Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Key phases during stepwise...
3. Morphological defects taking...
4. Spermiogenic defects: Insight...
5. Prospects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(10):2487-2503. doi:10.7150/ijbs.60384 This issue Cite
Review
Towards Post-Meiotic Sperm Production: Genetic Insight into Human Infertility from Mouse Models
Muhammad Azhar1, Saba Altaf1, Islam Uddin1, Jinbao Cheng2, Limin Wu1, Xianhong Tong1, Weibing Qin3 ![]() , Jianqiang Bao1
, Jianqiang Bao1 ![]()
1. Division of Life Sciences and Medicine, The First Affiliated Hospital of USTC, University of Science and Technology of China, Anhui, China.
2. The 901th hospital of Joint logistics support Force of PLA, Anhui, China.
3. NHC Key Laboratory of Male Reproduction and Genetics, Family Planning Research Institute of Guangdong Province, China.
Received 2021-3-12; Accepted 2021-5-16; Published 2021-6-16
Abstract

Declined quality and quantity of sperm is currently the major cause of patients suffering from infertility. Male germ cell development is spatiotemporally regulated throughout the whole developmental process. While it has been known that exogenous factors, such as environmental exposure, diet and lifestyle, et al, play causative roles in male infertility, recent progress has revealed abundant genetic mutations tightly associated with defective male germline development. In mammals, male germ cells undergo dramatic morphological change (i.e., nuclear condensation) and chromatin remodeling during post-meiotic haploid germline development, a process termed spermiogenesis; However, the molecular machinery players and functional mechanisms have yet to be identified. To date, accumulated evidence suggests that disruption in any step of haploid germline development is likely manifested as fertility issues with low sperm count, poor sperm motility, aberrant sperm morphology or combined. With the continually declined cost of next-generation sequencing and recent progress of CRISPR/Cas9 technology, growing studies have revealed a vast number of disease-causing genetic variants associated with spermiogenic defects in both mice and humans, along with mechanistic insights partially attained and validated through genetically engineered mouse models (GEMMs). In this review, we mainly summarize genes that are functional at post-meiotic stage. Identification and characterization of deleterious genetic variants should aid in our understanding of germline development, and thereby further improve the diagnosis and treatment of male infertility.
Keywords: Spermiogenesis, spermatogenesis, infertility, genetically engineered mouse model (GEMM), oligoasthenoteratozoospermia (OAT)
1. Introduction
Infertility is a rapidly rising concern among couples at reproductive age worldwide. Currently, around 6% of the married population among 15-44 years of age suffer from infertility [1]. It has thus become a severe health problem that elicits many social consequences. Almost 50% of fertility-related problems are caused by male factors [2], such as Y-chromosome microdeletions, Klinefelter syndrome, obstruction and genetic abnormalities [3]. In the clinics, some abnormalities can easily be screened by common cytogenetic and biochemical techniques available in most reproductive centers. However, nearly half of cases of male infertility are idiopathic and the genetic variants responsible for the compromised male fertility remain elusive.
In mammals, testis is mainly composed of tortuous, elongated seminiferous tubules in which spermatogenesis is taking place along the well-organized epithelia [4]. In general, the production of competent sperm after birth in mammals necessitates three successive processes: (i) mitotic proliferation of spermatogonial stem cells (SSCs) and subsequent differentiation into advanced spermatogonia (differentiated spermatogonia); (ii) meiotic division: one-time duplication of chromosomes followed by two times of cell division; (iii) spermiogenesis, known as haploid spermatid development, is a process whereby the round spermatids morphologically transform into nuclei-condensed spermatozoa. These normal-looking sperm must be released from seminiferous tubules and transit through further convoluted epididymis to undergo functional maturation in order to be fertilization-competent [5].
At haploid stage, probably the most characteristic feature that distinguishes spermatid development from other somatic cell differentiation is the unique cellular structures formed, such as the acrosome and sperm tail [6]. At the molecular level, spermatid chromatin undergoes drastic chromatin remodeling and genome-wide histone removal followed by protamine deposition, a process termed as histone-to-protamine transition, leading to the production of highly condensed sperm heads with much less cytosolic contents. It has thus been conventionally thought that sperm function as a “carrier” that solely delivers the paternal genetic DNA into oocyte which initiates fertilization [7]. Recent progress in the field argued that the rich non-coding RNA molecules buried inside the sperm nucleus execute a significant impact on the subsequent development in the offspring epigenetically (i.e., independent of the DNA sequence) [8].
Therefore, understanding the mechanisms by which gene expression is specifically regulated to facilitate the spermatid elongation and genome-wide re-organization prepared for the sperm-egg fertilization, has significant implications for reproduction and human health. In the past, a network of genes, either germline-specific or non-specific, has been identified and characterized to be essential for male fertility based on high-throughput sequencing in the patient's pedigree or GEMMs. However, it must be pointed out that, although the testicular structure and germline developmental process resemble each other to some extent between mice and humans, there are considerable differences in both species. For example, the meiosis persists for ~25 days in men but only ~14 days in male mice. Following meiosis, the haploid spermatid differentiation takes ~16 days in men but is shorter (~9 days) in mice. Thus, we must be cautious when interpreting the causative factors and mechanisms underlying human infertility by taking advantage of the findings from mouse studies [9]. In this review, we will particularly summarize those genes that are functionally important for post-meiotic male germ cell development in mice, which will aim in our understanding of human fertility.
2. Key phases during stepwise spermiogenesis
In mice, spermiogenesis entails the dramatic morphological transformation of round spermatids into hook-shaped mature spermatozoa. Unlike somatic cells, the motile sperm are capable of fertilizing an egg through their highly specialized cell structures [10]. Three major morphological changes take place in the process of spermiogenesis, including acrosome biogenesis, head reshaping, and development of a flagellum (Fig. 1). Right after meiosis, pairs of individual round spermatid are connected through intercellular bridges (Cytoplasmic bridges) through which haploid gene products transit and are shared across the cell membrane. On basis of the acrosome morphology, it can be divided into four phases during haploid spermatid development (Fig. 1) [11]. The first phase is called Golgi phase in which the proacrosomal vesicles fuse to form a single large acrosomal vacuole, which contains the granules in contact with the nuclear envelope (Step 1-3). Soon after meiosis, Golgi-derived vesicles traffick through microtubules and F-actin microfilaments, and subsequently aggregate and fuse to form the acrosome sac. Round spermatids consist of a centrosome with two centrioles - one is named distal centriole (mother centriole) based on the distal location while the other is called proximal centriole (daughter centriole). During sperm flagellar formation, the centrioles undergo dynamic remodeling and serve as the nucleators of the cilia axoneme, of which the process is explicitly reviewed elsewhere [12]. Upon occurrence of acrosomal sac, the centriolar pair migrates to the other pole opposite to the acrosomal vesicle wherein the axoneme starts to assemble. These distal centrioles will develop into flagellum while the proximal centrioles and pericentriolar matrix will eventually develop into head-tail coupling apparatus, which connects the sperm head and tail axoneme. In the second phase, also known as cap phase (Step 4-7), both the tail axoneme and acrosomal sac continue to grow. Meanwhile, the growing acrosomal sac becomes flatten and forms a cap tightly adhered to the anterior nuclear envelope through the F-actin-containing acroplaxome. The acrosome-acroplaxome complex descends caudally to the tail assembly direction in concert with the elongation of the nucleus. This process is followed by the acrosomal phase (phase 3), the acroplaxome attached to the acrosome continues to descend along the axoneme direction and spreads over the anterior nuclear envelope, in accompany with the developing of microtubule-containing ring-like manchette and the aligning of mitochondrion to the axoneme (Step 8-12). The outer dense fibers (ODFs) start to assemble and align along the developing axoneme. Upon completion of nuclear elongation, the acrosomes enter the maturation phase (phase 4) (Step 13-16) [13] when the manchette starts to dissemble and the majority of histones are step-wisely replaced by protamines whereby the chromatin condensation continues further. This process necessitates genome-wide chromatin remodeling as well as massive single-stranded breaks (SSBs) and/or double-stranded breaks (DSBs) [14].
Schematic diagram illustrating a total of 16 steps of haploid germline development in mice. Spermiogenesis is categorized into four phases (Golgi phase, Cap phase, Acrosome phase and Maturation phase) according to the acrosome morphology. Conventionally, spermatids are divided into 16 steps on basis of acrosome and head morphology, and the criteria is commonly leveraged to pinpoint the specific step of spermiogenic arrest through H&E staining of the sections from GEMMs. In literature, spermatids are often classified into four groups based on nuclear morphology: round spermatids (Steps 1~8), elongating/condensing spermatids (Steps 9~11), elongated/condensed spermatids (Step 12~14) and spermatozoa (Step 15~16). Representative genes essential for spermatid development are listed at the bottom.
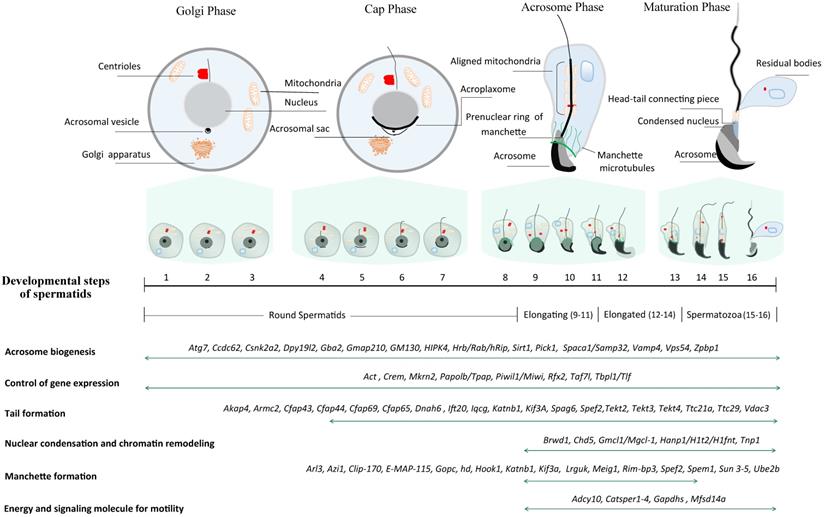
Currently, little is known about how the histones are globally substituted and how the SSBs/DSBs are repaired in such a narrow time window, although much knowledge has gained from GEMMs that the defect in any of these steps is closely linked to the compromised male fertility. In addition, it is noteworthy to point out that the acrosomal development unique to germ cells is successive and inherently connected to nuclear elongation and condensation. Secondly, since there is only one copy of allele owing to the haploid genome per se, spermatids are thus not capable of repairing the DSBs via homology-directed repair (HDR) mechanism. It is, therefore, conceivable that any disturbance at this stage would not only cause decreased fertility but also induce pathogenic mutations that can be carried over to progeny [15]. Finally, we must bear in mind that, although the sperm released into the lumen in the testicular tubules is morphologically identical to mature sperm, they must undergo further functional maturation during transit in the lengthy epididymis wherein the sperm might exchange information with extracellular vesicles in the fluid, in order to be capacitation-competent [16].
3. Morphological defects taking place during spermiogenesis
3.1. Acrosome assembly
As described above, the acrosome is a unique glycoprotein-rich organelle derived from Golgi complex, and thus can be readily visualized by the Periodic acid-Schiff (PAS) and PNA (Peanut agglutination assay) staining, or by electron microscopy [17]. The acrosome is also enriched in protease enzymes, such as acrosin, esterases, acid phosphatases, aryl sulfatases, and glycohydrolases, Sp56/AM67, SP-10, and Hyal-PH20, which are essential for sperm-oocyte fertilization. The acrosin and SP-10 are associated with the acrosomal matrix and released after the acrosomal reaction in large part [13]. SP56 is localized to the acrosomal matrix and has a specific affinity for mouse Zona pellucida sperm-binding protein 3 [18]. To date, Hyal-PH20 is the only ideal acrosomal membrane protein. It is a glycosylphosphatidylinositol-anchored protein and localized to both the inner acrosomal membrane and plasma membrane of sperm head. Interestingly, it has been shown to play in sperm-egg interaction that is unrelated to its enzymatic activity [19].
The sperm head shaping is an important event of spermiogenesis and is coordinated by nuclear condensation (Fig. 1). Two distinct subcellular structures are involved in the sculpting of the head into species-specific morphology (e.g., hook-like shape in mice, and oval shape in humans) -acroplaxome and manchette [20]. The anchoring of the acrosome to the nuclear surface is mediated via the acroplaxome, also named prenuclear theca, which is a thin layer of cytoskeletal elements between the nuclear and acrosomal membranes. In 2002, phospholipase C zeta (PLCζ) has been discovered [21], residing in the post-acrosomal sheath of perinuclear theca and is essential for oocyte activation [22]. The acroplaxome is comprised of myosin, F-actin and keratin-containing components essential for acrosome development. The acroplaxome provides mechanical support for transmitting forces around the anterior nucleus to facilitate the elongation of sperm head [23]. The presence of keratin-5 and F-actin is important in the acroplaxome for force generation and transfer [24]. On the other hand, it has been reported that some inhibitors related to microtubule dynamics interfere with the developing acrosomal vesicles resulting in abnormal spreading of acrosome over the nuclear membrane [25]. As a result, spermatozoa carrying defective acrosomes are not capable of penetrating zona pellucida of oocytes, leading to fertilization failure [26].
3.2. Manchette assembly
The second key structure that is essential for sperm head shaping is manchette, which is a transient skirt-like microtubular structure surrounding the posterior nucleus present in elongating spermatids (Fig. 1). It is composed of up to 1000 microtubules and consists of α and β tubulin heterodimers. The timing of manchette appearance is strictly step dependent. The short microtubules appear first around step 7 and then assemble at step 8 [27]. The microtubules are closely wrapping around posterior nuclear surface till step 14 when the manchette begins disassembling [28]. Through intra-manchette transport, it is thought to assist in the nucleocytoplasmic exchange, mechanical reshaping and elongation of sperm nucleus, as well as assembly of sperm tail. As a result, the excess cytosol is removed, and the nucleus becomes highly condensed. Any interference with the manchette structure or function can potentially give rise to the abnormal sperm head shape. Till now, a dozen of genes have been identified and are associated with manchette formation and development, which have been explicitly reviewed elsewhere [29]. Likewise, many mutagenic agents including inhibitors of microtubule dynamics, e.g., alkylating agent, can also induce manchette malfunction. Genetic deletion or mutation studies unveiled the aberrant manchette morphology, abnormal nuclear condensation/elongation and impaired fertility in those manchette-related gene mutation mouse models [30-32].
3.3. Flagellar assembly
The most distinctive feature of spermatids probably lies in the formation of a highly structured flagellum - the sperm tail, which is tightly relevant to sperm motility (Fig. 1 and 2) [10]. Morphologically, the flagellum is highly conserved in its ultrastructure and can be divided into four parts: i) neck, ii) midpiece, iii) principal piece and iv) end piece (Fig. 2). The neck is also known as connecting piece or head-tail coupling apparatus, which anchors the tail to the head tightly through the capitulum and the segmented column as magnified under transmission electronic microscopy (TEM). This ultrastructure has been elaborately discussed by several review papers elsewhere [30, 33]. The midpiece is comprised of mitochondria and 9 ODFs, which surround the 9+2 axoneme. Two ODFs are replaced in the principal piece by the longitudinal columns of the fibrous sheath (FS) connected by transverse ribs. The end piece contains only the axoneme surrounded by plasma membrane [34]. Upon completion of nuclear elongation and condensation, the fully developed sperm migrate close to the lumen of seminiferous tubules and are ready to be released into the lumen in a process called spermiation [10].
Schematic illustration of the ultrastructure of mature mouse sperm. Sperm flagellum is structurally divided into three parts - midpiece, principal piece, and end piece; Each part comprises the core axoneme, which is composed of the canonical “9+2” arrangement of microtubules, in the center. The vertical arrow points to the #1 pair of microtubules while the horizontal dash line parallels the central pair of microtubules, in the cross mark. The mitochondrial sheath and the outer dense fibers (ODFs) wrap around the mid-piece and principal piece, respectively. The connection part between sperm head and tail is the neck, also termed head-tail coupling apparatus or connecting piece (right panel) [34, 150].
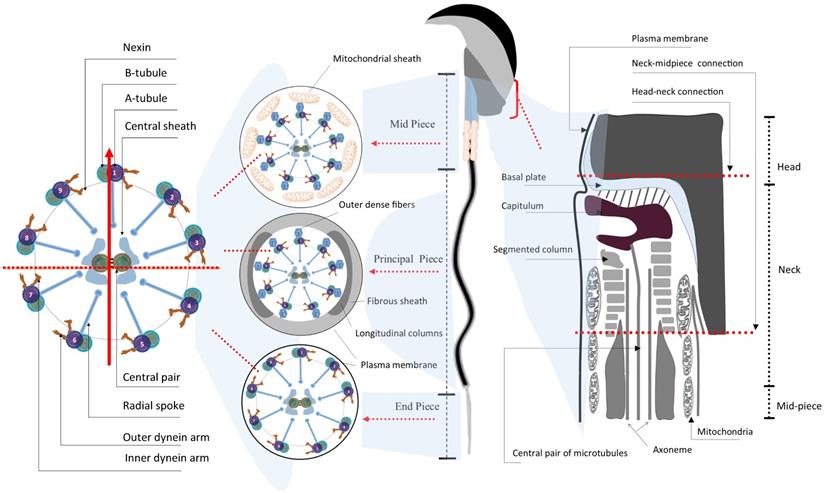
As the acrosome begins to form, the pair of centrioles move to the opposite pole of acrosome to initiate axoneme formation (Fig. 1). The axoneme arises from the centrioles and extends out into the cytoplasm [24]. The axoneme is comprised of characteristic “9+2” microtubules wherein a central pair of microtubules are wrapped by nine doublet microtubules. Notably, these microtubules are post-translationally modified including tyrosinated, acylated, and polyglutamylated modification [30, 33]. The outer doublets contain dynein motor arms that generate forces to endow waveform motion of the flagellum. Defects in post-translational modifications of tubulins or perturbations in microtubular arrangement can impair axoneme assembly, which can be visualized by staining acetylated tubulin or by electron microscopy [30, 33]. At the last step, prior to fertilization, the sperm must acquire its fertilizing capability in a process known as capacitation during transit in the female reproductive tract. Capacitation is linked to hyperactive motility acquisition, characterized by an increase in the amplitude and asymmetry of the flagellar bending, and genetic mutations causing defective capacitation are linked to male infertility [35].
Until now, whereas it remains challenging to decipher which signaling pathways are involved in the axoneme assembly and elongation in vivo, growing studies have discovered a large number of genes in humans with the mechanistic and phenotypic studies further characterized in mouse models, showing they are tightly associated with axoneme formation. Most often, mutations or disruptions to those genes can induce a severe type of morphological abnormality commonly observed in the clinics - multiple morphological abnormalities in flagella (MMAF), leading to male sterility, which has been elegantly summarized and reviewed in details [36].
4. Spermiogenic defects: Insight view from mouse models
Post-meiotic spermatids develop clonally in many species ranging from fruit flies to humans and remain connected to one another via intercellular bridges. These bridges work as a channel for sharing protein products or even small subcellular organelles such as chromatid body (CB) and mitochondria [37]. In this way, the bridges enable haploid spermatids to be functionally diploid [38]. Exogenous interruption or genetic mutations often result in aberrant spermiogenesis, and consequently, lead to the reduction of sperm number or motility, or morphologically or functionally defective sperm. On basis of these varied abnormalities, the most common phenotypes with clinical outcome can be generally categorized into four groups as discussed below.
4.1. Oligozoospermia
Oligozoospermia is a common type of human infertility, which refers to the low sperm count in the semen. According to the WHO 2010 criteria, the average sperm count is more than 60 million per ml, and a count of less than 15 million per ml is considered oligozoospermia [39]. It is further sub-categorized into three classes: mild oligozoospermia (10-15 million/ml), moderate oligozoospermia (5-10 million sperm/ml), severe oligozoospermia (< 5 million sperm/ml) [40]. In addition, it is now known that the disturbance in the post-meiotic spermatid development will likely cause aberrant spermatid apoptosis or premature spermatid sloughing off the seminiferous tubular epithelia, resulting in the reduction of spermatid number and hence impaired male fertility. This syndrome is more often connected to teratozoospermia and asthenozoospermia.
Until now, few genes are reported to be responsible for solely low sperm count. Fibroblast growth factors (FGFs) family contains 22 known members [41], which are involved in important cellular processes such as differentiation, migration, mitogenesis, and cell survival [42]. FGF action is mediated through high-affinity binding to tyrosine kinase receptors, which include FGF receptor 1 to FGF receptor 5 (FGFR1-FGFR5) [43, 44]. The role of Fgfr1 in male infertility and spermiogenesis has been assessed by dominant-negative transgenic mouse models, showing that Fgfr1 contributes to sperm production and function as the sperm daily-out has been examined low [45]. Recently, whole-exome sequencing (WES) efforts revealed a handful of genetic variants that are potentially linked to human oligozoospermia, such as HAUS7 [46], M1AP [47], MAGEB4 [48], RPL10L [49], and ZMYND15 [50] (Table 1).
Genes involved in spermiogenesis in mice and their corresponding mutation in humans.
| Gene Name | Knockout Mice Phenotype | Species | Phenotype | Reference(s) |
|---|---|---|---|---|
| Bromodomain and WD repeat domain containing 1 (BRWD1) | Low sperm count, deformed spermatid heads, abnormal acrosome morphogenesis, defective chromatin condensation | Human, Mouse | OAT | [151, 152] |
| Coactivator associated arginine methyltransferase 1 (Carm1) | Low sperm count, poor motility, abnormal morphology includes headless sperm with a bent midpiece | Mouse | OAT | [153] |
| Coiled-coil domain containing 62 (Ccdc62) | Aberrant sperm phenotypes, impaired sperm motility, deformed acrosome, retention of cytoplasmic fragments on the sperm head and neck, | Mouse | OAT | [154] |
| UDP-GalNAc: polypeptide N acetylgalactosaminyltransferase3 (Galnt3) | Low sperm count, deformed acrosome, | Mouse | OAT | [149] |
| Germ cell-less 1, spermatogenesis associated (Gmcl1/mGCL-1) | Low sperm count with poor motility, abnormal head, blunt acrosome, round/narrowed heads, insufficient chromatin condensation, multiple heads and flagella | Mouse | OAT | [155] |
| Homeodomain interacting protein kinase 4 (Hipk4) | Low sperm count, poor motility, aberrant head, abnormal tail (bent and coiled), ectopic localization of acrosome components | Human, Mouse | OAT | [148] |
| Intraflagellar transport 20 (Ift20) | Low sperm count, poor motility, swollen head and abnormal tail (kinked tail, short tails or tailless) | Mouse | OAT | [145] |
| Intraflagellar Transport 140 (IFT140) | Low sperm count, poor motility, amorphous sperm heads, short/bent flagella | Human, Mouse | OAT | [143, 144] |
| Katanin regulatory subunit B1 (KATNB1) | Low sperm count, poor motility, abnormal manchette, knob-like head | Human, Mouse | OAT | [32, 156] |
| Leucine-rich repeats and guanylate kinase-domain containing isoform 1 (Lrguk-1) | Low sperm count, poor motility, misshapen heads, shortened tails, acrosome and the acroplaxome detached from the nuclear membrane in elongated spermatids, abnormal grass skirt-like manchette structure | Mouse | OAT | [141] |
| Makorin-2 (MKRN2) | Low sperm count, poor motility, aberrant morphology, spermiation failure, misarrangement of ectoplasmic specialization in testes | Human, Mouse | OAT | [157] |
| Oxysterol-binding Protein (OSBP)-related Protein 4 (Orp4) | Abnormal head, disturbed mitochondrial distribution | Mouse | OAT | [147] |
| Septin4 (Sept4) | Poor motility, abnormal morphology, defective annulus or bent neck, defects in mitochondrial architecture and acrosome | Mouse | OAT | [158] |
| Septin12 (SEPT12) | Low sperm count, poor motility, distinctive sperm morphology, sloughing of round spermatids, mitochondria with deformed acrosome | Human,Mouse | OAT | [159, 160] |
| Serine protease inhibitor Kazal-type 2 (Spink2) | Missing proacrosomal vesicles fusion, disarrangement of the Golgi apparatus, and bent flagellum | Mouse | OAT | [161] |
| TATA-box binding protein associated factor 7 like (Taf7l) | Low sperm count, poor motility, abnormal morphology, compromised fertility | Mouse | OAT | [162] |
| Vacuolar protein sorting-associated protein (Vps13b) | Disorganization of the Golgi apparatus, degeneration of spermatid, deformed head | Mouse | OAT | [163] |
| H1.7 linker histone (Hanp1/H1t2/H1fnt) | Poor motility, aberrant chromatin packaging, delayed nuclear condensation, detachment of acrosome | Mouse | OAT | [164] |
| Serine/threonine kinase 36 (Stk36) | Poor motility, club-shaped head with large gap between acrosome and the nucleus, absence of central pair of flagellar axonemes | Mouse | OAT | [31] |
| Adenylate cyclase 10 (ADCY10) | Poor and lack of progressive sperm motility, deficit in cyclic adenosine monophosphate (cAMP) | Human, Mouse | Asthenozoospermia | [53, 69] |
| A-kinase anchoring protein 4 (AKAP4) | Poor motility, diameter of principal piece reduced, distal flagellum splits apart into several filaments, incomplete development of fibrous sheath | Human, Mouse | Asthenozoospermia | [165, 166] |
| Cation channel sperm associated 1-4 (CATSPER1-4) | Hyperactivated motility failure | Human, Mouse | Asthenozoospermia | [54, 71-73, 167] |
| Polypeptide N-Acetylgalactosaminyltransferase Like 5 (GALNTL5) | Poor motility, lack of Acrosomal Component, ubiquitin-proteasome systems aberrant distribution | Human, Mouse | Asthenozoospermia | [56, 168] |
| Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic (GAPDHS) | Poor motility, sluggish movement, bending of flagellar in the middle piece, declined concentration of ATP | Human, Mouse | Asthenozoospermia | [76, 77] |
| IQ motif containing G (IQCG) | Poor motility, missing flagella, short and disorganized structure of tail, deformed nucleus | Human, Mouse | Asthenozoospermia | [62, 169] |
| Mouse dynein heavy chain 7 (Mdhc7)/(DNAH1) | Poor motility, lack of progressive movement and straight-line velocity, missing FS, longer ODF 1 and 9 with asymmetric mitochondrial line, Length of midpiece increased 2-3 times | Human, Mouse | Asthenozoospermia | [120, 170] |
| Sperm Associated Antigen 17 (SPAG17) | Poor motility, irregular nuclear shape with detached acrosomes, altered manchette microtubules, reduced chromatin condensation | Human, Mouse | Asthenozoospermia | [58, 171] |
| Tecktin-2 (TEKT2) | Bent flagella, loss of the inner dynein arm structures | Human, Mouse | Asthenozoospermia | [64, 66] |
| Tecktin-3 Tekt3 | Poor motility, defective flagellar pattern with abnormal bending between 90° and 180°, midpiece thinning | Mouse | Asthenozoospermia | [67] |
| Tecktin-4 Tekt4 | Poor motility, defect in flagellar bending restricted to the end piece, twitching motion of principal piece, disorganized sub-mitochondrial reticulum | Mouse | Asthenozoospermia | [65] |
| Ttransition protein 1 (TNP1) | Poor and lack of progressive motility, DNA breaks, abnormal pattern of chromatin condensation, less condensed chromatin, large rod-like structures | Human, Mouse | Asthenozoospermia | [86, 172, 173] |
| Voltage-dependent anion channel 3 (VDAC3) | Poor motility, loss of one outer doublet, axonemal defects, aberrant appearance of mitochondria along the midpiece | Human, Mouse | Asthenozoospermia | [78, 174] |
| Sperm associated antigen 6 (Spag6) | Low sperm count, poor motility, loss of the sperm head, midpiece fragmentation, truncated flagella, lack of central pair of microtubules, alterations in the outer dense fibers and fibrous sheath | Mouse | Asthenozoospermia | [175] |
| Aurora Kinase C (AURKC) | Defective nuclear chromatin condensation with defective acrosome, blunted heads | Human, Mouse | Macrozoospermia | [99, 100] |
| ADP ribosylation factor like GTPase 3 (Arl3) | Abnormal head shape, lasso-like coiled tail | Mouse | Teratozoospermia | [176] |
| 5-azacytidine induced gene 1 (Azi1) | Defective manchette structure, misorientation of elongated spermatids, most of the sperm lack flagella while the remaining axoneme truncated | Mouse | Teratozoospermia | [177] |
| Cadherin 5 (CHD5) | Abnormal head morphology (nuclear deformation, partial failure of chromatid condensation) | Human, Mouse | Teratozoospermia | [178, 179] |
| CAP-GLY domain containing linker protein 170 (Clip-170) | Abnormal hook-shape head, absence of manchette microtubules along with the nuclear envelope | Mouse | Teratozoospermia | [180] |
| Epithelial Microtubule-Associated Protein Of 115 (E-map-115) | Abnormal head (Nuclei deformation, microtubules of manchette reduced) | Mouse | Teratozoospermia | [181] |
| Hypodactylous (hd) | Detached centrosome, asymmetric and ectopic perinuclear ring of manchette, abnormal assembly of ODFs resulting in decapitated tails | Mouse | Teratozoospermia | [182] |
| Hook microtubule tethering protein 1 (HOOK1) | Elongated manchette with conically shaped, club-shaped nucleus, positional abnormalities of ODFs and FS result in lasso-like tail formation | Human, Mouse | Teratozoospermia | [183, 184] |
| Kinesin family member 3A (Kif3a) | Low sperm count, malformed head, detached perinuclear ring, abnormal manchette, absence of axonemal and flagellar structures, displaced mitochondria, microtubules, ODFs, FS | Mouse | Teratozoospermia | [95] |
| Meiosis/Spermiogenesis Associated 1 (Meig1) | Disrupted microtubular organelle and flagellar formation, disrupted manchette | Mouse | Teratozoospermia | [96] |
| Polyamine Modulated Factor 1 Binding Protein 1 (PMFBP1) | Flagella without heads, retention of cytoplasm, abnormal axoneme assembly and misarranged mitochondria inside flagellum | Human, Mouse | Teratozoospermia | [90] |
| RIMS binding protein 3 (Rim-bp3) | Severe malformation of the sperm head and deformed nucleus with enlarged perinuclear space, misplaced acrosome | Mouse | Teratozoospermia | [185] |
| Spermatogenesis associated 6 (Spata6) | Acephalic spermatozoa | Mouse | Teratozoospermia (acephalic sperm) | [92] |
| spermatogenesis and centriole associated 1 like (Spatc1l) | Acephalic spermatozoa | Mouse | Teratozoospermia (acephalic sperm) | [91] |
| Sperm flagellar 2 (Spef2) | Short and bent tail at all steps of spermatid elongation, absence/twin tails at steps 15-16, missing central pair of the axonemal microtubules and defective assembly of tail accessory structures | Mouse | Teratozoospermia | [98] |
| Spermatid maturation 1 (Spem1) | Poor motility, deformed head, completely bent tail, retained cytoplasm | Mouse | Teratozoospermia | [186] |
| Serine/threonine kinase 33 (Stk33) | Head-dislocation, Misarrangement of microtubules and mitochondria | Mouse | Teratozoospermia | [97] |
| Ubiquitin conjugating enzyme E2 B (UBE2B) | Aberrant head morphology, ectopic manchette, middle piece deformation | Human, Mouse | Teratozoospermia | [187, 188] |
| Armadillo Repeat Containing 2 (ARMC2) | Poor motility, short, coiled, and absent flagella, complete deficiency of motility | Human, Mouse | MMAF | [131] |
| Cilia and Flagella Associated Protein 43-44 (CFAP43-44) | Poor motility, disorganization in fibrous sheaths and periaxonemal structures, distorted cytoskeletal components, lack of central microtubules with disorganized ODFs | Human, Mouse | MMAF | [124, 189] |
| Cilia and Flagella Associated Protein 65 (CFAP65) | Lack of progressive motility, absent or short flagella, lack of central microtubules with disorganized ODFs | Human, Mouse | MMAF | [126, 190] |
| Cilia and Flagella Associated Protein 69 (CFAP69) | Shortening of midpiece and principal piece, shorter flagella, abnormal head morphology | Human, Mouse | MMAF | [127] |
| Dynein Axonemal Heavy Chain 17 (DNAH17) | Impaired sperm flagellar assembly with disorganized axonemal structure | Human, Mouse | MMAF | [123] |
| Glutamine Rich 2 (QRICH2) | Sperm flagella with short, bent, coiled and other irregular shapes, displacement of the central microtubules and ODF, abnormal 9 + 2 structure | Human, Mouse | MMAF | [119, 134] |
| Tetratricopeptide Repeat Domain 21A (TTC21A) | Poor motility, aberrant structure of connecting piece, lack of central microtubules with disorganized ODFs, extra peripheral microtubule doublets, absent dynein arms | Human, Mouse | MMAF | [136] |
| Tetratricopeptide Repeat Domain 29 (TTC29) | Poor motility (lower progressive motility), axonemal disorganization, lack of central microtubules with disorganized ODFs, | Human, Mouse | MMAF | [137] |
| Autophagy related 7 (Atg7) | Round-headed sperm, lack of acrosome | Mouse | Globozoospermia | [116] |
| Casein kinase 2 alpha 2 (Csnk2a2) | Abnormal head with bent flagella, deformed acrosome that detached from the nucleus | Mouse | Globozoospermia/Oligozoospermia | [191] |
| Dpy-19 like 2 (DPY19L2) | Round-headed sperm, midpiece defect, lack/deformed acrosome, 70% show absence of mitochondria while the remaining have a disorganized sheath, 20% have coiled tail | Human, Mouse | Globozoospermia | [105, 111] |
| Glucosylceramidase beta 2 (Gba2) | Low sperm count, poor motility, larger and round-headed acrosome, disordered mitochondria, condensed pyknotic nuclei | Mouse | Globozoospermia | [192] |
| Golgi matrix protein 130 kD (Gm130) | Lack of acrosome, round-headed sperm, aberrant arrangement of mitochondria | Mouse | Globozoospermia | [193] |
| Golgi-associated microtubule-binding protein 210 (Gmap210) | Low sperm count, deformed acrosome, abnormal mitochondria sheath, dispersed microtubules and ODFs in the cytoplasm, ectopic perinuclear ring and elongated manchette | Mouse | Globozoospermia | [107] |
| Golgi associated PDZ and coiled-coil motif containing (Gopc) | Poor motility, round-headed, coiled tail, fragmented acrosome and disarrangement of mitochondria, some acrosomes completely lost | Mouse | Globozoospermia | [115] |
| Hrb (Rab or hRip) | Round-headed spermatids, lack of acrosome due to failure of proacrosomic vesicles to fuse | Mouse | Globozoospermia | [108] |
| Heat shock protein 90 beta family member 1 (Hsp90b1) | Poor motility, large round-headed sperm, lack/abnormal acrosome, disorganization of mitochondria, high amount of cytoplasm around the nucleus | Mouse | Globozoospermia | [194] |
| Major facilitator superfamily domain containing 14A (Mfsd14a) | Low sperm count, round-headed spermatids, deformed head with anomalous nuclear condensation, failure of acrosome formation | Mouse | Globozoospermia | [109] |
| Protein interacting with (PICK1) | Low sperm count, poor motility, abnormal head (irregular shape and malformation of acrosome with mislocalization) | Human, Mouse | Globozoospermia | [103, 195] |
| Sirtuin 1 (Sirt1) | Low sperm count, round-headed spermatozoa, Abnormal acrosome biogenesis, disrupted autophagic flux | Mouse | Globozoospermia | [117] |
| Sperm acrosome associated 1 (SPACA1/SAMP32) | Round-headed spermatozoa, small acrosomes of deformed shape, aberrant assembly of mitochondria inside large cytoplasmic droplets, ectopic localization of nuclei and flagella | Human, Mouse | Globozoospermia | [196] |
| VPS54 subunit of GARP complex (Vps54) | Defective cytoskeletal structure and head, failure of acrosome to attach to the nucleus, irregularity of mid-piece and tail | Mouse | Globozoospermia | [110] |
| Sad1 And UNC84 Domain Containing 3 (Sun3) | Low sperm count with missing/mislocalized/fragmented acrosome, absence of manchette microtubules | Mouse | Globozoospermia-like | [197] |
| Sad1 and UNC84 domain containing 4 (SUN4) | Aberrant roundish spermatids, severely impaired head, spermatozoa encircled by their tail, coiled tail, disorganized manchette | Human, Mouse | Globozoospermia-like | [29, 198] |
| Sad1 And UNC84 Domain Containing 5 (SUN5) | Acephalic spermatozoa | Human, Mouse | Globozoospermia-like (acephalic sperm) | [89, 93] |
| Zona pellucida binding protein (ZPBP1) | Poor motility, round-headed sperm, coiled tail around the nucleus, lack of acrosome, disorganized mitochondria, and detached flagella | Human, Mouse | Globozoospermia | [199, 200] |
| Fibroblast Growth Factor Receptor1 (Fgfr-1) | Low sperm count, impaired sperm capacitation | Mouse | Oligozoospermia | [45] |
4.2. Asthenozoospermia
Spermatozoa own highly specialized structures, which discriminate them from other somatic cells, especially the long tails, which are required for sperm motility. Defective microtubular assembly of sperm flagellum or functional suppression directly dampen sperm motility and thus cause reduced male fertility known as asthenozoospermia. It is one of the common types of male infertility in the clinics [51]. Mouse model studies have shown that the structural integrity of the flagella is of extreme importance for normal sperm motility, and defects in the axoneme assembly render poor sperm motility [52]. Not surprisingly, WES has identified a large number of genetic variants relevant to asthenozoospermia, while each variant is likely responsible for just a small fraction of patients, particularly including ADCY10 [53], CATSPER1-4 [54], EIF4G1 [55], GALNTL5 [56], NSUN7 [57], PLA2G6 [54], SPAG17 [58], and TRPC5 [59] (Table 1).
As aforementioned, the functional unit of flagella is the axoneme, which consists of structural microtubules and motor proteins that cooperate to produce waveforms and generate progressive motion in a synchronized and structured manner [60]. Inner dynein arms (IDA) and outer dynein arms (ODA) are ATPase complexes consisting of multiple motor proteins and reflect distinctive functions in flagellar motility. Dynein axonemal heavy chain family proteins (DNAH1, DNAH2, DNAH17), IQ motif containing G (IQCG), and tektin protein family are structural constituents of axoneme. In patients with asthenozoospermia from different ethnic backgrounds, common deleterious mutations have been found in the genes encoding inner dynein complex and nexin-dynein regulatory complex. Nevertheless, those null mouse models have immotile sperm elicited by disassembly of the axoneme and consequently, are unable to fertilize eggs [61-63]. Tektin is a family of filament-forming cytoskeleton proteins that comprise tubulin and dynein heterodimers in axoneme. It has been assumed that these constitutive proteins contribute to axonemal microtubular stability and structural complexity [64]. In humans, five tektins (TEKT1, 2, 3, 4, and 5) are present in the flagella of spermatozoa. Disruption to these proteins led to a low level of ATP and clumsy movement of flagella [64-67].
Capacitation is a cAMP-dependent bicarbonate-induced process by which sperm develop the capacity to fertilize eggs. A key enzyme in the PKA pathway is a soluble adenylyl cyclase (sAC) in the sperm tail encoded by the Adcy10. Adenylate cyclase uses ATP as a substrate to generate cAMP, further catalyzed by PKA to phosphorylate downstream proteins [68]. Male mice deficient in sAC are infertile owing to a complete lack of sperm motility [69]. Noticeably, homozygous frameshift variants of ADCY10 have been reported in asthenozoospermia patients [53]. Another core messenger during capacitation is Ca2+ [70], although the nature of the source responsible for the inflow of Ca2+ remains uncertain. CATSPER1-4, four cation channel-like proteins, have pore-lining residues forming alpha subunits and three auxiliary subunits, and their amino acid sequences imitate those of the conventional four-repeat voltage-gated Ca2+ channel with a single repeat. They are sperm-specific heterotetrametric Ca2+ selective channel proteins located on the middle and principal piece of flagella that regulate the calcium entry into the sperm, which is critical for the activation of sperm capacitation and motility. Mouse knockout models exhibit drastically reduced sperm motility and are thus sterile only in the males. Recurrent genetic mutations were recently discovered in CATSPER family members in human patients with asthenozoospermia through Exome-sequencing, suggesting highly conservative roles of CATSPER members in sperm activation in mammals [54, 71-73].
Synchronized motion of the sperm tail involves energy production, commonly through oxidative phosphorylation, and signaling from its environment, and thus disturbances in the supply of energy and signaling transduction may also cause motility defects. It is generally speculated that oxidative phosphorylation is restricted to the midpiece harboring mitochondria, whilst glycolysis is confined to the principal piece [74, 75]. Several glycolysis isozymes are present in mammalian sperm, including Gapdhs, Adcy10, and Vdac3. Glyceraldehyde 3-phosphate dehydrogenase-S (Gapds) is the only Gapdhs isozyme tightly bound to the fibrous sheath. Voltage‐dependent anion channel 3(Vdac3) is a non-specific hollow porin in the outer mitochondrial membrane of sperm flagella. ATP levels significantly dropped in the Gapdhs- and Vdac3-null mice leading to non-progressive and weak motility of sperm flagella [69, 76-78]. However, for majority of patients with severe asthenozoospermia, the hereditary pathogenic variants, if present, remain largely undetermined, and further screening and functional studies are required to fully understand the pathogenesis of severe asthenozoospermia.
4.3. Teratozoospermia
Teratozoospermia is defined as a condition of male infertility characterized by the presence of abnormal morphology of sperm head or tail (Fig. 3), which is often linked to poor sperm motility. In patients with teratozoospermia, spermatozoa with irregular morphology account for more than 85% in the semen (Fig. 3). Teratozoospermia can be classified into polymorphic and monomorphic. Polymorphic teratozoospermia is thought to present an abundance of spermatozoa with heterogeneous forms of abnormalities. In contrast, monomorphic teratozoospermia exhibit a particular homogeneous morphological defect. Three well-described representative types of monomorphic teratozoospermia, i.e., macrozoospermia and globozoospermia, acephalic sperm syndrome have been reported thus far [26, 79, 80].
Schematic representation of morphological abnormalities of sperm in mice and humans. A. Comparison of normal sperm head morphology between mouse and human; B. Common types of aberrant morphologies of sperm in mice and humans.
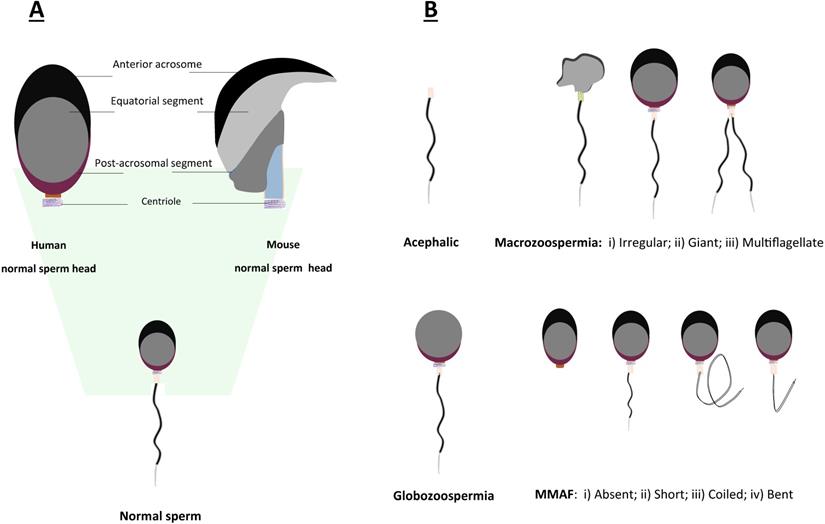
Nuclear elongation and chromatin condensation occur concomitantly, accompanied by the displacement of nuclear DNA-wrapped histones by protamines. Meanwhile, during this histone-to-protamine transition process, multiple histone variants are expressed, such as tH2A, tH2B, H1t, Hanp1, ssH2B, and Hils1, which are essential for sperm head maturation. Therefore, not surprisingly, genetic ablation of those histone variants usually caused severe defect in sperm condensation and male sterility [81-85]. In condensing spermatids, Tnp1 is loaded onto the nucleosomes, promoting protamines deposition and processing, and removal of histone proteins. Mice lacking Tnp1/2 are sterile, presenting poor motility and irregular chromatin condensation [86, 87]. Inside the sperm nucleus, the chromatin undergoes global modifications and remodeling during condensation, of which the process, however, remains largely unknown. We thus envision that further attempts to investigate the molecular changes throughout nuclear condensation will likely provide insights into the molecular mechanisms underlying patients with teratozoospermia.
4.3.1. Acephalic sperm syndrome
One rare subtype but highly severe condition is decaudated sperm (acephalic sperm or headless sperm). This syndrome is most often caused by the defective formation of the head-tail coupling apparatus (Fig. 2). Acephalic spermatozoa syndrome has been reported in humans for decades and is characterized by semen often containing sperm flagella without heads [88]. They are headless tails with a bulbous drop of residual cytoplasm but sometimes they are analyzed erroneously as a globozoospermia-like condition. A few acephalic spermatozoa-related genes, such as Sun5, Pmfbp1, Spata6, and Spatc1l, have been reported. Ablation of these genes in mice led to the production of acephalic sperm [89-92]. Sun5 has been identified to localize on the sperm nuclear membrane facing the acrosome and to bind the acrosome, and SUN5 mutations are most often found in patients suffering from acephalic sperm syndrome so far [93].
Teratozoospermia is commonly a severe consequence of defective flagellar assembly or destructive function of flagella. As discussed above, both intramanchette transport (IMT) and intraflagellar transport (IFT) are functionally similar to each other and both are important for the assembly and maintenance of cilia during the tail formation. By IMT, target cargo proteins are transported from nucleus to cytosol, of which the process is essential not only for nuclear condensation but also for tail assembly [94]. This is exemplified by protein-coding genes involved in cargo transport, such as Kif31, Meig1, Spef2 and Stk33. Deletion of Kif31 and Meig1 caused a substantial lethal effect on manchette formation and sperm tail production, resulting in deformed head (disrupted structure of manchette) and tail (axoneme disorganization, failure of flagellar formation) [95, 96]. Spef2 and Stk33 KO mice exhibited a similar phenotype with the irregular formation of manchette and ultimately highly disorganized tail structure [97, 98].
4.3.2. Macrozoospermia
Macrozoospermia is a very rare syndrome found in less than 1% of infertile males. It can be described as the presence of an enlarged sperm head resulting from the failure of nucleus condensation (Fig. 3). Currently, AURKC is the only well-known gene that manifests this kind of phenotype resulting in male infertility. Two brothers underwent genetic study of the AURKC gene presenting a relatively large-headed sperm phenotype. Aurkc deficient sperm of mice showed defects including heterogeneous chromatin condensation, and blunt heads with loose acrosomes [99, 100].
4.3.3. Globozoospermia
Globozoospermia is a rare type of teratozoospermia that accounts for less than 0.1% of male infertility. Any disruption to the acrosome formation might result in malformation of the acrosome and the condition is characterized by the presence of a round-shaped sperm head as well as the lack of acrosome [74]. The complexity of acrosome biogenesis, which lasts about two weeks in mice and one month in humans, makes it susceptible to endogenous or exogenous insults that may be detrimental for male fertility. Animal studies have unveiled a few genes and their pathways related to globozoospermia in haploid spermatids (Table 1), including Dpy19l2 [101], Dnah6 [102], Pick1 [103], and Spata16 [104], that account for the genetic causes of globozoospermia. In particular, DPY19L2 variants appear to be responsible for globozoospermia in most patients [105].
As described earlier, the fusion of Golgi-derived vesicles culminates in acrosome formation, of which the process has been tightly regulated and involves numerous vital players. Pick1 (protein interacting with C kinase 1) facilitates the vesicle trafficking from Golgi complex to acrosomes through protein trafficking. By gold particle labeling, it has been shown to localize in Golgi-related vesicles in round spermatids. Gmap210 is a Golgi membrane receptor for Ift20 and is critical for Golgi apparatus assembly [106, 107]. In these gene knockout mouse models, ultrastructural analysis through TEM unveiled oblong head along with deformed acrosome, and abnormal formation of mitochondria sheath. Another gene, Hrb, encodes a protein related to nucleoporins essential for acrosome formation and deficient Hrb mice had round-headed spermatids and a complete lack of acrosome [108]. Dpy19l2 (a transmembrane protein found in the inner nuclear membrane), Vps54 (Golgi-associated retrograde protein complex from the endosome for tethering of vesicles to the trans-Golgi network), and Mfsd14a (transmembrane protein) are responsible for the detachment of acroplaxome from the nucleus. Dpy19l2 KO led to the intrusion of a layered structure present at the nuclear-acrosome junction, Vps54 deficient mice lack the true acrosome, and Mfsd14a-null mice failed to form an acrosome surround the nucleus [109-111].
Growing evidence showed that the autophagic molecular machinery, evolutionarily conserved from yeast to mammals, is involved in the transfer and fusion of Golgi-derived vesicles to the acrosome during acrosome biogenesis. The core members for autophagosome formation include ubiquitin-like conjugation proteins, Atg5/Atg7/Atg12 and microtubule-associated protein 1A/1B-light chain 3 (LC3) lipids. Atg7 is homologous to the ubiquitin-activating enzyme E1 essential for the conjugation structures [112, 113]. Atg7 mutant mice have aberrant round-headed spermatozoa with abnormal acrosome formation, which results from the failure of proacrosomal vesicle fusion and membrane trafficking [113, 114]. In round spermatids, Gopc partially colocalized with sp56 on the acrosome but failed to localize to the acrosome in spermatids deficient in Atg7 [115, 116]. Atg5 is also involved in the correct formation of acrosome, and upon knockout, Atg5-null sperm exhibit disorganized mitochondrion, misshapen heads and tails [114]. Interestingly, Sirt1 also appears to be involved in acrosome biogenesis via the autophagy pathway. The germline-specific Sirt1 KO mice have round-headed sperm with aberrant acrosome [117].
4.3.4. Multiple morphological abnormalities of the sperm flagella
The syndrome of multiple morphological abnormalities of the sperm flagella (MMAF) is considered as a specific subtype of asthenoteratozoospermia (Fig. 3) [52]. MMAF is characterized by mosaic anomalies of flagella involving coiled, twisted, irregular, shortened, or absent flagella [118, 119]. Other forms of asthenozoospermia, including MS defects, primary ciliary dyskinesia-related defects, ion channel defects and annulus instability should be distinguished from MMAF, and they are in general confined to the mid-piece that continuously displays mitochondrial ultrastructural changes or the lack of mitochondria [36]. Recent studies by WES screening have discovered a myriad of genetic variants as causes of MMAF patients, such as Dynein axonemal heavy chain family proteins (DNAH1 [120], DNAH2 [121], DNAH6 [102], DNAH8 [122] and DNAH17 [123], and Cilia and flagella associated protein family (CFAP43 and CFAP44 [124], CFAP58 [125], CFAP65 [126], CFAP69 [127], CFAP70 [128], CFAP74 [63] and CFAP251 [129]). Apart from these two families, many other genes are also disclosed through WES, particularly AK7 [130], ARMC2 [131], DZIP1 [132], FSIP2 [133], QRICH2 [134], SPEF2 [135], TTC21A [136], TTC29 [137] and WDR66 [138] (Table 1). We expect that more other genetic variants concerning MMAF would be uncovered owing to the declining cost and advance of high throughput sequencing. The genetic mutations and featured phenotypes of mouse models for genetic variants discovered in MMAF patients have been explicitly discussed in several recent review articles [36, 139].
4.4. Oligoasthenoteratozoospermia
Oligoasthenoteratozoospermia (OAT) is the most common but severe form of pathogenic condition of male infertility characterized by a combination of semen abnormalities as described above, including low sperm count, poor motility, and abnormal morphology [140]. Noteworthily, OAT syndrome patients have sometimes been categorized into other types of defects in the clinics. Recent studies in both mouse models and human patients have revealed abundant genetic variants that account for OAT. However, the causative genetic mutations underlying at least 50% of OAT patients remain elusive. As mentioned above, any defect taking place throughout the prolonged development of haploid spermatids is likely to elicit OAT (see Table 1).
4.4.1. Intraflagellar transport (IFT)
As partially described above, numerous genes involved in intraflagellar transport relating to flagellar assembly exert vital roles during sperm head reshaping and tail formation. As a result, genetic mutations and functional disruption will cause OAT syndrome. Katnb1 and Lrguk-1 are predominantly expressed in germ cells and important for depolarization of microtubules during sperm head and tail axoneme development. Genetic inactivation of both genes resulted in OAT phenotype due to manchette dysfunction and tail defects [141, 142]. Ift20 and Ift140 are localized to the Golgi complex and involved in cargo protein transport among Golgi complex, microtubules and flagella, whereby the excess residual cytosol is removed during flagellar assembly. There is no wonder that genetic disruption of Ift20 and Ift140 led to male infertility and OAT phenotype because of severe morphological defects in the sperm head and tails [143-145]. Likewise, IFT27 has been shown to bind IFT25 and is essential for flagellar formation even they are not required for ciliogenesis in somatic cells [146].
4.4.2. Acrosome formation
A handful of genes, such as Vps13b, GalNAc-T3, Orp4, and Hipk4, have been identified in the proper organization of Golgi apparatus and fusion of proacrosomal vesicles. Vps13b is involved in the sorting and transport of proacrosomal vesicles to the nuclear dense lamina in early spermatids. Oxysterol-binding protein (OSBP)-related protein 4 (Orp4) is a cytosolic receptor with a high affinity for oxysterols and transport cholesterol among intracellular organelles. Galnt3 encodes UDP-GalNAc polypeptide N-acetylgalactosaminyltransferase 3 that facilitates the fusion of proacrosomal vesicles. Hipk4 (homeodomain interacting protein kinase 4) functions as a critical regulator of the acrosome-acroplaxome complex. Genetic deletions of these genes all gave rise to OAT phenotype, mostly exhibiting fragmented and unfused acrosomes [147-149].
5. Prospects
Male germline development is a unique and sophisticated process in that it undergoes successive mitosis, meiosis and haploid differentiation. Abundant genes are spatiotemporally regulated and highly expressed in meiotic spermatocytes and round spermatids to guide correct spermatogenesis. As such, our current mechanistic understanding of male germline development is gained largely from mouse models. However, we must keep in mind that there are still limitations to using mouse models in that the germline in both species differs in many aspects, such as the different spermatogenic stem cell markers and differential chromatin condensation. In the clinics, while defective meiosis usually caused severe azoospermia, i.e., absence of sperm, any disturbance during the haploid stage most often caused either severe oligozoospermia, asthenozoospermia, teratozoospermia, or a combination (OAT). Luckily, the patients suffering from these kinds of syndromes can resolve to intracytoplasmic sperm injection (ICSI) approach, which is commonly adopted in the reproductive center and has been a powerful means to overcome fertility issues. However, we must bear in mind that the sperm in those patients with decreased fertility might be genetically defective, and thus in vitro assisted reproductive techniques might cause the defective DNA mutations to be carried over to the offspring. Additionally, due to the lack of the long-term health record of babies born through in vitro fertilization (IVF) techniques, it is still not well-known or controversial if IVF techniques themselves, e.g., the culture medium, ROS exposure in culture, choice of sperm, etc, could somehow have an unexpected impact on progeny development in the future. Thus far, whole-genome screening by next-generation sequencing (e.g., exome-sequencing) combined with mouse models, have been validated as an effective way to investigate the causative roles and mechanisms of genetic variants of interest, and hence the landscape of genetic variants yet to-be-identified will undergo rapid expanding. This will lead to improved diagnosis and treatment for patients suffering sterility. In conjugation with the pre-implantation genetic testing (PGT), we envision that the transmission of more deleterious genetic mutations causing congenital diseases will be blocked from one generation to the next, further reducing the birth defects and improving human health worldwide.
Acknowledgements
Funding
Ministry of Science and Technology of China (2019YFA0802600); National Natural Science Foundation of China (Grant Number: 31970793); The open project of NHC Key Laboratory of Male Reproduction and Genetics (No. KF201901); the Fundamental Research Funds for the Central Universities” (WK2070000156).
Author Contributions
M.A. drafted the initial manuscript. J.B. and W.Q. revised the manuscript with comments from S.A., I.U., J.C., L.W. and X.T.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lindsay TJ, Vitrikas KR. Evaluation and treatment of infertility. American family physician. 2015;91:308-14
2. Fertilisation H, Authority E. facts and figures 2006: fertility problems and treatment, October 2008. London: HEFA. 2010
3. Punab M, Poolamets O, Paju P, Vihljajev V, Pomm K, Ladva R. et al. Causes of male infertility: a 9-year prospective monocentre study on 1737 patients with reduced total sperm counts. Human reproduction. 2017;32:18-31
4. Cheng CY, Mruk DD. The biology of spermatogenesis: the past, present and future. The Royal Society. 2010
5. Waclawska A, Kurpisz M. Key functional genes of spermatogenesis identified by microarray analysis. Systems biology in reproductive medicine. 2012;58:229-35
6. Russell LD, Ettlin RA, Hikim APS, Clegg ED. Histological and histopathological evaluation of the testis. Wiley Online Library. 1993
7. Cannarella R, Condorelli RA, Mongioì LM, La Vignera S, Calogero AE. Molecular Biology of Spermatogenesis: Novel Targets of Apparently Idiopathic Male Infertility. Int J Mol Sci. 2020 21
8. Sharma U. Paternal contributions to offspring health: role of sperm small RNAs in intergenerational transmission of epigenetic information. Frontiers in cell and developmental biology. 2019;7:215
9. Muciaccia B, Boitani C, Berloco BP, Nudo F, Spadetta G, Stefanini M. et al. Novel stage classification of human spermatogenesis based on acrosome development. Biology of reproduction. 2013;89:60 1-10
10. Lehtiniemi T, Kotaja N. The Genetics of Postmeiotic Male Germ Cell Differentiation from Round Spermatids to Mature Sperm. Genetics of Human Infertility: Karger Publishers. 2017 p. 101-15
11. Abou-Haila A, Tulsiani DR. Mammalian sperm acrosome: formation, contents, and function. Archives of biochemistry and biophysics. 2000;379:173-82
12. Avidor-Reiss T, Carr A, Fishman EL. The sperm centrioles. Molecular and Cellular Endocrinology. 2020: 110987.
13. Khawar MB, Gao H, Li W. Mechanism of acrosome biogenesis in mammals. Frontiers in cell and developmental biology. 2019;7:195
14. Champroux A, Torres-Carreira J, Gharagozloo P, Drevet J, Kocer A. Mammalian sperm nuclear organization: resiliencies and vulnerabilities. Basic and clinical andrology. 2016;26:1-22
15. Cavé T, Desmarais R, Lacombe-Burgoyne C, Boissonneault G. Genetic Instability and Chromatin Remodeling in Spermatids. Genes. 2019;10:40
16. Gervasi MG, Visconti PE. Molecular changes and signaling events occurring in spermatozoa during epididymal maturation. Andrology. 2017;5:204-18
17. de Kretser DM, O'Donnell L. Phenotypic assessment of male fertility status in transgenic animal models. Spermatogenesis: Springer. 2013 p. 531-48
18. Kim K-S, Cha MC, Gerton GL. Mouse sperm protein sp56 is a component of the acrosomal matrix. Biology of reproduction. 2001;64:36-43
19. Ito C, Toshimori K. Acrosome markers of human sperm. Anatomical science international. 2016;91:128-42
20. Kierszenbaum AL, Tres LL. The acrosome-acroplaxome-manchette complex and the shaping of the spermatid head. Archives of histology and cytology. 2004;67:271-84
21. Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM. et al. PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development. 2002;129:3533-44
22. Kashir J, Nomikos M, Swann K, Lai FA. PLCζ or PAWP: revisiting the putative mammalian sperm factor that triggers egg activation and embryogenesis. Mhr: Basic science of reproductive medicine. 2015;21:383-8
23. Wong EW, Mruk DD, Lee WM, Cheng CY. Par3/Par6 polarity complex coordinates apical ectoplasmic specialization and blood-testis barrier restructuring during spermatogenesis. Proceedings of the National Academy of Sciences. 2008;105:9657-62
24. Kierszenbaum AL, Tres LL, Rivkin E, Kang-Decker N, Van Deursen JM. The acroplaxome is the docking site of Golgi-derived myosin Va/Rab27a/b-containing proacrosomal vesicles in wild-type and Hrb mutant mouse spermatids. Biology of reproduction. 2004;70:1400-10
25. Huang W-P, Ho H-C. Role of microtubule-dependent membrane trafficking in acrosomal biogenesis. Cell and tissue research. 2006;323:495-503
26. De Braekeleer M, Nguyen MH, Morel F, Perrin A. Genetic aspects of monomorphic teratozoospermia: a review. Journal of assisted reproduction and genetics. 2015;32:615-23
27. Moreno RD, Schatten G. Microtubule configurations and post-translational α-tubulin modifications during mammalian spermatogenesis. Cell motility and the cytoskeleton. 2000;46:235-46
28. O'Donnell L, O'Bryan MK. Microtubules and spermatogenesis. Seminars in cell & developmental biology: Elsevier. 2014 p. 45-54
29. Pasch E, Link J, Beck C, Scheuerle S, Alsheimer M. The LINC complex component Sun4 plays a crucial role in sperm head formation and fertility. Biology open. 2015;4:1792-802
30. Sperry AO. The dynamic cytoskeleton of the developing male germ cell. Biology of the Cell. 2012;104:297-305
31. Nozawa YI, Yao E, Gacayan R, Xu S-M, Chuang P-T. Mammalian Fused is essential for sperm head shaping and periaxonemal structure formation during spermatogenesis. Developmental biology. 2014;388:170-80
32. O'Donnell L, Rhodes D, Smith SJ, Merriner DJ, Clark BJ, Borg C. et al. An essential role for katanin p80 and microtubule severing in male gamete production. PLoS genetics. 2012 8
33. Ikegami K, Setou M. Unique post-translational modifications in specialized microtubule architecture. Cell structure and function. 2010: 1002190051-.
34. Lehti MS, Sironen A. Formation and function of sperm tail structures in association with sperm motility defects. Biology of reproduction. 2017;97:522-36
35. Puga Molina LC, Luque GM, Balestrini PA, Marín-Briggiler CI, Romarowski A, Buffone MG. Molecular basis of human sperm capacitation. Frontiers in cell and developmental biology. 2018;6:72
36. Wang W-L, Tu C-F, Tan Y-Q. Insight on multiple morphological abnormalities of sperm flagella in male infertility: what is new? Asian Journal of Andrology. 2020;22:236
37. Greenbaum MP, Iwamori T, Buchold GM, Matzuk MM. Germ cell intercellular bridges. Cold Spring Harbor perspectives in biology. 2011;3:a005850
38. Morales CR, Wu XQ, Hecht NB. The DNA/RNA-binding protein, TB-RBP, moves from the nucleus to the cytoplasm and through intercellular bridges in male germ cells. Developmental biology. 1998;201:113-23
39. Cooper TG, Noonan E, Von Eckardstein S, Auger J, Baker H, Behre HM. et al. World Health Organization reference values for human semen characteristics. Human reproduction update. 2010;16:231-45
40. Padubidri V, Daftary S. Disorders of menstruation. Shaw's Textbook of Gynaecology. 2011;14:256-7
41. Ornitz DM, Itoh N. Fibroblast growth factors. Genome biology. 2001;2:reviews3005. 1
42. Goldfarb M. Functions of fibroblast growth factors in vertebrate development. Cytokine & growth factor reviews. 1996;7:311-25
43. Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P. et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271:171-82
44. Powers C, McLeskey S, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocrine-related cancer. 2000;7:165-97
45. Cotton L, Gibbs GM, Sanchez-Partida LG, Morrison JR, de Kretser DM, O'Bryan MK. FGFR-1 signaling is involved in spermiogenesis and sperm capacitation. Journal of cell science. 2006;119:75-84
46. Li L, Sha Y-W, Su Z-Y, Mei L-B, Ji Z-Y, Zhang Q. et al. A novel mutation in HAUS7 results in severe oligozoospermia in two brothers. Gene. 2018;639:106-10
47. Tu C, Wang Y, Nie H, Meng L, Wang W, Li Y. et al. An M1AP homozygous splice-site mutation associated with severe oligozoospermia in a consanguineous family. Clinical Genetics. 2020;97:741-6
48. Okutman O, Muller J, Skory V, Garnier JM, Gaucherot A, Baert Y. et al. A no-stop mutation in MAGEB4 is a possible cause of rare X-linked azoospermia and oligozoospermia in a consanguineous Turkish family. Journal of assisted reproduction and genetics. 2017;34:683-94
49. Tu C, Meng L, Nie H, Yuan S, Wang W, Du J. et al. A homozygous RPL10L missense mutation associated with male factor infertility and severe oligozoospermia. Fertility and Sterility. 2020
50. Hu TY, Zhang H, Meng LL, Yuan SM, Tu CF, Du J. et al. Novel homozygous truncating variants in ZMYND15 causing severe oligozoospermia and their implications for male infertility. Human Mutation. 2020
51. Curi S, Ariagno J, Chenlo P, Mendeluk G, Pugliese N, Sardi S. et al. Asthenozoospermia: analysis of a large population. Journal of andrology. 2000;21:137 -
52. Coutton C, Escoffier J, Martinez G, Arnoult C, Ray PF. Teratozoospermia: spotlight on the main genetic actors in the human. Human reproduction update. 2015;21:455-85
53. Akbari A, Pipitone GB, Anvar Z, Jaafarinia M, Ferrari M, Carrera P. et al. ADCY10 frameshift variant leading to severe recessive asthenozoospermia and segregating with absorptive hypercalciuria. Human reproduction. 2019;34:1155-64
54. Visser L, Westerveld GH, Xie F, van Daalen SK, van der Veen F, Lombardi MP. et al. A comprehensive gene mutation screen in men with asthenozoospermia. Fertility and sterility. 2011;95:1020-4 e9
55. Sha Y, Liu W, Huang X, Li Y, Ji Z, Mei L. et al. EIF4G1 is a novel candidate gene associated with severe asthenozoospermia. Molecular genetics & genomic medicine. 2019;7:e807
56. Hagiuda J, Takasaki N, Oya M, Ishikawa H, Narimatsu H. Mutation of GALNTL5 gene identified in patients diagnosed with asthenozoospermia. Human Fertility. 2019:1-8
57. Ren H, Zhong R, Ding X, Chen Z, Jing Y. Investigation of polymorphisms in exon7 of the NSUN7 gene among Chinese Han men with asthenospermia. Genetics and Molecular Research. 2015;14:9261-8
58. Xu X, Sha YW, Mei LB, Ji ZY, Qiu Pp, Ji H. et al. A familial study of twins with severe asthenozoospermia identified a homozygous SPAG17 mutation by whole-exome sequencing. Clinical genetics. 2018;93:345-9
59. Zhu G, Xie C, Yang Z, Wang Y, Chen D, Wang X. Expression of TRPC5 is decreased in the sperm of patients with varicocele-associated asthenozoospermia. Biomedical reports. 2018;8:529-34
60. Porter ME, Sale WS. The 9+ 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. The Journal of cell biology. 2000;151:F37-F42
61. Whitfield M, Thomas L, Bequignon E, Schmitt A, Stouvenel L, Montantin G. et al. Mutations in DNAH17, encoding a sperm-specific axonemal outer dynein arm heavy chain, cause isolated male infertility due to asthenozoospermia. The American Journal of Human Genetics. 2019;105:198-212
62. Li R-K, Tan J-L, Chen L-T, Feng J-S, Liang W-X, Guo X-J. et al. Iqcg is essential for sperm flagellum formation in mice. PLoS One. 2014 9
63. Sha Y, Wei X, Ding L, Ji Z, Mei L, Huang X. et al. Biallelic mutations of CFAP74 may cause human primary ciliary dyskinesia and MMAF phenotype. Journal of Human Genetics. 2020;65:961-9
64. Tanaka H, Iguchi N, Toyama Y, Kitamura K, Takahashi T, Kaseda K. et al. Mice deficient in the axonemal protein Tektin-t exhibit male infertility and immotile-cilium syndrome due to impaired inner arm dynein function. Molecular and cellular biology. 2004;24:7958-64
65. Roy A, Lin Y-N, Agno JE, DeMayo FJ, Matzuk MM. Absence of tektin 4 causes asthenozoospermia and subfertility in male mice. The FASEB Journal. 2007;21:1013-25
66. Zuccarello D, Ferlin A, Garolla A, Pati MA, Moretti A, Cazzadore C. et al. A possible association of a human tektin-t gene mutation (A229V) with isolated non-syndromic asthenozoospermia: case report. Human reproduction. 2008;23:996-1001
67. Roy A, Lin YN, Agno JE, DeMayo FJ, Matzuk MM. Tektin 3 is required for progressive sperm motility in mice. Molecular Reproduction and Development: Incorporating Gamete Research. 2009;76:453-9
68. Buffone MG, Wertheimer EV, Visconti PE, Krapf D. Central role of soluble adenylyl cyclase and cAMP in sperm physiology. Biochimica et biophysica acta. 2014;1842:2610-20
69. Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM. et al. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proceedings of the National Academy of Sciences. 2004;101:2993-8
70. Yanagimachi R. Fertility of mammalian spermatozoa: its development and relativity. Zygote. 1994;2:371-2
71. Quill TA, Sugden SA, Rossi KL, Doolittle LK, Hammer RE, Garbers DL. Hyperactivated sperm motility driven by CatSper2 is required for fertilization. Proceedings of the National Academy of Sciences. 2003;100:14869-74
72. Carlson AE, Quill TA, Westenbroek RE, Schuh SM, Hille B, Babcock DF. Identical phenotypes of CatSper1 and CatSper2 null sperm. Journal of Biological Chemistry. 2005;280:32238-44
73. Jin J, Jin N, Zheng H, Ro S, Tafolla D, Sanders KM. et al. Catsper3 and Catsper4 are essential for sperm hyperactivated motility and male fertility in the mouse. Biology of reproduction. 2007;77:37-44
74. Yan W. Male infertility caused by spermiogenic defects: lessons from gene knockouts. Molecular and cellular endocrinology. 2009;306:24-32
75. WELCH JE, BROWN PL, O'BRIEN DA, MAGYAR PL, BUNCH DO, MORI C. et al. Human glyceraldehyde 3-phosphate dehydrogenase-2 gene is expressed specifically in spermatogenic cells. Journal of andrology. 2000;21:328-38
76. Miki K, Qu W, Goulding EH, Willis WD, Bunch DO, Strader LF. et al. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proceedings of the National Academy of Sciences. 2004;101:16501-6
77. Elkina Y, Kuravsky M, Bragina E, Kurilo L, Khayat S, Sukhomlinova M. et al. Detection of a mutation in the intron of Sperm-specific glyceraldehyde-3-phosphate dehydrogenase gene in patients with fibrous sheath dysplasia of the sperm flagellum. Andrologia. 2017;49:e12606
78. Sampson MJ, Decker WK, Beaudet AL, Ruitenbeek W, Armstrong D, Hicks MJ. et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. Journal of Biological Chemistry. 2001;276:39206-12
79. Perrin A, Morel F, Moy L, Colleu D, Amice V, De Braekeleer M. Study of aneuploidy in large-headed, multiple-tailed spermatozoa: case report and review of the literature. Fertility and sterility. 2008;90:1201. e13-. e17
80. Perrin A, Coat C, Nguyen M, Talagas M, Morel F, Amice J. et al. Molecular cytogenetic and genetic aspects of globozoospermia: a review. Andrologia. 2013;45:1-9
81. Wang T, Gao H, Li W, Liu C. Essential Role of Histone Replacement and Modifications in Male Fertility. Frontiers in Genetics. 2019 10
82. Tanaka H, Iguchi N, Isotani A, Kitamura K, Toyama Y, Matsuoka Y. et al. HANP1/H1T2, a novel histone H1-like protein involved in nuclear formation and sperm fertility. Molecular and cellular biology. 2005;25:7107-19
83. Yan W, Ma L, Burns KH, Matzuk MM. HILS1 is a spermatid-specific linker histone H1-like protein implicated in chromatin remodeling during mammalian spermiogenesis. Proc Natl Acad Sci U S A. 2003;100:10546-51
84. Boissonneault G, Lau YF. A testis-specific gene encoding a nuclear high-mobility-group box protein located in elongating spermatids. Mol Cell Biol. 1993;13:4323-30
85. Moss SB, Challoner PB, Groudine M. Expression of a novel histone 2B during mouse spermiogenesis. Dev Biol. 1989;133:83-92
86. Yu YE, Zhang Y, Unni E, Shirley CR, Deng JM, Russell LD. et al. Abnormal spermatogenesis and reduced fertility in transition nuclear protein 1-deficient mice. Proceedings of the National Academy of Sciences. 2000;97:4683-8
87. Adham IM, Nayernia K, Burkhardt-Göttges E, Topaloglu Ö, Dixkens C, Holstein AF. et al. Teratozoospermia in mice lacking the transition protein 2 (Tnp2). Molecular Human Reproduction. 2001;7:513-20
88. Perotti M, Gioria M. Fine structure and morphogenesis of" headless" human spermatozoa associated with infertility. Cell biology international reports. 1981;5:113 -
89. Shang Y, Zhu F, Wang L, Ouyang Y-C, Dong M-Z, Liu C. et al. Essential role for SUN5 in anchoring sperm head to the tail. elife. 2017;6:e28199
90. Zhu F, Liu C, Wang F, Yang X, Zhang J, Wu H. et al. Mutations in PMFBP1 cause acephalic spermatozoa syndrome. The American Journal of Human Genetics. 2018;103:188-99
91. Kim J, Kwon JT, Jeong J, Kim J, Hong SH, Kim J. et al. SPATC 1L maintains the integrity of the sperm head-tail junction. EMBO reports. 2018;19:e45991
92. Yuan S, Stratton CJ, Bao J, Zheng H, Bhetwal BP, Yanagimachi R. et al. Spata6 is required for normal assembly of the sperm connecting piece and tight head-tail conjunction. Proceedings of the National Academy of Sciences. 2015;112:E430-E9
93. Zhu F, Wang F, Yang X, Zhang J, Wu H, Zhang Z. et al. Biallelic SUN5 mutations cause autosomal-recessive acephalic spermatozoa syndrome. The American Journal of Human Genetics. 2016;99:942-9
94. Kierszenbaum AL, Rivkin E, Tres LL. Cytoskeletal track selection during cargo transport in spermatids is relevant to male fertility. Spermatogenesis. 2011;1:221-30
95. Lehti MS, Kotaja N, Sironen A. KIF3A is essential for sperm tail formation and manchette function. Molecular and cellular endocrinology. 2013;377:44-55
96. Zhang Z, Shen X, Gude DR, Wilkinson BM, Justice MJ, Flickinger CJ. et al. MEIG1 is essential for spermiogenesis in mice. Proceedings of the National Academy of Sciences. 2009;106:17055-60
97. Martins LR, Bung RK, Koch S, Richter K, Schwarzmüller L, Terhardt D. et al. Stk33 is required for spermatid differentiation and male fertility in mice. Developmental biology. 2018;433:84-93
98. Lehti MS, Zhang F-P, Kotaja N, Sironen A. SPEF2 functions in microtubule-mediated transport in elongating spermatids to ensure proper male germ cell differentiation. Development. 2017;144:2683-93
99. Dieterich K, Rifo RS, Faure AK, Hennebicq S, Amar BB, Zahi M. et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nature genetics. 2007;39:661-5
100. Kimmins S, Crosio C, Kotaja N, Hirayama J, Monaco L, Hoog C. et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Molecular endocrinology. 2007;21:726-39
101. Shang Y-L, Zhu F-X, Yan J, Chen L, Tang W-H, Xiao S. et al. Novel DPY19L2 variants in globozoospermic patients and the overcoming this male infertility. Asian journal of andrology. 2019;21:183
102. Li L, Sha YW, Xu X, Mei LB, Qiu PP, Ji ZY. et al. DNAH 6 is a novel candidate gene associated with sperm head anomaly. Andrologia. 2018;50:e12953
103. Xiao N, Kam C, Shen C, Jin W, Wang J, Lee KM. et al. PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. The Journal of clinical investigation. 2009;119:802-12
104. Dam AH, Koscinski I, Kremer JA, Moutou C, Jaeger A-S, Oudakker AR. et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. The American Journal of Human Genetics. 2007;81:813-20
105. Celse T, Cazin C, Mietton F, Martinez G, Martinez D, Thierry-Mieg N. et al. Genetic analyses of a large cohort of infertile patients with globozoospermia, DPY19L2 still the main actor, GGN confirmed as a guest player. Human Genetics. 2020:1-15
106. Follit JA, San Agustin JT, Xu F, Jonassen JA, Samtani R, Lo CW. et al. The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS genetics. 2008 4
107. Kierszenbaum AL, Rivkin E, Tres LL, Yoder BK, Haycraft CJ, Bornens M. et al. GMAP210 and IFT88 are present in the spermatid golgi apparatus and participate in the development of the acrosome-acroplaxome complex, head-tail coupling apparatus and tail. Developmental Dynamics. 2011;240:723-36
108. Kang-Decker N, Mantchev GT, Juneja SC, McNiven MA, van Deursen JM. Lack of acrosome formation in Hrb-deficient mice. Science. 2001;294:1531-3
109. Doran J, Walters C, Kyle V, Wooding P, Hammett-Burke R, Colledge B. Mfsd14a (Hiat1) gene disruption causes globozoospermia and infertility in male mice. 2016.
110. Paiardi C, Pasini ME, Gioria M, Berruti G. Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis. 2011;1:52-62
111. Pierre V, Martinez G, Coutton C, Delaroche J, Yassine S, Novella C. et al. Absence of Dpy19l2, a new inner nuclear membrane protein, causes globozoospermia in mice by preventing the anchoring of the acrosome to the nucleus. Development. 2012;139:2955-65
112. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nature cell biology. 2007;9:1102-9
113. Wang H, Wan H, Li X, Liu W, Chen Q, Wang Y. et al. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res. 2014;24:852-69
114. Huang Q, Liu Y, Zhang S, Yap YT, Li W, Zhang D. et al. Autophagy core protein ATG5 is required for elongating spermatid development, sperm individualization and normal fertility in male mice. Autophagy. 2020:1-15
115. Ito C, Suzuki-Toyota F, Maekawa M, Toyama Y, Yao R, Noda T. et al. Failure to assemble the peri-nuclear structures in GOPC deficient spermatids as found in round-headed spermatozoa. Archives of histology and cytology. 2004;67:349-60
116. Wang H, Wan H, Li X, Liu W, Chen Q, Wang Y. et al. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell research. 2014;24:852-69
117. Liu C, Song Z, Wang L, Yu H, Liu W, Shang Y. et al. Sirt1 regulates acrosome biogenesis by modulating autophagic flux during spermiogenesis in mice. Development. 2017;144:441-51
118. Amiri-Yekta A, Coutton C, Kherraf Z-E, Karaouzène T, Le Tanno P, Sanati MH. et al. Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations. Human Reproduction. 2016;31:2872-80
119. Shen Y, Zhang F, Li F, Jiang X, Yang Y, Li X. et al. Loss-of-function mutations in QRICH2 cause male infertility with multiple morphological abnormalities of the sperm flagella. Nature communications. 2019;10:1-15
120. Khelifa MB, Coutton C, Zouari R, Karaouzène T, Rendu J, Bidart M. et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. The American Journal of Human Genetics. 2014;94:95-104
121. Li Y, Sha Y, Wang X, Ding L, Liu W, Ji Z. et al. DNAH2 is a novel candidate gene associated with multiple morphological abnormalities of the sperm flagella. Clinical genetics. 2019;95:590-600
122. Liu C, Miyata H, Gao Y, Sha Y, Tang S, Xu Z. et al. Bi-allelic DNAH8 variants lead to multiple morphological abnormalities of the sperm flagella and primary male infertility. The American Journal of Human Genetics. 2020;107:330-41
123. Zhang B, Khan I, Liu C, Ma A, Khan A, Zhang Y. et al. Novel loss-of-function variants in DNAH17 cause multiple morphological abnormalities of the sperm flagella in humans and mice. Clinical Genetics. 2021;99:176-86
124. Tang S, Wang X, Li W, Yang X, Li Z, Liu W. et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. The American Journal of Human Genetics. 2017;100:854-64
125. Sha Y, Sha Y, Liu W, Zhu X, Weng M, Zhang X. et al. Biallelic mutations of CFAP58 are associated with multiple morphological abnormalities of the sperm flagella. Clinical Genetics. 2020
126. Zhang X, Shen Y, Wang X, Yuan G, Zhang C, Yang Y. A novel homozygous CFAP65 mutation in humans causes male infertility with multiple morphological abnormalities of the sperm flagella. Clinical Genetics. 2019;96:541-8
127. Dong FN, Amiri-Yekta A, Martinez G, Saut A, Tek J, Stouvenel L. et al. Absence of CFAP69 causes male infertility due to multiple morphological abnormalities of the flagella in human and mouse. The American Journal of Human Genetics. 2018;102:636-48
128. Beurois J, Martinez G, Cazin C, Kherraf Z-E, Amiri-Yekta A, Thierry-Mieg N. et al. CFAP70 mutations lead to male infertility due to severe astheno-teratozoospermia. A case report. Human Reproduction. 2019;34:2071-9
129. Li W, He X, Yang S, Liu C, Wu H, Liu W. et al. Biallelic mutations of CFAP251 cause sperm flagellar defects and human male infertility. Journal of human genetics. 2019;64:49-54
130. Lorès P, Coutton C, El Khouri E, Stouvenel L, Givelet M, Thomas L. et al. Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia. Human Molecular Genetics. 2018;27:1196-211
131. Coutton C, Martinez G, Kherraf Z-E, Amiri-Yekta A, Boguenet M, Saut A. et al. Bi-allelic mutations in ARMC2 lead to severe astheno-teratozoospermia due to sperm flagellum malformations in humans and mice. The American Journal of Human Genetics. 2019;104:331-40
132. Lv M, Liu W, Chi W, Ni X, Wang J, Cheng H. et al. Homozygous mutations in DZIP1 can induce asthenoteratospermia with severe MMAF. Journal of Medical Genetics. 2020
133. Martinez G, Kherraf Z-E, Zouari R, Fourati Ben Mustapha S, Saut A, Pernet-Gallay K. et al. Whole-exome sequencing identifies mutations in FSIP2 as a recurrent cause of multiple morphological abnormalities of the sperm flagella. Human Reproduction. 2018;33:1973-84
134. Kherraf ZE, Cazin C, Coutton C, Amiri-Yekta A, Martinez G, Boguenet M. et al. Whole exome sequencing of men with multiple morphological abnormalities of the sperm flagella reveals novel homozygous QRICH2 mutations. Clinical genetics. 2019;96:394-401
135. Liu W, Sha Y, Li Y, Mei L, Lin S, Huang X. et al. Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). Journal of Medical Genetics. 2019;56:678-84
136. Liu W, He X, Yang S, Zouari R, Wang J, Wu H. et al. Bi-allelic mutations in TTC21A induce asthenoteratospermia in humans and mice. The American Journal of Human Genetics. 2019;104:738-48
137. Liu C, He X, Liu W, Yang S, Wang L, Li W. et al. Bi-allelic mutations in TTC29 cause male subfertility with asthenoteratospermia in humans and mice. The American Journal of Human Genetics. 2019;105:1168-81
138. Kherraf Z-E, Amiri-Yekta A, Dacheux D, Karaouzène T, Coutton C, Christou-Kent M. et al. A homozygous ancestral SVA-insertion-mediated deletion in WDR66 induces multiple morphological abnormalities of the sperm flagellum and male infertility. The American Journal of Human Genetics. 2018;103:400-12
139. Jiao S-Y, Yang Y-H, Chen S-R. Molecular genetics of infertility: loss-of-function mutations in humans and corresponding knockout/mutated mice. Human Reproduction Update. 2021;27:154-89
140. Tüttelmann F, Ruckert C, Röpke A. Disorders of spermatogenesis. medizinische genetik. 2018;30:12-20
141. Liu Y, DeBoer K, de Kretser DM, O'Donnell L, O'Connor AE, Merriner DJ. et al. LRGUK-1 is required for basal body and manchette function during spermatogenesis and male fertility. PLoS Genet. 2015;11:e1005090
142. Pleuger C, Fietz D, Hartmann K, Weidner W, Kliesch S, O'Bryan MK. et al. Expression of katanin p80 in human spermatogenesis. Fertility and Sterility. 2016;106:1683-90 e1
143. Wang X, Sha Yw, Wang Wt, Cui Yq, Chen J, Yan W. et al. Novel IFT140 variants cause spermatogenic dysfunction in humans. Molecular Genetics & Genomic Medicine. 2019;7:e920
144. Zhang Y, Liu H, Li W, Zhang Z, Zhang S, Teves ME. et al. Intraflagellar transporter protein 140 (IFT140), a component of IFT-A complex, is essential for male fertility and spermiogenesis in mice. Cytoskeleton. 2018;75:70-84
145. Zhang Z, Li W, Zhang Y, Zhang L, Teves ME, Liu H. et al. Intraflagellar transport protein IFT20 is essential for male fertility and spermiogenesis in mice. Molecular biology of the cell. 2016;27:3705-16
146. Zhang Y, Liu H, Li W, Zhang Z, Shang X, Zhang D. et al. Intraflagellar transporter protein (IFT27), an IFT25 binding partner, is essential for male fertility and spermiogenesis in mice. Developmental biology. 2017;432:125-39
147. Charman M, Colbourne TR, Pietrangelo A, Kreplak L, Ridgway ND. Oxysterol-binding protein (OSBP)-related protein 4 (ORP4) is essential for cell proliferation and survival. Journal of Biological Chemistry. 2014;289:15705-17
148. Crapster JA, Rack PG, Hellmann ZJ, Elias JE, Perrino JJ, Behr B. et al. HIPK4 is essential for murine spermiogenesis. bioRxiv. 2019: 703637.
149. Miyazaki T, Mori M, Yoshida CA, Ito C, Yamatoya K, Moriishi T. et al. Galnt3 deficiency disrupts acrosome formation and leads to oligoasthenoteratozoospermia. Histochemistry and cell biology. 2013;139:339-54
150. Wu B, Gao H, Liu C, Li W. The coupling apparatus of the sperm head and tail. Biology of Reproduction. 2020;102:988-98
151. Guo T, Tu C-F, Yang D-H, Ding S-Z, Lei C, Wang R-C. et al. Bi-allelic BRWD1 variants cause male infertility with asthenoteratozoospermia and likely primary ciliary dyskinesia. Human Genetics. 2021:1-13
152. Philipps DL, Wigglesworth K, Hartford SA, Sun F, Pattabiraman S, Schimenti K. et al. The dual bromodomain and WD repeat-containing mouse protein BRWD1 is required for normal spermiogenesis and the oocyte-embryo transition. Developmental biology. 2008;317:72-82
153. Bao J, Rousseaux S, Shen J, Lin K, Lu Y, Bedford MT. The arginine methyltransferase CARM1 represses p300• ACT• CREMτ activity and is required for spermiogenesis. Nucleic acids research. 2018;46:4327-43
154. Li Y, Li C, Lin S, Yang B, Huang W, Wu H. et al. A nonsense mutation in Ccdc62 gene is responsible for spermiogenesis defects and male infertility in repro29/repro29 mice. Biology of Reproduction. 2017;96:587-97
155. Kimura T, Ito C, Watanabe S, Takahashi T, Ikawa M, Yomogida K. et al. Mouse germ cell-less as an essential component for nuclear integrity. Molecular and cellular biology. 2003;23:1304-15
156. O'Donnell L, McLachlan RI, Merriner DJ, O'Bryan MK, Jamsai D. KATNB 1 in the human testis and its genetic variants in fertile and oligoasthenoteratozoospermic infertile men. Andrology. 2014;2:884-91
157. Qian X, Wang L, Zheng B, Shi Z-M, Ge X, Jiang C-F. et al. Deficiency of Mkrn2 causes abnormal spermiogenesis and spermiation, and impairs male fertility. Scientific reports. 2016;6:39318
158. Kissel H, Georgescu M-M, Larisch S, Manova K, Hunnicutt GR, Steller H. The Sept4 septin locus is required for sperm terminal differentiation in mice. Developmental cell. 2005;8:353-64
159. Lin Y-H, Lin Y-M, Wang Y-Y, Yu I-S, Lin Y-W, Wang Y-H. et al. The expression level of septin12 is critical for spermiogenesis. The American journal of pathology. 2009;174:1857-68
160. Kuo YC, Lin YH, Chen HI, Wang YY, Chiou YW, Lin HH. et al. SEPT12 mutations cause male infertility with defective sperm annulus. Human mutation. 2012;33:710-9
161. Lee B, Park I, Jin S, Choi H, Kwon JT, Kim J. et al. Impaired spermatogenesis and fertility in mice carrying a mutation in the Spink2 gene expressed predominantly in testes. Journal of Biological Chemistry. 2011;286:29108-17
162. Cheng Y, Buffone MG, Kouadio M, Goodheart M, Page DC, Gerton GL. et al. Abnormal sperm in mice lacking the Taf7l gene. Molecular and cellular biology. 2007;27:2582-9
163. Da Costa R, Bordessoules M, Guilleman M, Carmignac V, Lhussiez V, Courot H. et al. Vps13b is required for acrosome biogenesis through functions in Golgi dynamic and membrane trafficking. Cellular and Molecular Life Sciences. 2020;77:511-29
164. Martianov I, Brancorsini S, Catena R, Gansmuller A, Kotaja N, Parvinen M. et al. Polar nuclear localization of H1T2, a histone H1 variant, required for spermatid elongation and DNA condensation during spermiogenesis. Proceedings of the National Academy of Sciences. 2005;102:2808-13
165. Miki K, Willis WD, Brown PR, Goulding EH, Fulcher KD, Eddy EM. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Developmental biology. 2002;248:331-42
166. Baccetti B, Collodel G, Estenoz M, Manca D, Moretti E, Piomboni P. Gene deletions in an infertile man with sperm fibrous sheath dysplasia. Human Reproduction. 2005;20:2790-4
167. Avenarius MR, Hildebrand MS, Zhang Y, Meyer NC, Smith LL, Kahrizi K. et al. Human male infertility caused by mutations in the CATSPER1 channel protein. The American Journal of Human Genetics. 2009;84:505-10
168. Takasaki N, Tachibana K, Ogasawara S, Matsuzaki H, Hagiuda J, Ishikawa H. et al. A heterozygous mutation of GALNTL5 affects male infertility with impairment of sperm motility. Proceedings of the National Academy of Sciences. 2014;111:1120-5
169. Harris TP, Schimenti KJ, Munroe RJ, Schimenti JC. IQ motif-containing G (Iqcg) is required for mouse spermiogenesis. G3: Genes, Genomes, Genetics. 2014;4:367-72
170. Woolley DM, Neesen J, Vernon GG. Further studies on knockout mice lacking a functional dynein heavy chain (MDHC7). 2. A developmental explanation for the asthenozoospermia. Cell motility and the cytoskeleton. 2005;61:74-82
171. Kazarian E, Son H, Sapao P, Li W, Zhang Z, Strauss III JF. et al. SPAG17 is required for male germ cell differentiation and fertility. International journal of molecular sciences. 2018;19:1252
172. Miyagawa Y, Nishimura H, Tsujimura A, Matsuoka Y, Matsumiya K, Okuyama A. et al. Single-nucleotide polymorphisms and mutation analyses of the TNP1 and TNP2 genes of fertile and infertile human male populations. Journal of andrology. 2005;26:779-86
173. Meistrich ML, Mohapatra B, Shirley CR, Zhao M. Roles of transition nuclear proteins in spermiogenesis. Chromosoma. 2003;111:483-8
174. Nuraini T, Sumarsih T, Paramita R, Saleh MI, Narita V, Moeloek N. et al. Mutations in exons 5, 7 and 8 of the human voltage-dependent anion channel type 3 (VDAC3) gene in sperm with low motility. Andrologia. 2012;44:46-52
175. Sapiro R, Kostetskii I, Olds-Clarke P, Gerton GL, Radice GL, Strauss III JF. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Molecular and cellular biology. 2002;22:6298-305
176. Qi Y, Jiang M, Yuan Y, Bi Y, Zheng B, Guo X. et al. ADP-ribosylation factor-like 3, a manchette-associated protein, is essential for mouse spermiogenesis. Molecular human reproduction. 2013;19:327-35
177. Hall EA, Keighren M, Ford MJ, Davey T, Jarman AP, Smith LB. et al. Acute versus chronic loss of mammalian Azi1/Cep131 results in distinct ciliary phenotypes. PLoS genetics. 2013 9
178. Zhuang T, Hess RA, Kolla V, Higashi M, Raabe TD, Brodeur GM. CHD5 is required for spermiogenesis and chromatin condensation. Mechanisms of development. 2014;131:35-46
179. Peyser A, Bristow S, Puig O, Pollock A, Mills A, Niknazar M. et al. Do mutations in CHD5 cause male infertility? Fertility and Sterility. 2017;108:e140-e1
180. Akhmanova A, Mausset-Bonnefont A-L, van Cappellen W, Keijzer N, Hoogenraad CC, Stepanova T. et al. The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes & development. 2005;19:2501-15
181. Komada M, McLean DJ, Griswold MD, Russell LD, Soriano P. E-MAP-115, encoding a microtubule-associated protein, is a retinoic acid-inducible gene required for spermatogenesis. Genes & development. 2000;14:1332-42
182. Liška F, Gosele C, Rivkin E, Tres L, Cardoso MC, Domaing P. et al. Rat hd mutation reveals an essential role of centrobin in spermatid head shaping and assembly of the head-tail coupling apparatus. Biology of reproduction. 2009;81:1196-205
183. Mochida K, Tres LL, Kierszenbaum AL. Structural and biochemical features of fractionated spermatid manchettes and sperm axonemes of the azh/azh mutant mouse. Molecular reproduction and development. 1999;52:434-44
184. Chen H, Zhu Y, Zhu Z, Zhi E, Lu K, Wang X. et al. Detection of heterozygous mutation in hook microtubule-tethering protein 1 in three patients with decapitated and decaudated spermatozoa syndrome. Journal of medical genetics. 2018;55:150-7
185. Zhou J, Du Y-R, Qin W-H, Hu Y-G, Huang Y-N, Bao L. et al. RIM-BP3 is a manchette-associated protein essential for spermiogenesis. Development. 2009;136:373-82
186. Zheng H, Stratton CJ, Morozumi K, Jin J, Yanagimachi R, Yan W. Lack of Spem1 causes aberrant cytoplasm removal, sperm deformation, and male infertility. Proceedings of the National Academy of Sciences. 2007;104:6852-7
187. Suryavathi V, Khattri A, Gopal K, Rani DS, Panneerdoss S, Gupta NJ. et al. Novel variants in UBE2B gene and idiopathic male infertility. Journal of andrology. 2008;29:564-71
188. Escalier D, Bai XY, Silvius D, Xu PX, Xu X. Spermatid nuclear and sperm periaxonemal anomalies in the mouse Ube2b null mutant. Molecular Reproduction and Development: Incorporating Gamete Research. 2003;65:298-308
189. Coutton C, Vargas AS, Amiri-Yekta A, Kherraf Z-E, Mustapha SFB, Le Tanno P. et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nature communications. 2018;9:1-18
190. Li W, Wu H, Li F, Tian S, Kherraf Z-E, Zhang J. et al. Biallelic mutations in CFAP65 cause male infertility with multiple morphological abnormalities of the sperm flagella in humans and mice. Journal of Medical Genetics. 2020;57:89-95
191. Xu X, Toselli PA, Russell LD, Seldin DC. Globozoospermia in mice lacking the casein kinase II α′ catalytic subunit. Nature genetics. 1999;23:118-21
192. Yildiz Y, Matern H, Thompson B, Allegood JC, Warren RL, Ramirez DM. et al. Mutation of β-glucosidase 2 causes glycolipid storage disease and impaired male fertility. The Journal of clinical investigation. 2006;116:2985-94
193. Han F, Liu C, Zhang L, Chen M, Zhou Y, Qin Y. et al. Globozoospermia and lack of acrosome formation in GM130-deficient mice. Cell death & disease. 2018;8:e2532-e
194. Audouard C, Christians E. Hsp90b1 knockout targeted to male germline: a mouse model for globozoospermia. Fertility and sterility. 2011;95:1475-7 e4
195. Liu G, Shi Q-W, Lu G-X. A newly discovered mutation in PICK1 in a human with globozoospermia. Asian journal of andrology. 2010;12:556
196. Fujihara Y, Satouh Y, Inoue N, Isotani A, Ikawa M, Okabe M. SPACA1-deficient male mice are infertile with abnormally shaped sperm heads reminiscent of globozoospermia. Development. 2012;139:3583-9
197. Gao Q, Khan R, Yu C, Alsheimer M, Jiang X, Ma H. et al. The testis specific LINC component SUN3 is essential for sperm head shaping during mouse spermiogenesis. Journal of Biological Chemistry. 2020: jbc. RA119. 012375.
198. Khosronezhad N, Colagar AH, Mortazavi SM. The Nsun7 (A11337)-deletion mutation, causes reduction of its protein rate and associated with sperm motility defect in infertile men. Journal of assisted reproduction and genetics. 2015;32:807-15
199. Yatsenko AN, O'Neil DS, Roy A, Arias-Mendoza PA, Chen R, Murthy LJ. et al. Association of mutations in the zona pellucida binding protein 1 (ZPBP1) gene with abnormal sperm head morphology in infertile men. Molecular human reproduction. 2012;18:14-21
200. Lin Y-N, Roy A, Yan W, Burns KH, Matzuk MM. Loss of zona pellucida binding proteins in the acrosomal matrix disrupts acrosome biogenesis and sperm morphogenesis. Molecular and cellular biology. 2007;27:6794-805
Author contact
![]() Corresponding authors: Jianqiang Bao, Division of Life Sciences and Medicine, The First Affiliated Hospital of USTC, University of Science and Technology of China, Anhui, China. Email: jqbaoedu.cn; https://orcid.org/0000-0003-1248-2687. Or Weibing Qin, NHC Key Laboratory of Male Reproduction and Genetics, Family Planning Research Institute of Guangdong Province, Guangdong, China. Email: guardqincom
Corresponding authors: Jianqiang Bao, Division of Life Sciences and Medicine, The First Affiliated Hospital of USTC, University of Science and Technology of China, Anhui, China. Email: jqbaoedu.cn; https://orcid.org/0000-0003-1248-2687. Or Weibing Qin, NHC Key Laboratory of Male Reproduction and Genetics, Family Planning Research Institute of Guangdong Province, Guangdong, China. Email: guardqincom