Fourth Military Medical University, Xi'an 710032, China.
5. Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Xi'an 710032, China.
*These authors contributed equally to this work.
Received 2021-3-3; Accepted 2021-5-8; Published 2021-5-27
Abstract

The phenotypic transformation of hepatic myofibroblasts (MFs) is involved in the whole process of the progression and regression of liver fibrosis. Notch signaling has been demonstrated to modulate the fibrosis. In this study, we found that Notch signaling in MFs was overactivated and suppressed with the progression and regression of hepatic fibrosis respectively, by detecting Notch signaling readouts in MFs. Moreover, we inactivated Notch signaling specifically in MFs with Sm22αCreER-RBPjflox/flox mice (RBPjMF-KO), and identified that MFs-specific down-regulation of Notch signaling significantly alleviated CCl4-induced liver fibrosis during the progression and regression. During the progression of liver fibrosis, MFs-specific blockade of Notch signaling inhibited the activation of HSCs to MFs and increases the expression of MMPs to reduce the deposition of ECM. During the regression of fibrosis, blocking Notch signaling in MFs increased the expression of HGF to promote proliferation in hepatocytes and up-regulated the expression of pro-apoptotic factors, Ngfr and Septin4, to induce apoptosis of MFs, thereby accelerating the reversal of fibrosis. Collectively, the MFs-specific disruption of Notch signaling attenuates liver fibrosis by modulating fibrosis progression and regression, which suggests a promising therapeutic strategy for liver fibrosis.
Keywords: Myofibroblasts, hepatic stellate cells, Notch signaling, liver fibrosis progression, liver fibrosis regression
Introduction
Liver fibrosis is a highly conservative response to liver injury, which occurs in almost all types of liver injury. Liver fibrosis can be observed in chronic viral hepatitis, alcoholic liver disease, non-alcoholic fatty liver disease, cholestatic and autoimmune liver disease [1, 2]. Under the continuous stimulation of harmful factors, the non-parenchymal cells, mainly hepatic stellate cells (HSCs), can be activated into myofibroblasts (MFs) and start expressing α-smooth muscle actin (α-SMA) and collagens to produce excessive extracellular matrix (ECM), thereby resulting in liver fibrosis [3, 4]. However, when harmful stimuli are evacuated, the liver can undergo rapid regeneration, which is mainly manifested by the proliferation of hepatocytes under the action of mitogens, including Wnt2a, Wnt9b and hepatocyte growth factor (HGF) [5, 6]. Moreover, a variety of cells including MFs and macrophages in the liver can produce matrix metalloproteinases (MMPs) that degrade ECM and fibrotic septum. During liver fibrosis regression, MFs convert into different cell fates, including inactivation, senescence and apoptosis [7-9], on the absence of pro-fibrotic factors, such as transforming growth factor-β (TGF-β), platelet-derived growth factor β (PDGF-β) and interleukin-17A (IL-17A), etc. [10-12]. Additionally, the activation of death receptor-mediated pathways, the increase of pro-apoptotic proteins and the decrease of pro-survival proteins are considered to play causative roles in MFs apoptosis [7]. MFs also increase the expression of Fas receptors or tumor necrosis factor receptor (TNFR) family members, which triggers caspase8/caspase3-dependent cell apoptosis. Moreover, overexpression of pro-apoptotic proteins such as p53 or Bax in MFs lead to caspase-mediated programmed cell death [13-15]. In addition, MFs are also the main source of tissue inhibitors of metalloproteinases (TIMPs). The loss of MF during liver fibrosis regression can lead to decreased TIMPs expression, which in turn contributes to increased activity of MMPs and subsequent degradation of ECM [6, 16].
Notch signaling is a highly evolutionarily conserved pathway that regulates cell differentiation, proliferation and apoptosis in many kinds of tissues [17, 18]. Activation of Notch signaling relies on binding of Notch receptors (Notch1, -2, -3, and-4) and ligands (Jagged1, -2 and Dll1, -3, -4) between neighboring cells. This ligand receptor interaction triggers sequential receptor cleavage within the transmembrane domain, resulting in the release of the Notch intracellular domain (NICD). NICD then transfers into the nucleus and combines with transcription factor CSL (C promoter-binding factor 1[CBF-1]/suppressor of hairless [Su(H)]/Lin-12 and Glp-1 [LAG-1], also known as j-kappa recombination signal-binding protein [RBPj] in mice), which is inhibited by a corepressor complex under normal conditions. Association of NICD and CSL replaces the corepressors with a coactivating complex containing Mastermind-like (MAML) protein and activates the transcription of downstream genes, such as Hes and Hey family members [19, 20].
Notch signaling pathway participated in fibrosis in a variety of tissues and organs, such as skin, kidney, lung and liver [21-24]. It was initially found in vitro that rat HSCs express Notch receptors and begin expressing Jagged1 upon activation and differentiation to MFs [25]. Afterwards, other in vivo studies showed that Notch signaling pathway can also cooperate with other signaling pathways to mediate the activation process of HSCs to MFs, such as TGF-β, Wnt and Hedgehog signaling pathways [26, 27]. Our previous studies found that myeloid-specific disruption of RBPj ameliorates hepatic fibrosis by inhibiting the activation of HSCs to MFs and liver inflammation and reducing the expression of pro-fibrotic factors, including TGF-β and PDGF-β [28]. Moreover, endothelial-specific Notch signaling activation can aggravate liver fibrosis [29]. The in vivo dependence of HSCs activation on Notch signaling indicates that inhibition of this pathway in the liver can prevent fibrosis or significantly ameliorate fibrosis [30]. However, the overall consequence of MFs-specific Notch inactivation during fibrosis progression and regression has not been evaluated. In this study, we report that MFs-specific disruption of Notch signaling attenuates liver fibrosis by inhibiting fibrosis progression and altering MFs expression spectrum that expedited fibrosis regression.
Materials and Methods
Animal experiments
C57BL/6 wild-type mice were from the Laboratory Animal Center of Air Force Military Medical University, Sm22αCreERT2 mice (Model Animal Institute of Nanjing University) were crossed with RBPjflox/flox mice (Jackson Labs). The littermates were genotyped by PCR to obtain Sm22αCreERT2 (control, Ctrl) and Sm22αCreERT2-RBPjflox/flox mice. Male Sm22αCreERT2-RBPjflox/flox mice of 6-8 weeks old were injected with carbon tetrachloride (CCl4, mixed with olive oil, 0.6 mL/kg, twice a week, Sigma-Aldrich) and tamoxifen (100 mg/kg, 5 times started from the fifth week, Sigma-Aldrich, St. Louis, MO) intraperitoneally (IP) to obtain RBPjMF-KO, with olive oil as a control. Progressive liver fibrosis mice model and regressive liver fibrosis mice model: liver fibrosis progression mice were induced by intraperitoneal CCl4 injection for 6weeks as previously described [31]. For fibrosis regression study, the CCl4 injections were terminated after 6 weeks injection for 5 days as previously described [31].
All the animals were maintained in the specific pathogen free (SPF) level standards environment. All the operations of the experimental animals were reviewed the Animal Experiment Administration Committee of the Fourth Military Medical University (Xi'an, China) and were under China National “Regulations on the Management of Experimental Animals”. The study complied with the guideline outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals of the National Academy of Science and published by the National Institute of Health.
Histopathology
Mice liver samples were fixed in 4% paraformaldehyde (PFA) or in 10% buffered formalin. PFA-fixed samples were embedded with optimal cutting temperature (OCT) compound and sectioned at 8-μm thickness for immunofluorescence (IF) as described previously. Immunofluorescence (IF) was performed using fluorescent antibodies and counterstained with Hoechst 33258 (Sigma-Aldrich). Formalin-fixed samples were paraffin-embedded, sectioned, and subjected to hematoxylin and eosin (HE) staining and immunohistochemistry (IHC) routinely. Apoptotic cells were stained with a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) kit (Promega, Madison, WI). Specific antibodies shown in Supplementary Table 2. Digital images were taken under a fluorescence microscope (BX51; Olympus, Japan) or laser-scanning confocal fluorescence microscope (FV-1000; Olympus, Japan). Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were carried out as described previously.
Cell isolation, culture, and identification
Primary HSCs/MFs were isolated from mice by in situ perfusion followed by finely minced with a tissue homogenizer in perfusion buffer containing Dnase I (Roche, Basel, Switzerland)/Collagenase IV (Sigma-Aldrich) and subsequent density gradient centrifugation as described previously. The obtained cells were identified by α-SMA and Collagen1 IF staining and purity was more than 95%. The isolated primary HSCs/MFs were then cultured in low glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco, USA), supplemented with 10% fetal bovine serum (FBS) (Biological Industries, Israel), and maintained in 37 °C incubator with 5% CO2. Culture medium was replaced every 24 h. Human HSCs cell line LX-2 (kindly provided by YM-Li, State Key Laboratory of Cancer Biology, FMMU, China) was cultured in high glucose DMEM (Gibco, USA), supplemented with 10% FBS and maintained in 37 °C incubator with 5% CO2. GSI (Sigma Aldrich) and TGF-β (Pepro Tech) were added at concentrations of 100 μM and 0.1 ng/mL respectively.
Gene expression profiling
The total RNA of fresh isolated and purified MFs from RBPjMF-KO and control mice was extracted by using TRIzol (Invitrogen), according to the manufacturer's protocol. Guangzhou GENE DENOVO Biology (Co. Ltd.) was commissioned to carry out quality inspection and full transcriptome expression profile sequencing service, using Digital Gene Expression Tag Profiling mode sequencing. Bioinformatics analysis such as KEGG signal pathway enrichment analysis, GO enrichment analysis, Heat Map and other bioinformatics analysis were carried out by using OmicShare Tools platform (www.omicshare.com/tools).
qPCR
Total RNA extraction and reverse transcription were performed by using TRIzol and PrimeScrip RT reagent kit (TaKaRa Biotechnology, Dalian, China) according to standard procedures. qPCR was performed using a TB Green Premix Ex Taq Kit (TaKaRa Biotechnology) and an ABI PRISM 7500 Real-time PCR system (Life Technologies, Waltham, MA), with β-Actin as a reference control. The primers are showed in Supplementary Table 1.
Western blot analysis
Total Protein was extracted from liver tissue or MFs with RIPA (Beyotime) lysis buffer containing phosphatase inhibitor (phenylmethylsulphonyl fluoride, PMSF), and protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo Scientific, USA). Following separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were blotted onto polyvinylidene fluoride (PVDF) membranes, and probed with specific antibodies shown in Supplementary Table 2. Protein bands were detected using an enhanced chemiluminescence (ECL) system, with β-Actin or GAPDH as a loading control.
Hydroxyproline test
Hydroxyproline content was quantified to determine the extent of liver fibrosis by using a kit (Sigma- Aldrich) according to the manufacturer's instruction. Briefly, collected flash frozen liver samples were weighed, homogenized and hydrolyzed in concentrated hydrochloric acid (HCl, 12N) in pressure-tight vials with PTFE-lined cap at 120°C for 3 hours. Equivalent volume of supernatant was transferred to a 96 well plate and was evaporated to dryness under a vacuum. Chloramine T/oxidation buffer mixture was added to each well and incubated at room temperature for 5 minutes. Next, diluted 4-(Dimethylamino) benzaldehyde reagent was added to each sample and was incubated at 60 °C for 90 minutes. Absorbance at the wavelength of 560 nm was determined, and the content of hydroxyproline in the liver was calculated on the basis of the sample weight by comparing the absorbance to a hydroxyproline standard curve.
Statistics analysis
The Image-Pro Plus 6.0 program was used to carry out morphological quantitative. Statistical analysis was performed with the GraphPad Prism 6 software. All the results were presented as the mean ± standard deviation (SD). Comparisons between groups were performed using unpaired, two-tailed, Student's t-test. P < 0.05 was considered statistically significant.
Results
Notch signaling pathway is activated in liver fibrosis progression and suppressed in liver fibrosis regression
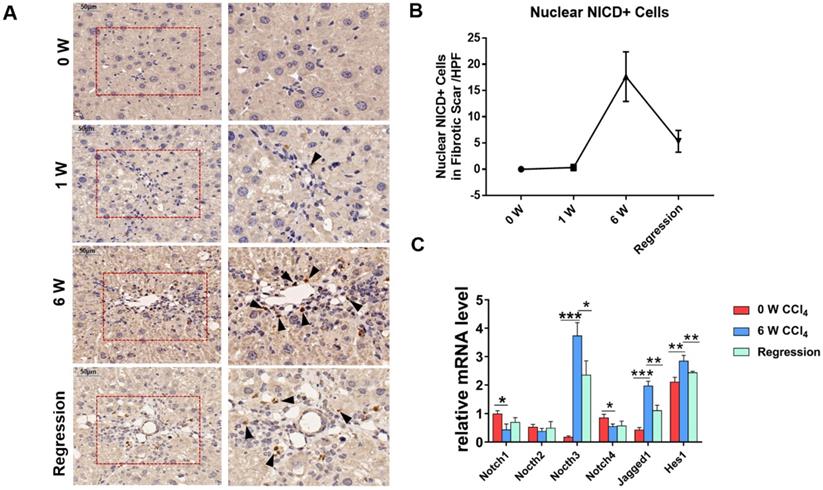
To investigate whether Notch signaling mediates the liver fibrosis, we detected the expression of Notch signaling readouts in MFs. IHC staining showed the translocation of NICD into the MFs nucleus in fibrosis septum area (Figure 1A). Statistical analyses indicated that the number of nuclear NICD positive cells in the fibrous scar area increases with progression, and decreases with regression (Figure 1B). Then, we isolated the HSCs/MFs of wild-type mice in different stages of liver fibrosis. The results of qPCR showed that the expression of Notch3, Jagged1 increased with the progression of hepatic fibrosis and decreased with the reversal of hepatic fibrosis, consistent with previous reports [32-34]. Although the expression of Notch1 and Notch4 decreased during the progression of hepatic fibrosis, the expression pattern of Hes1, a target gene downstream of Notch signaling, was similar to Notch3 and Jagged1, fluctuated with the progression and regression of hepatic fibrosis (Figure 1C). These results indicated that Notch signaling closely correlates with liver fibrosis and may be involved in the progression and regression of liver fibrosis.
The Notch signaling pathway in MFs participates in the regulation of liver fibrosis progression and regression. The samples of olive oil or CCl4 injected C57BL/6 mice for 0, 1, 6 weeks and regression for 5 days were subjected to the following assays. (A) NICD immunohistochemical staining of liver fibrous scar area in different stages of liver fibrosis. (B) The number of nucleus NICD positive cells in the fibrous scar area increases with progression, and decreases with regression. (C) Changes in the expression of Notch signaling related molecules in liver HSCs/MFs at different stages of liver fibrosis. (Bars = means ± SD, n = 6, *P <0.05, **P < 0.01, ***P < 0.001).
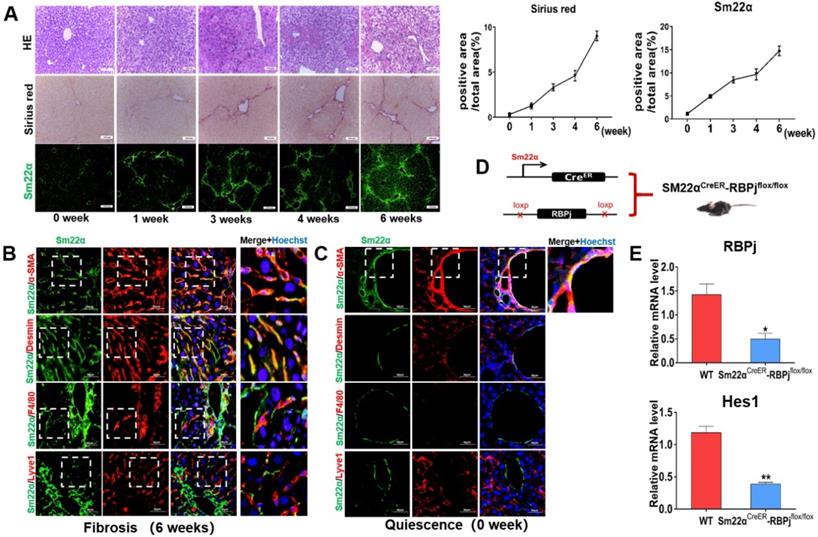
Induction of liver fibrosis mice and MFs-specific Notch disruption mice (RBPjMF-KO)
Liver fibrosis was induced by repeated intraperitoneal injection of CCl4 for 6 weeks [29]. HE and Sirius red staining of liver tissues showed that the area of centrilobular necrosis was increased accompanied by bridging fibrous septa in the livers of CCl4-treated mice (Figure 2A). In a systemic approach to obtain the MFs-specific Notch disruption mice, we detected the expression of transgelin (Smooth muscle 22α, Sm22α) in liver (Figure 2A). Previous studies have shown that Sm22α is not expressed in quiescent HSCs, but gradually up-regulated during the activation of HSCs [32-34]. In our research, Sm22α IF staining showed that the expression of Sm22α increased gradually with the increase of CCl4 injection times, and was abundant at 3-4 weeks (Figure 2A). In order to precisely identify the expression of Sm22α in hepatic nonparenchymal cells, we compared the expression of Sm22α with α-SMA, Desmin, F4/80 and Lyve1, other kinds of nonparenchymal cells markers in the liver. The results of co-localization staining showed that in the fibrotic liver, Sm22α had a high proportion (96%) of co-localization with α-SMA and Desmin (Figure 2B). But in normal liver, Sm22α is only co-located with α-SMA around large vessels (Figure 2C). This indicates that during hepatic fibrosis, Sm22α is mainly expressed in MFs while Sm22α is only expressed in perivascular smooth muscle cells in normal liver. Based on the expression of Sm22α, we hybridized Sm22αCreER mice and RBPjflox/flox mice and screened out Sm22αCreER-RBPjflox/flox from the descendants (Figure 2D). In CCl4- and tamoxifen-induced Sm22αCreER-RBPjflox/flox mice, the expression of RBPj and Hes1, downstream molecules of canonical Notch signaling, was down-regulated significantly in MFs isolated from Sm22αCreER-RBPjflox/flox mice (Figure 2E), indicating that we obtained MFs-specific Notch disruption mice (RBPjMF-KO).
Establishment and identification of RBPjMF-KO models of liver fibrosis. (A) Representative hematoxylin-eosin (HE)-stained liver sections from mouse exposed to CCl4 for 1, 3or 6 weeks for evaluation of steatosis. Sirius red staining for evaluation of collagen deposition. Sm22α immunofluorescence staining for evaluation of activation of HSCs. (B) In liver fibrosis, Sm22α is mainly expressed in MFs. (n=6). (C) In normal liver, Sm22α is only expressed in vascular smooth muscle cells (n=6). (D) Induction schedules of genetically modified mouse models. (E) qPCR showed the expression changes of Notch signaling downstream molecules, including RBPj and Hes1, in isolated MFs of Sm22αCreER- RBPjflox/flox mice after CCl4 and tamoxifen induced. (Bars = means ± SD, n=3, *P < 0.05, **P < 0.01, ***P < 0.001).
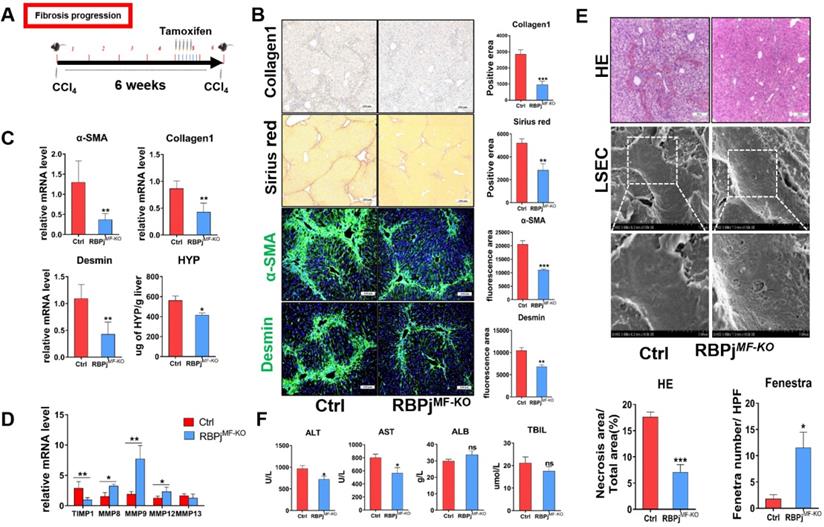
MFs-specific Notch signaling disruption inhibits the progression of hepatic fibrosis via decreasing ECM deposition and liver tissue destruction
In fibrosis progression mouse models, CCl4 and tamoxifen were injected as previously reported (Figure 3A). Induction of liver fibrosis progression in RBPjMF-KO and control mice showed that RBPjMF-KO mice manifested significantly alleviative fibrogenesis and decreased ECM deposition, as evidenced by less intensely stained Collagen1 and Sirius Red (Figure 3B) and reduced tissue hydroxyproline (HYP) level (Figure 3C). IF staining showed Notch inactivation in MFs down-regulated α-SMA and Desmin expression in liver (Figure 3B). qPCR showed that the mRNA level of Collagen1, α-SMA and Desmin also decreased in RBPjMF-KO liver (Figure 3C). Additionally, mRNA level of MMP8/9/12 elevated and TIMP1 reduced in RBPjMF-KO liver (Figure 3D). To confirm the condition of liver tissue destruction, liver sections from RBPjMF-KO and control mice were analyzed with HE staining and SEM. In RBPjMF-KO mice of fibrosis progression, less necrosis area and more liver sinusoid fenestrae were observed, suggesting alleviated liver tissues destruction and liver sinusoid capillarization (Figure 3E). Additionally, serum ALT and AST level of RBPjMF-KO mice was lower than control mice though ALB and TBIL level didn't change significantly (Figure 3F). These data collectively indicated that blocking Notch signaling in MFs inhibits the progression of hepatic fibrosis.
Blocking of Notch signaling in MFs inhibits the progression of CCl4-induced liver fibrosis. (A) Knockout induction schedule for liver fibrosis progression models. (B) Liver Collagen1, Sirius red, α-SMA, Desmin staining showed ECM was significantly reduced. (C) Changes in mRNA expression of liver tissue fibrosis-related molecules and changes in liver hydroxyproline level. (D) Changes in the expression of TIMP1 and MMPs in liver tissues. (E) HE staining of liver tissue and SEM of LSEC. The statistical results of HE staining showed that the destruction of liver tissue was reduced; the statistical results of fenestrae showed that the degree of capillarization of hepatic sinusoid was reduced. (F) The serum ALT and AST level of RBPjMF-KO mice decreased. (Ctrl=5, KO=6, *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant).
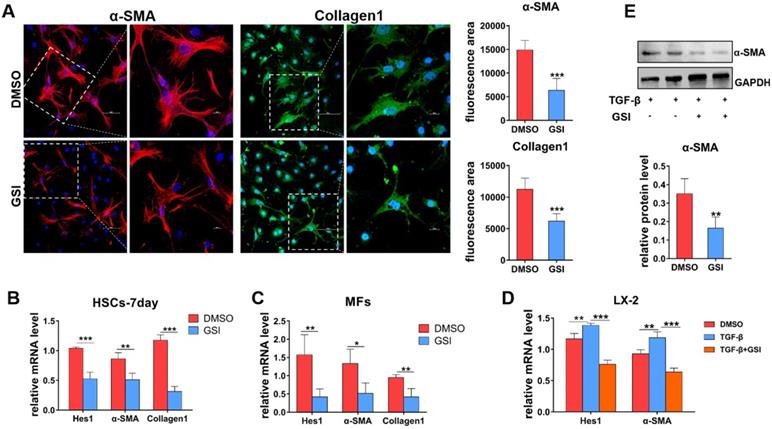
Blocking of Notch signaling inhibits the activation of HSCs to MFs in vitro
To further delineate the role of Notch signaling in liver fibrosis progression, primary HSCs and hepatic MFs isolated from wild-type and CCl4-induced mice were cultured in vitro. Then, gamma(γ)-secretase inhibitor (GSI), a canonical Notch signaling repressor, was used to treat primary MFs and spontaneously activated HSCs (HSCs-7day, cultured for 7 days in vitro). As shown by IF staining results (Figure 4A), α-SMA and Collagen1 decreased in MFs after GSI treatment for 24h. qPCR also showed that the expression of α-SMA and Collagen1 was significantly reduced in HSCs-7day and MFs (Figure 4B, C). In addition, human hepatic stellate cell line LX-2 was stimulated with TGF-β for 48h for activation then treated with GSI, and the expression of activation marker was evaluated (Figure 4D). qPCR and Western bloting (WB) showed that α-SMA expression was modestly decreased in GSI pre-treated cells compared with cells without GSI pre-administration (Figure 4D, E). Collectively, these data further confirm that Notch disruption inhibits the activation of HSCs to MFs, as well as reduced ECM deposition.
In-vitro experiments verify the effect of Notch signaling blocking on HSCs/MFs. (A) α-SMA and collagen1 IF staining of isolated primary liver MFs cultured in vitro and treated GSI for 24h from mice with hepatic fibrosis. (B) The mRNA level changes in isolated primary HSCs, cultured for 7 days and treated GSI for 24 hours, from normal mice. (C) The mRNA level changes of isolated MFs after GSI administration for 24 hours from mice with liver fibrosis. (D) The changes of mRNA level of human hepatic stellate cell line LX-2 treated GSI for 24 hours after activation administration via TGF-β. (E) Changes in LX-2 protein levels (n=6, *P < 0.05, **P < 0.01, ***P < 0.001).
MFs-specific Notch signaling disruption promotes the regression process of liver fibrosis via expediting fibrous septum degradation and hepatocyte proliferation
To further evaluate weather Notch signaling of MFs can mediate fibrosis regression models, we treated mice with CCl4 for 6 weeks to induce advanced liver fibrosis and allowed them to recover in the absence of CCl4 for 5 days [31] (Figure 5A). On gross observation, the livers of RBPjMF-KO mice were better-perfused, smoother, and lighter in liver/body weight than those of control mice. HE staining demonstrated a significantly decreased tissue destruction area in RBPjMF-KO mice comparing to control mice (Figure 5B), and RBPjMF-KO mice also displayed reduced serum ALT and AST level (Figure 5G), together suggesting that blockade of Notch signaling benefits the recovery of liver tissue necrosis. In addition to the decrease of serum HYP level of RBPjMF-KO mice (Figure 5D), fibrotic bridge and ECM deposition were markedly reduced in the liver section of RBPjMF-KO mice compared with control mice, shown by Sirius Red and Collagen1 IHC staining (Figure 5B). Moreover, qPCR showed that α-SMA, Collagen1 and TIMP1 mRNA level decreased in liver of RBPjMF-KO mice (Figure 5C), which indicates MFs-specific Notch inactivation expedited the degradation of fibrotic septa.
Blocking of Notch signaling in MFs promotes the resolution of liver fibrosis. (A) Induction schedule of liver fibrosis regression and RBPj knockout. (B) The general appearance of the liver, liver section HE, Sirius red and Collagen1 staining showed that ECM deposition was significantly reduced. (C) Changes in mRNA expression of fibrosis-related molecules in liver. (D) Changes in hydroxyproline level of liver tissues. (E) Liver section Ki67 staining showed that hepatocyte proliferation increased. (F) Detect the change of HGF mRNA level in vivo and the change of HGF mRNA level of MFs after treated GSI (100 μM for 24h) in vitro. (G) The serum liver function level was improved. (Bars = means ± SD, n=6, *P < 0.05, **P < 0.01, ***P < 0.001).
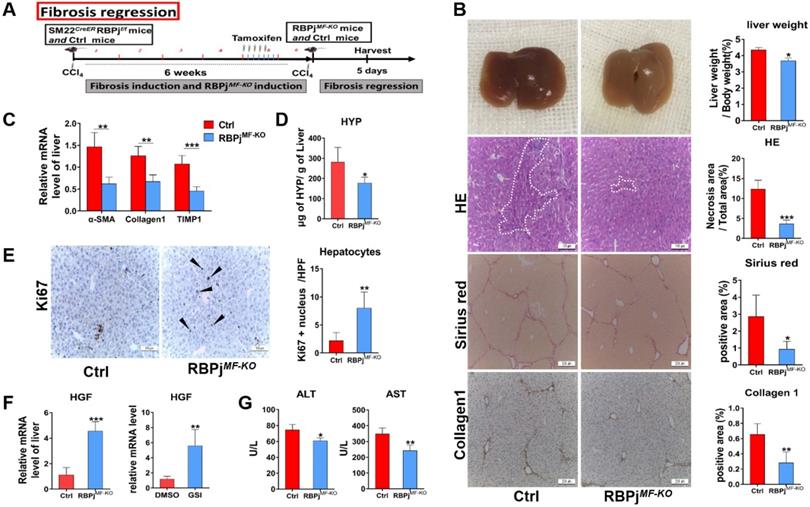
Next, we detected the condition of hepatocyte proliferation of control and RBPjMF-KO mice by anti-Ki67 IHC staining. Anti-Ki67 IHC showed that Notch inactivation in MFs significantly enhanced hepatocyte proliferation in the process of liver fibrosis regression (Figure 5E). To clarify what triggers hepatocytes proliferation, the expression of several hepatocyte mitogens, including Wnt2a, Wnt9b, and HGF, was determined by qPCR. We observed the mRNA level of HGF was up-regulated in RBPjMF-KO mice (Figure 5F). Moreover, we examined the mRNA level of HGF in apoptotic LX-2 (induced by being cultured with serum-free medium for 24h) in the attendance or absence of GSI administration. In line with this, HGF was remarkably increased in GSI-treated LX-2 as shown by qPCR (Figure 5F). These data indicated that Notch disruption in MFs may improve hepatocyte proliferation by enhancing the expression of HGF through other undefined signaling.
Notch signaling disruption enhances apoptosis of MFs during the process of liver fibrosis regression
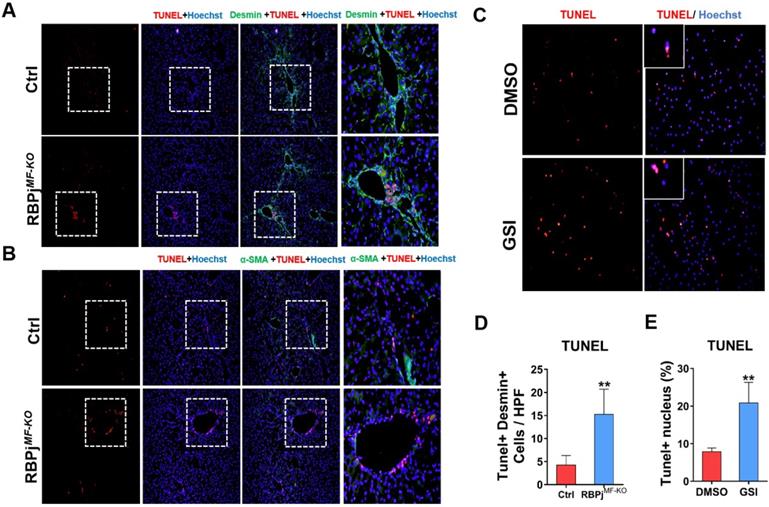
In the process of reversing liver fibrosis, MFs can undergo inactivation, aging or apoptosis to accomplish the fibrosis regression. Among them, the apoptosis of MFs makes a great contribution to the regression of liver fibrosis [6, 8]. To explore whether Notch signaling is involved in MFs apoptosis during fibrosis regression, we performed co-localization staining of liver MFs markers α-SMA/Desmin and TUNEL in the liver section of control and RBPjMF-KO mice (Figure 6A, B). The results showed that after Notch signaling was blocked, more positive apoptosis signals were observed in the α-SMA/Desmin positive area and perivascular area of liver (Figure 6D). After isolation from wild-type hepatic fibrosis mice, MFs were cultured in serum-free medium to induce apoptosis and treated with GSI for 24h, and we found TUNEL+ MFs significantly increased in GSI-treated cultures comparing to controls (Figure 6C, E). In summary, these data suggested that Notch signaling disruption favors regression of liver fibrosis through enhancing the apoptosis of MFs.
Blocking of Notch signaling in MFs promotes MFs apoptosis during liver fibrosis regression. (A) Liver TUNEL and Desmin co-localized staining, Desmin positive cells increased apoptosis. (B) Liver TUNEL and α-SMA co-localized staining. The expression of α-SMA reversed significantly, but the cell apoptosis in perivascular and portal area increased obviously. (C) TUNEL staining of culturing primary MFs after 24h treatment with GSI (100 μM for 24h) during the induction of apoptosis in vitro. (D) Statistical results of apoptosis in co-stained with TUNEL and Desmin. (E) Statistical results of apoptotic MFs after pro-apoptosis induction in vitro. (Bars = means ± SD, n=6, *P < 0.05, **P < 0.01, ***P < 0.001).
Notch signaling blocking in MFs promotes Bax-mediated apoptosis of MFs by up-regulating Ngfr and Septin4
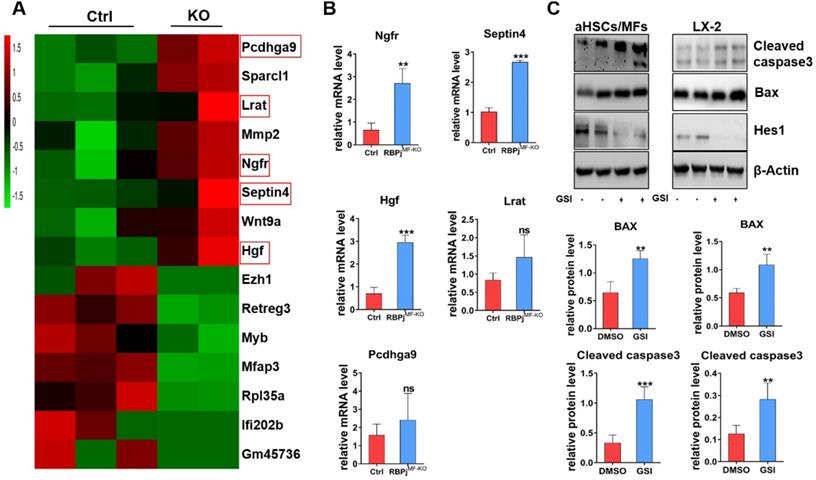
To further investigate the mechanism of Notch signaling in regulating MFs apoptosis during the regression of liver fibrosis, we isolated the hepatic MFs in the reversal stage of liver fibrosis of RBPjMF-KO and control mice and performed transcriptome sequencing. Comparison of gene expression profiles of MFs and further analysis of RNA-sequencing data showed that several apoptosis-related molecules, including nerve growth factor receptor (Ngfr) and Septin4, were up-regulated remarkably in RBPjMF-KO mice (Figure 7A). qPCR confirmed up-regulation of Ngfr and Septin4 in MFs from RBPjMF-KO mice (Figure 7B). Ngfr is known to mediate cell apoptosis, which is a member of tumor necrosis factor receptor (TNFR) superfamily [35-37]. According to previous research [38, 39], overexpression of Ngfr in liver promotes the MFs apoptosis during regression of liver fibrosis. Septin4 belongs to Septin family and is involved in apoptosis, vesicle trafficking and other cellular events [40, 41]. Using the Septin4-/- mice, Iwaisako et al. found that loss of Septin4 could aggravate the Collagens deposition [42]. In our research, Ngfr and Septin4 were overexpressed after Notch inactivation in MFs (Figure 7A, B). Additionally, WB showed that Bax and Cleaved caspase3 were up-regulated after Notch signaling blockade in MFs and LX-2 during undergoing apoptosis (induced by being cultured with serum-free medium for 24h) (Figure 7C), which indicated that Notch downstream molecules may expedite the Ngfr and Septin4 expression in other mechanism to aggravate Bax-mediate apoptosis of MFs. Moreover, RNA-sequencing results showed that HGF was increased in RBPjMF-KO (Figure 7A, B), which validated our previous data that Notch disruption in MFs enhances hepatocyte proliferation via up-regulating the expression of HGF during liver fibrosis regression (Figure 5E, F).
In-vitro experiments verify that the blockade of Notch signaling in MFs promotes Bax-mediated apoptosis of MFs by potentially up-regulating Ngfr and Septin4. (A) Heatmap comparison of related genes with RNA-sequencing data derived from RBPjMF-KO and control MFs. (B) qPCR results of differentially expressed genes indicated that Ngfr, Septin4, HGF are consistent with the RNA-seq data, which may be involved in the mechanism of increased MFs apoptosis and increased hepatocyte proliferation. (n=3, *P < 0.05, ** P < 0.01, *** P < 0.001). (C) Primary isolated aHSCs/MFs and activated LX-2 cell line were cultured with serum-free medium for 24h to induce apoptosis and treated with GSI (100 μM for 24h) simultaneously. Protein levels of Hes1, BAX and Cleaved caspase3 were determined by WB and quantitatively compared. (Bars = means ± SD, n=6, *P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
Notch signaling has been suspected for some time to play a role in liver fibrosis [43]. Alagille syndrome patients (mutations in Jagged1 gene in 94% of patients or “Notch2” in 1-2%) were reported to rarely go onto cirrhosis and exhibit less ECM deposition than other chronic liver diseases, representing an early clue toward a role for Notch signaling pathway in the liver fibrotic response [30, 43, 44]. Rat HSCs express Notch receptors in vitro and begin expressing Jagged1 upon activation and differentiation to MFs [25]. Additionally, MFs activation and subsequent collagen deposition in the lung has also been found to be partially dependent on Notch activity, suggesting that the role for Notch in fibrosis is not limited to the liver [45]. In the present study, MFs-specific Notch signaling is activated in liver fibrosis progression and depressed in liver fibrosis regression, which indicates that Notch signaling in MFs may participate in the regulation of liver fibrosis progression and regression (Figure 1). Then, we precisely confirmed the expression of one of the MFs markers- Sm22α, and utilized Sm22αCreER-RBPjflox/flox mice after conditional induction to obtain a MFs-specific loss-of-function model of Notch signaling- RBPjMF-KO (Figure 2). Indeed, Sm22αCreER model has been included in MFs-related research [46, 47], however, the expression and pinpoint of Sm22α during the progression of liver fibrosis has not been determined. Based on our data (Figure 2A, B), Sm22αCreER mice can be used as a reliable model for the study of hepatic MFs.
The GSI (pharmacological Notch signaling inhibitor) intraperitoneal injection alleviated liver fibrosis in rats [48]. However, because of GSI's indiscriminate effect to all types of cells in the liver, it has not been elucidated which kind of cell Notch signaling plays an important role in this process. Our previous studies found that myeloid-specific disruption of RBPj ameliorated hepatic fibrosis and LSEC-specific Notch signaling activation aggravated liver fibrosis and inhibited hepatocyte proliferation [28, 29]. Additionally, our recent research revealed that LSEC-specific Notch signaling disruption alleviated hepatic sinusoidal fibrosis (unpublished). However, the effects of MFs-specific Notch signaling on liver fibrosis are still ambiguous. Therefore, these prompted us to investigate the exact role and mechanism of MFs-specific Notch signaling to mediate liver fibrosis progression and regression. In the present study, specific blocking of Notch signaling of MFs inhibited the activation of HSCs to MFs both in cultures and RBPjMF-KO mice, showing decreased expression of α-SMA and Desmin, the markers of HSCs-MFs activation (Figure 3, 4).
Another important effector mediating fibrosis is MMPs, which consist of a family of enzymes with different substrate affinities to matrix components [43, 49]. These fibrinolytic mediators, especially MMP12 and MMP13, were mainly secreted by macrophages [50], other different MMPs were derived from MFs and neutrophils [49]. In this study, the expression of MMP8, MMP9 and MMP12 increased in the liver of RBPjMF-KO mice (Figure 3D). This is probably due to the increased MMPs expression of MFs. The blockage of Notch signaling in MFs not only reduced the deposition of ECM by inhibiting the activation of HSCs to MFs but also up-regulated the expression of MMPs to promote the degradation of ECM and thus to inhibit the progress of liver fibrosis. Besides, due to the Sm22α expression of hepatic vascular smooth muscle cells (VMSCs) in normal mice (Figure 2C), we also analyzed the phenotype of Sm22αCreER-RBPjMF-KO and control mice without CCl4 administration. The results showed that the blockage of Notch signaling restricted to hepatic VMSCs had no effect on the liver of healthy mice (supplementary materials. Figure S1), which lays a theoretical foundation for the safety of targeting MFs-related Notch signaling to treat liver fibrosis in the future.
After the insults were terminated, liver fibrosis will be gradually reversed as mainly manifested of degradation of ECM and regeneration of liver tissues, which involves the interaction of many kinds of cells in the liver, such as LSECs, macrophages, MFs [6]. The deactivation of MFs is key to fibrosis regression, and three mechanisms have been proposed: senescence, apoptosis and inactivation [6, 51]. Indeed, in the present study, the regression of hepatic fibrosis was significantly improved in RBPjMF-KO mice (Figure 5), which was mainly characterized by the decrease of extracellular matrix deposition (Figure 5B, D), the promotion of hepatocyte proliferation (Figure 5E) and the recovery of liver function (Figure 5G). These results were partly explained by the fact that Notch signaling regulated the expression of TIMP1 and HGF in MFs during fibrosis regression. Besides, liver section IF counterstaining and in vitro experiments showed that the MFs-specific Notch signaling disruption promoted the apoptosis of MFs (Figure 6), validating the conclusion of liver fibrosis regression acceleration in RBPjMF-KO mice.
During fibrosis regression, senescence describes a phenotype of MFs with reduced fibrogenic gene expression and cell cycle exit, which confers susceptibility to natural killer (NK) cells mediated apoptosis [7]. Driven by the withdrawal of anti-apoptotic signaling pathways as well as by NK, γδ T and CD8+ T cells, MFs undergo apoptosis [11, 51]. Many signaling pathways have also been confirmed to stimulate apoptosis in activated HSCs-MFs. These include CEBP/β signaling, farnesoid X receptor (FXR or bile acid receptor), cannabinoid receptor 2 (CB2), adiponectin, Septin4 and TRAIL (also known as TNFSF10) [10, 40]. Nerve growth factor (Ngf) can be released from infiltrating inflammatory cells or regenerating hepatocytes and is pro-apoptotic for activated HSCs-MFs [8, 38, 52]. According to our RNA-sequencing results, blockade of Notch signaling in MFs upregulated the expression of Ngfr and Septin4. As a member of TNFR superfamily molecules, Ngfr mediated canonical cell apoptosis [53, 54]. Previous study reported that in the fibrotic liver of mice with systemic Ngfr knockout, the apoptosis of MFs was significantly decreased and the regression of hepatic fibrosis was slowed down [39]. Overexpression of Septin4 can ameliorate hepatic fibrosis in mice as well [55]. So, in this study, the overexpression of Ngfr and Septin4 is likely the reason why Notch blockade in MFs promoted BAX-mediated apoptosis of MFs itself. However, the mechanism of Notch blockage leading to the increase of Ngfr and Septin4 remains to be explored.
In summary, the Notch signaling is closely associated with the liver fibrosis progression and regression. MFs-specific Notch signaling blockade can alleviate the liver fibrosis by suspending the progression of hepatic fibrosis, but also improving the resolution of hepatic fibrosis. As a major global healthcare burden, the discovery of key therapeutic targets with high relevance to liver fibrotic disease and the subsequent development of effective antifibrotic therapies directed against these targets continues to be a research priority. Our study establishes a relationship between MFs-specific Notch signaling and liver fibrosis progression and regression through Notch-mediated cytological and molecular changes, and sheds insights into therapeutic development against liver fibrosis targeting Notch signaling in MFs.
Abbreviations
MFs: myofibroblasts; HSCs: hepatic stellate cells; α-SMA: α-smooth muscle actin; ECM: extracellular matrix; HGF: hepatocyte growth factor; MMPs: matrix metalloproteinases (MMPs); TGF-β: transforming growth factor-β; PDGF-β: platelet-derived growth factor β; IL-17A: interleukin-17A; TNFR: tumor necrosis factor receptor; TIMPs: tissue inhibitors of metalloproteinases; NICD: Notch intracellular domain; RBPj: j-kappa recombination signal-binding protein; Sm22α: smooth muscle 22α; HYP: hydroxyproline; GSI: gamma(γ)-secretase inhibitor; Ngfr: nerve growth factor receptor; VSMCs: vascular smooth muscle cells; CCl4: carbon tetrachloride; IF: immunofluorescence; HE: hematoxylin and eosin; IHC: immunohistochemistry; SEM: scanning electron microscopy.
Supplementary Material
Supplementary figure and table.
Acknowledgements
This work was supported by grants from the MOST (2016YFA0102100 [The National Key Research and Development Program of China: Stem Cell and Translational Research]), and the National Natural Science Foundation of China (31730041, 81422009, 81401940, 81770560, 81800533, 81870430, 31671523). The experiments were fulfilled at the Laboratory of Department of Hepatobiliary Surgery (Xi-Jing Hospital) and Postgraduate Innovative Research Center of the Fourth Military Medical University.
Author Contributions
Zhensheng Yue carried out animal and vitro experiments and analyzed the data; Zijian Jiang analyzed the data; Bai Ruan and Juanli Duan helped in primary cell isolation and culture; Ping Song collected the specimen and performed staining; Jingjing Liu helped in western blotting.; Hua Han directed the study and gave critical discussion about data; Lin Wang designed project and directed study and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425-36
2. Bataller R, Brenner DA. Liver fibrosis. Journal of Clinical Investigation. 2005;115:209-18
3. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411
4. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555-66
5. Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S60-3
6. Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63:1038-9
7. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C. et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657-67
8. Ramachandran P, Iredale JP, Fallowfield JA. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin Liver Dis. 2015;35:119-31
9. Campana L, Iredale JP. Regression of Liver Fibrosis. Semin Liver Dis. 2017;37:1-10
10. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425-56
11. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S. et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538-49
12. Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D. et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765-76 e3
13. Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427-36
14. Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554-61
15. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-69
16. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD. et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795-808
17. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770-6
18. Yamamoto S, Schulze KL, Bellen HJ. Introduction to Notch signaling. Methods Mol Biol. 2014;1187:1-14
19. Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci. 2013;126:2135-40
20. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-33
21. Gharaee-Kermani M, Hu B, Thannickal VJ, Phan SH, Gyetko MR. Current and emerging drugs for idiopathic pulmonary fibrosis. Expert Opin Emerg Drugs. 2007;12:627-46
22. Kuma A, Tamura M, Otsuji Y. [Mechanism of and Therapy for Kidney Fibrosis]. J UOEH. 2016;38:25-34
23. Desbois AC, Cacoub P. Systemic sclerosis: An update in 2016. Autoimmun Rev. 2016;15:417-26
24. Toosi AE. Liver Fibrosis: Causes and Methods of Assessment, A Review. Rom J Intern Med. 2015;53:304-14
25. Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology. 2009;50:1617-24
26. Xie G, Karaca G, Swiderska-Syn M, Michelotti GA, Kruger L, Chen Y. et al. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology. 2013;58:1801-13
27. Zhang K, Zhang YQ, Ai WB, Hu QT, Zhang QJ, Wan LY. et al. Hes1, an important gene for activation of hepatic stellate cells, is regulated by Notch1 and TGF-beta/BMP signaling. World J Gastroenterol. 2015;21:878-87
28. He F, Guo FC, Li Z, Yu HC, Ma PF, Zhao JL. et al. Myeloid-Specific Disruption of Recombination Signal Binding Protein J kappa Ameliorates Hepatic Fibrosis by Attenuating Inflammation Through Cylindromatosis in Mice. Hepatology. 2015;61:303-14
29. Duan JL, Ruan B, Yan XC, Liang L, Song P, Yang ZY. et al. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration. Hepatology. 2018;68:677-90
30. Adams JM, Jafar-Nejad H. The Roles of Notch Signaling in Liver Development and Disease. Biomolecules. 2019;9:608
31. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N. et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060-72
32. Jiang F, Parsons CJ, Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J Hepatol. 2006;45:401-9
33. De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA. et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132:1937-46
34. Schumacher EC, Gotze S, Kordes C, Benes V, Haussinger D. Combined Methylome and Transcriptome Analysis During Rat Hepatic Stellate Cell Activation. Stem Cells Dev. 2017;26:1759-70
35. Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945-8
36. Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci U S A. 1993;90:7859-63
37. Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554-8
38. Trim N, Morgan S, Evans M, Issa R, Fine D, Afford S. et al. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am J Pathol. 2000;156:1235-43
39. Kendall TJ, Hennedige S, Aucott RL, Hartland SN, Vernon MA, Benyon RC. et al. p75 Neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology. 2009;49:901-10
40. Koren E, Fuchs Y. The ARTS of Cell Death. J Cell Death. 2019;12:1179066019836967
41. Garcia-Fernandez M, Kissel H, Brown S, Gorenc T, Schile AJ, Rafii S. et al. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Gene Dev. 2010;24:2282-93
42. Iwaisako K, Hatano E, Taura K, Nakajima A, Tada M, Seo S. et al. Loss of Sept4 exacerbates liver fibrosis through the dysregulation of hepatic stellate cells. J Hepatol. 2008;49:768-78
43. Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology. 2015;61:382-92
44. Fabris L, Cadamuro M, Guido M, Spirli C, Fiorotto R, Colledan M. et al. Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Am J Pathol. 2007;171:641-53
45. Liu T, Hu B, Choi YY, Chung M, Ullenbruch M, Yu H. et al. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol. 2009;174:1745-55
46. Klein S, Rick J, Lehmann J, Schierwagen R, Schierwagen IG, Verbeke L. et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut. 2017;66:145-55
47. Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15-22
48. Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S. et al. Inhibition of Notch signaling by a gamma-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS One. 2012;7:e46512
49. Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim Biophys Acta. 2013;1832:876-83
50. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A. et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186-95
51. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181-94
52. Oakley F, Trim N, Constandinou CM, Ye W, Gray AM, Frantz G. et al. Hepatocytes express nerve growth factor during liver injury: evidence for paracrine regulation of hepatic stellate cell apoptosis. Am J Pathol. 2003;163:1849-58
53. Wajant H. Death receptors. Essays in biochemistry. 2003;39:53-71
54. Bhardwaj A, Aggarwal BB. Receptor-mediated choreography of life and death. J Clin Immunol. 2003;23:317-32
55. He X, Bao J, Chen J, Sun X, Wang J, Zhu D. et al. Adenovirus-mediated over-expression of Septin4 ameliorates hepatic fibrosis in mouse livers infected with Schistosoma japonicum. Parasitol Int. 2015;64:487-92
Author contact
![]() Corresponding authors: Lin Wang, Department of Hepatobiliary Surgery, Xi-Jing Hospital, Fourth Military Medical University, Chang-Le Xi Street #127, Xi'an 710032, China, E-mail: fierywangcom; Hua Han, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi'an 710032, China, E-mail, huahanedu.cn.
Corresponding authors: Lin Wang, Department of Hepatobiliary Surgery, Xi-Jing Hospital, Fourth Military Medical University, Chang-Le Xi Street #127, Xi'an 710032, China, E-mail: fierywangcom; Hua Han, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi'an 710032, China, E-mail, huahanedu.cn.