Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
m6A writers, erasers,...
Aberrant m6A...
Effects and underlying...
Mutants of m6A sites...
m6A as biomarkers of...
m6A as a therapeutic...
Conclusions and perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(9):2323-2335. doi:10.7150/ijbs.60115 This issue Cite
Review
N6-methyladenosine (m6A) in pancreatic cancer: Regulatory mechanisms and future direction
Jian Li1,2#, Fangjuan Wang3#, Yongkang Liu2 ![]() , Huaizhi Wang4
, Huaizhi Wang4 ![]() , Bing Ni1
, Bing Ni1 ![]()
1. Department of Pathophysiology, College of High Altitude, Army Medical University (Third Military Medical University), Chongqing 400038, PR China.
2. Department of General Surgery, Air Force Hospital of Western Theater Command, Chengdu 610021, PR China.
3. Department of Cardiology, Southwest Hospital, Army Medical University (Third Military Medical University), Chongqing 400038, PR China.
4. Institute of Hepatopancreatobiliary Surgery, Chongqing General Hospital, University of Chinese Academy of Sciences, Chongqing 401120, PR China.
#These authors contributed equally to this work.
Received 2021-3-7; Accepted 2021-5-21; Published 2021-6-4
Abstract

N6-methyladenosine (m6A), the most abundant RNA modification in eukaryotes, plays a pivotal role in regulating many cellular and biological processes. Aberrant m6A modification has recently been involved in carcinogenesis in various cancers, including pancreatic cancer. Pancreatic cancer is one of the deadliest cancers. It is a heterogeneous malignant disease characterized by a plethora of diverse genetic and epigenetic events. Increasing evidence suggests that dysregulation of m6A regulatory factors, such as methyltransferases, demethylases, and m6A-binding proteins, profoundly affects the development and progression of pancreatic cancer. In addition, m6A regulators and m6A target transcripts may be promising early diagnostic and prognostic cancer biomarkers, as well as therapeutic targets. In this review, we highlight the biological functions and mechanisms of m6A in pancreatic cancer and discuss the potential of m6A modification in clinical applications.
Keywords: m6A, pancreatic cancer, RNA modification, clinical application
Introduction
Pancreatic cancer is one of the deadliest malignances with high invasiveness, early metastasis, lack of specific symptoms, and a 5-year survival rate of around 10% in the USA [1]. In the past 20 years, the morbidity of pancreatic cancer has increased 6-fold in China [2]. Pancreatic cancer risk factors include family history, smoking, type 2 diabetes, and obesity. Pancreatic ductal adenocarcinoma (PDAC) is the primary pathological type of pancreatic cancer. More than 85% of PDAC cases are complicated by distant metastasis, and patients present with poor clinical outcomes [3]. Research efforts in PDAC have traditionally focused on genetic abnormalities, including chromosome gain/loss and somatic mutations. Previous research on these genetic alterations showed common mechanisms of pancreatic cancer tumorigenesis, such as activation of KRAS mutations or inactivation of tumor suppressor genes TP53, SMAD4, and CDKN2A [4]. KRAS, which functions as a signal transducer between cell membrane-based growth factor signaling and the MAPK pathways, is the most frequently mutated oncogene (~ 90% of pancreatic cancer ) [5, 6]. Somatic mutations in TP53 tumor suppressor genes are also frequently observed in pancreatic cancer. The protein encoded by TP53 plays a crucial role in multicellular organisms, where it prevents tumor formation [5, 6]. Currently, surgical resection is the only treatment for pancreatic cancer, and adjuvant chemotherapy after surgical resection is an essential part of multimodality pancreatic cancer treatment. Unfortunately, although the prognosis of advanced pancreatic cancer has been enhanced by 5-fluorouracil/leucovorin with irinotecan and oxaliplatin (FOLFIRINOX) and gemcitabine/nab-paclitaxel, the development of chemoresistance in patients still results in poor clinical outcomes [7]. It is worth noting that in addition to the restrictive contribution of genetic variation, the acquisition of chemoresistant phenotypes is usually largely reversible. The dynamic characteristics of cell plasticity and chemoresistance indicate that epigenetic alterations may be involved in the regulation of pancreatic cancer phenotypic heterogeneity [8]. Importantly, in contrast to genetic defects, epigenetic alterations are reversible; therefore, they can be used as potential bona fide targets.
Accumulating evidence has revealed that epigenetic deregulation is critically associated with the pathophysiology of pancreatic cancer [9]. N6-methyladenosine (m6A), methylated adenosine at the N6 position, is a new frontier of this field. Since its discovery in the 1970s [10], m6A has been identified as the most abundant form of internal mRNA modification in eukaryotes. Transcriptome-wide research has shown that m6A may affect more than 7000 transcripts in humans [11]. m6A modifications can regulate the generation and function of transfer RNA (tRNA), ribosomal RNA (rRNA), and various non-coding RNAs (ncRNAs), such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) [12-14]. Owing to improvements in molecular biology and sequencing, m6A modification has gained renewed interest in the past couple of decades and is currently the most widely studied type of RNA modification. Accumulating evidence has revealed that m6A affects almost every step of RNA metabolism, including alternative splicing, nuclear export, stability, translation, and decay [12, 15, 16]. m6A are clustered in the 3′ untranslated region (UTR) near the stop codons of mRNAs and the m6A position has a consensus sequence of RRm6ACH (where R = G or A, and H = A, C or U) [11, 17]. Recent studies have found that m6A modification is involved in various physiological behaviors, and its dysregulation may be implicated in the mechanisms associated with a variety of diseases [18-20]. Increasing evidence suggests that m6A modification pathways are also implicated in the carcinogenesis of various malignancies, including pancreatic cancer [21-25]. Aberrant regulation of m6A modification in coding and non-coding RNAs found in pancreatic cancer is crucial for multiple biological processes such as tumorigenesis, chemoresistance, and progression [24, 25]. Here, we provide a comprehensive review of m6A modifications and highlight the potential molecular mechanisms of m6A in pancreatic cancer. We further emphasize the prospects for using m6A modification as a new biomarker and therapeutic target for pancreatic cancer.
m6A writers, erasers, and readers
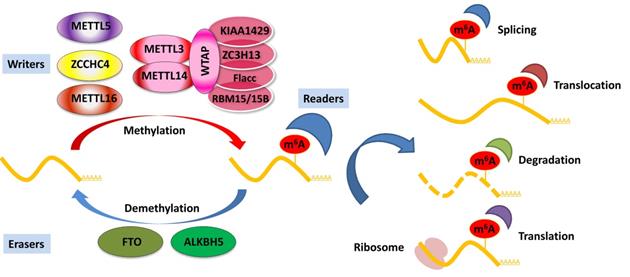
The installation of m6A modification is a reversible process modulated by the dynamic balance of m6A writer and eraser enzymes. The m6A writer complex, traditionally identified as a highly conserved multicomponent m6A methyltransferase complex (MTC), consists of methyltransferase-like 3 and 14 proteins (METTL3 and METTL14) and their cofactors Wilms' tumor 1-associating protein (WTAP). Several co-factors identified to interact with the MTC to affect m6A deposition include vir-like m6A methyltransferase-associated (KIAA1429), RNA-binding motif protein 15/15B (RBM15/15B), zinc finger CCCH domain-containing protein 13 (ZC3H13), and Fl(2)d-associated complex component (Flacc) [26-28]. In addition, certain m6A methyltransferases do not exert their function via the MTC, including methyltransferase-like 16 (METTL16), zinc finger CCHC-type containing 4 (ZCCHC4), and methyltransferase-like 5 (METTL5) [29-31]. METTL16 has recently been identified as an independent RNA methyltransferase and is responsible for m6A of mRNA in the 3′ UTR and A43 of the U6 small nuclear RNA (snRNA) during splicing [29, 32, 33]. The demethylation of m6A is mediated by m6A erasers, mainly including fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) [34, 35]. FTO was identified as the first demethylase in nuclear RNA in 2011 [34], and the notion of reversible m6A modification was described. Both FTO and ALKBH5 belong to the same protein family. However, studies have shown that FTO can act as a demethylase for both internal m6A and 5′ cap N6, 2Odimethyladenosine (m6Am) in mRNA [36, 37]. Unlike FTO, ALKBH5 seems to be an m6A-specific demethylase that catalyzes the direct removal of m6A modification [35]. In addition, ALKBH3 was recently identified as a novel demethylase of m6A in tumor progression via RNA demethylation and enhanced protein synthesis [38]. ALKBH3 has also been shown to be an antitumor target and can be a potential diagnostic marker for cancer [39]. The dynamic balance between m6A methylation and demethylation is essential for normal biological processes.
Deposited on native RNA transcripts, m6A modification requires m6A-binding proteins (readers) to perform specific cellular bioprocesses. There are currently three main types of reader proteins, including the YT521-B homology (YTH) domain family proteins, heterogeneous nuclear ribonucleoproteins, and IGFBP family proteins. YTH domain-containing proteins, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, were the first five characterized readers possessing a conserved domain for m6A recognition [40]. YTHDF2 was the first identified among these and is the most studied m6A-binding protein. Nuclear YTHDF2 preserves the m6A modification of mRNA located in the 5′ UTR and influences mRNA translation under heat shock stress [41]. In addition, YTHDF2 destabilizes m6A-containing RNA by recruiting the CCR4-NOT deadenylase complex in mammalian cells [42]. Another m6A reader protein, YTHDF1, positively interacts with translation machinery and increases translation efficiency, eventually promoting protein synthesis [43]. Furthermore, YTHDF3 can enhance protein synthesis in synergy with YTHDF1, and regulate m6A-modified mRNA decay mediated through YTHDF2 [44]. These three YTHDF proteins play crucial roles in modulating the translation and decay of m6A-modified mRNA in the cytoplasm [44]. Heterogeneous nuclear ribonucleoproteins (hnRNPC, hnRNPG, and hnRNPA2B1) and IGFBP family proteins (IGFBP1-3) can act as m6A switch readers via remodeling specific m6A-dependent RNA structures and affect RNA-protein interactions for biological regulation [45-47]. Notably, a number of novel m6A reader proteins have been identified in recent studies. Eukaryotic initiation factor 3 (eIF3) directly binds to the m6A site in the 5′ UTR of RNA, which is sufficient to recruit the 43S complex to initiate cap-independent translation [48]. Fragile X mental retardation protein (FMRP) has been recently identified as an indirect reader and can regulate the stability of its m6A-modified mRNA targets via YTHDF2 [49]. Indeed, the aforementioned m6A reader proteins have pleiotropic functions and are implicated in regulating RNA splicing, translocation, stability, and translation, which affect various bioprocesses and are crucial in mammals (Figure 1).
Regulation of m6A modification on mRNA. The m6A modification is added by writers, multicomponent methyltransferase complex (including METTL3, METTL14, WTAP, KIAA1429, ZC3H13, Flacc, and RBM15/15B) or METTL16, ZCCHC4, and METTL5 alone. m6A could be reversibly removed by m6A eraser proteins (FTO and ALKBH5) or recognized by m6A binding proteins (readers) to influence RNA splicing, translocation, stabilization, and translation.
Aberrant m6A regulation in cancers
Increasing evidence has demonstrated that m6A modification is closely associated with tumor initiation and progression [12]. Alterations in m6A levels are critical for cancer stem cell formation, cancer initiation, cancer metabolism, epithelial-mesenchymal transition (EMT), drug resistance, and cancer relapse [12, 50, 51]. Studies have reported that METTL3 overexpression suppresses the self-renewal and oncogenesis of glioblastoma stem cells (GSCs) by increasing m6A levels and decreasing the expression of ADAM19, which plays crucial roles in GSCs [52]. Another study showed that downregulation of METTL3 decreased m6A levels and restrained cancer migration, invasion, and EMT both in vitro and in vivo, and further confirmed that Snail, a key transcription factor of EMT, participates in m6A-mediated EMT [53]. Notably, the global m6A abundance and expression of m6A modulators are highly heterogeneous, which indicates that the effects of m6A may vary in different cancer environments. Recently, it has been reported that the global m6A profile in a number of tumors abnormally decreases or increases, which may be related to the development and clinical outcome of cancer. For example, m6A levels were higher in approximately 70% of pancreatic cancer tissues than in pair-matched adjacent tissues, and higher levels of m6A were significantly correlated with decreased survival [24]. Another group found that the level of m6A RNA was significantly elevated in human gastric cancer tissues compared with normal control tissues [54]. Conversely, it has been reported that global m6A modification was substantially reduced in bladder cancer tissues, especially in advanced-stage bladder cancer patients. In addition, lower m6A modification content was related to poor clinical outcomes in patients with bladder cancer [55].
In addition, different components of m6A regulators have been shown to play either oncogenic or tumor-suppressive roles during tumorigenesis. For example, most studies support the oncogenic role of METTL3 in human cancers [13]; however, METTL3 exerts a tumor-suppressive role in endometrial cancer [56]. Although METTL14, another writer protein, was identified mainly as a tumor suppressor in cancers [13], it functions as an oncogene in acute myeloid leukemia [57] and breast cancer [58]. The pleiotropic roles of METTL14 and other m6A writers are often inconsistent. Furthermore, m6A demethylase ALKBH5 plays a tumor-promoting role in the majority of studies [13]; in contrast, ALKBH5 acts as a tumor suppressor in bladder cancer [59] and pancreatic cancer [22, 23]. Even the same m6A regulator can play a controversial role in the same tumor. For instance, a different role of METTL3 in glioblastoma (GBM) has been shown in different studies [52, 60, 61]. This may be due to the different sources of original samples used, different m6A sites, and m6A modified RNAs in different groups, resulting in tumor heterogeneity. Further research is required to clarify the enigmatic role of m6A modifications and m6A modulators in different cancer types and ultimately reconcile these seemingly contradictory findings in the future.
Effects and underlying mechanisms of m6A in pancreatic cancer
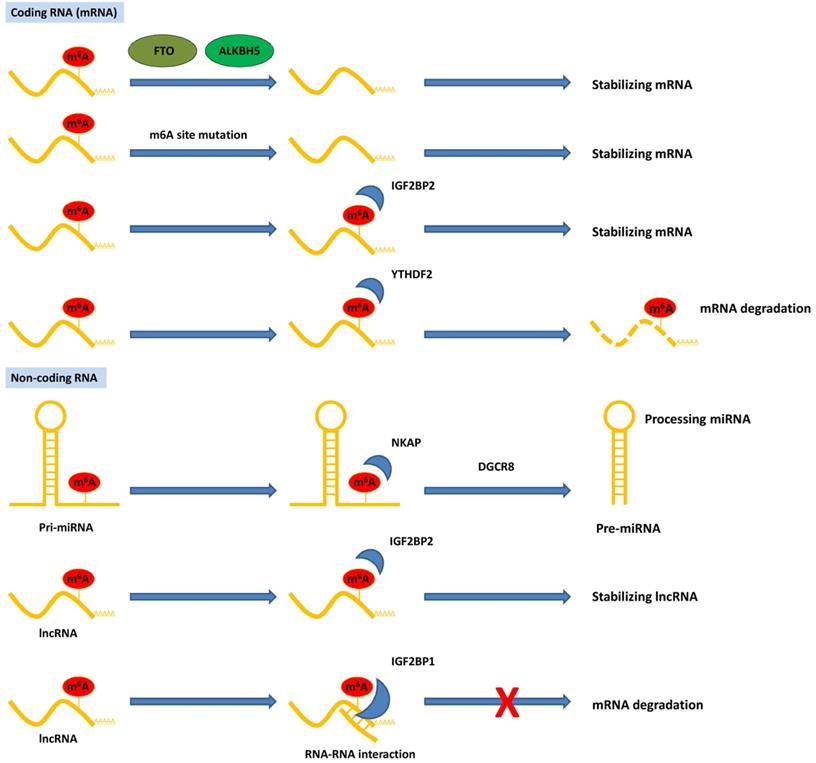
Although studies on the role of m6A in pancreatic cancer are in their early stages, emerging data have suggested that RNA m6A methylation is closely involved in pancreatic cancer progression, including carcinogenesis, proliferation, migration, invasion, EMT, and therapy resistance. Aberrant regulation of m6A and its modulators, including writers, erasers, and readers, plays a substantial role in pancreatic cancer by targeting various RNAs and signaling pathways (Table 1). The potential mechanism of m6A modification of coding and non-coding RNAs in pancreatic cancer is summarized in Figure 2. Herein, we briefly review recent studies on m6A modifications in pancreatic cancer.
Roles of m6A regulators in pancreatic cancer
| m6A regulators | Expression | Roles in cancer | Biological function | Target/signaling | References |
|---|---|---|---|---|---|
| Writer | |||||
| METTL3 | ↑ | oncogenic | promotes chemoresistance | MAPK cascades, ubiquitin-dependent process, and RNA splicing and regulation of cellular process | 21 |
| METTL3 | ↑ | oncogenic | promotes proliferation, migration, and invasion | -- | 62 |
| METTL3 | ↑ | oncogenic | promotes initiation and progression | processing pri-miR-25 | 25 |
| METTL14 | ↑ | oncogenic | promotes growth and metastasis; increases apoptosis induced by cisplatin | PERP/increase mRNA turnover | 24 |
| METTL14 | ↑ | oncogenic | inhibits apoptosis induced by cisplatin and autophagy | mTOR signaling pathway | 63 |
| WTAP | ↑ | oncogenic | promotes metastasis and chemoresistance to gemcitabine | stabilizing Fak mRNA | 67 |
| METTL16 | ↓ | antitumor | inhibits proliferation | p21 pathway | 69 |
| Eraser | |||||
| ALKBH5 | ↓ | antitumor | inhibits cell motility | KCNK15-AS1 | 71 |
| ALKBH5 | ↓ | antitumor | inhibits proliferation, migration, and invasion | PER1-ATM-CHK2-P53/CDC25C signaling | 22 |
| ALKBH5 | ↓ | antitumor | inhibits proliferation, migration, and invasion | WIF-1/Wnt signaling | 23 |
| FTO | ↑ | oncogenic | promotes proliferation and inhibits apoptosis | MYC, bHLH/ regulate mRNA stability | 72 |
| Reader | |||||
| YTHDF2 | ↑ | Oncogenic/ antitumor | promotes proliferation and suppresses migration, invasion, and EMT | YAP and TGF-β/Smad signaling | 76 |
| YTHDF2 | ↑ | oncogenic | promotes proliferation, migration, and invasion | PER1/inhibit mRNA degradation | 22 |
| YTHDF2 | ↑ | oncogenic | promotes proliferation and migration | PIK3CB | 77 |
| IGF2BP2 | ↑ | oncogenic | involved in apoptosis and ubiquitination | PKC signaling pathway | 81 |
| IGF2BP2 | ↑ | oncogenic | promotes proliferation and aerobic glycolysis | GLUT1 | 83 |
| IGF2BP2 | ↑ | oncogenic | promotes proliferation | PI3K/AKTpathway | 90 |
| IGF2BP2 | ↑ | oncogenic | promotes proliferation | lncRNA DANCR | 87 |
| IGF2BP1 | ↑ | oncogenic | promotes proliferation | AKT signaling pathway | 89 |
| IGF2BP1 | ↑ | oncogenic | promotes proliferation and metastasis | ELF3 | 91 |
| IGF2BP1 | ↑ | oncogenic | promotes proliferation | c-myc | 92 |
| IGF2BP3 | ↑ | oncogenic | promotes migration and invasion | ARF6 and ARHGEF4 | 86 |
| hnRNPC | ↑ | oncogenic | promotes proliferation | miR-183-3p | 93 |
Functions of m6A modification on coding and non-coding RNAs in pancreatic cancer. The m6A modification recognized by reader proteins affects RNAs fate and exerts post-transcriptional regulation.
Dysregulation of m6A writers in pancreatic cancer
Aberrant expression of m6A writers in pancreatic cancer has been shown to play a crucial role in chemoresistance and progression. It has been reported that the m6A writer METTL3 is significantly overexpressed in pancreatic cancer and is linked to cancer aggressiveness and patient survival. Furthermore, METTL3 knockdown decreases m6A modifications and inhibits pancreatic cancer cell proliferation and migration [62]. Taketo et al. showed that downregulation of METTL3 increased pancreatic cancer cell sensitivity to anticancer reagents, such as 5-fluorouracil, gemcitabine, cisplatin, and irradiation. Using cDNA expression analysis, METTL3 was involved in MAPK cascades, ubiquitin-dependent processes, RNA splicing, and cellular processes regulation [21]. Furthermore, Zhang et al. reported that oncogenic miR-25 was excessively maturated by cigarette smoke condensate (CSC) through m6A modification, which is mediated by the upregulation of METTL3 expression in pancreatic duct epithelial cells. Interestingly, they found a coincidence of CSC-induced upregulation of miR-25 and METTL3, but not METTL14 and WTAP [25]. However, the prognostic value was based on a small patient specimen size, and large-scale patient cohorts from multiple centers are needed to confirm the prognostic role of METTL3 in pancreatic cancer.
METTL14, another vital component of the m6A writer complex, has been identified as a tumor suppressor in multiple types of malignancies. In contrast, Wang et al. reported that upregulation of METTL14 directly targets the downstream PERP mRNA (p53 effector related to PMP-22) in an m6A-dependent manner, promoting the growth and metastasis of pancreatic cancer. Functionally, methylation of the target adenosine results in enhanced PERP mRNA turnover, thereby hindering PERP mRNA and protein expression [24]. Another study showed that high METTL14 expression in pancreatic cancer tissues is associated with clinicopathological variables. Loss of METTL14 increased apoptosis induced by cisplatin in pancreatic cancer cells, and autophagy was enhanced through an mTOR signaling-dependent pathway [63]. WTAP, a specific WT1-binding protein, has gained increasing attention owing to its important roles in tumorigenesis [64-66]. In pancreatic cancer, Li et al. showed that high nuclear expression of WTAP was significantly correlated with poor prognosis, as well as several pathological characteristics [67]. Further studies have shown that WTAP promotes metastasis and chemoresistance to gemcitabine via stabilizing Fak mRNA, and a specific FAK inhibitor, GSK2256098, could restore WTAP-mediated chemoresistance and metastasis in pancreatic cancer [68]. However, as a regulatory factor of the m6A methyltransferase complex, there are still limited studies investigating the m6A modification-related function of WTAP in pancreatic cancer, which has to be clarified in the future.
At present, the research of m6A writers has mainly focused on METTL3 or METLL14; however, few studies have been mediated by METTL16. Our group recently revealed that METTL16 was significantly downregulated in pancreatic cancer and low METTL16 expression was a poor prognostic factor. In addition, METTL16 inhibited the p21 pathway by mediating m6A modification, resulting in a tumor-suppressive role in the proliferation of pancreatic cancer cells. Therefore, METTL16 may be a new therapeutic target for pancreatic cancer [69]. However, more investigations are urgently warranted to fully characterize the function of METTL16 in pancreatic and other cancers.
Dysregulation of m6A erasers in pancreatic cancer
Aberrant regulation of m6A erasers has also been demonstrated in pancreatic cancer. In a retrospective multicohort study, Cho et al. reported that ALKBH5 expression was positively associated with the prognosis of pancreatic cancer, and multivariate analysis showed that ALKBH5 is an independent prognostic factor [70]. A recent study found that ALKBH5 was decreased in pancreatic cancer cells and inhibited pancreatic cancer motility by demethylating the lncRNA KCNK15-AS1 [71]. In addition, Guo et al. reported that knockdown of ALKBH5 increased pancreatic cancer cell proliferation, migration, and invasion in vitro and in vivo, whereas ALKBH5 overexpression restrained pancreatic cancer progression. Mechanistically, ALKBH5 inhibited PER1-ATM-CHK2-P53/CDC25C signaling in an m6A-YTHDF2-dependent manner, and P53-induced ALKBH5 activation acted as a feedback loop modulating m6A modification in pancreatic cancer [22]. Gemcitabine resistance usually develops within weeks after starting treatment, limiting its overall efficacy as a first-line chemotherapy for pancreatic cancer. Another group showed that ALKBH5 is downregulated in the gemcitabine-treated patient-derived xenograft (PDX) model, and its upregulation enhanced PDAC cells to chemotherapy. Furthermore, ALKBH5 impaired the Wnt pathway and its downstream targets via demethylation of m6A-modified Wnt inhibitory factor 1 (WIF-1) transcripts [23]. Based on the above knowledge, we could reach a firm conclusion that ALKBH5 acts as a tumor suppressor in pancreatic cancer.
Another m6A demethylase, FTO, previously linked with obesity and type II diabetes, was gradually discovered to be involved in diverse cancers. Tang et al. reported that FTO was overexpressed in pancreatic cancer, and knockdown of FTO decreased proliferation and promoted apoptosis of pancreatic cancer cells. Functionally, FTO has been shown to interact with the MYC proto-oncogene and bHLH transcription factor and regulate its stability via decreased m6A modification [72]. Previous evidence has shown that FTO is strongly involved in the pathophysiology of various types of malignancies [73-75]. The role of FTO in pancreatic cancer is not well understood and needs to be clarified in the future.
Dysregulation of m6A readers in pancreatic cancer
Most of the biological functions of m6A are mediated by multiple m6A readers, which are also involved in pancreatic cancer. Chen et al. showed that YTHDF2 was overexpressed in pancreatic cancer, which was correlated with the later stages of pancreatic cancer. Furthermore, YTHDF2 orchestrated two cellular processes, including promoting proliferation and suppressing migration and invasion in pancreatic cancer cells, a phenomenon called the “migration-proliferation dichotomy”. Mechanistically, loss of YTHDF2 noticeably increased total YAP expression but suppressed TGF-β/Smad signaling [76]. Another study showed that demethylase ALKBH5 suppressed pancreatic cancer progression by post-transcriptional activation of PER1 in an m6A-YTHDF2-dependent manner. PER1 mRNA was a novel target of YTHDF2, and downregulation of YTHDF2 enhanced PER1 mRNA expression [22]. In addition, a recent study revealed that the rs142933486 G>T polymorphism in PIK3CB is significantly correlated with the clinical severity of PDAC patients by decreasing the PIK3CB m6A modification and causing a YTHDF2-mediated increase in its mRNA and protein levels. Interestingly, YTHDF2 predominantly binds to PIK3CB [G] compared to PIK3CB [T] in pancreatic cancer cells [77].
In addition to the YTH domain family proteins, IGF2BP proteins have also been identified as m6A readers. The primary role of IGF2BP2 is to modulate cell metabolism; however, emerging studies have shown that IGF2BP2 is involved in various types of cancers [78-80]. Dahlem et al. reported that IGF2BP2 was markedly overexpressed in pancreatic intraepithelial neoplasia (PanIN), a well-known precursor of PDAC, implying that IGF2BP2 might be a diagnostic marker for early-stage pancreatic cancer. In addition, increased IGF2BP2 expression was associated with a poor prognosis in pancreatic cancer. Strict correlation analysis showed 22 highly positive genes and 9 genes negatively associated with IGF2BP2, and these genes were thought to be involved in apoptosis, ubiquitination, and the protein kinase C (PKC) signaling pathway. Interestingly, higher IGF2BP2 expression was detected in circulating tumor cells than normal hematological cells and normal tissues from the tumor origin [81]. Similarly, another study showed that IGF2BP2 was correlated with clinical outcome and multiple biological processes involved in cancer, of which the most significant processes were associated with cancer cell cycle, immortalization, and tumor immunity [82]. Another study also found that IGF2BP2 promoted cell proliferation and aerobic glycolysis in PDAC by directly binding and stabilizing GLUT1 mRNA [83]. In addition, Schaeffer et al. reported that IGF2BP3 overexpression was associated with poor survival in PDAC [84]. Another group demonstrated that IGF2BP3 and IGF2BP3-bound transcripts were localized in cytoplasmic RNA granules, and IGF2BP3 promoted pancreatic cancer cell migration and invasion by regulating the localized translation of IGF2BP3 target transcripts in cell protrusions [85]. Furthermore, they found that loss of KIF20A suppressed the accumulation of IGF2BP3-containing stress granules in cell protrusions and restrained local protein expression from certain IGF2BP3-bound transcripts, ARF6 and ARHGEF4 [86].
Emerging evidence has shown that m6A readers can regulate ncRNAs [13]. Recently, NF-κB associated protein (NKAP) was identified as a reader of m6A in pri-miR-25 maturation, and mature miR-25 could promote pancreatic cancer progression [25]. LncRNA differentiation antagonizes non-protein coding RNA (DANCR) involved in the tumorigenesis of different cancer types [87]. IGF2BP2 acts as a reader for the m6A modification of DANCR and promotes pancreatic cancer cell proliferation [88]. In turn, ncRNAs also regulate m6A reader proteins. Wan et al. revealed that IGF2BP1 is overexpressed and correlated with poor survival in pancreatic cancer. Additionally, IGF2BP1 is a new target of miR-494, and re-expression of miR-494 can partially reverse the oncogenic role of IGF2BP1 [89]. Xu et al. showed that IGF2BP2 was identified as a direct target of miR-141, and the miR-141/IGF2BP2 axis promoted pancreatic cancer cell proliferation by activating the PI3K/Akt pathway in vitro and in vivo [90]. Another study reported that lncRNA NEAT1 was overexpressed and correlated with poor prognosis in patients with pancreatic cancer. Further, NEAT1 could increase the combination of E74 like ETS transcription factor 3 (ELF3) mRNA and IGF2BP1, therefore enhanced the stability of ELF3 mRNA [91]. A recent study showed that LINC00261 is a tumor suppressor with clinical significance in pancreatic cancer. Mechanistically, LINC00261 could decrease c-myc mRNA stability by sequestering IGF2BP1 [92]. Furthermore, the rs7495G allele in the hnRNPC gene promotes hnRNPC expression by disrupting a putative binding site for miR-183-3p in pancreatic cancer [93]. Dysregulation of m6A readers aberrantly regulates the expression of various RNAs and their downstream pathways, which play a crucial role in pancreatic cancer.
Mutants of m6A sites and m6A regulators in pancreatic cancer
Mutations in tumor-promoting and tumor-suppressing genes are common during tumor development. Nevertheless, little is known about the role of mutations at the m6A site in cancer. m6A site mutations may influence RNA m6A modification, leading to aberrant post-transcriptional regulation and participation in tumorigenesis. For example, a recent study showed that m6A at the point-mutated transited codon 273 (G>A) of p53 pre-mRNA enhanced its splicing via methylation of METTL3, resulting in overexpression of the p53 R273H mutant protein, which induced drug resistance in cancer cells [94]. In pancreatic cancer, it was reported that the missense variant rs142933486 in the 20th exon of PIK3CB was clearly correlated with the clinical outcome. Further study identified that this variant was a G>T change and was coincidently located 3 bp from a predicted m6A site. PIK3CB [T] expression decreased PIK3CB m6A levels and enhanced its mRNA and protein expression. Functionally, upregulation of PIK3CB potentiates the proliferation and migration of PTEN-deficient pancreatic cancer cells by targeting the AKT signaling pathway [77].
Given the pivotal role of m6A modification in various biological processes, it is rational to assume that genetic variants in m6A regulators, including its writers, erasers, and readers, might be involved in oncogenesis. The m6A eraser FTO, identified by genome-wide association studies (GWAS), is a dangerous gene related to the risk of obesity and body mass index (BMI) [95, 96]. In a case-control study, the FTO polymorphism rs9939609 was linked to the risk of pancreatic cancer in Japanese population [97]. Furthermore, there was a significant association between pancreatic cancer and endometrial cancer; however, no statistical significance was found in other malignancies through a meta-analysis [98]. Overall, rs9939609 in the FTO gene might be a potential biomarker for early diagnosis or gene therapy targeting pancreatic cancer. Interestingly, another study showed that FTO gene mutations might be positively correlated with pancreatic cancer only in overweight people. Stratification analysis revealed that both heterozygous and homozygous mutations of the FTO IVS1-27777 C>A and IVS1-23525 T>A SNPs were correlated with a decreased risk of pancreatic cancer among participants with BMIs <25 kg/m2 but were correlated with an increased risk among participants with BMIs >25 kg/m2 [99]. In addition, to investigate all SNPs in 22 m6A modification genes, Ying et al. recently conducted a two-stage case-control study in a Chinese population and found that rs7495 in the 3′ UTR of hnRNPC, an m6A reader, was significantly linked to an increased risk of PDAC. Mechanistically, the rs7495G allele promoted hnRNPC expression by disrupting a putative binding site for miR-183-3p. [93].
m6A as biomarkers of pancreatic cancer
Studies have shown that most m6A regulators are dysregulated in pancreatic cancer, and their expression was found to be correlated with clinical outcomes, suggesting the potential to become novel biomarkers for the early diagnosis and prognosis of pancreatic cancer. PanIN is a well-known precursor of PDAC, and early detection of PanIN would help block the progression of PanIN to PDAC. A recent study revealed that IGF2BP2 was significantly overexpressed in human PanIN [81], which is associated with a high risk of developing pancreatic cancer. Consistent with this result, Huang et al. reported that IGF2BP2 protein levels were gradually elevated from normal pancreas and PanIN to PDAC in mice [83]. These findings highlight that IGF2BP2 has potential value for the early diagnosis of pancreatic cancer. In addition, the detection of circulating tumor cells (CTCs) is a blood-based, non-invasive approach that can be used for the early diagnosis of cancers [100, 101]. A recent study showed that m6A levels in CTCs were significantly elevated in lung cancer patients, suggesting that the examination of m6A in CTCs might be a novel method for cancer diagnosis [102]. However, the m6A modification of CTCs in pancreatic cancer is not well understood. Further studies should elucidate whether the dysregulation of m6A modification and m6A modulators is an early event in pancreatic cancer tumorigenesis, which is crucial for developing a potential approach for utilizing m6A and m6A regulatory factors for early cancer diagnosis.
Recently, several studies have investigated the prognostic value of m6A-related mRNA signature and m6A regulators in pancreatic cancer using database analysis [82, 103-106]. For instance, Meng et al. provided an mRNA signature that may enhance the prognostic prediction of patients with pancreatic cancer based on the genetic status of m6A regulators. In addition, they generated a 16-mRNA signature score system via least absolute shrinkage and selection operator (LASSO) Cox regression analysis, and a high-risk score was clearly associated with poor prognosis [104]. Similarly, 283 candidate m6A-related genes and 4 m6A regulators, including RBM15, METTL14, FTO, and ALKBH5, differed clearly among different stages of the American Joint Committee on Cancer (AJCC) staging system [103]. Additionally, another study showed that m6A regulator-based sample clusters, including 19 m6A regulators, were associated with overall survival (OS), and LASSO regression identified a six-m6A-regulator-signature prognostic model, including METTL3, KIAA1429, HNRNPC, YTHDF1, IGF2BP2, and IGF2BP3 [103]. Furthermore, our group found that METTL16 was significantly decreased in pancreatic cancer and was correlated with patient survival, indicating the prognostic value of METTL16 in pancreatic cancer [69]. As mentioned before, upregulation of ALKBH5 or IGF2BP2 were both significantly associated with poor survival in several studies, highlighting the prognostic value of ALKBH5 and IGF2BP2 in pancreatic cancer. Overall, the m6A regulatory factors and m6A-related mRNA signature, which correlate with clinical outcomes, can be implicated in the malignant progression of pancreatic cancer.
m6A as a therapeutic target of pancreatic cancer
Based on the critical roles of m6A modification and m6A modulators in pancreatic cancer, m6A exhibits great potential as a novel therapeutic target. As mentioned above, downregulation of METTL3 suppressed proliferation, migration, and invasion and enhanced the sensitivity of pancreatic cancer cells to anticancer reagents [21]. Downregulation of METTL14 suppresses pancreatic cancer cell growth and metastasis to the liver and increases apoptosis induced by cisplatin [24, 63]. Furthermore, overexpression of ALKBH5 inhibits proliferation, migration, and invasion both in vitro and in vivo [22, 23]. Thus, these results provide a strong rationale for m6A regulators to be potential therapeutic targets for pancreatic cancer therapy in the future. Recently, FTO has been the most attractive target for developing specific inhibitors targeting m6A modulators for cancer treatment. Meclofenamic acid (MA), a nonsteroidal anti-inflammatory drug, is a selective FTO inhibitor that competes with FTO binding sites and suppresses m6A demethylase activity [107]. Another MA-derived inhibitor, FB23-2, that directly binds to FTO has been developed to impair proliferation and increase the differentiation of human acute myeloid leukemia cells in vitro and in vivo [108]. Su et al. reported that R-2-hydroxyglutarate (R-2HG) inhibited FTO demethylase activity and increased m6A modification in leukemia cells, which decreased the stability of MYC/CEBPA transcripts, exhibiting broad anticancer activity in vitro and in vivo [109]. Additionally, this group recently showed that R-2HG impaired aerobic glycolysis by targeting the FTO/m6A/PFKP/LDHB axis in leukemia [110]. Of note, Peng et al. showed that Entacapone, an FDA-approved drug, could directly bind to FTO and inhibit FTO activity [111]. FTO has been found to promote proliferation and decrease the apoptosis of pancreatic cancer cells [72], and the aforementioned FTO inhibitors might provide new therapeutic opportunities for pancreatic cancer patients.
Although immune checkpoint blockade (ICB) therapy is at the forefront of immunotherapy for various cancers, many patients do not respond or develop resistance to ICB [112, 113]. In recent years, the critical role of m6A modification in regulating the immune response to anti-PD-1 therapy has been reported. Wang et al. found that inhibition of m6A modification by knockdown of METTL3 and METTL14 enhanced the immune response to anti-PD-1 treatment in mice [114]. Another study showed that ALKBH5 enhanced anti-PD-1 therapy response by regulating Mct4/Slc16a3 expression and lactate content and the composition of tumor-infiltrating Tregs and myeloid-derived suppressor cells in the tumor microenvironment [115]. In addition, Han et al. discovered that the knockdown of YTHDF1 increased the efficacy of PD-L1 ICB therapy [111]. Interestingly, using a small-molecule ALKBH5 inhibitor could enhance the efficacy of cancer immunotherapy, suggesting future translational applications [115]. Thus, the crucial role of m6A modification in regulating immune response may contribute to cancer immunotherapy, and further research is required to provide new directions for efficient pancreatic cancer treatment. Although targeting m6A appears to be a promising new therapeutic strategy, its side effects cannot be ignored. Since m6A plays a broad and critical role in almost all aspects of RNA metabolism, the application of specific agonists or inhibitors of m6A regulatory proteins may disturb normal physiological processes, resulting in severe outcomes.
Conclusions and perspectives
RNA m6A modification has gained increasing attention as a new frontier of epigenetic research, and its involvement in a variety of biological processes and disease progression has been recently reported. This review summarizes recent advances in understanding the regulatory mechanisms and future direction of m6A in pancreatic cancer. The specific mechanism for m6A modification in pancreatic cancer, it should be noted, is complex and even contradictory among studies. For example, it seems inconsistent that the m6A writer MELLT3, which increases the m6A level, acts as an oncogene in pancreatic cancer, while the m6A eraser FTO, which reduces the m6A level, is also an oncogene in pancreatic cancer. It is hypothesized that if the writer and the eraser act on the same site of a particular RNA, they may conversely modify m6A and play opposite roles in cancer. This phenomenon has also been reported in a number of cancers, including colorectal cancer [116, 117] and breast cancer [118, 119]. These seemingly contradictory findings may be attributed to various factors, such as intratumoral heterogeneity, different tumor origins, and ethnic groups. For instance, the rs9939609 polymorphism in FTO was significantly correlated with cancer risk in Asians, while no consistent association was found in Caucasians and mixed ethnicities [98]. In addition, the m6A reader is a crucial effector of post-transcriptional regulation, which may explain the seemingly inconsistent role between m6A writers and m6A erasers. Furthermore, there are still potential m6A regulatory factors that have not been discovered thus far that may also be involved in m6A modification. Therefore, additional investigations are warranted to characterize the existence of other regulators implicated in m6A fully.
Accumulated studies have revealed the importance of m6A regulators as potential early diagnosis and prognosis biomarkers for pancreatic cancer. For example, overexpression of METTL3 has been associated with poor prognosis in pancreatic cancer [62]. Several studies have reported that IGF2BP2 was markedly overexpressed in PanIN and associated with clinical outcomes, implying that IGF2BP2 might be a potential diagnostic and prognostic biomarker for pancreatic cancer [81-83]. Intriguingly, we also found that both m6A writer (METTL3 and METTL14) and eraser (FTO) are abnormally upregulated and have a carcinogenic effect in pancreatic cancer. Therefore, to a certain extent, global m6A signatures may be unreliable for pancreatic cancer diagnosis, and the m6A modification of target genes or sites may be used as better biomarkers. However, it is reported that the main m6A detection methods currently available cannot accurately detect the m6A profile in the whole transcriptome, so it is difficult to fully understand the correlation between m6A modification and tumors [120]. In addition, current detection methods still require a large amount of RNA and cannot accurately detect RNA modifications in rare and precious samples. Thus, a novel detection approach with reduced sample volume, high precision, and low cost is urgently needed. This will help promote the use of m6A target transcripts or m6A sites as potential novel biomarkers for pancreatic cancer. Of note, the biological function of m6A modification at specific sites still remains largely unclear. With the advancement of editing tools based on CRISPR, different m6A editing systems have been reported recently, which may greatly promote research on the effect of specific m6A modifications in the near future. For instance, the fusion of dCas9 or dCas13 with m6A writers or erasers can edit specific m6A sites guided by single-guide RNA (sgRNA) and the m6A protospacer adjacent motif (PAM) locus [121, 122]. The m6A modification editing tool represents a revolutionary advancement in the study of m6A functions and seems to provide unprecedented opportunities for m6A research. Future research on m6A modification will focus on accurately identifying m6A sites using m6A editing tools to edit m6A and then conducting functional experiments at these specific m6A sites.
Apart from m6A, other RNA modifications have also been disclosed in pancreatic cancer, such as 5-methylcytosine (m5C), 3-methylcytosine (m3C), 1-methyladenosine (m1A), and 27-methylguanine (m27G). Yang et al. recently revealed that m5C methyltransferase NSUN6 is downregulated in pancreatic cancer and suppressed pancreatic cancer cell proliferation and tumor growth in xenograft mouse models. Notably, NSUN6 has also been reported to play an essential role in predicting pancreatic cancer recurrence and patient survival time [123]. Another study showed that ALKBH3 is a 1-methyladenosine (m1A) and 3-methylcytidine (m3C) demethylase of transfer RNA (tRNA) and can increase cancer cell proliferation, migration, and invasion [124]. Interestingly, a previous study reported that ALKBH3 was overexpressed in pancreatic cancer and was associated with advanced tumor status, pathological stage, and VEGF intensity. Thus, we have good reason to speculate that ALKBH3 may play an important role in pancreatic cancer by regulating RNA modification [125]. In addition, it has been reported that m1A, m27G, and Asm are the most important features distinguishing cell lines derived from poorly differentiated pancreatic cancer from well-differentiated pancreatic cancer [126]. Since the tumor differentiation state is related to the degree of cancer malignancy, m1A, m27G, and Asm may function as valuable biomarkers for pancreatic cancer. Notably, it has been reported that various RNA modifications, such as m6A and m5C, could regulate the same RNA and coordinately promote translation [127]. Furthermore, Chen et al. recently identified potential cross-talk between m6A and m5C methylation at the multiomic level, which is also involved in onco-immunogenic features and patient survival across 33 cancer types [122]. These results highlight multiple cross-talks of RNA modifications in cancers, which provide novel and essential insights into the epigenetic regulation of cancer and opens up new avenues for developing related therapeutic targets.
Although emerging evidence has shown that m6A modification plays essential and diverse biological roles in the development and progression of pancreatic cancer, m6A studies in pancreatic cancer are still incipient. We know very little about the detailed mechanism of m6A modification in pancreatic cancer, and the conclusions of some of the studies above are sometimes inconsistent in this field. Further research is needed to clarify the heterogeneity and complexity of m6A modifications and m6A modulators in the development of pancreatic cancer. More efforts are also needed in the future to identify specific m6A for the early diagnosis of cancer and to develop specific inhibitors to target m6A regulatory factors. The rapid development of m6A mapping methods and m6A editing tools will greatly promote the research of m6A at the single-nucleotide level, which may significantly promote the development of this exciting field. In general, research in this field is progressing rapidly. It is expected that research on m6A modification in pancreatic cancer will be greatly expanded in the near future.
Acknowledgements
This work was supported by grants from the National Key Research and Development Project (No. 2016YFA0502203).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. 2020;395:2008-20
2. He Y, Zheng R, Li D, Zeng H, Zhang S, Chen W. Pancreatic cancer incidence and mortality patterns in China, 2011. Chin J Cancer Res. 2015;27:29-37
3. Avula LR, Hagerty B, Alewine C. Molecular mediators of peritoneal metastasis in pancreatic cancer. Cancer Metastasis Rev. 2020;39:1223-43
4. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6
5. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73-85
6. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
7. Zeng S, Pottler M, Lan B, Grutzmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci. 2019;20:4504
8. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
9. Hessmann E, Johnsen SA, Siveke JT, Ellenrieder V. Epigenetic treatment of pancreatic cancer: is there a therapeutic perspective on the horizon? Gut. 2017;66:168-79
10. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971-5
11. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201-6
12. Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303-22
13. Huang H, Weng H, Chen J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37:270-88
14. Zhang L, Hou C, Chen C, Guo Y, Yuan W, Yin D. et al. The role of N(6)-methyladenosine (m(6)A) modification in the regulation of circRNAs. Mol Cancer. 2020;19:105
15. Roundtree IA, Luo GZ, Zhang ZJ, Wang X, Zhou T, Cui YQ. et al. YTHDC1 mediates nuclear export of N-6 - methyladenosine methylated mRNAs. Elife. 2017;6:e31311
16. Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N. et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl. 2015;54:1587-90
17. Kane SE, Beemon K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol. 1985;5:2298-306
18. Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C. et al. The role of m(6)A modification in physiology and disease. Cell Death Dis. 2020;11:960
19. Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019;79:1285-92
20. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176
21. Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T. et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621-9
22. Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y. et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 2020;19:91
23. Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y. et al. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol Cancer. 2020;19:3
24. Wang M, Liu J, Zhao Y, He R, Xu X, Guo X. et al. Upregulation of METTL14 mediates the elevation of PERP mRNA N(6) adenosine methylation promoting the growth and metastasis of pancreatic cancer. Mol Cancer. 2020;19:130
25. Zhang J, Bai R, Li M, Ye H, Wu C, Wang C. et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10:1858
26. Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG. et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep. 2014;8:284-96
27. Moindrot B, Cerase A, Coker H, Masui O, Grijzenhout A, Pintacuda G. et al. A Pooled shRNA Screen Identifies Rbm15, Spen, and Wtap as Factors Required for Xist RNA-Mediated Silencing. Cell Rep. 2015;12:562-72
28. Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH. et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415-29
29. Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C. et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004-14
30. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R. et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88-94
31. van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P. et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47:7719-33
32. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP. et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169:824-35 e14
33. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y. et al. S-Adenosylmethionine Synthesis Is Regulated by Selective N(6)-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354-63
34. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885-7
35. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
36. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP. et al. Reversible methylation of m(6)Am in the 5' cap controls mRNA stability. Nature. 2017;541:371-5
37. Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ. et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15:340-7
38. Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K. et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271
39. Beharry AA, Lacoste S, O'Connor TR, Kool ET. Fluorescence Monitoring of the Oxidative Repair of DNA Alkylation Damage by ALKBH3, a Prostate Cancer Marker. J Am Chem Soc. 2016;138:3647-50
40. Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42:13911-9
41. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591-4
42. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M. et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626
43. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
44. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ. et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315-28
45. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560-4
46. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299-308
47. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
48. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O. et al. 5' UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999-1010
49. Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W. et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 2018;27:3936-50
50. Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D. et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol Cancer. 2020;19:104
51. Chen Z, Wu L, Zhou J, Lin X, Peng Y, Ge L. et al. N6-methyladenosine-induced ERRgamma triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics. 2020;10:3382-96
52. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G. et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622-34
53. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J. et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10:2065
54. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J. et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193-205
55. Gu C, Wang Z, Zhou N, Li G, Kou Y, Luo Y. et al. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N(6)-methyladenosine of Notch1. Mol Cancer. 2019;18:168
56. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K. et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074-83
57. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L. et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell. 2018;22:191-205 e9
58. Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S. et al. Cross-talk among writers, readers, and erasers of m(6)A regulates cancer growth and progression. Sci Adv. 2018;4:eaar8263
59. Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J. et al. ALKBH5 Inhibited Cell Proliferation and Sensitized Bladder Cancer Cells to Cisplatin by m6A-CK2alpha-Mediated Glycolysis. Mol Ther Nucleic Acids. 2021;23:27-41
60. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522-33
61. Li F, Yi Y, Miao Y, Long W, Long T, Chen S. et al. N(6)-Methyladenosine Modulates Nonsense-Mediated mRNA Decay in Human Glioblastoma. Cancer Res. 2019;79:5785-98
62. Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang J. et al. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathol Res Pract. 2019;215:152666
63. Kong F, Liu X, Zhou Y, Hou X, He J, Li Q. et al. Downregulation of METTL14 increases apoptosis and autophagy induced by cisplatin in pancreatic cancer cells. Int J Biochem Cell Biol. 2020;122:105731
64. Little NA, Hastie ND, Davies RC. Identification of WTAP, a novel Wilms' tumour 1-associating protein. Hum Mol Genet. 2000;9:2231-9
65. Jin DI, Lee SW, Han ME, Kim HJ, Seo SA, Hur GY. et al. Expression and roles of Wilms' tumor 1-associating protein in glioblastoma. Cancer Sci. 2012;103:2102-9
66. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO. et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171-4
67. Li BQ, Huang S, Shao QQ, Sun J, Zhou L, You L. et al. WT1-associated protein is a novel prognostic factor in pancreatic ductal adenocarcinoma. Oncol Lett. 2017;13:2531-8
68. Li BQ, Liang ZY, Seery S, Liu QF, You L, Zhang TP. et al. WT1 associated protein promotes metastasis and chemo-resistance to gemcitabine by stabilizing Fak mRNA in pancreatic cancer. Cancer Lett. 2019;451:48-57
69. Xie F, Liu S, Wang H. M6A Methyltransferase METTL16 Suppresses Pancreatic Cancer Proliferation Through p21 Pathways. Pancreas. 2020;49:1437
70. Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee KH. et al. ALKBH5 gene is a novel biomarker that predicts the prognosis of pancreatic cancer: A retrospective multicohort study. Ann Hepatobiliary Pancreat Surg. 2018;22:305-9
71. He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P. et al. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell Physiol Biochem. 2018;48:838-46
72. Tang X, Liu S, Chen D, Zhao Z, Zhou J. The role of the fat mass and obesity-associated protein in the proliferation of pancreatic cancer cells. Oncol Lett. 2019;17:2473-8
73. Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L. et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46
74. Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y. et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782
75. Zhuang C, Zhuang C, Luo X, Huang X, Yao L, Li J. et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1alpha signalling axis. J Cell Mol Med. 2019;23:2163-73
76. Chen J, Sun Y, Xu X, Wang D, He J, Zhou H. et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16:2259-71
77. Tian J, Zhu Y, Rao M, Cai Y, Lu Z, Zou D. et al. N(6)-methyladenosine mRNA methylation of PIK3CB regulates AKT signalling to promote PTEN-deficient pancreatic cancer progression. Gut. 2020;69:2180-92
78. Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX. et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18:174
79. Pu J, Wang J, Qin Z, Wang A, Zhang Y, Wu X. et al. IGF2BP2 Promotes Liver Cancer Growth Through an m6A-FEN1-Dependent Mechanism. Front Oncol. 2020;10:578816
80. Cao J, Mu Q, Huang H. The Roles of Insulin-Like Growth Factor 2 mRNA-Binding Protein 2 in Cancer and Cancer Stem Cells. Stem Cells Int. 2018;2018:4217259
81. Dahlem C, Barghash A, Puchas P, Haybaeck J, Kessler SM. The Insulin-Like Growth Factor 2 mRNA Binding Protein IMP2/IGF2BP2 is Overexpressed and Correlates with Poor Survival in Pancreatic Cancer. Int J Mol Sci. 2019;20:3204
82. Gao W, Cheng L, He S, Li W, Zhou C, Zhou B. et al. Multiomics integrative analysis for gene signatures and prognostic values of m(6)A regulators in pancreatic adenocarcinoma: a retrospective study in The Cancer Genome Atlas project. Aging (Albany NY). 2020;12:20587-610
83. Huang S, Wu Z, Cheng Y, Wei W, Hao L. Insulin-like growth factor 2 mRNA binding protein 2 promotes aerobic glycolysis and cell proliferation in pancreatic ductal adenocarcinoma via stabilizing GLUT1 mRNA. Acta Biochim Biophys Sin (Shanghai). 2019;51:743-52
84. Schaeffer DF, Owen DR, Lim HJ, Buczkowski AK, Chung SW, Scudamore CH. et al. Insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) overexpression in pancreatic ductal adenocarcinoma correlates with poor survival. BMC Cancer. 2010;10:59
85. Taniuchi K, Furihata M, Hanazaki K, Saito M, Saibara T. IGF2BP3-mediated translation in cell protrusions promotes cell invasiveness and metastasis of pancreatic cancer. Oncotarget. 2014;5:6832-45
86. Taniuchi K, Furihata M, Saibara T. KIF20A-mediated RNA granule transport system promotes the invasiveness of pancreatic cancer cells. Neoplasia. 2014;16:1082-93
87. Thin KZ, Liu X, Feng X, Raveendran S, Tu JC. LncRNA-DANCR: A valuable cancer related long non-coding RNA for human cancers. Pathol Res Pract. 2018;214:801-5
88. Hu X, Peng WX, Zhou H, Jiang J, Zhou X, Huang D. et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2020;27:1782-94
89. Wan BS, Cheng M, Zhang L. Insulin-like growth factor 2 mRNA-binding protein 1 promotes cell proliferation via activation of AKT and is directly targeted by microRNA-494 in pancreatic cancer. World J Gastroenterol. 2019;25:6063-76
90. Xu X, Yu Y, Zong K, Lv P, Gu Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J Exp Clin Cancer Res. 2019;38:497
91. Feng Y, Gao L, Cui G, Cao Y. LncRNA NEAT1 facilitates pancreatic cancer growth and metastasis through stabilizing ELF3 mRNA. Am J Cancer Res. 2020;10:237-48
92. Zhai S, Xu Z, Xie J, Zhang J, Wang X, Peng C. et al. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene. 2021;40:277-91
93. Ying P, Li Y, Yang N, Wang X, Wang H, He H. et al. Identification of genetic variants in m(6)A modification genes associated with pancreatic cancer risk in the Chinese population. Arch Toxicol. 2021;95:1117-1128
94. Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD. et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134-45
95. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889-94
96. Coto E, Tavira B, Gomez J, Tranche S, Corte CD. Effect of the FTO rs9930506 Polymorphism on the Main Comorbidities of the Cardiorenal Metabolic Syndrome in an Elderly Spanish Cohort. Cardiorenal Med. 2014;4:82-7
97. Lin Y, Ueda J, Yagyu K, Ishii H, Ueno M, Egawa N. et al. Association between variations in the fat mass and obesity-associated gene and pancreatic cancer risk: a case-control study in Japan. BMC Cancer. 2013;13:337
98. Huang X, Zhao J, Yang M, Li M, Zheng J. Association between FTO gene polymorphism (rs9939609 T/A) and cancer risk: a meta-analysis. Eur J Cancer Care (Engl). 2017;26:e12464
99. Tang H, Dong X, Hassan M, Abbruzzese JL, Li D. Body mass index and obesity- and diabetes-associated genotypes and risk for pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:779-92
100. Maly V, Maly O, Kolostova K, Bobek V. Circulating Tumor Cells in Diagnosis and Treatment of Lung Cancer. In vivo. 2019;33:1027-37
101. Wu CH, Lin SR, Yu FJ, Wu DC, Pan YS, Hsieh JS. et al. Development of a high-throughput membrane-array method for molecular diagnosis of circulating tumor cells in patients with gastric cancers. Int J Cancer. 2006;119:373-9
102. Huang W, Qi CB, Lv SW, Xie M, Feng YQ, Huang WH. et al. Determination of DNA and RNA Methylation in Circulating Tumor Cells by Mass Spectrometry. Anal Chem. 2016;88:1378-84
103. Geng Y, Guan R, Hong W, Huang B, Liu P, Guo X. et al. Identification of m6A-related genes and m6A RNA methylation regulators in pancreatic cancer and their association with survival. Ann Transl Med. 2020;8:387
104. Meng Z, Yuan Q, Zhao J, Wang B, Li S, Offringa R. et al. The m(6)A-Related mRNA Signature Predicts the Prognosis of Pancreatic Cancer Patients. Mol Ther Oncolytics. 2020;17:460-70
105. Tang R, Zhang Y, Liang C, Xu J, Meng Q, Hua J. et al. The role of m6A-related genes in the prognosis and immune microenvironment of pancreatic adenocarcinoma. PeerJ. 2020;8:e9602
106. Xu F, Zhang Z, Yuan M, Zhao Y, Zhou Y, Pei H. et al. M6A Regulatory Genes Play an Important Role in the Prognosis, Progression and Immune Microenvironment of Pancreatic Adenocarcinoma. Cancer Invest. 2021;39:39-54
107. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373-84
108. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu HJ. et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35:677-691
109. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y. et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell. 2018;172:90-105 e23
110. Qing Y, Dong L, Gao L, Li C, Li Y, Han L. et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. 2021;81:922-939.e9
111. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q. et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Transl Med. 2019;11:eaau7116
112. Shi H, Lan J, Yang J. Mechanisms of Resistance to Checkpoint Blockade Therapy. Adv Exp Med Biol. 2020;1248:83-117
113. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P. et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35:559-72 e7
114. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R. et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39:e104514
115. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H. et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159-70
116. Shen XP, Ling X, Lu H, Zhou CX, Zhang JK, Yu Q. Low expression of microRNA-1266 promotes colorectal cancer progression via targeting FTO. Eur Rev Med Pharmacol Sci. 2018;22:8220-6
117. Li J, Liang L, Yang Y, Li X, Ma Y. N(6)-methyladenosine as a biological and clinical determinant in colorectal cancer: progression and future direction. Theranostics. 2021;11:2581-93
118. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z. et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11-9
119. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047-56
120. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767-72
121. Liu XM, Zhou J, Mao Y, Ji Q, Qian SB. Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol. 2019;15:865-71
122. Wilson C, Chen PJ, Miao Z, Liu DR. Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. 2020;38:1431-40
123. Yang R, Liang X, Wang H, Guo M, Shen H, Shi Y. et al. The RNA methyltransferase NSUN6 suppresses pancreatic cancer development by regulating cell proliferation. EBioMedicine. 2021;63:103195
124. Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J. et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019;47:2533-45
125. Yamato I, Sho M, Shimada K, Hotta K, Ueda Y, Yasuda S. et al. PCA-1/ALKBH3 contributes to pancreatic cancer by supporting apoptotic resistance and angiogenesis. Cancer Res. 2012;72:4829-39
126. Lagies S, Schlimpert M, Braun LM, Kather M, Plagge J, Erbes T. et al. Unraveling altered RNA metabolism in pancreatic cancer cells by liquid-chromatography coupling to ion mobility mass spectrometry. Anal Bioanal Chem. 2019;411:6319-28
127. Li Q, Li X, Tang H, Jiang B, Dou Y, Gorospe M. et al. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J Cell Biochem. 2017;118:2587-98
Author contact
![]() Corresponding authors: Bing Ni, E-mail: nibingedu.cn. Tel.: +86-23-68772348; fax: +86 23 68772348. ORCID: 0000-0002-4297-5346; Huaizhi Wang, E-mail: wanghuaizhiac.cn. Tel.: +86-23-13996950719; fax: +86 23 63390599. Yongkang Liu, E-mail: Liuyk100com. Tel.:+86-23-18008027560; fax: +86 28 86590311.
Corresponding authors: Bing Ni, E-mail: nibingedu.cn. Tel.: +86-23-68772348; fax: +86 23 68772348. ORCID: 0000-0002-4297-5346; Huaizhi Wang, E-mail: wanghuaizhiac.cn. Tel.: +86-23-13996950719; fax: +86 23 63390599. Yongkang Liu, E-mail: Liuyk100com. Tel.:+86-23-18008027560; fax: +86 28 86590311.