Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Aβ mediates tau toxicity
Joint action of Aβ and tau...
Conclusions and future directions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(9):2181-2192. doi:10.7150/ijbs.57078 This issue Cite
Review
Interaction between Aβ and Tau in the Pathogenesis of Alzheimer's Disease
Huiqin Zhang1, Wei Wei1, Ming Zhao2, Lina Ma1, Xuefan Jiang2, Hui Pei1, Yu Cao1 ![]() , Hao Li1
, Hao Li1 ![]()
1. Institute of Geriatrics, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing 100091, China.
2. Beijing University of Chinese Medicine, Beijing 100029, China.
Received 2020-12-12; Accepted 2021-4-23; Published 2021-5-27
Abstract

Extracellular neuritic plaques composed of amyloid‑β (Aβ) protein and intracellular neurofibrillary tangles containing phosphorylated tau protein are the two hallmark proteins of Alzheimer's disease (AD), and the separate neurotoxicity of these proteins in AD has been extensively studied. However, interventions that target Aβ or tau individually have not yielded substantial breakthroughs. The interest in the interactions between Aβ and tau in AD is increasing, but related drug investigations are in their infancy. This review discusses how Aβ accelerates tau phosphorylation and the possible mechanisms and pathways by which tau mediates Aβ toxicity. This review also describes the possible synergistic effects between Aβ and tau on microglial cells and astrocytes. Studies suggest that the coexistence of Aβ plaques and phosphorylated tau is related to the mechanism by which Aβ facilitates the propagation of tau aggregation in neuritic plaques. The interactions between Aβ and tau mediate cognitive dysfunction in patients with AD. In summary, this review summarizes recent data on the interplay between Aβ and tau to promote a better understanding of the roles of these proteins in the pathological process of AD and provide new insights into interventions against AD.
Keywords: Alzheimer's disease, amyloid-β, tau, interaction, phosphorylation
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease that is characterized by the progressive deterioration of cognition and memory [1]. AD is defined pathologically by an extensive loss of neurons and two characteristic protein deposits in the brain, extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs), which are caused by tau hyperphosphorylation [1, 2]. Several hypotheses related to the pathogenesis of AD were studied [3], and the amyloid-β (Aβ) cascade and the hyperphosphorylation of tau protein are the two main hypotheses. The Aβ cascade hypothesis states that Aβ, deposited in the form of neuroinflammatory plaques, induces AD by damaging neuronal cells [4]. Aβ facilitates the development of AD and initiates a deleterious cascade involving tau pathology and neurodegeneration [5]. However, the development of many AD drugs examined in clinical trials and laboratory studies focused on the reduction of amyloid deposition and the clearance of Aβ oligomers and have not yielded satisfactory effects [6-8]. Aβ may not be the only disease-causing protein during the course of AD. Intraneuronal tangles containing hyperphosphorylated tau are a hallmark of AD pathology [9], and tau is a mediator of Aβ cytotoxicity [7]. Therefore, the pathological changes to tau in AD also attracted attention. However, extensive studies focused solely on the neurotoxicity of Aβ or tau have not shown significant efficacy in the treatment of AD [8-11]. Therefore, focusing solely on the role of Aβ or tau in AD lesions while ignoring the interaction between Aβ and tau may not be entirely correct. Aβ and tau may interact via intermediate materials, such as kinases (e.g., GSK-3β, CDK-5 and ERK) [12]. The removal of Aβ or tau alone does not completely terminate the interaction pathway, which continues to play a role in the acceleration of the pathological process [11, 13]. The pathological process likely involves an interplay between Aβ and tau in which these proteins amplify toxic effects rather than a strictly hierarchical mode of interaction [13]. Therefore, suppression of the interaction may be of more practical significance than simply focusing on the neurotoxicity of Aβ or tau alone [14]. The present review describes the following interactions between Aβ and tau. A) Aβ drives tau pathology by inducing tau hyperphosphorylation, and hyperphosphorylated tau mediates toxicity in neurons. B) Tau mediates Aβ toxicity, and Aβ toxicity is critically dependent on the presence of tau. C) Aβ and tau target cellular processes or organelles synergistically and may amplify each other's toxic effects. D) Aβ and tau may coexist in pathological locations [11]. We hope that this review provides a better understanding of the role of Aβ and tau in the pathological development of AD to promote further research on the mechanism of the interaction between Aβ and tau and the development of more effective therapies for AD.
Aβ mediates tau toxicity
Aβ accelerates the phosphorylation of tau protein
Tau is a microtubule (MT)-binding protein, and tau phosphorylation at multiple sites controls its binding to MTs [15]. The identified phosphorylation sites are located in the N-terminal region (Ser46 [16], Thr123 [17], Ser198, Ser199, Ser202, Ser208, Ser210, Thr212, Ser214, Thr217, Thr231, and Ser235), the repeat region (Ser262 and Ser356), and the C-terminal region (Ser396, Ser400, Thr403, Ser404, Ser409, Ser412, Ser413, and Ser422 [18]) (Figure 1). These sites are phosphorylated by several enzymes, including A-kinase, C-kinase, cyclin-dependent kinase-5 (CDK-5), CaM kinase II, glycogen synthase kinase-3β (GSK-3β) and MAPKs [12, 19] (Table 1). Under pathological conditions, these kinases contribute to the hyperphosphorylation of tau protein, which subsequently leads to the dissociation of tau protein from MTs and the formation of NFTs [20, 21]. Hyperphosphorylated tau protein results in abnormal increases in cytoskeletal proteins, axoplasmic transport disorders and neuronal degeneration [22]. Tau hyperphosphorylation is the product of deregulated Ser/Thr kinases, such as CDK-5 and GSK-3β [23]. Tau protein is prephosphorylated by A-kinase, C-kinase, CDK-5, and CaM kinase II and subsequently phosphorylated by GSK-3β. Ser262/Ser356 is first prephosphorylated by CDK-5, CaM kinase II and C-kinase, and this prephosphorylation makes it more accessible to GSK-3β for further phosphorylation [24]. The prephosphorylation of Ser262/Ser356 may cause tau to assume a more favourable conformation such that Thr231, Ser235, Ser396, Ser404 [19, 24] and other sites may be rapidly recognized by GSK-3β in the subsequent phosphorylation step [25]. Aβ accelerates the hyperphosphorylation of tau by mediating the activation of CDK-5 [23] and GSK-3β [26]. GSK-3β, which is inextricably related to Aβ, is an important factor in the phosphorylation of tau, and it aggravates tau-induced neurotoxicity [15, 27] (Table 1). Tau phosphorylation at Ser262 is equally critical for Aβ42-induced tau toxicity [11]. In conclusion, aggregated Aβ induces tau hyperphosphorylation by enhancing the activity of GSK-3β and CDK-5, which are involved in the phosphorylation of tau at multiple Ser/Thr sites [28] (Figure 1). Hyperphosphorylated tau protein detaches from MTs and is more likely to form NFTs [23, 27, 30], which causes neurofibrillary degeneration. Therefore, CDK-5 and GSK-3β kinase inhibitors may help inhibit Aβ-mediated tau hyperphosphorylation. For example, inhibitors of GSK-3β and CDK-5, such as lithium, AZD1080 and roscovitine, significantly lowered the levels of tau phosphorylation and significantly reduced the levels of aggregated, insoluble tau [31-33]. However, their clinical efficacy has not been established [34]. This result further suggests that proteins other than Aβ act on tau hyperphosphorylation or phosphorylation caused by kinases other than CDK-5 and GSK-3β, that Aβ is not the only protein that activates CDK-5 and GSK-3β, and that other tau phosphorylation sites should be further explored. Further studies have demonstrated that mitogen-activated protein kinases (MAPKs) and GSK-3β were involved in the formation of PHF-tau in the AD brains [35-37]. Similarly, Aβ-induced tau phosphorylation in neurons is mediated via activation of both MAPK and GSK-3β [12, 36-38]. MAPKs, including ERK1/2, SAPKs and p38, are activated by tyrosine phosphorylation and are involved in the phosphorylation of tau in AD [36-38]. Therefore, further exploration is needed to develop new drugs that act at different specific phosphorylation sites of tau via various mechanisms.
Tau phosphorylation sites and associated kinases. The identified tau phosphorylation sites are located in the N-terminal region (Ser46 [16], Thr123 [17], Ser198, Ser199, Ser202, Ser208, Ser210, Thr212, Ser214, Thr217, Thr231, and Ser235), the repeat region (Ser262 and Ser356), and the C-terminal region (Ser396, Ser400, Thr403, Ser404, Ser409, Ser412, Ser413, and Ser422 [18]). Ser199, Ser202, Thr231, Ser235, Ser262, Ser396, and Ser404 are phosphorylated by various activated kinases, namely A-kinase, C-kinase, CaM kinase II, CDK-5 and Gsk-3β, and Ser356 is phosphorylated by C-kinase, CaM kinase II, CDK-5 and Gsk-3β. A-kinase, C-kinase, CaM kinase II and CDK-5 are primarily involved in the prephosphorylation of tau. This prephosphorylation increases the affinity of GSK-3β to these sites, which ultimately leads to tau hyperphosphorylation.
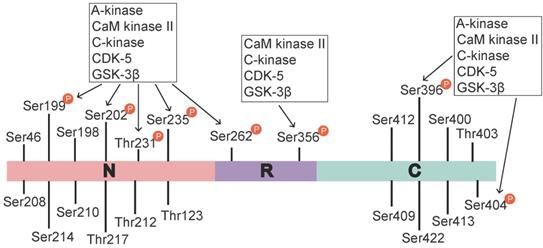
Sites on tau phosphorylated by different kinases
| Phosphorylation stage | Kinase | Phosphorylation sites | Whether Aβ is involved | References |
|---|---|---|---|---|
| Prephosphorylation | A-kinase | Ser262, Ser293, Ser305, Ser324, Ser356 | No | [18, 19, 24] |
| C-kinase | Ser305 | No | [24] | |
| CaM kinase II | Ser416/Ser262 | No | [19, 24] | |
| CDK-5 | Ser195, Ser202, Thr231, Ser235, Ser396, Ser404 | Yes | [19, 23, 24, 28] | |
| Phosphorylation | GSK-3β | Ser199*, Ser202, Thr231*, Ser235, Ser262, Ser396*, Ser404* | Yes | [19, 23, 24, 28] |
| MAPK | Thr181, Ser202*, Thr205*, Ser396*, Ser404*, Ser422, Ser199*, Thr50* | Yes | [36-38] | |
| Prephosphorylation + Phosphorylation | A-kinase + GSK-3β | Ser199*, Ser202*, Thr231, Ser235, Ser262, Ser396*, Ser404* | Yes | [24, 25] |
| C-kinase + GSK-3β | Ser199, Ser202, Thr231*, Ser235*, Ser262, Ser396*, Ser404d* | Yes | [24, 25] | |
| CaM kinase II + GSK-3β | Ser199*, Ser202*, Thr231*, Ser235*, Ser262*, Ser396*, Ser404* | Yes | [24, 25] | |
| CDK-5 + GSK-3β | Ser199, Ser202, Thr231*, Ser235*, Ser262, Ser396, Ser404d | Yes | [23-26] |
Note: * Phosphorylation of these sites is significantly enhanced.
Aβ also interferes with tau oligomerization
In addition to its driving effect on tau phosphorylation, Aβ also interferes with tau oligomerization and aggregation. Tau oligomers, which are an intermediate form of tau that occurs prior to NFT formation, are toxic [39]. Hyperphosphorylated tau dislodges from MTs, and its affinity for other tau monomers induces binding between individual tau proteins to form tau oligomers, which are detergent-soluble aggregates, by acting on the phosphorylation sites. This process is mediated by Aβ-activated CDK-5 and GSK-3β. These tau oligomers potentiate neuronal damage, which leads to neurodegeneration [40, 41]. Aβ also triggers caspase-3 (CASP3) cleavage of tau at Asp421 in the C-terminus to yield an N-terminal caspase cleavage product (amino acids 1-421) [42] (Figure 2). Truncated tau lacking its C-terminal 20 amino acids assembles more rapidly into filaments than full-length tau [42]. Aβ also induces the generation of a 17-kDa tau fragment via the activation of calpain-1 in hippocampal neurons. The expression of this tau fragment leads to neurite degeneration and cell death in neurons and nonneuronal cell types [43]. Cleaved tau protein self-aggregates and misfolds, and these self-aggregated and misfolded products are further assembled into soluble oligomers [42]. Tau oligomers may act as “seeds” by inducing endogenous tau misfolding and redistribution to the somatodendritic compartment, which suggests the existence of a unifying mechanism for the propagation of amyloid proteins [44, 45]. Oligomers in this aggregation process are the most neurotoxic form of tau and may be transmitted between neuronal cells [39]. Tau oligomers in neurons induce neuroinflammatory factors, which bind to astrocytes and microglia and induce apoptosis [39, 46]. Therefore, the inhibition of tau oligomer formation is also worth further examination. For example, a targeted inhibitor of caspase-3 may be developed to reduce tau cleavage.
Reciprocal toxicity between Aβ and tau. Aβ precursor protein (APP) is cleaved by β/γ secretases to form Aβ. Aβ activates GSK-3β and CDK-5 to phosphorylate tau protein and activate caspase-3 and calpain 1 to hydrolyse tau protein and form tau oligomers. Phosphorylated tau protein interacts with Fyn. Aβ-activated Fyn also accelerates tau phosphorylation and binds to tau. Phosphorylated Fyn acts on NR2B to form the NMDAR-PSD95-Fyn complex. NMDARs are activated to increase Ca2+, which affects the function of mitochondria. Aβ and phosphorylated tau induce the fusion and fission of mitochondria by acting on Drp1, which induces the dysfunction of mitochondrial dynamics and ultimately leads to reactive oxygen species (ROS) overproduction and apoptosis.
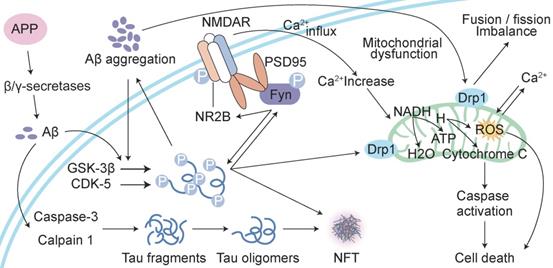
Aβ toxicity is dependent on tau
Extracellular deposits of fibrillar Aβ form senile plaques and accelerate tau hyperphosphorylation, which induces neurotoxicity. However, whether the neurotoxicity of Aβ is dependent on tau is not clear. One study showed that the absence of tau in glutamate N-methy1-1-D-aspartate (NMDA) receptor subunit GluN2B-bearing spines, where tau is typically located, prevented the toxic effect of the binding of Aβ to NMDA receptors (NMDARs) [47]. A recent study proposed that the phosphorylation of tau at Tyr 18 by Fyn kinase also blocks Aβ toxicity [48, 49]. Aβ may be the initiator of tau pathology and play a role in downstream neuronal injury [50, 51]. Tau mediates Aβ toxicity by interacting with Fyn kinase via its amino-terminal projection domain (PD) [50]. Aβ promotes Fyn phosphorylation, and phosphorylated tau and Fyn are transferred to postsynaptic membrane receptors, which are denoted by NMDARs. Fyn phosphorylates the NR subunit 2 (NR2) to facilitate the interaction with postsynaptic density protein 95 (PSD95) [47, 52], which leads to excitotoxic downstream signalling [47, 53]. Soluble Aβ binds to or near NMDARs, which indicates that NMDARs are potential targets of Aβ, and the NMDAR and PSD95 complex participates in NMDAR-mediated Aβ neurotoxicity [54, 55]. Therefore, the activation of Fyn, which migrates into dendritic spines via tau facilitation, is a key event in Aβ oligomer-induced synaptic toxicity [45, 56]. Some studies have also indicated that a reduction in tau levels prevented the cognitive impairment in AD transgenic mice overexpressing Aβ and that overexpression of Fyn can enhance their cognitive impairment [57, 58]. The absence of tau prevents Aβ toxicity and tau phosphorylation by GSK3 [59, 60] or another kinase (p38γ) [61] at a specific residue (threonine 205) [62]. P38γ regulates PSD95-tau-Fyn complexes by phosphorylating tau at Thr205, which interferes with the postsynaptic excitotoxic signalling complexes engaged by Aβ. Tau-dependent Aβ toxicity is modulated by site-specific tau phosphorylation, which inhibits postsynaptic PSD95-tau-Fyn complexes, which reveals an Aβ toxicity-limiting role of p38γ in AD [61]. A new study found that Aβ alone caused hyperactivity, and tau alone suppressed activity and promoted the silencing of many neurons [62]. Neuronal silencing dominates hyperactivity in the presence of both Aβ and tau, and this finding was corroborated in a recent in vitro study using extracellular field recordings of entorhinal cortex (EC) slices [62]. Tau blocks Aβ-dependent hyperactivity, which results in the profound silencing of circuits in the presence of Aβ and tau in the cortex [63]. This result suggests that tau and Aβ exert antagonistic effects on neural circuit activation. Caspase-cleaved tau may also be an important downstream event in Aβ-induced cascades, such as the poisonous 17-kDa tau fragment, which leads to neurite degeneration and cell death [43]. Therefore, Aβ-induced neurotoxicity may be mediated not only by Aβ overdeposition but also, at least in part, by protease activation, which leads to the generation of neurotoxic tau fragments, such as the 17-kDa neurotoxic tau fragment. Aβ may serve as the trigger, and tau may be the bullet [45, 53]. All of these results further support the notion that the toxicity of Aβ is dependent on tau (Figure 2). In summary, tau protein may play a key role in the Aβ-induced mechanism, and the recombinant expression of human tau protein in tau-depleted neurons restores the sensitivity of neurons to Aβ toxicity.
Aβ and tau work together to damage mitochondria
Another cellular compartment in which Aβ and tau work together to damage neurons is the mitochondrion [45]. Mitochondrial dysfunction is widely implicated in the aetiology of AD [64]. Aβ deposition, NFT formation, and neurodegeneration may be consequences of mitochondrial malfunctioning [65]. The overexpression of APP and treatment with amyloid-β-derived diffusible ligands induce mitochondrial fragmentation and abnormal distribution without cell death [66, 67], which suggests that Aβ-induced abnormal mitochondrial dynamics may play a role in the early pathogenesis of AD [68]. Aβ induces axonal and dendritic swelling and decreases axonal transport in hippocampal neurons [69], which contribute to an altered mitochondrial distribution. Excessive mitochondrial fission correlates with increased reactive oxygen species (ROS) production [66, 67, 70], which leads to the progression of extensive macromolecular oxidative damage and amyloid lesions caused by ROS [71]. Intracellular Aβ interacts with 3-hydroxyacyl-CoA dehydrogenase type-2 (HSD17B10, also known as ABAD), and this interaction promotes mitochondrial dysfunction and ROS leakage [72]. Another protein localized at the outer mitochondrial membrane, dynamin-1-like protein Drp1 (DNM1L), interacts with Aβ and phosphorylated tau, which likely leads to excessive mitochondrial fragmentation and neuronal damage [73]. Tau is a substrate for mitochondrial caspase-3 in vitro [41, 74], which contributes to mitochondrial fragmentation in AD brains. Caspase-cleaved tau and tau truncated at Asp421, which mimics the product obtained with caspase cleavage (T4C3), induce mitochondrial fragmentation and mitochondrial fission in a calcineurin-dependent manner [75]. The expression of caspase-cleaved tau fragments in neurons impairs mitochondrial function, and this impairment leads to changes in the mitochondrial membrane potential, the dysregulation of mitochondrial calcium levels and mitochondrial integrity and an increase in mitochondrial O2- [68, 75]. The introduction of tau in mature hippocampal neurons results in the degeneration of synapses via perturbation of mitochondrial transport and ATP levels at the synapse [76]. N-terminal tau fragments exert toxic effects on mitochondria and lead to mitochondrial dysfunction associated with impairments in oxidative phosphorylation by distorting the structure of the complex V enzyme and the level of adenine nucleotide translocator, which ultimately perturb the synthesis of ATP in mitochondria [77]. The dysregulation of Ca2+ and its secondary processes directly affect tau phosphorylation, APP processing and lysosome function, and in turn, tau and Aβ further aggravate Ca2+ dyshomeostasis [78]. Aβ stimulates neurons to cause a large amount of Ca2+ influx and intracellular Ca2+ overload then damage to mitochondria, which results in the impairment of neuronal productivity, activation of apoptosis-related proteins and factors and the initiation of apoptosis [79]. The dysregulation of Ca2+ homeostasis further promotes mitochondrial dysfunction, impairments in synaptic transmission and plasticity and oxidative stress, which contribute to the age-related cognitive impairment. Aβ and tau exert deleterious effects on mitochondria, and these effects result in ROS accumulation, ATP synthesis and Ca2+ dysregulation [80] (Figure 2).
Joint action of Aβ and tau in nonneuronal cells
Combined effects of Aβ and tau on microglia in AD
Microglial activation is a key neuropathological feature of AD, and clinical studies found reduced levels of glial cell activation in more benign cases of AD [81]. Soluble Aβ and subsequent plaques trigger microglial activation and the release of proinflammatory cytokines, such as interleukin-1β and tumour necrosis factor-α (TNF-α), ROS and reactive nitrogen species [82]. The neuroinflammatory reaction in the brain caused by activated glial cells primarily occurs around capillary sites where Aβ is deposited [83]. The expression of mutant tau also drives microglial activation, even prior to tangle formation [84] (Figure 3). Young APP/PS1-rTg4510 mice without plaques and tangles exhibit a synergistically enhanced tau aggregation phenotype, which may be an early consequence of microglial changes. This result directly implicate microglial changes in the progression of tau pathology [85]. Microglial depletion protected against the propagation of tau pathology from the EC to the dentate gyrus (DG) in PS19 transgenic mice and rescued tau-dependent DG hypoexcitability [86]. Glial tau pathology reportedly occurs in tauopathies [87] and AD [88]. Microglia take up and decompose seed-competent tau [89], albeit ineffectively [90], and tau uptake may reciprocally lead to microglial activation. Activated microglia may be directly involved in the repackaging of tau into exosomes or indirectly involved in the enhancement of tau phosphorylation via proinflammatory cytokine signalling [85, 91]. Microglia activated as a consequence of neuronal injury may also be related to the induction of neurodegeneration, which leads to tau hyperphosphorylation and aggregation [92]. Enhanced microglial activation and accelerated onset and progression of tau pathology may be caused by the absence of the microglial fractalkine receptor CX3CR1 [85, 91]. Tau associated with exosomes and phosphorylated at Thr181 (AT270+ tau) was identified in human cerebrospinal fluid (CSF) samples from patients with AD [93]. Therefore, microglia may phagocytose tau-containing neurons or synapses and secrete tau protein via exosomes [93, 94]. During this process, microglia may facilitate the spread of tau pathology via exosomal vesicles. Variation in the TREM2 gene is a major risk factor for AD. TREM2 deletion and the secondary loss of plaque-associated microglia enhance the seeding and spreading of tau pathology, which suggests that TREM2 mitigates tau pathology by directing microglia to scavenge plaques [95]. Microglia may also neutralize Aβ or toxic tau species to delay propagation and neurodegeneration. Briefly, activated microglia may play a role in Aβ toxicity and tau pathology and the formation of neuritic plaques and tau hyperphosphorylation, which contribute to microglial activation.
Interaction of Aβ and tau with microglia and astrocytes. In response to Aβ or tau, microglia and astrocytes are converted into a reactive state, which triggers the inflammatory cascade. These cells reciprocally activate each other via this cascade, which leads to neuronal injury. Inflammatory cytokines (such as interleukin-1β and TNF-α) induce the neuronal release of tau, and activated microglia take up extracellular tau. Cytokines also accelerate tau phosphorylation to ultimately induce the formation of neurofibrillary tangles (NFTs). Transcription factor EB (TFEB) activates astrocytes to take up extracellular tau. This process consists of a cycle of Aβ deposition, tau phosphorylation, release and uptake, and the roles of cytokines to ultimately lead to cell death and the NFT-mediated exacerbation of neurodegenerative changes.
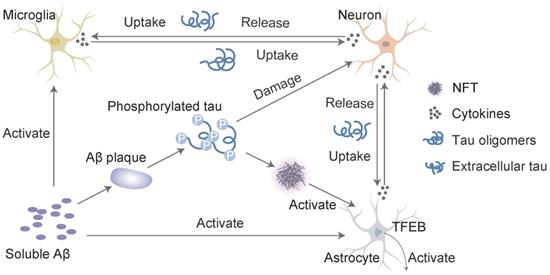
Interaction of Aβ and tau with astrocytes in AD
Astrocytes are abundant in the brain and perform many functions in the central nervous system (CNS), such as modulating synaptic formation, maintaining neuronal homeostasis via metabolic support [96], and comprising part of the blood-brain barrier (BBB) [97]. Astrocytes may be activated in the preclinical stages of AD [98]. One of the earliest neuropathological changes in AD is the accumulation of astrocytes at sites of Aβ deposition [99]. Activated astrocytes surround amyloid plaques and NFTs, which are the neuropathological hallmarks of advanced AD [100]. Aβ deposition and the NFTs formed by tau hyperphosphorylation may promote the activation and accumulation of astrocytes. Reactive astrocytes release proinflammatory mediators and cytotoxic molecules in neuroinflammation, and these effects exacerbate the pathology of AD [101]. However, astrocytes also express genes involved in phagocytosis, which may attenuate the pathology via the uptake and clearance of protein aggregates [102]. This phagocytic capacity was demonstrated for Aβ [103, 104]. Insights from animal models indicate that impairing astrocyte activation via the ablation of glial fibrillary acidic protein (GFAP) and vimentin resulted in an increased plaque load [105]. Aβ is taken up and trafficked to lysosomes for degradation in astrocytes [103]. The impairment of lysosome function with ageing [106] or the loss of presenilin is likely the underlying mechanism for the accumulation of Aβ and phagocytosed amyloid material within astrocytes [107], which may be the key to promoting amyloid plaque progression [100]. Transcription factor EB (TFEB) plays an important role in this process. Astrocytic TFEB expression attenuates amyloid plaque accumulation by enhancing Aβ uptake from the interstitial fluid (ISF) and facilitating clearance via lysosomal degradation in the brain. TFEB directly stimulates astrocytes to degrade Aβ, which reduces the pathogenesis of amyloid plaques (Figure 3). In a transgenic model of tau spreading, astrocytes took up hyperphosphorylated tau as the synapses degenerated [108]. The accumulation of abnormally phosphorylated tau in astrocytes is named ageing-related tau astrogliopathy (ARTAG) [109]. ARTAG may play a role in mediating the formation of pathological tau inclusions in astrocytes or indicate astrocyte involvement in the early stages of transsynaptic tau spreading [109]. TFEB in astroglia stimulated the uptake and clearance of aberrant extracellular tau, which prevented the neuronal spreading of tau pathology in a mouse model of tauopathy [110]. In conclusion, Aβ and tau may share a common site of action in astrocytes, which promotes the expression of TFEB activated by Aβ deposition and the over-deposition of hyperphosphorylated tau. Damage to astrocytes reduces TFEB expression, which results in reduced clearance and increased aggregation of Aβ and tau. In contrast, Aβ over-deposition and tau hyperphosphorylation accelerate astrocyte injury by releasing neuroinflammatory cytokines. However, excessive damage to astrocytes worsens the inadequate removal of Aβ and tau, which further aggravates self-injury and the progression of AD.
Aβ and tau may be involved in damage to the BBB, endothelial cells, and pericytes during the pathological process of AD. Aβ and tau activate microglia and astrocytes. Activated microglia disrupts chemokine and cytokine secretion from the BBB [111]. Microglia-mediated BBB alterations in AD are also reflected by the destruction of pericytes and basement membrane (BM) by microglia. Activated microglia also promotes pericyte apoptosis in vitro via the upregulation of NADPH oxidase in pericytes [112]. Activated astrocytes secrete vascular permeability factors, such as vascular endothelial growth factors (VEGFs), nitric oxide (NO) [113], and endothelin (ET) [114], which aggravate the destruction of the BBB. Aβ and tau may exert indirect or direct effects on BBB components. Currently available data are insufficient. Therefore, the specific mechanism must be further examined, and a further understanding may help identify new methods for the treatment and prevention of AD.
Aβ plaques facilitate neuritic plaque tau aggregation and propagation
Widespread deposition of Aβ plaques in the neocortex and a hierarchically organized pattern of NFTs (composed largely of tau aggregates) in limbic and cortical association areas are the neuropathological hallmarks of AD [5]. Aβ and p-τ pathology spread throughout the brain in a hierarchical pattern. However, prior to NFT formation in the transentorhinal region, an accumulation of p-τ is observed in the locus coeruleus, raphenuclei, substantia nigra, dorsal nucleus of the vagus nerve, and basal nucleus of Meynert [115-117]. Aβ plaques and NFTs are found in neocortical brain regions after a certain stage [118, 119]. This finding suggests that neurodegenerative lesions spread or propagate from one brain region to another [120]. Nevertheless, the mechanisms of Aβ and tau pathology interactions and whether other proteins are involved in this process are not clear and should be the focus of our attention and thinking.
A recent study showed that cellular prion protein (PrPC) was a receptor for toxic Aβ species and α-synuclein oligomers [121]. PrPC occurs in the neuropil [122, 123], and it was detected in amyloid plaques and neurons in patients with AD [124, 125]. PrPC is enriched in postsynaptic densities, which leads to Fyn kinase activation, and activated Fyn kinase phosphorylates the GluN2B subunit of NMDA receptors [126] and interacts with the phosphorylation of tau [127, 128]. Aβ oligomers initiate PrPC-Fyn-related phosphorylation of tau [129]. PrPC may be directly or indirectly involved in the interplay between Aβ and p-τ pathology propagation. Soluble p-τ may be released at synapses [130] and may be present in the extracellular space, which creates the conditions for the binding of soluble p-τ to PrPC. Aβ and PrPC combine with an interaction partner to activate Fyn [126, 131], which increases the levels of p-τ via Pyk2-related phosphorylation of tau [132, 133]. Tau appears to spread into neocortical regions almost solely in people with coexistent Aβ pathology [134, 135]. Tau-knockout neurons are resistant to neuritic degeneration induced by synthetic or human-derived Aβ species, and tau overexpression exacerbates Aβ-induced damage [136]. During this process, the binding of Aβ to PrPC [137], Fyn activation, and τ-protein phosphorylation may provide alternative explanations for the finding that Aβ plaques accelerate tau phosphorylation propagation via a PrPC-related mechanism, which may encourage the deposition of tau in areas of the brain where Aβ aggregates. These correlations at the lesion site again demonstrate direct and indirect interactions between Aβ plaques and tau aggregation.
Relationships between Aβ, tau and cognition
Aβ and p-tau interact to cause neuronal loss and synaptic damage, which lead to cognitive decline in patients with AD [68, 138]. The accumulation of plaques and tangles induce changes in the behavioural symptoms of patients with AD, which are a direct consequence of the damage and destruction of synapses that mediate memory and cognition [53]. Previous longitudinal PET studies tracked Aβ [139, 140] and tau [141, 142] accumulation. Clinical studies observed greater cognitive decline in clinically healthy older individuals with abnormalities of CSF Aβ and phospho-tau [143, 144]. PET and CSF data indicate that synergy between Aβ and tau is associated with brain dysfunction [145, 146] and cognitive decline [144, 147]. Aβ and tau in the inferior temporal neocortex interact and potentiate tauopathy and cognitive decline. One clinical study indicated that memory progressively declined in individuals with posterior cingulate hypometabolism, which is the region most strongly linked to the tau-Aβ interaction [145]. During AD progression, Aβ and tau proteins are readily excreted from the brain into the peripheral blood through disruption of the BBB and receptor-mediated mechanisms [148, 149]. Aβ and tau are detected in plasma using highly sensitive and accurate analytical techniques. Single-molecule array (SIMOA) and xMAP technology determined that Aβ42 levels, which are positively correlated with CSF Aβ42 levels but negatively correlated with the CSF t-tau levels, were higher at the mild cognitive impairment (MCI) stage than the AD stage, and the t-tau levels were positively associated with the burden of brain tau deposition on tau PET across the AD spectrum; i.e., the Aβ42 levels are elevated during the MCI stage and reach a plateau prior to the demented stage, whereas the t-tau levels increased with AD-associated tau pathology [150, 151]. These findings are also consistent with laboratory findings, which suggested that tau was likely the primary mechanism of Aβ-related neurotoxicity [136] and previous work identifying other imaging markers of neurodegeneration [145, 152, 153], which indicated that Aβ pathology alone may be insufficient to drive imminent cognitive decline [154, 155]. Previous autopsy studies [156] also support the notion that Aβ pathology precedes and accelerates neocortical tau pathology, which together precipitate cognitive decline. These findings also explain why the clinical treatment of Aβ has not yielded satisfactory results. Tau and amyloid pathology may begin independently, but the spread of tau beyond the mesial temporal lobe is associated with and may be dependent on amyloid accumulation [157]. Therefore, higher plasma Aβ42 and t-tau levels at the MCI stage are predictors of greater risk for the development of cognitive decline at the predementia stage of AD. The combination of the two plasma biomarkers with other markers may help identify subjects with MCI who are at risk for developing AD, which may be beneficial for the administration of early and preventative pharmacological interventions.
Conclusions and future directions
Aβ and tau proteins play well-established roles in AD and form the two hallmark pathologies that are visible in postmortem AD brains as amyloid plaques and NFTs, respectively. Although these two proteins play important roles in the pathogenesis of AD, treatments that target Aβ or tau alone have not achieved good clinical results [6-11]. Related experimental and clinical studies were performed, which support the hypothesis that the interplay between Aβ and tau amplifies the toxic effects of each protein [13-15, 27, 47, 48]. Some efforts have been made to interfere with the interaction between Aβ and tau protein, such as studies on GSK-3β and CDK-5 inhibitors [31-33]. However, the exact clinical efficacy is not certain [34]. The p38/JNK MAPK pathway also mediates cortical neuronal apoptosis [158], and ERK is related to memory and learning [159]. Inhibition of ERK1/2, p38 and JNK kinases may be efficacious for the treatment of AD through attenuation of Aβ-induced neurotoxicity via activation of the Erk1/2, p38 and JNK pathways [158, 159]. However, the pharmacological inhibition of ERK1/2 in mice and SH-SY5Y cells did not reduce the basal levels of phospho-tau or hypothermia-induced tau hyperphosphorylation [160, 161]. This result may be due to the multiple mechanisms involved in the interaction between Aβ and tau protein, and inhibitors cannot simultaneously inhibit the pathways of multiple targets. Clinical drugs for the treatment of AD primarily include cholinesterase inhibitors (donepezil, rivastigmine, and galantamine), which are effective for cognition in mild-to-moderate AD, and NMDAR antagonists (memantine), which are effective for moderate-to-severe AD [162]. Combination therapy (cholinesterase inhibitors and memantine) may be beneficial for moderate-to-severe dementia [153]. However, neither cholinesterase inhibitors nor memantine are effective in patients with mild cognitive impairment [162]. AD is a multifactorial disease that likely is the result of different interactions of genomic, epigenetic, interatomic, and environmental aspects [163, 164]. Therefore, a multidisciplinary approach is needed. We need to focus not only on pharmacological therapy but also on the complex biopsychosocial aspects of caring for patients with AD, such as by developing psychological interventions. Moreover, the development of new drugs that can treat AD via multiple pathways should be further examined.
Abbreviations
Aβ: amyloid‑β; AD: Alzheimer's disease; NFTs: neurofibrillary tangles; MT: microtubule; CDK-5: cyclin-dependent kinase-5; GSK-3β: glycogen synthase kinase-3β; MAPKs: mitogen-activated protein kinases; CASP3: caspase-3; PHF: paired helical filament; PD: projection domain; NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartic acid receptor; NR2: NR subunit 2; EC: entorhinal cortex; PSD-95: postsynaptic density protein 95; APP: amyloid precursor protein; ROS: reactive oxygen species; DNM1L: dynamin-1-like protein Drp1; TNF-α: tumour necrosis factor-α; DG: dentate gyrus; TREM2: triggering receptor expressed on myeloid cells 2; CNS: central nervous system; BBB: blood-brain barrier; GFAP: glial fibrillary acidic protein; CSF: cerebrospinal fluid; TFEB: transcription factor EB; ISF: interstitial fluid; ARTAG: ageing-related tau astrogliopathy; BM: basement-membrane; NADPH: nicotinamide adenine dinucleotide 2'-phosphate reduced tetrasodium salt; NADH: nicotinamide adenine dinucleotide; β-APP: amyloid-β precursor protein; VEGFs: vascular endothelial growth factors; NO: nitric oxide; ET: endothelin; PrPC: prion protein; PET: positron emission tomography; SIMOA: single-molecule array; MCI: mild cognitive impairment.
Acknowledgements
This review was funded by the National Science and Technology Major Project for “Essential new drug research and development” (No. 2019ZX09301114), the National Natural Science Foundation of China (No. 81873350) and the Beijing Natural Science Foundation (No. 7202174).
Author Contributions
H.Z., W.W. and Y.C. wrote the manuscript. H.L., Y.C., L.M. and H.P. revised the manuscript. X.J. and M.Z. revised the figures. All the authors provided intellectual input and read the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Selkoe D. Alzheimer's disease: genes, proteins, and therapy. Physiological reviews. 2001;81:741-66
2. Mielke M, Vemuri P, Rocca W. Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Clinical epidemiology. 2014;6:37-48
3. Hodson R. Alzheimer's disease. Nature. 2018;559:S1
4. Selkoe D. Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer's disease. Handbook of clinical neurology. 2008;89:245-60
5. Busche M, Hyman B. Synergy between amyloid-β and tau in Alzheimer's disease. Nature neuroscience. 2020;23:1183-93
6. Panza F, Lozupone M, Logroscino G, Imbimbo B. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nature reviews Neurology. 2019;15:73-88
7. Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534-42
8. Salloway S, Sperling R, Fox N, Blennow K, Klunk W, Raskind M. et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. The New England journal of medicine. 2014;370:322-33
9. Kosik K, Joachim C, Selkoe D. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:4044-8
10. Ittner L, Götz J. Amyloid-β and tau-a toxic pas de deux in Alzheimer's disease. Nature reviews Neuroscience. 2011;12:65-72
11. Iijima K, Gatt A, Iijima-Ando K. Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease. Human molecular genetics. 2010;19:2947-57
12. Zheng W, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115:201-11
13. Grundke-Iqbal I, Iqbal K, Tung Y, Quinlan M, Wisniewski H, Binder L. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:4913-7
14. Manczak M, Reddy P. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. Journal of Alzheimer's disease: JAD. 2013;36:285-95
15. Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Archives of biochemistry and biophysics. 1998;357:299-309
16. Iqbal K, Grundke-Iqbal I, Smith A, George L, Tung Y, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:5646-50
17. Brion J, Hanger D, Bruce M, Couck A, Flament-Durand J, Anderton B. Tau in Alzheimer neurofibrillary tangles. N- and C-terminal regions are differentially associated with paired helical filaments and the location of a putative abnormal phosphorylation site. The Biochemical journal. 1991:127-33
18. Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K. et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. The Journal of biological chemistry. 1995;270:823-9
19. Singh T, Haque N, Grundke-Iqbal I, Iqbal K. Rapid Alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS letters. 1995;358:267-72
20. Iqbal K, Liu F, Gong C. Tau and neurodegenerative disease: the story so far. Nature reviews Neurology. 2016;12:15-27
21. Lee S, Hall G, Shea T. Potentiation of tau aggregation by cdk5 and GSK3β. Journal of Alzheimer's disease: JAD. 2011;26:355-64
22. Iqbal K, Liu F, Gong C, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Current Alzheimer research. 2010;7:656-64
23. Hernandez P, Lee G, Sjoberg M, Maccioni R. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): involvement of lipid rafts. Journal of Alzheimer's disease: JAD. 2009;16:149-56
24. Singh T, Grundke-Iqbal I, Wu W, Chauhan V, Novak M, Kontzekova E. et al. Protein kinase C and calcium/calmodulin-dependent protein kinase II phosphorylate three-repeat and four-repeat tau isoforms at different rates. Molecular and cellular biochemistry. 1997;168:141-8
25. Goedert M, Spillantini M, Cairns N, Crowther R. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159-68
26. Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H. et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. The American journal of pathology. 2008;172:786-98
27. Jackson G, Wiedau-Pazos M, Sang T, Wagle N, Brown C, Massachi S. et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509-19
28. Zempel H, Thies E, Mandelkow E, Mandelkow E. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:11938-50
29. Oddo S, Caccamo A, Shepherd J, Murphy M, Golde T, Kayed R. et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409-21
30. Lewis J, Dickson D, Lin W, Chisholm L, Corral A, Jones G. et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science (New York, NY). 2001;293:1487-91
31. Medina M, Avila J. Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer's disease. Current pharmaceutical design. 2010;16:2790-8
32. Peineau S, Nicolas C, Bortolotto Z, Bhat R, Ryves W, Harwood A. et al. A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Molecular brain. 2009;2:22
33. Mazanetz M, Fischer P. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nature reviews Drug discovery. 2007;6:464-79
34. Liu J, Yang J, Xu Y, Guo G, Cai L, Wu H. et al. Roscovitine, a CDK5 Inhibitor, Alleviates Sevoflurane-Induced Cognitive Dysfunction via Regulation Tau/GSK3β and ERK/PPARγ/CREB Signaling. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;44:423-35
35. Drewes G, Lichtenberg-Kraag B, Döring F, Mandelkow E, Biernat J, Goris J. et al. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. The EMBO journal. 1992;11:2131-8
36. Swatton J, Sellers L, Faull R, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain. The European journal of neuroscience. 2004;19:2711-9
37. Feijoo C, Campbell D, Jakes R, Goedert M, Cuenda A. Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. Journal of cell science. 2005;118:397-408
38. Ferrer I, Blanco R, Carmona M, Puig B, Barrachina M, Gómez C. et al. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson's disease and Dementia with Lewy bodies. Journal of neural transmission (Vienna, Austria: 1996). 2001;108:1383-96
39. Shafiei S, Guerrero-Muñoz M, Castillo-Carranza D. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Frontiers in aging neuroscience. 2017;9:83
40. Hawkins B, Krishnamurthy S, Castillo-Carranza D, Sengupta U, Prough D, Jackson G. et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. The Journal of biological chemistry. 2013;288:17042-50
41. Sengupta U, Guerrero-Muñoz M, Castillo-Carranza D, Lasagna-Reeves C, Gerson J, Paulucci-Holthauzen A. et al. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biological psychiatry. 2015;78:672-83
42. Gamblin T, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet A. et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10032-7
43. Bloom G. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology. 2014;71:505-8
44. Mirbaha H, Holmes B, Sanders D, Bieschke J, Diamond M. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. The Journal of biological chemistry. 2015;290:14893-903
45. Campion D, Pottier C, Nicolas G, Le Guennec K, Rovelet-Lecrux A. Alzheimer disease: modeling an Aβ-centered biological network. Molecular psychiatry. 2016;21:861-71
46. Nilson A, English K, Gerson J, Barton Whittle T, Nicolas Crain C, Xue J. et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. Journal of Alzheimer's disease: JAD. 2017;55:1083-99
47. Ittner L, Ke Y, Delerue F, Bi M, Gladbach A, van Eersel J. et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387-97
48. Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J. et al. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Molecular neurodegeneration. 2017;12:41
49. Avila J. Our Working Point of View of Tau Protein. Journal of Alzheimer's disease: JAD. 2018;62:1277-85
50. Roberson E, Scearce-Levie K, Palop J, Yan F, Cheng I, Wu T. et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science (New York, NY). 2007;316:750-4
51. Amadoro G, Corsetti V, Ciotti M, Florenzano F, Capsoni S, Amato G. et al. Endogenous Aβ causes cell death via early tau hyperphosphorylation. Neurobiology of aging. 2011;32:969-90
52. Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:435-40
53. Roberson E, Halabisky B, Yoo J, Yao J, Chin J, Yan F. et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:700-11
54. Decker H, Jürgensen S, Adrover M, Brito-Moreira J, Bomfim T, Klein W. et al. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-β peptide oligomers. Journal of neurochemistry. 2010;115:1520-9
55. De Felice F, Velasco P, Lambert M, Viola K, Fernandez S, Ferreira S. et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. The Journal of biological chemistry. 2007;282:11590-601
56. Petersen R, Parisi J, Dickson D, Johnson K, Knopman D, Boeve B. et al. Neuropathologic features of amnestic mild cognitive impairment. Archives of neurology. 2006;63:665-72
57. Kaufman A, Salazar S, Haas L, Yang J, Kostylev M, Jeng A. et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Annals of neurology. 2015;77:953-71
58. Nygaard H. Targeting Fyn Kinase in Alzheimer's Disease. Biological psychiatry. 2018;83:369-76
59. Angulo S, Orman R, Neymotin S, Liu L, Buitrago L, Cepeda-Prado E. et al. Tau and amyloid-related pathologies in the entorhinal cortex have divergent effects in the hippocampal circuit. Neurobiology of disease. 2017;108:261-76
60. Illenberger D, Schwald F, Gierschik P. Characterization and purification from bovine neutrophils of a soluble guanine-nucleotide-binding protein that mediates isozyme-specific stimulation of phospholipase C beta2. European journal of biochemistry. 1997;246:71-7
61. Godemann R, Biernat J, Mandelkow E, Mandelkow E. Phosphorylation of tau protein by recombinant GSK-3beta: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS letters. 1999;454:157-64
62. Ittner A, Chua S, Bertz J, Volkerling A, van der Hoven J, Gladbach A. et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer's mice. Science (New York, NY). 2016;354:904-8
63. Busche M, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N. et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nature neuroscience. 2019;22:57-64
64. Silva D, Esteves A, Oliveira C, Cardoso S. Mitochondria: the common upstream driver of amyloid-β and tau pathology in Alzheimer's disease. Current Alzheimer research. 2011;8:563-72
65. Frank S, Gaume B, Bergmann-Leitner E, Leitner W, Robert E, Catez F. et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Developmental cell. 2001;1:515-25
66. Wang X, Su B, Lee H, Li X, Perry G, Smith M. et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:9090-103
67. Wang X, Su B, Siedlak S, Moreira P, Fujioka H, Wang Y. et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19318-23
68. von Bernhardi R, Eugenín J. Alzheimer's disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxidants & redox signaling. 2012;16:974-1031
69. Shah S, Nolan R, Davis E, Stokin G, Niesman I, Canto I. et al. Examination of potential mechanisms of amyloid-induced defects in neuronal transport. Neurobiology of disease. 2009;36:11-25
70. Yu T, Robotham J, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2653-8
71. Lakatos A, Derbeneva O, Younes D, Keator D, Bakken T, Lvova M. et al. Association between mitochondrial DNA variations and Alzheimer's disease in the ADNI cohort. Neurobiology of aging. 2010;31:1355-63
72. Lustbader J, Cirilli M, Lin C, Xu H, Takuma K, Wang N. et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science (New York, NY). 2004;304:448-52
73. Manczak M, Calkins M, Reddy P. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Human molecular genetics. 2011;20:2495-509
74. Quintanilla R, Matthews-Roberson T, Dolan P, Johnson G. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. The Journal of biological chemistry. 2009;284:18754-66
75. Chung C, Song Y, Kim I, Yoon W, Ryu B, Jo D. et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiology of disease. 2001;8:162-72
76. Thies E, Mandelkow E. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:2896-907
77. Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G. et al. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochimica et biophysica acta. 2008;1777:1289-300
78. Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G. et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. Journal of Alzheimer's disease: JAD. 2010;21:445-70
79. Crouch P, Cimdins K, Duce J, Bush A, Trounce I. Mitochondria in aging and Alzheimer's disease. Rejuvenation research. 2007;10:349-57
80. Kang D, Roh S, Woo J, Liu T, Bu J, Jung A. et al. The Interface between Cytoskeletal Aberrations and Mitochondrial Dysfunction in Alzheimer's Disease and Related Disorders. Experimental neurobiology. 2011;20:67-80
81. Perez-Nievas B, Stein T, Tai H, Dols-Icardo O, Scotton T, Barroeta-Espar I. et al. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain: a journal of neurology. 2013;136:2510-26
82. Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell. 2019;179:292-311
83. Carrano A, Hoozemans J, van der Vies S, van Horssen J, de Vries H, Rozemuller A. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neuro-degenerative diseases. 2012;10:329-31
84. Yoshiyama Y, Higuchi M, Zhang B, Huang S, Iwata N, Saido T. et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337-51
85. Maphis N, Xu G, Kokiko-Cochran O, Jiang S, Cardona A, Ransohoff R. et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain: a journal of neurology. 2015;138:1738-55
86. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T. et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature neuroscience. 2015;18:1584-93
87. Ballatore C, Lee V, Trojanowski J. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nature reviews Neuroscience. 2007;8:663-72
88. Nishimura M, Tomimoto H, Suenaga T, Namba Y, Ikeda K, Akiguchi I. et al. Immunocytochemical characterization of glial fibrillary tangles in Alzheimer's disease brain. The American journal of pathology. 1995;146:1052-8
89. Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct Evidence of Internalization of Tau by Microglia In vitro and In vivo. Journal of Alzheimer's disease: JAD. 2016;50:77-87
90. Hopp S, Lin Y, Oakley D, Roe A, DeVos S, Hanlon D. et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. Journal of neuroinflammation. 2018;15:269
91. Bhaskar K, Konerth M, Kokiko-Cochran O, Cardona A, Ransohoff R, Lamb B. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19-31
92. Shi Y, Manis M, Long J, Wang K, Sullivan P, Remolina Serrano J. et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. The Journal of experimental medicine. 2019;216:2546-61
93. Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S. et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. The Journal of biological chemistry. 2012;287:3842-9
94. Danzer K, Kranich L, Ruf W, Cagsal-Getkin O, Winslow A, Zhu L. et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Molecular neurodegeneration. 2012;7:42
95. Leyns C, Gratuze M, Narasimhan S, Jain N, Koscal L, Jiang H. et al. TREM2 function impedes tau seeding in neuritic plaques. Nature neuroscience. 2019;22:1217-22
96. Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiological reviews. 2018;98:239-389
97. Abbott N, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews Neuroscience. 2006;7:41-53
98. Carter S, Schöll M, Almkvist O, Wall A, Engler H, Långström B. et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2012;53:37-46
99. Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X. et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. 2020:1-13
100. Nagele R, D'Andrea M, Lee H, Venkataraman V, Wang H. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain research. 2003;971:197-209
101. Heneka M, Carson M, El Khoury J, Landreth G, Brosseron F, Feinstein D. et al. Neuroinflammation in Alzheimer's disease. The Lancet Neurology. 2015;14:388-405
102. Cahoy J, Emery B, Kaushal A, Foo L, Zamanian J, Christopherson K. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:264-78
103. Basak J, Verghese P, Yoon H, Kim J, Holtzman D. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. The Journal of biological chemistry. 2012;287:13959-71
104. Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A. et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:9607-20
105. Kraft A, Hu X, Yoon H, Yan P, Xiao Q, Wang Y. et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:187-98
106. Wolfe D, Lee J, Kumar A, Lee S, Orenstein S, Nixon R. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. The European journal of neuroscience. 2013;37:1949-61
107. Wyss-Coray T, Loike J, Brionne T, Lu E, Anankov R, Yan F. et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nature medicine. 2003;9:453-7
108. de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz D, Kopeikina K. et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685-97
109. Kovacs G, Robinson J, Xie S, Lee E, Grossman M, Wolk D. et al. Evaluating the Patterns of Aging-Related Tau Astrogliopathy Unravels Novel Insights Into Brain Aging and Neurodegenerative Diseases. Journal of neuropathology and experimental neurology. 2017;76:270-88
110. Martini-Stoica H, Cole A, Swartzlander D, Chen F, Wan Y, Bajaj L. et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. The Journal of experimental medicine. 2018;215:2355-77
111. Shigemoto-Mogami Y, Hoshikawa K, Sato K. in vitroActivated Microglia Disrupt the Blood-Brain Barrier and Induce Chemokines and Cytokines in a Rat Model. Frontiers in cellular neuroscience. 2018;12:494
112. Ding X, Zhang M, Gu R, Xu G, Wu H. Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2017;255:777-88
113. Jiang Z, Li C, Arrick D, Yang S, Baluna A, Sun H. Role of nitric oxide synthases in early blood-brain barrier disruption following transient focal cerebral ischemia. PloS one. 2014;9:e93134
114. Hung V, Yeung P, Lai A, Ho M, Lo A, Chan K. et al. Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:1687-96
115. Attems J, Thomas A, Jellinger K. Correlations between cortical and subcortical tau pathology. Neuropathology and applied neurobiology. 2012;38:582-90
116. Braak H, Thal D, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. Journal of neuropathology and experimental neurology. 2011;70:960-9
117. Grinberg L, Rüb U, Ferretti R, Nitrini R, Farfel J, Polichiso L. et al. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathology and applied neurobiology. 2009;35:406-16
118. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica. 1991;82:239-59
119. Thal D, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791-800
120. Sepulcre J, Grothe M, d'Oleire Uquillas F, Ortiz-Terán L, Diez I, Yang H. et al. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nature medicine. 2018;24:1910-8
121. Ferreira D, Temido-Ferreira M, Vicente Miranda H, Batalha V, Coelho J, Szegö É. et al. α-synuclein interacts with PrP to induce cognitive impairment through mGluR5 and NMDAR2B. Nature neuroscience. 2017;20:1569-79
122. Esiri M, Carter J, Ironside J. Prion protein immunoreactivity in brain samples from an unselected autopsy population: findings in 200 consecutive cases. Neuropathology and applied neurobiology. 2000;26:273-84
123. Rezaie P, Pontikis C, Hudson L, Cairns N, Lantos P. Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer's disease, diffuse Lewy body disease, and in normal brain: an immunohistochemical study. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2005;53:929-40
124. Takahashi R, Tobiume M, Sato Y, Sata T, Gouras G, Takahashi H. Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer's disease brain. Neuropathology: official journal of the Japanese Society of Neuropathology. 2011;31:208-14
125. Velayos J, Irujo A, Cuadrado-Tejedor M, Paternain B, Moleres F, Ferrer V. The cellular prion protein and its role in Alzheimer disease. Prion. 2009;3:110-7
126. Um J, Nygaard H, Heiss J, Kostylev M, Stagi M, Vortmeyer A. et al. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nature neuroscience. 2012;15:1227-35
127. Lau D, Hogseth M, Phillips E, O'Neill M, Pooler A, Noble W. et al. Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer's disease. Acta neuropathologica communications. 2016;4:49
128. Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N. et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. The Journal of biological chemistry. 2012;287:32040-53
129. Larson M, Sherman M, Amar F, Nuvolone M, Schneider J, Bennett D. et al. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:16857-71a
130. Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B. et al. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell reports. 2015;11:1176-83
131. De Mario A, Castellani A, Peggion C, Massimino M, Lim D, Hill A. et al. The prion protein constitutively controls neuronal store-operated Ca(2+) entry through Fyn kinase. Frontiers in cellular neuroscience. 2015;9:416
132. Chen R, Chang W, Lin Y, Cheng P, Chen Y. Alzheimer's amyloid-β oligomers rescue cellular prion protein induced tau reduction via the Fyn pathway. ACS chemical neuroscience. 2013;4:1287-96
133. Li C, Götz J. Pyk2 is a Novel Tau Tyrosine Kinase that is Regulated by the Tyrosine Kinase Fyn. Journal of Alzheimer's disease: JAD. 2018;64:205-21
134. Price J, Morris J. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Annals of neurology. 1999;45:358-68
135. Elobeid A, Soininen H, Alafuzoff I. Hyperphosphorylated tau in young and middle-aged subjects. Acta neuropathologica. 2012;123:97-104
136. Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe D. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5819-24
137. Gomes L, Hipp S, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper M. et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta neuropathologica. 2019;138:913-41
138. Nussbaum J, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T. et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651-5
139. Villemagne V, Burnham S, Bourgeat P, Brown B, Ellis K, Salvado O. et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. The Lancet Neurology. 2013;12:357-67
140. Landau S, Fero A, Baker S, Koeppe R, Mintun M, Chen K. et al. Measurement of longitudinal β-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2015;56:567-74
141. Jack C, Wiste H, Schwarz C, Lowe V, Senjem M, Vemuri P. et al. Longitudinal tau PET in ageing and Alzheimer's disease. Brain: a journal of neurology. 2018;141:1517-28
142. Harrison T, La Joie R, Maass A, Baker S, Swinnerton K, Fenton L. et al. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Annals of neurology. 2019;85:229-40
143. Schindler S, Jasielec M, Weng H, Hassenstab J, Grober E, McCue L. et al. Neuropsychological measures that detect early impairment and decline in preclinical Alzheimer disease. Neurobiology of aging. 2017;56:25-32
144. Clark L, Berman S, Norton D, Koscik R, Jonaitis E, Blennow K. et al. Age-accelerated cognitive decline in asymptomatic adults with CSF β-amyloid. Neurology. 2018;90:e1306-e15
145. Hanseeuw B, Betensky R, Schultz A, Papp K, Mormino E, Sepulcre J. et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Annals of neurology. 2017;81:583-96
146. Schultz A, Chhatwal J, Hedden T, Mormino E, Hanseeuw B, Sepulcre J. et al. Phases of Hyperconnectivity and Hypoconnectivity in the Default Mode and Salience Networks Track with Amyloid and Tau in Clinically Normal Individuals. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2017;37:4323-31
147. Albert M, Zhu Y, Moghekar A, Mori S, Miller M, Soldan A. et al. Predicting progression from normal cognition to mild cognitive impairment for individuals at 5 years. Brain: a journal of neurology. 2018;141:877-87
148. Tarasoff-Conway J, Carare R, Osorio R, Glodzik L, Butler T, Fieremans E. et al. Clearance systems in the brain-implications for Alzheimer disease. Nature reviews Neurology. 2015;11:457-70
149. Ovod V, Ramsey K, Mawuenyega K, Bollinger J, Hicks T, Schneider T. et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2017;13:841-9
150. Park J, Han S, Yi D, Byun M, Lee J, Jang S. et al. Plasma tau/amyloid-β1-42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer's disease. Brain: a journal of neurology. 2019;142:771-86
151. Hanon O, Vidal J, Lehmann S, Bombois S, Allinquant B, Tréluyer J. et al. Plasma amyloid levels within the Alzheimer's process and correlations with central biomarkers. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2018;14:858-68
152. Burnham S, Bourgeat P, Doré V, Savage G, Brown B, Laws S. et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer's disease pathophysiology (SNAP) or Alzheimer's disease pathology: a longitudinal study. The Lancet Neurology. 2016;15:1044-53
153. Machulda M, Hagen C, Wiste H, Mielke M, Knopman D, Roberts R. et al. [Formula: see text]Practice effects and longitudinal cognitive change in clinically normal older adults differ by Alzheimer imaging biomarker status. The Clinical neuropsychologist. 2017;31:99-117
154. Mormino E, Betensky R, Hedden T, Schultz A, Amariglio R, Rentz D. et al. Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA neurology. 2014;71:1379-85
155. Sperling R, Mormino E, Schultz A, Betensky R, Papp K, Amariglio R. et al. The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Annals of neurology. 2019;85:181-93
156. Nelson P, Alafuzoff I, Bigio E, Bouras C, Braak H, Cairns N. et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. Journal of neuropathology and experimental neurology. 2012;71:362-81
157. Jack C, Knopman D, Jagust W, Petersen R, Weiner M, Aisen P. et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. The Lancet Neurology. 2013;12:207-16
158. Haddad J. Mitogen-activated protein kinases and the evolution of Alzheimer's: a revolutionary neurogenetic axis for therapeutic intervention? Progress in neurobiology. 2004;73:359-77
159. Zhu X, Lee H, Raina A, Perry G, Smith M. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neuro-Signals. 2002;11:270-81
160. Xue M, Liu X, Zhang Y, Gao F. Nicotine exerts neuroprotective effects against β-amyloid-induced neurotoxicity in SH-SY5Y cells through the Erk1/2-p38-JNK-dependent signaling pathway. International journal of molecular medicine. 2014;33:925-33
161. Li Y, Chen G, Zhao J, Nie X, Wan C, Liu J. et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces microglial nitric oxide production and subsequent rat primary cortical neuron apoptosis through p38/JNK MAPK pathway. Toxicology. 2013;312:132-41
162. O'Brien J, Holmes C, Jones M, Jones R, Livingston G, McKeith I. et al. Clinical practice with anti-dementia drugs: A revised (third) consensus statement from the British Association for Psychopharmacology. Journal of psychopharmacology (Oxford, England). 2017;31:147-68
163. Briggs R, Kennelly S, O'Neill D. Drug treatments in Alzheimer's disease. Clinical medicine (London, England). 2016;16:247-53
164. Hepp Rehfeldt S, Majolo F, Goettert M, Laufer S. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer's Diseases. International journal of molecular sciences. 2020 21
Author contact
![]() Corresponding authors: Hao Li, Institute of Geriatrics, Xiyuan Hospital of China Academy of Chinese Medical Sciences, No.1 Xiyuan Caochang Road, Haidian District, Beijing 100091, China. Tel.: +86 10 6283 5631, E-mail: 65090075edu.cn; xyhplihao1965com; Yu Cao, Institute of Geriatrics, Xiyuan Hospital of China Academy of Chinese Medical Sciences, No.1 Xiyuan Caochang Road, Haidian District, Beijing 100091, China. Tel.: +86 10 6283 5631, E-mail: yu.cao-xiyuancom.
Corresponding authors: Hao Li, Institute of Geriatrics, Xiyuan Hospital of China Academy of Chinese Medical Sciences, No.1 Xiyuan Caochang Road, Haidian District, Beijing 100091, China. Tel.: +86 10 6283 5631, E-mail: 65090075edu.cn; xyhplihao1965com; Yu Cao, Institute of Geriatrics, Xiyuan Hospital of China Academy of Chinese Medical Sciences, No.1 Xiyuan Caochang Road, Haidian District, Beijing 100091, China. Tel.: +86 10 6283 5631, E-mail: yu.cao-xiyuancom.