Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Microorganisms and pancreatic...
Viruses and pancreatic cancer...
Bacteria and pancreatic cancer...
Prospects for use of...
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(10):2666-2682. doi:10.7150/ijbs.59117 This issue Cite
Review
Microorganisms in chemotherapy for pancreatic cancer: An overview of current research and future directions
Si-Yuan Lu1,2,3,4*, Jie Hua1,2,3,4*, Jin Xu1,2,3,4*, Miao-Yan Wei1,2,3,4, Chen Liang1,2,3,4, Qing-Cai Meng1,2,3,4, Jiang Liu1,2,3,4, Bo Zhang1,2,3,4, Wei Wang1,2,3,4, Xian-Jun Yu1,2,3,4 ![]() , Si Shi1,2,3,4
, Si Shi1,2,3,4 ![]()
1. Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai, China.
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China.
3. Shanghai Pancreatic Cancer Institute, Shanghai, China.
4. Pancreatic Cancer Institute, Fudan University, Shanghai, China.
*These authors contributed equally to this work.
Received 2021-2-6; Accepted 2021-6-8; Published 2021-6-26
Abstract

Pancreatic cancer is a malignant tumor of the digestive system with a very high mortality rate. While gemcitabine-based chemotherapy is the predominant treatment for terminal pancreatic cancer, its therapeutic effect is not satisfactory. Recently, many studies have found that microorganisms not only play a consequential role in the occurrence and progression of pancreatic cancer but also modulate the effect of chemotherapy to some extent. Moreover, microorganisms may become an important biomarker for predicting pancreatic carcinogenesis and detecting the prognosis of pancreatic cancer. However, the existing experimental literature is not sufficient or convincing. Therefore, further exploration and experiments are imperative to understanding the mechanism underlying the interaction between microorganisms and pancreatic cancer. In this review, we primarily summarize and discuss the influences of oncolytic viruses and bacteria on pancreatic cancer chemotherapy because these are the two types of microorganisms that are most often studied. We focus on some potential methods specific to these two types of microorganisms that can be used to improve the efficacy of chemotherapy in pancreatic cancer therapy.
Keywords: Pancreatic cancer, Oncolytic viruses, Bacteria, Mycoplasma, Chemotherapy
Introduction
Pancreatic cancer is one of the most destructive and lethal malignant neoplasms. According to data from the authoritative American Medical Association, the numbers of new male and female clinical cases of pancreatic cancer rank as the tenth and ninth highest, respectively, among all new tumor cases, and more critically, the number of deaths from pancreatic cancer ranks 4th among all deaths from carcinoma. However, more than 80% of pancreatic cancer patients have already lost the opportunity to receive surgery when they are diagnosed. This is a possible explanation for the extremely low five-year survival rate (less than 9%) among pancreatic cancer patients [1]. Since the Food and Drug Administration (FDA) approved the use of gemcitabine in 1996, this drug has been widely used to treat breast cancer, lymphoma, ovarian cancer and other tumors and is the cornerstone of pancreatic cancer chemotherapy [2]. Unfortunately, patients often develop gemcitabine resistance within a few weeks of chemotherapy. Changes in drug metabolism, reduced apoptosis, and the effects of the tumor stroma are all fundamental causes of drug resistance [2]. Therefore, many scientists have begun to develop new methods of pancreatic cancer therapy, including the application of nanotechnology [3], strategies targeting tumor metabolism [4, 5], immunotherapy [6], stem cell therapy, strategies targeting the stroma [7], and strategies targeting signaling pathways [8]. Currently, the pancreatic cancer tumor microenvironment is a popular research topic. Many investigators have noted that microorganisms are closely related to the occurrence and progression of pancreatic cancer [9-11]. Other studies have shown that microorganisms are involved in chemotherapy resistance or can aid in its effect [12-14]. While the underlying mechanisms are still being elucidated, the role of microorganisms in chemotherapy deserves to be explored further. This article primarily addresses the effects of microorganisms on carcinogenesis and chemotherapy in pancreatic cancer. Summarizing the benefits and detriments of these microorganisms may aid in the identification of new directions and different research ideas to improve the survival rates of pancreatic cancer patients.
Microorganisms and pancreatic cancer tumorigenesis
Many studies have demonstrated the potential role of microorganisms in pancreatic cancer carcinogenesis. Chronic inflammation primarily induced by microorganisms may be a vital mechanism, and microorganisms can also change the immune microenvironment and regulate the hallmarks of pancreatic cancer [15]. The relationship between microorganisms and pancreatic cancer is summarized in Figure 1; viruses, bacteria, and fungi may all contribute to the carcinogenesis of pancreatic cancer.
The relationship between microorganisms and pancreatic cancer. Viruses, bacteria, and fungi may all contribute to the carcinogenesis of pancreatic cancer. Among the pathogenic pathogens, the viruses primarily include hepatitis B and hepatitis C viruses; the bacteria primarily include oral and gastrointestinal bacteria. In addition, bacteria have complex interactions with risk factors for pancreatic cancer.
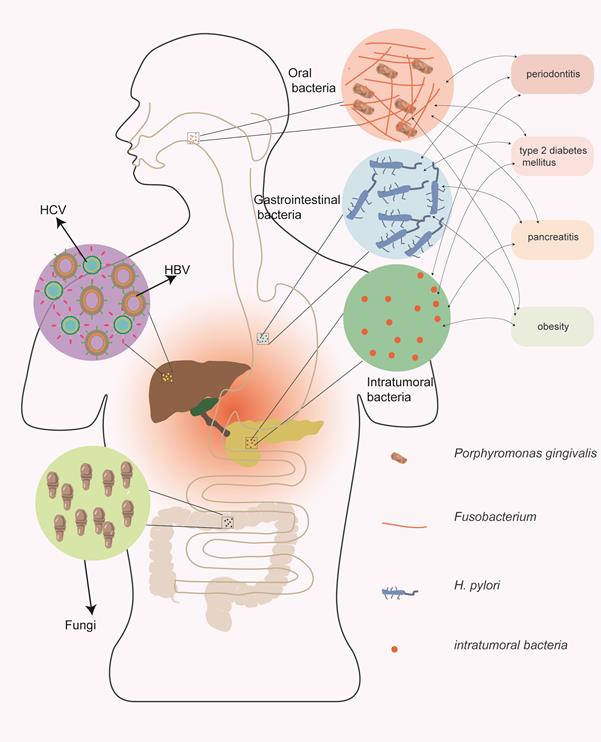
Virus and pancreatic cancer tumorigenesis
Many viruses are thought to be related to cancer carcinogenesis, including human papilloma virus (related to cervical cancer), Epstein-Barr virus (related to nasopharyngeal carcinoma) and hepatitis virus (related to liver cancer). Interestingly, hepatitis virus may also be correlated with pancreatic cancer, particularly hepatitis B virus (HBV) and hepatitis C virus (HCV) [16]. HBV and HCV, typical hepatotropic viruses, can not only appear in the liver but can also be detected in the pancreas [16]. Some researchers detected HBV in the pancreatic acinar and pancreatic juice of HBV patients, and a correlation with pancreatitis was found [17]. In addition, studies have shown that people with HBV or HCV have a higher risk of pancreatic cancer than those without hepatitis [18, 19]. The potential mechanisms by which HBV and HCV promote pancreatic cancer occurrence may include persistent chronic inflammation and changes in tissue elasticity [17, 20]. Some investigators have proposed that the HBx protein expressed by HBV may induce pancreatic cancer carcinogenesis through the PI3K/AKT signaling pathway. However, this induction may only explain a small part of the underlying mechanism, and more investigations are still needed to explore the inner relationships between viruses and pancreatic cancer.
Bacteria and pancreatic cancer tumorigenesis
Bacteria are critical carcinogens and have been investigated in many tumors, including pancreatic cancer. Oral bacteria, including Porphyromonas gingivalis (P. gingivalis) [21] and Fusobacterium [22], gastric and intestinal bacteria, including H. pylori [23] and some intratumoral bacteria, may play a significant role in the occurrence of pancreatic cancer. These bacteria, which may reflux to the pancreas along the digestive tract, have been detected in pancreatic cancer specimens during many studies. A high level of P. gingivalis was correlated with a 2-fold increased risk of pancreatic cancer occurrence [22]. Similarly, H. pylori was found to be positively related to pancreatic cancer, and Helicobacter DNA was confirmed in tumor tissues but not in normal tissues [24]. In addition, some pancreatic cancer specimens were found to have elevated bacterial abundance compared to normal tissues, indicating their effects on pancreatic carcinogenesis [25]. Some regulatory mechanisms in inflammation and immune microenvironments were elucidated when researchers explored the correlation between bacteria and pancreatic cancer tumorigenesis. The bacterial pathogens remaining in the oral and gastrointestinal tracts can always lead to local inflammation and induce the production of inflammatory factors, including interleukins, tumor necrosis factor, and some kinases [26], further causing the activation of tumor-related signaling pathways and the development of important tumor hallmarks [10]. In addition, some studies have shown that gut bacteria can upregulate TLR receptors in pancreatic cancer and induce immune tolerance [27], playing an important role in regulating the tumor microenvironment.
Some risk factors for pancreatic cancer may also interact with bacteria in the digestive tract, including periodontitis, type 2 diabetes mellitus, pancreatitis and obesity. Periodontitis, which is always caused by P. gingivalis and Fusobacterium, has been shown to be a critical risk factor for pancreatic cancer [28]. While periodontitis pathogens destroy the oral health environment and cause chronic inflammation of the oral cavity, they may also promote p53 and K-ras mutations [29], promoting the occurrence of pancreatic cancer. Intestinal bacteria may also influence the pathophysiological process of type 2 diabetes [30]. Low levels of beneficial bacteria may reportedly cause intestinal inflammation and insulin resistance [30]. In addition, intestinal bacteria can also affect the metabolism of short-chain fatty acids [31], inhibit bile acid synthesis and disrupt the intestinal barrier [32], promoting the development of type 2 diabetes to some extent. Obesity, which is currently thought to be a risk factor for pancreatic cancer, is also regulated by intestinal bacteria. Some studies have indicated that the number of Bacteroidetes in obese mice is decreased and the level of Firmicutes is increased [33]. Interestingly, a similar phenomenon was observed in human subjects [34]. The diversity of the bacteria in obese patients is lower than that of normal-weight people [35], suggesting that bacteria may be involved in the pathogenesis of obesity. Another risk factor affected by bacteria includes pancreatitis. Although there may be no bacterial infection during the early stage of acute pancreatitis, bacterial infection can induce persistent inflammation and even pancreatic necrosis in the late stage of pancreatitis [36]. In addition, the microbiota composition of chronic pancreatitis patients changes significantly compared to that of people without pancreatitis [37]. Downregulated Actinobacteria abundance and upregulated Escherichia-Shigella abundance are observed, and the diversity and coordination of gut microbiota are decreased [37].
Fungi and pancreatic cancer tumorigenesis
Fungi, a component of the gut microbiota, have not received much attention from researchers. Although fungi are much lower than bacteria in terms of flora number and abundance, they may play an equally important role as bacteria in pancreatic cancer oncogenesis [38]. Aykut et al found that the number of fungi in pancreatic cancer tissues and mouse models was 3000 times higher than that in normal tissues, with a remarkable enrichment effect on Malassezia spp. [39]. Interestingly, scientists also found Malassezia spp. in the oral cavity, which also confirmed the important role of oral pathogens in the occurrence of pancreatic cancer [40]. Further studies suggested that the mechanisms by which fungi promote pancreatic cancer progression may lie in the MBL-C3 pathways, as verified in Malassezia spp. [39]. In addition, the interaction between bacteria and fungi is also worth exploring. Some researchers have shown that fungi may participate in the bacterial immune response by activating C3 and that bacteria may also regulate MBL-C3 pathways through the immune response [41]. The mutual regulation of bacteria and fungi will make it more complicated to identify the carcinogenic mechanisms.
Viruses and pancreatic cancer chemotherapy
Viruses are pathogens that cause various detrimental effects on physical and mental health. Of the nearly one million vertebrate viruses, approximately 320,000 can infect mammalian cells [42]. Research on the mutual effect of common viruses that influence pancreatic cancer chemotherapy has not yet been reported. However, oncolytic viruses, either as genetically engineered or naturally occurring viruses, have become subjects of note for their interaction with chemotherapy. Bischoff et al reported the first oncolytic virus, dl1520, which can replicate selectively in p53-deficient human tumor cells [43]. This discovery caused a great upsurge in research on oncolytic viruses in the scientific community, which led to the exploration of a series of oncolytic viruses for targeting various tumors. Furthermore, oncolytic virus therapy has recently been recognized as a promising new therapeutic approach to cancer treatment [44-56]. Surprisingly, many oncolytic viruses that act against pancreatic cancer are being researched, and when used in combination with gemcitabine, they can significantly assist in the killing of pancreatic cancer cells and dramatically improve the effect of chemotherapy. This part of the review will describe the latest results of combining oncolytic viruses with chemotherapy (as represented by gemcitabine) according to the category of virus in use, with a focus on the underlying mechanisms to explore better combination therapies.
Adenoviruses and pancreatic cancer chemotherapy
Adenoviruses are small, non-enveloped double-stranded icosahedral DNA viruses, and there are >57 serotypes of adenoviruses that are classified into subtypes A-G based on their respective agglutination properties [57, 58]. Intriguingly, the adenovirus can function as both a pathogen and a therapeutic tool [59-61]. Every oncolytic adenovirus is essentially produced by deleting viral genes, including E1B55K, E1B19K, and E1ACR2, and the majority of oncolytic adenoviruses are based on adenovirus serotype 5 (Ad5), which has been shown to be safe in cancer patients and to eliminate cancer cells with limited toxicity to healthy cells specifically [62]. Studies have shown that most oncolytic adenoviruses can produce a sensitization effect when combined with gemcitabine and can enhance the killing effect on pancreatic cancer cells [63]. The first studied oncolytic adenovirus was onyx-015, which carried a deletion of the E1B55K gene [43]. The E1B55K gene is found within the E1B region of the viral genome. It can combine with the p53 gene and inactivate it. Due to its deletion, the oncolytic adenovirus can replicate in tumor cells but cannot replicate in normal cells. This property guarantees the safety of the oncolytic adenovirus [43]. Experiments using cell lines and tumor-bearing nude mice showed that onyx-015 efficiently lyses pancreatic cancer cells deficient in p53 expression, and some scientists found that infected pancreatic cancer cells can express viral antigens on the surface, resulting in a host immune response to reinforce antitumor immunity [43]. However, Hecht and Mulvihill et al. evaluated the effects of ONYX-015 combined with gemcitabine on pancreatic cancer cells, and the results were not satisfactory [64, 65]. This poor result may have occurred because the deletion of E1B55K weakened the ability of the virus to replicate and spread [66, 67]. As a result, scientists began to explore new excision sites, and on this basis, they developed new oncolytic adenovirus mutants accompanied by different gene deletions, for example, AdΔE1B19K (E1B19K deleted), AdΔΔ (E1ACR2 and E1B19K deleted), AdΔCR2 (E1ACR2 deleted), dl312 (E1A and E3B deleted) and dl922-947 (E1ACR2 and E3B deleted) [63, 68, 69]. These newly developed oncolytic viruses have shown exciting effectiveness. Oncolytic adenoviruses with E1B19K gene deletion can achieve valid targeting at a dose lower than the clinically toxic dose when used in combination with chemotherapy. The enhanced antitumor effect is due to the enhanced drug-induced apoptosis induced by synergism between the virus and gemcitabine. Gemcitabine induces tumor cell death through classical apoptosis, and adenovirus activates tumor cell death through nonapoptotic pathways [68]. Interestingly, scientists have found that oncolytic adenovirus mutants such as dl922-947, from which E1ACR2 was deleted, are more efficient than ONYX-015, but they are more toxic [69-73]. AdΔΔ is an oncolytic mutant with both E1ACR2 and E1B19K deleted. Compared with wild-type adenovirus, ONYX-015 and dl922-947, AdΔΔ retains the ability to sensitize cancer cells in combination with cytotoxic drugs, and when combined with gemcitabine, it not only induces more apoptosis to effectively kill tumor cells but also has lower toxicity than when it is used as a monotherapy [63, 68, 74]. However, to treat pancreatic cancer more briefly and effectively in combination with chemotherapy through systemic administration, adenovirus mutants must be further modified for the following reasons: (1) coxsackie virus receptors, natural virus receptors and adenovirus receptors, which are expressed at a high level in human erythrocytes, can bind to the fiber knob of the virus[75]; and (2) the high-affinity binding to numerous blood factors and Kupffer cells in the liver can quickly clear the virus [62, 76]. To improve the tumor-killing and chemotherapy synergistic effects, some researchers modified AdΔΔ to achieve increased targeting. Compared with normal tissue cells, most cancer cells can express integrin avβ6, which is a useful target. Therefore, they added the A20FMDV2 peptide to target the Arg-Gly-Asp (RGD) domain of integrin avβ6 selectively and deleted E3gp19k, resulting in a new mutant, Ad-5-3Δ-A20T, which was more lethal than AdΔΔ against pancreatic cancer when combined with gemcitabine. Its primary mechanisms lie in its increased recruitment of immune cells to infected tumor cells and its enhancement of gemcitabine-dependent apoptosis [77]. To date, the most promising mutants for pancreatic cancer treatment are LOAd703 and VCN-01. LOAd703 has a deleted EIACR2 region to improve tumor selectivity and has an E2F binding site inserted upstream of E1A to control virus replication; in addition, its most critical feature is an insertion of the trimerized, membrane-bound human CD40 ligand (TMZ-CD40L) and the full-length human 4-1BB ligand (4-1BBL) [78-80]. CD40L activates apoptosis in cancer cells and increases myeloid and T cell infiltration to increase the antitumor effect. 4-1BBL can increase the infiltration of lymphocytes into tumors and enhance the CD40/CD40L synergistic antitumor immune response [78, 81, 82]. VCN-01 also has a deleted E1ACR2 region and an inserted E2F-binding site [83, 84]. The replacement of the KKTK binding site in VCN-01 with an RGD domain prevents the effects of adenovirus on hepatocytes. The addition of PH20 hyaluronidase facilitates the spread of adenovirus in the interstitium of connective tissue and the extracellular matrix and promotes the infiltration of immune cells [85-89]. Currently, relevant clinical trials of VCN-01 and LOAd703 are ongoing, and researchers have added gemcitabine to their list of intervention measures.
Herpes simplex virus type 1 and pancreatic cancer chemotherapy
Herpes simplex viruses (HSVs) can be divided into type 1 and type 2. HSV-1 is an enveloped double-stranded DNA virus that has the following merits and can be used as an oncolytic virus [90-92]: (1) It has a wide range of infection targets and can infect almost all types of human cells and various tumor cell types [92]. (2) Almost all HSV-1 gene sequences have been discovered, and they are long and suitable for the insertion of foreign genes [93, 94]. (3) HSV-1 infectivity is higher than that of adenovirus and adeno-associated virus [95]. (4) Oncolytic viruses based on HSV-1 are very safe. Although they are associated with some adverse events, there are sensitive anti-HSV drugs, such as ganciclovir and acyclovir. Because HSV-1 has good oncolytic activity, many scientists have used it for treating pancreatic cancer, and in combination therapy with chemotherapy, it has achieved beneficial synergistic effects. G207 is a second-generation mutant of HSV-1 with a deletion of the γ134.5 gene and an insertion of the E. coli. lacZ gene in the infected cell polypeptide 6 (ICP-6)-coding region [96, 97]. The insertion of the E. coli lacZ gene enables G207 to replicate more efficiently than HSV-1 in nondividing cells [96, 97]. To explore the therapeutic effect of G207 in pancreatic cancer, Lee et al. conducted experiments on three pancreatic cancer cell lines, ASPC-1, MIA Paca-2 and BxPC-3, in vitro. Their study confirmed the infection, replication and killing abilities of G207 and was the first to demonstrate the great potential of G207 for treating pancreatic cancer [98]. In addition, scientists have begun to explore chemotherapy combination strategies. Martuza and Samuel et al. found that when HSV-1-based viruses such as G207 and NV1020 are combined with traditional chemotherapy, their killing effect on pancreatic cancer cells is greatly enhanced, providing a promising clinical treatment strategy[99]. However, some of the oncolytic HSVs mentioned above have some limitations in terms of their tumor-killing ability and replication efficiency. With the development of science and technology, many new mutants have emerged. Myb34.5 is a typical mutant used in pancreatic cancer studies in which the expression of ICP6 is defective and the expression of γ134.5 is driven by the B-myb promoter [100]. In an experimental model of pancreatic cancer, mice that received an intratumoral injection of Myb34.5 had a good prognosis. Their tumors were necrotic and showed bleeding, and their cancer cells died due to apoptosis [101]. The synergy between Myb34.5 and chemotherapy drugs is intriguing. The combined use of low-dose virus and gemcitabine can significantly kill MIA Paca-2 pancreatic cancer cells in vitro and is much more effective than the use of gemcitabine alone [101]. Interestingly, the killing effect of Myb34.5 on tumors depends on virus replication rather than the host immune response, according to some studies [101]. HF10 is an oncolytic virus derived from HSV-1 that is currently a popular topic of research. It has a strong tumor-killing ability but does not damage normal tissues [102]. Previous clinical studies have shown that HF10 increases the number of CD4+, CD8+ and natural killer cells in tumors, which may slow tumor growth and prolong survival rates [95, 102, 103]. Recently, in a clinical trial on 12 patients with unresectable pancreatic cancer, HF10 combined with gemcitabine and erlotinib showed a higher antitumor effect than HF10 alone. Among the 12 patients, 3 had partial remission, 4 had stable disease, and the total effective rate was 78% [104]. Although the mechanism by which HF10 interacts with chemotherapy is still unclear, the role of HF10 in combination therapy for pancreatic cancer deserves further exploration.
Vaccinia viruses and pancreatic cancer chemotherapy
Vaccinia viruses have been widely known since their application in preventing smallpox. Recently, due to their tropism for cancer cells, easy genetic modification and other biological characteristics, vaccinia viruses have become a popular choice for use as oncolytic viruses [105, 106]. They have shown good antitumor activity and the ability to replicate selectively in tumors in in vivo and in vitro models of pancreatic cancer. More specifically, many scientists have discovered their beneficial synergistic effects when combined with gemcitabine for treating pancreatic cancer. ING4 is a protein that can inhibit angiogenesis and increase the sensitivity of cancer cells to chemotherapy; undoubtedly, it is a highly effective protein that can be used to treat pancreatic cancer [107, 108]. Wu et al. constructed a new oncolytic virus, VV-ING4, which contains the gene encoding ING4, and this oncolytic virus had a stronger cytotoxic effect than the original virus and could induce pancreatic cancer cell apoptosis and G2/M phase arrest. When VV-ING4 and gemcitabine were used synergistically in in vitro experiments, they significantly inhibited the replication of SW1990 and PANC-1 pancreatic cancer cells. This result may have occurred because VV-ING4 improves chemotherapy sensitivity and promotes the penetration of gemcitabine [109]. Because sensitizing proteins are a hot research topic, research into the antigenicity of tumors is another direction for scientists to identify novel therapeutic strategies. Survivin, a tumor-associated antigen, is overexpressed in most pancreatic cancer cells and lacks expression in most differentiated and mature cells. Increased survivin expression is related to increased activity of cancer cells, resistance to cancer treatments and tumor progression. A modified vaccinia virus Ankara (MVA-survivin) expressing survivin was constructed by Ishizaki et al., who also explored its effect on pancreatic cancer models in combination with gemcitabine [110]. The experimental results showed that when MVA-survivin and gemcitabine were used in combination to treat Pan02 tumors, the tumors regressed significantly, and the survival time was prolonged. This finding may be related to the enhancement of related antitumor immunity [110].
Interestingly, in research on the combined application of GLV-1h68 (an oncolytic vaccinia virus) and chemotherapy drugs in pancreatic cancer, different scientists have obtained slightly different results. Yu et al. found that the tumor-killing effect was significantly enhanced when GLV-1h68 was combined with gemcitabine to treat PANC-1 tumors [111]. However, in another study on GLV-1h68 combined with nab-paclitaxel plus gemcitabine to treat pancreatic cancer, scientists found that the cytotoxicity of GLV-1h68 combined with gemcitabine treatment was not increased compared with that of the single treatment. In addition, in GLV-1h68 combined with nab-paclitaxel plus gemcitabine triple therapy, the cytotoxicity to BxPC-3 and MIA Paca-2 tumor cells was significantly reinforced [112]. Therefore, we hypothesize that vaccinia viruses do have beneficial synergistic effects in combination with chemotherapy for treating pancreatic cancer, and determining the most effective combination strategy is worthy of further exploration.
Other oncolytic viruses and pancreatic cancer chemotherapy
In addition to the commonly used oncolytic viruses that have been introduced above, there are several oncolytic viruses that have shown to have increasing potential for treating pancreatic cancer, including Newcastle disease virus, measles virus, myxoma virus and vesicular stomatitis virus.
Newcastle disease virus belongs to the Paramyxoviridae family and is a naturally occurring negative-sense single-stranded RNA virus that has been studied for many years. Its genome contains 6 genes encoding eight proteins that can efficiently and selectively kill many types of human tumor cells [113]. Early clinical trials have found that several naturally occurring Newcastle disease viruses have the ability to kill pancreatic cancer tumors, but the effect is minimal. Therefore, some scientists continue to use genetic engineering techniques to transform Newcastle disease viruses to achieve better oncolytic effects [114, 115]. Buijs et al. [116] evaluated the response of 11 different human pancreatic cancer cell lines to Newcastle disease virus infection and interferon treatment and found that improving the anti-interferon properties of Newcastle disease virus may enhance its oncolytic effect. At present, the combination of Newcastle disease virus and chemotherapy for treating pancreatic cancer has not been explored, and it may achieve surprising results [117]. Interestingly, a combination of measles virus and chemotherapy has been reported. Some scientists found that the combined application of a small amount of measles virus and a subtherapeutic concentration of gemcitabine could reduce the mass of pancreatic cancer cells by more than 50%, and measles virus and gemcitabine were shown to have synergistic effects, greatly improving the oncolytic activity [118]. To explore the synergy of measles virus and gemcitabine, Bossow et al. designed a completely redirected measles virus, namely, MV-PNP-anti-PSCA. It can target prostate stem cell antigen (PSCA; which can be expressed in pancreatic cancer) and carries the gene encoding the prodrug-converting enzyme PNP. This new oncolytic measles virus can specifically infect pancreatic cancer cells and shows high oncolytic activity in cell lines that express PSCA. Importantly, in pancreatic cancer cells resistant to gemcitabine, cross-resistance to this mutant has not been detected. This finding is undoubtedly good news for pancreatic cancer patients who are resistant to chemotherapy [119]. Myxoma virus is a member of the poxvirus family. Although it is considered nearly harmless to normal human cells, it has a good killing effect on pancreatic cancer cells. Resistance to gemcitabine is related to increased levels of activated Akt, and the upregulation of phosphorylated Akt enhances productive infection by myxoma virus, suggesting that myxoma virus may be a potential alternative therapy for pancreatic cancer, especially for those resistant to gemcitabine [120-122]. In vivo and in vitro studies have shown that the use of myxoma virus and gemcitabine sequential therapy on the Hs766T and Pan02 pancreatic cancer cell lines improves the overall survival rate of mice. In Hs766T tumor cells, treating with gemcitabine followed by oncolytic virus is more effective than either agent as a monotherapy. In the Pan02 cell line, treating with oncolytic virus followed by gemcitabine had better results than either agent as a monotherapy [123]. Vesicular stomatitis virus is also very popular for treating pancreatic cancer. Vesicular stomatitis virus is a non-segmented, negative-strand RNA virus and a promising oncolyte that can kill pancreatic cancer cells by inducing apoptosis [124]. VSV-ΔM51-GFP is a new type of vesicular stomatitis virus that can be used as a chemotherapy sensitizer. VSV-ΔM51-GFP has a methionine deletion at amino acid 51 of the matrix protein and a green fluorescent protein (GFP) open reading frame (ORF) inserted at position 5 of the viral genome, which can activate both the endogenous and exogenous apoptotic pathways to induce pancreatic tumor cell apoptosis; compared to other viruses, it is a more effective oncolytic agent [125]. When VSV-ΔM51-GFP was combined with gemcitabine for pancreatic cancer treatment, a significant improvement in the therapeutic effect was observed. This finding suggests that vesicular stomatitis virus has a promising future in combination with gemcitabine [126].
Underlying mechanisms of synergism between oncolytic viruses and chemotherapy
Although substantial evidence indicates that oncolytic viruses can have a good synergistic effect with chemotherapy, the specific molecular basis has not yet been summarized. In this section, we prioritize concrete mechanisms that may involve direct oncolysis, increasing the sensitivity of cancer cells to chemotherapy and activating antitumor immunity and the apoptosis of tumor cells. These important mechanisms are summarized in Fig. 2.
Some underlying mechanisms by which oncolytic viruses influence the treatment effect of chemotherapy. 1) Oncolytic viruses can enhance the apoptosis of tumor cells induced by chemotherapy. Moreover, they can also induce tumor cell apoptosis by themselves. 2) Oncolytic viruses can activate antitumor immunity and enhance its efficacy. They can cause the infiltration of T cells, myeloid cells and other immune cells in the tumor to enhance the antitumor activity of chemotherapy. 3) Oncolytic viruses can increase the sensitivity of tumor cells to chemotherapy, thereby making tumor cells easier to kill. 4) Oncolytic viruses can replicate and multiply only in tumor cells, thereby directly killing tumor cells and complementing the killing effect of chemotherapy. Different oncolytic viruses contain different synergistic mechanisms, but they all improve the tumoricidal effect of chemotherapy to varying degrees.
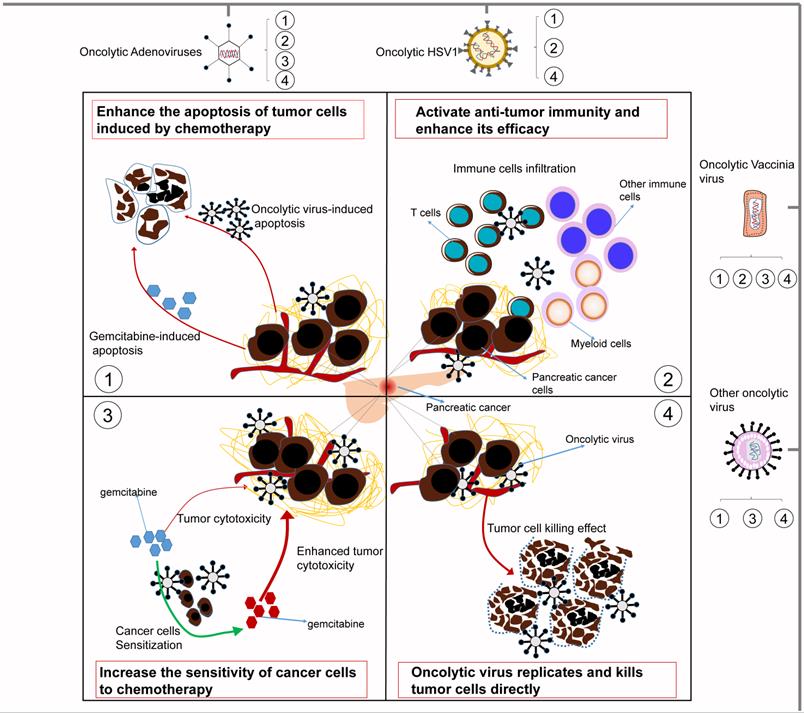
Direct tumor-killing effect
The oncolytic effect of oncolytic viruses is their foremost property. Oncolytic viruses can selectively invade pancreatic cancer tumor cells, leading to their cracking and death, which in turn leads to increasing positive feedback for lysis. Normal cells have complete antiviral immunity, and oncolytic viruses are quickly cleared when they try to invade normal cells. However, the antiviral capabilities of tumor cells are defective, and they may have defects in the PI3K/AKT signaling pathway or tumor suppressor genes such as P53 and RB [127], which often makes tumor cells more sensitive to oncolytic viruses.
Enhance tumor sensitivity to chemotherapy
The biggest flaw of chemotherapy is its easily induced tolerance, and oncolytic viruses solve this drawback to a certain extent. Deleting the E1B19K gene can make pancreatic cancer cells sensitive to chemotherapy-induced death [68]. Oncolytic viruses primarily act on cell cycle regulation, increase the DNA damage induced by chemotherapy and exert sensitization effects. They can attenuate the activation of Chk1 and the DNA repair factor Mre11 [128]. Another important mechanism is to prevent the drug-induced accumulation of Claspin, which is a required protein for Chk1 activation. These processes lead to a virus-mediated reduction in the DNA damage response (DDR) and eventually to sensitization [128]. Another sensitization mechanism is related to the extracellular matrix (ECM). The ECM is often distributed on the surface of pancreatic cancers, and its primary components are collagen, fibronectin and elastin [129], which greatly block the effective arrival of chemotherapy. Chemotherapy-mediated drug resistance is also related to ECM-mediated signal transduction, and oncolytic viruses can selectively eliminate abnormal ECM, which can offset ECM-mediated drug resistance and indirectly increase tumor cell sensitivity to chemotherapy [123].
Antitumor immunity boosted by the oncolytic virus
Over the past few years, the immune response has been considered to be an obstacle to oncolytic virotherapy. However, we are now aware of the great importance of the immune system in oncolytic virotherapy despite its clearing of oncolytic viruses. Typically, pancreatic cancer generates an immunosuppressive microenvironment that includes immune suppressive cytokines such as interleukin-10 (IL-10) and transforming growth factor β1 (TGFβ1) and immunosuppressive cells, including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), leading to the inhibition of the immune response [130]. The emergence of oncolytic viruses changes this situation. Oncolytic virus infection can induce direct cell lysis [131] and immunogenic cell death (ICD), which includes pyroptosis [132, 133], necroptosis [134, 135] and autophagic cell death [132, 133]. These processes can release cellular damage-associated molecular patterns (DAMPs, for example, heat shock proteins, high mobility group box 1 (HMGB1) protein, calreticulin, and IFN-1), pathogen-associated molecular patterns (PAMPs) and tumor-associated antigens (TAAs) [48]. These substances can promote the maturation of antigen-presenting cells (APCs), such as dendritic cells (DCs), and activate the immune response of CD4+ T cells and CD8+ T cells. Activated T cells can produce cytotoxic effects, thereby mediating effective antitumor immune responses [136, 137]. In addition, the released cytokines, such as IL-6, IL-8, and IFN-1, can directly activate NK cells and have a direct killing effect on cancer cells [138, 139]. Surprisingly, oncolytic viruses can also inhibit the immune escape of pancreatic cancer cells, which is one of the most important hallmarks of cancer. As mentioned above, the tumor immune microenvironment (TME) is a prominent cause of immune escape. Oncolytic viruses can change the cytokines in the TME, such as IL-10 and TGF-β, and the types of recruited immune cells, such as Tregs and myeloid-derived suppressor cells (MDSCs) [140-142], which results in the hyperresponsiveness of tumor cells to the immune system.
Induced apoptosis by oncolytic virus
Chemotherapy drugs such as gemcitabine can induce apoptosis in pancreatic cancer tumor cells by damaging DNA and terminating chain synthesis [143]. Interestingly, oncolytic viruses can enhance the apoptotic ability of gemcitabine through various additional apoptosis-inducing pathways. In cell cycle regulation, the P53 and RB pathways are two classic pathways that ensure the normal progression of the cell cycle. The occurrence of P53 mutations in pancreatic cancer cells is the key factor in their unlimited replication capacity. Oncolytic viruses often act on the P53 pathway to promote the upregulation of Bax and the downregulation of Bcl2 and further activate caspase8/9/3 to cause apoptosis [109, 144]. During this process, both the mitochondrial-dependent apoptosis pathway and the death receptor-dependent signaling pathway play a significant role. In addition, there are some other ways of inducing apoptosis that also play an important role. For example, oncolytic viruses can negatively regulate the signal transduction of the NF-κB pathway and other tumor suppressor factors to promote apoptosis [145, 146]. Some oncolytic viruses, such as H-1PV, can even establish an apoptotic pathway independent of caspase3 by activating lysosomal proteases and cytosolic relocation [147].
Bacteria and pancreatic cancer chemotherapy
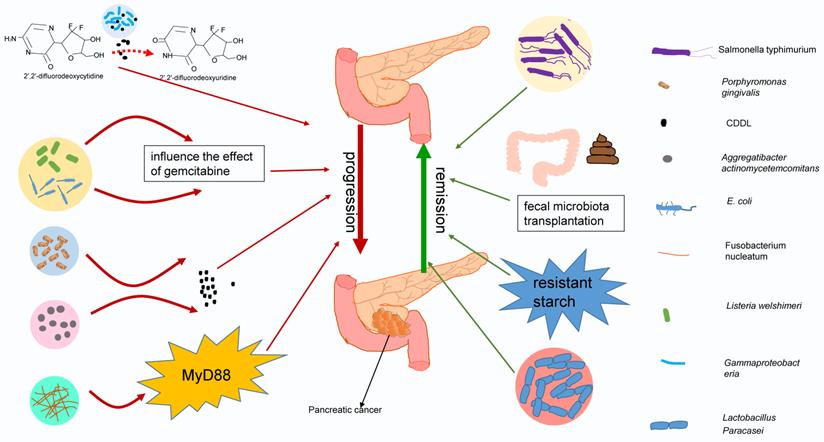
Recently, many studies have shed light on the potential link between the microbiota and pancreatic cancer [12, 148-150]. As an important component of the microbiota, bacteria are inextricably related to the response to chemotherapy (typically gemcitabine) by pancreatic cancer. Some studies have proposed that bacteria in tumors can modulate the chemotherapeutic effect on pancreatic cancer, leading to negative or beneficial effects. Chemotherapy can in turn act on the tumor or intestinal flora, causing various problems. Interestingly, antibiotics also play an important role in chemotherapy for pancreatic cancer by affecting intestinal bacteria. The effects of bacteria on chemotherapy in pancreatic cancer are summarized in Fig. 3. The interactions among chemotherapy, bacteria and antibiotics are summarized in Fig. 4. These findings may provide a basis for using tumor-related bacteria to develop new strategies for treating pancreatic cancer.
Different bacteria have different effects on chemotherapy for pancreatic cancer. Gammaproteobacteria, E. coli, Listeria welshimeri, Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, and Fusobacterium nucleatum may have negative effects on pancreatic cancer treatment. They act directly on chemotherapy or indirectly change chemotherapy drugs by secreting enzymes or through signaling pathways, deteriorating the treatment effect of pancreatic cancer. However, Salmonella typhimurium and Lactobacillus paracasei may have beneficial effects on chemotherapy, and fecal microbiota transplantation or resistant starch may improve the treatment efficacy against pancreatic cancer by adjusting the intestinal flora.
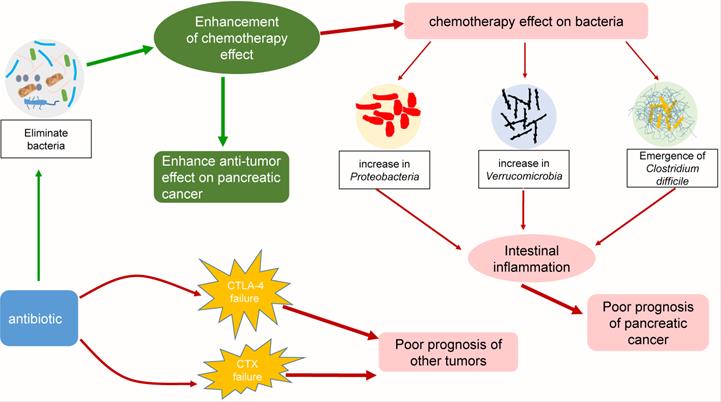
Some interactions among chemotherapy, bacteria and antibiotics. Chemotherapy can affect the composition of the intestinal flora and increase the ratio of Proteobacteria, Verrucomicrobia, and Clostridium difficile. These bacteria can cause intestinal inflammation, leading to poor prognosis in pancreatic cancer patients. The addition of antibiotics enhanced the efficacy of chemotherapy by eliminating bacteria, but it also exacerbated the changes in the intestinal flora. Interestingly, while other tumors were being treated with CTLA-4 or CTX in combination with antibiotics, similar consequences were observed.
The negative effect of bacteria on pancreatic cancer chemotherapy
Previous studies on the relationship between bacteria and pancreatic cancer have focused on the mechanism by which bacteria give rise to the occurrence and progression of pancreatic cancer [15]. Currently, the focus of many studies has begun to shift to the influence of bacteria on chemotherapy in pancreatic cancer. Some evidence shows that the presence of bacteria in the tumor microenvironment will modulate the effectiveness of cancer treatment. Bacteria can metabolize chemotherapy drugs, change their chemical structure, and affect their activity and local concentration [151, 152]. Relatively recently, Geller et al. discovered that bacteria can disrupt the metabolism of gemcitabine by pancreatic cancer [150]. These researchers examined tumor specimens from 113 pancreatic cancer patients and found bacteria in 76% of tumor specimens, primarily Gammaproteobacteria, which can express the long form of CDD (cytidine deaminase) and can metabolize the active form of gemcitabine (2′,2′-difluorodeoxycytidine) into the inactive form, 2′,2′-difluorodeoxyuridine. Interestingly, scientists have observed a similar phenomenon in mycoplasma, which can encode the CDD to inhibit the antitumor activity of gemcitabine in mycoplasma-infected mouse tumor cells, and this inhibition can be relieved by tetrahydrouridine (a cytidine deaminase inhibitor) or antibiotics such as tetracycline [153], suggesting that these bacteria may be related to chemotherapy resistance in pancreatic cancer. An animal model designed to detect the influences of Escherichia coli (E. coli) and Listeria welshimeri on the efficacy of commonly used chemotherapeutics can be used to support this conjecture partially. The tumor volumes of mice treated with gemcitabine plus bacteria were significantly larger than those of mice treated with gemcitabine alone, indicating that the presence of bacteria had an adverse effect on gemcitabine chemotherapy [151]. Surprisingly, some scientists have found that bacteria from other tissues may also be related to chemotherapy resistance. Elevated levels of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans have been detected in patients receiving pancreatic cancer treatment. More specifically, these two bacteria can also express CDD, which indicates that they may be related to chemotherapy resistance in pancreatic cancer [12, 154]. Even Fusobacterium nucleatum (F. nucleatum), a bacterium that can cause colorectal cancer to be resistant to oxaliplatin and 5-fluorouracil by affecting the TLR4/MyD88 pathway, has been found to be more abundant in the cancer tissues of patients with pancreatic cancer than in nonpatient controls [155]. According to the literature, F. nucleatum leads to the loss of miR-18a* and miR-4802 through the TLR4/MYD88 pathway, which leads to the activation of ULK1 and ATG7 and further induces the autophagy pathway, thereby triggering drug resistance in colon cancer. F. nucleatum can also cause the immunosuppression of the pancreatic cancer microenvironment through the same MyD88 pathway. Thus, some scientists speculate that this bacterium may also be related to drug resistance in pancreatic cancer based on the similar signaling pathways involved [156, 157].
The beneficial effects of bacteria on pancreatic cancer chemotherapy
The role of bacteria in pancreatic cancer chemotherapy is not always deleterious, and bacteria can sometimes have a beneficial effect. In a mouse model of pancreatic cancer treated with gemcitabine and bevacizumab, adding Salmonella typhimurium improved the therapeutic effect in pancreatic cancer-bearing mice [158]. A large cohort study drew a similar conclusion. Researchers found that the diversity of the tumor microbiota in long-term survivors (>5 years) was higher than that in short-term survivors [159]. On this basis, many scientists have also begun to investigate strategies that employ bacteria to improve the pancreatic cancer prognosis, and probiotics have become the focus of exploration in new therapies. Lactobacillus paracasei is a gram-positive lactic acid bacterium located in the human intestine. Adding Lactobacillus paracasei to a mouse model treated with gemcitabine can improve the efficacy of the chemotherapy and increase tolerance [160]. Further research showed that it primarily elevated IFN to transfer the Th2 immune phenotype into the Th1 immune phenotype, subsequently enhancing the antitumor ability [161]. In addition, some components derived from probiotics also seem to exhibit antitumor activity. The ferrichrome extracted from Lactobacillus paracasei can inhibit the proliferation of pancreatic cancer cells and restrain pancreatic cancer cells resistant to 5‑fluorouracil in a mouse model, indicating that the antitumor activity of probiotics may come from the active ingredients in probiotics [162]. In addition, probiotics seem to have the ability to reduce adverse postoperative reactions. Postoperative patients taking Enterococcus faecalis and Clostridium butyricum seem to have fewer infection complications [163]. Some methods that can modulate the intestinal flora may also modify the effects of chemotherapy in pancreatic cancer, such as fecal microbiota transplantation, alterations in lifestyle or diet, and the use of prebiotics. Using resistant starch as a prebiotic can facilitate the growth of bacteria involved in butyrate production and delay tumor deterioration in mice bearing pancreatic cancer [164]. The application of prebiotics and other modulation methods in treating pancreatic cancer are being explored.
The effect of chemotherapy on intestinal bacteria and the exploration of antibiotic therapy
The physical condition of cancer patients is generally poor, and the addition of chemotherapy undoubtedly aggravates the patient condition [165, 166]. The side effects of chemotherapy include digestive system reactions such as diarrhea and vomiting, myelosuppression, immune impairment, and liver and kidney damage. The most common reactions are digestive system reactions, which strongly implies that chemotherapy affects the gastrointestinal flora. Panebianco et al. evaluated whether gemcitabine treatment can affect the intestinal bacterial composition of mice that were xenografted with pancreatic cancer [167]. The results showed that the proportion of gram-positive Firmicutes (from approximately 39 to 17%) and gram-negative Bacteroidetes (from 38 to 17%) in mouse intestines in the tumor-bearing control group decreased significantly, while the proportions of Proteobacteria (E. coli and Aeromonas hydrophila) and Verrucomicrobia (Akkermansia muciniphila) increased dramatically [167]. Gram-positive Firmicutes and gram-negative Bacteroidetes are usually the dominant species in the gut of tumor-bearing control mice, while Proteobacteria and Verrucomicrobia are usually minor components [168-170]. According to the analysis, the increase in Proteobacteria and Verrucomicrobia was related to inflammation of the intestine [171, 172]. Researchers analyzed the serum metabolites of mice and found that those receiving gemcitabine had highly significant decreases in creatinine, which has anti-inflammatory and immunosuppressive properties [173, 174]. In addition, the application of antibiotics to chemotherapy patients has also received attention from researchers. Previously, Geller et al. found that in a colorectal cancer model, when combined with gemcitabine, the antibiotic ciprofloxacin could eliminate the chemotherapy resistance caused by the bacteria in the tumor, significantly enhancing the treatment effect [150]. This finding suggests that the combination of antibiotics and chemotherapy may be extremely beneficial in pancreatic cancer therapy. To explore the application of antibiotics during chemotherapy, some scientists retrospectively analyzed the indexes of 169 patients with advanced cancer (including pancreatic cancer) who received gemcitabine treatment. The patients were divided into two groups: an antibiotic-free group (treated with a gemcitabine-containing regimen but not treated with antibiotics) and an antibiotic treatment group (treated with a gemcitabine-containing regimen plus antibiotics). The effective rate, progression-free survival and overall survival of each group were evaluated. The results showed that the median progression-free survival and median overall survival of the antibiotic treatment group were longer than those of the antibiotic-free group, which indicates that adding antibiotics can improve the efficacy of chemotherapy in patients with advanced cancer [175]. However, antibiotics also cause some side effects, such as adverse gastrointestinal events. Corty et al. used gemcitabine to treat 430 patients with metastatic pancreatic cancer. They used the Anderson-Gill survival model to compare the risk of adverse events between patients receiving antibiotics and those not receiving antibiotics. The results showed that receiving antibiotics was related to an increased risk of gemcitabine-associated, dose-limiting adverse events, including adverse gastrointestinal and hematological events [176]. The survival period of patients who discontinued treatment due to adverse events was shorter than that of patients who continued treatment until the disease progressed. This finding indicates that antibiotics must be used cautiously in pancreatic cancer treatment and may actually be detrimental [176]. Interestingly, the application of antibiotics in treating other cancers also supports this hypothesis. In mouse sarcoma, melanoma, and colon cancer models, a cocktail of the antibiotics ampicillin, colistin, and streptomycin renders cytotoxic lymphocyte antigen 4 (CTLA-4) treatment ineffective [177]. Similarly, Viaud et al. found that pretreating mice receiving cyclophosphamide (CTX) with the gram-positive bacteria-targeting antibiotic vancomycin failed to activate the antitumor immune response, resulting in treatment failure [178]. To conclude, chemotherapy can influence intestinal flora homeostasis. In addition, chemotherapy combined with antibiotics has immeasurable potential of showing its effectiveness against pancreatic cancer, but monitoring for possible side effects and adverse effects regarding the effectiveness of chemotherapy drugs during administration is indispensable.
Prospects for use of microorganisms in pancreatic cancer chemotherapy
Research on the relationship between microbes and tumors has made noticeable progress, which will surely bring new promise for the treatment of pancreatic cancer. Oncolytic viruses, which are microorganisms that can be manipulated by humans, have unpredictable clinical prospects in pancreatic cancer treatment. As we mentioned above, many oncolytic viruses have significant synergistic effects when used in combination with chemotherapy to treat pancreatic cancer. However, there are still many directions worth exploring. For example, how can genetic modification achieve greater tumor selectivity and immunogenicity? How can we minimize the toxicity and maximize the activity of oncolytic viruses? How can we reduce virus loss before reaching the target site? When we combine chemotherapy and oncolytic viruses, which order of administration and dosage have the best effect? In the future, oncolytic viruses that interfere with the tumor microenvironment may reduce the number of resistant cases and the number of cancer stem cells. At present, most oncolytic viruses are delivered by intratumoral injection, and some experiments are guided through endoscopic ultrasound (EUS) [104]. Determining which is the most appropriate delivery method is also warranted. Many clinical trials of oncolytic virus therapy are ongoing (representative clinical trials of oncolytic viruses with gemcitabine as a treatment for pancreatic cancer are listed in Table 1), and perhaps in the near future, oncolytic virus therapy will become the most promising treatment for pancreatic cancer. Our previous discussions show that bacteria in the tumor and gastrointestinal tract may have a vital impact on the effect of chemotherapy, so targeting relevant bacteria as a therapeutic strategy may induce different effects. The use of antibiotics in combination with chemotherapy has advantages and disadvantages. How to optimize these combinations to bring about better results warrants further exploration. Similarly, the use of chemotherapy to treat pancreatic cancer may induce a series of adverse effects by affecting the intestinal bacteria. An improved understanding of these concepts may lead to a better prognosis for patients with pancreatic cancer. Pharmacomicrobiomics, which can be used to evaluate the interaction between drugs and microorganisms, has become a popular topic in research and the clinic [152, 179]. The use of bacteria as biomarkers for treating pancreatic cancer with chemotherapy is a potential strategy that can be studied in the future. In this way, we can monitor changes in treatment effects and the occurrence of chemotherapy resistance by monitoring changes in bacteria, which can ultimately be used to guide dose adjustments in parallel.
Representative clinical trials of oncolytic viruses with gemcitabine as treatment for pancreatic cancer
| Virus category | Virus names | Study phase | Structural modification | Chemotherapy intervention | Outcome measures | Clinical Trial ID/reference |
|---|---|---|---|---|---|---|
| oncolytic adenovirus | LOAd703 | I/II | EIACR2 deletion, CD40L and 4-1BBL insertion | yes | Overall Response Rate Overall Survival | NCT02705196 |
| oncolytic adenovirus | VCN-01 | I | EIACR2 deletion, PH20 hyaluronidase addition | yes | Recommended Phase 2 Dose (RP2D) of VCN-01 | NCT02045589 |
| oncolytic adenovirus | VCN-01 | I | EIACR2 deletion, PH20 hyaluronidase addition | yes | Safety and Tolerability, Presence of VCN-01 in tumor | NCT02045602 |
| oncolytic adenovirus | ONYX-015 | I/II | E1B55K gene deletion | yes | treatment effect | [65] |
| herpesviruses | OrienX010 | I | Recombinant hGM-CSF | no | preliminary efficacy | NCT01935453 |
| herpesviruses | HF10 | I | UL56 deletion | yes | Dose limiting toxicity (DLT) Adverse events (AEs) | NCT03252808 |
| herpesviruses | HF10 | I | UL56 deletion | yes | safety assessment | [104] |
| herpesviruses | T-VEC | I | ICP34.5 and ICP47 Deletions, GM-CSF Insertion | no | Change in size of injected lesion(s),Overall response rate | NCT03086642 |
| herpesviruses | T-VEC | I | ICP34.5 and ICP47 Deletions, GM-CSF Insertion | no | Adverse Events, (HSV-1) Antibodies | NCT00402025 |
| Vaccinia virus | PANVAC-F plus PANVAC-V | I | None | no | MTD of falimarev. T cell proliferation, Cytokine production | NCT00669734 |
| Vaccinia virus | MVAp53 | I | Express WT murine p53 | no | Safety and tolerance, Immunogenicity | NCT01191684 |
| Vaccinia virus | p53MVA | I | Express p53 and pembrolizumab | no | Tolerability, | NCT02432963 |
| reoviruses | Reolysin | II | None | yes | clinical benefit rate, safety and tolerability | NCT00998322 |
Conclusions
Pancreatic cancer is a malignant neoplasm with poor prognosis, and gemcitabine is the primary chemotherapy drug for advanced pancreatic carcinoma. Currently, plenty of evidence suggests that there is an intricate interplay between microorganisms and pancreatic cancer. However, attention is primarily focused on how microorganisms lead to the occurrence and development of pancreatic cancer, and few studies have focused on elaborating on and summarizing the impact of microorganisms on pancreatic cancer chemotherapy. In this review, we provide new insights into the interplay of microorganisms and chemotherapy, summarizing some meaningful views and guidance for the development of new treatment methods and patterns. Our results indicate that the combination of oncolytic viruses and chemotherapy has great potential utility, and its mechanism possesses four characteristics: apoptosis of tumor cells, activation of antitumor immunity, increased sensitivity of cancer cells to chemotherapy and direct oncolysis. In addition, the use of bacteria in the treatment of pancreatic cancer has beneficial and negative effects on patient prognosis; chemotherapy may also interact with bacteria and cause various results, and antibiotics might have therapeutic potential in pancreatic cancer treatment. Surprisingly, fungi can also contribute to the occurrence of pancreatic cancer, but their influence and application during chemotherapy still require investigation. In addition, it should be noted that most of the studies in the above discussion were conducted in mouse models. Because there will likely be different effects during applications in humans, the results should be interpreted with caution. Overall, microorganisms are of paramount importance to pancreatic cancer chemotherapy, although the complex relationship between microorganisms, chemotherapy and pancreatic cancer remains unclear. In the near future, microorganisms will surely become highly important in pancreatic cancer treatment and new drug development.
Abbreviations
HSVs: Herpes simplex viruses; FDA: Food and Drug Administration; CDD: Cytidine deaminase; E. coli: Escherichia coli; F. nucleatum: Fusobacterium nucleatum; TLR4: Toll-like receptor 4; MYD88: myeloid differentiation factor 88; CTX: cyclophosphamide; CTLA4: cytotoxic lymphocyte antigen 4; Ad5: adenovirus serotype 5; RGD: Arg-Gly-Asp; TMZ-CD40L: trimerized, membrane-bound human CD40 ligand; 4-1BBL: 4-1BB ligand; ICP-6: infected cell polypeptide 6; PSCA: prostate stem cell antigen; GFP: green fluorescent protein; ORF: open reading frame; EUS: endoscopic ultrasound; DDR: DNA damage response; ECM: extracellular matrix; IL-10: interleukin-10; TGFβ1: transforming growth factor β1; Tregs: regulatory T cells; MDSCs: myeloid-derived suppressor cells; DAMPs: damage-associated molecular patterns; HMGB1: high mobility group box 1; PAMPs: pathogen-associated molecular patterns; TAAs: tumor-associated antigens; APCs: antigen-presenting cells; DCs: dendritic cells; and TME: tumor immune microenvironment.
Acknowledgements
Funding
This study was jointly funded by the National Natural Science Foundation of China (No. 81772555, 81802352 and 81902428), the National Science Foundation for Distinguished Young Scholars of China (No. 81625016), the Shanghai Sailing Program (No. 19YF1409400 and 20YF1409000), the Shanghai Rising-Star Program (No. 20QA1402100), the Shanghai Anticancer Association Young Eagle Program (No. SACA-CY19A06), the Clinical and Scientific Innovation Project of Shanghai Hospital Development Center (No. SHDC12018109 and SHDC12019109) and the Scientific Innovation Project of Shanghai Education Committee (No. 2019-01-07-00-07-E00057).
Author Contributions
SYL, JH, MYW, CL, QCM, and JL collected the literature and drafted the manuscript. JX and WW revised the manuscript. SS and XJY proposed the idea of the review and revised the manuscript. All the authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Binenbaum Y, Naara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat. 2015;23:55-68
3. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M. et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N Engl J Med. 2013;369:1691-703
4. Yu M, Zhou Q, Zhou Y, Fu Z, Tan L, Ye X. et al. Metabolic phenotypes in pancreatic cancer. PLoS One. 2015;10:e0115153
5. Blum R, Kloog Y. Metabolism addiction in pancreatic cancer. Cell death & disease. 2014;5:e1065
6. Lutz ER, Kinkead H, Jaffee EM, Zheng L. Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. OncoImmunology. 2014;3:e962401
7. Xie D, Xie K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015;2:133-43
8. Eltawil KM, Renfrew PD, Molinari M. Meta-analysis of phase III randomized trials of molecular targeted therapies for advanced pancreatic cancer. HPB. 2012;14:260-8
9. Ertz-Archambault N, Keim P, Von Hoff D. Microbiome and pancreatic cancer: A comprehensive topic review of literature. World J Gastroenterol. 2017;23:1899-908
10. Karpinski TM. The Microbiota and Pancreatic Cancer. Gastroenterol Clin North Am. 2019;48:447-64
11. McAllister F, Khan MAW, Helmink B, Wargo JA. The Tumor Microbiome in Pancreatic Cancer: Bacteria and Beyond. Cancer Cell. 2019;36:577-9
12. Choy ATF, Carnevale I, Coppola S, Meijer LL, Kazemier G, Zaura E. et al. The microbiome of pancreatic cancer: from molecular diagnostics to new therapeutic approaches to overcome chemoresistance caused by metabolic inactivation of gemcitabine. Expert Rev Mol Diagn. 2018;18:1005-9
13. Zhang X, Liu Q, Liao Q, Zhao Y. Pancreatic Cancer, Gut Microbiota, and Therapeutic Efficacy. J Cancer. 2020;11:2749-58
14. Wang Y, Yang G, You L, Yang J, Feng M, Qiu J. et al. Role of the microbiome in occurrence, development and treatment of pancreatic cancer. Mol Cancer. 2019;18:173
15. Wei MY, Shi S, Liang C, Meng QC, Hua J, Zhang YY. et al. The microbiota and microbiome in pancreatic cancer: more influential than expected. Mol Cancer. 2019;18:97
16. Fiorino S, Visani M, Acquaviva G, Fornelli A, Masetti M, Cuppini A. et al. Search for HBV and HCV Genome in Cancer Cells of Pancreatic Tumors. Pancreas. 2016;45:e12-4
17. Jin Y, Gao H, Chen H, Wang J, Chen M, Li G. et al. Identification and impact of hepatitis B virus DNA and antigens in pancreatic cancer tissues and adjacent non-cancerous tissues. Cancer lett. 2013;335:447-54
18. Fiorino S, Chili E, Ba Cchi-Reggiani L, Masetti M, Deleonardi G, Grondona AG. et al. Association between hepatitis B or hepatitis C virus infection and risk of pancreatic adenocarcinoma development: A systematic review and meta-analysis. Pancreatology. 2013;13:147-60
19. Wang Y, Yang S, Song F, Cao S, Lu Z. Hepatitis B virus status and the risk of pancreatic cancer: a meta-analysis. Eur J Cancer Prev. 2013;22:328-34
20. Fiorino S, Bacchi-Reggiani L, Pontoriero L, Gallo C, Chili E, Masetti M. et al. Tensegrity model hypothesis: may this paradigm be useful to explain hepatic and pancreatic carcinogenesis in patients with persistent hepatitis B or hepatitis C virus infection? JOP. 2014;15:151-64
21. Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM. et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2018;67:120-7
22. Michaud DS, Izard J, Wilhelm-Benartzi CS, You D-H, Grote VA, Tjønneland A. et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut. 2013;62:1764-70
23. Raderer M, Wrba F, Kornek G, Maca T, Koller DY, Weinlaender G. et al. Association between Helicobacter pylori infection and pancreatic cancer. Oncology. 1998;55:16-9
24. Nilsson HO, Stenram U, Ihse I, Wadström T. Helicobacter species ribosomal DNA in the pancreas, stomach and duodenum of pancreatic cancer patients. World J Gastroenterol. 2006;12:3038-43
25. Pushalkar S, Hundeyin M, Zambirinis CP, Miller G. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018;8:403-16
26. Hoare A, Soto C, Rojas-Celis V, Bravo D. Chronic Inflammation as a Link between Periodontitis and Carcinogenesis. Mediators of inflammation. 2019;2019:1029857
27. Riquelme E, Maitra A, McAllister F. Immunotherapy for pancreatic cancer: more than just a gut feeling. Cancer Discov. 2018;8:386-8
28. Michaud DS, Joshipura K, Giovannucci E, Fuchs CS. A prospective study of periodontal disease and pancreatic cancer in US male health professionals. J Natl Cancer Inst. 2007;99:171-5
29. Öğrendik M. Periodontal pathogens in the etiology of pancreatic cancer. Gastrointest Tumors. 2017;3:125-7
30. Ma Q, Li Y, Li P, Wang M, Wang J, Tang Z. et al. Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed Pharmacother. 2019;117:109138
31. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99-103
32. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut microbes. 2012;3:279-88
33. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070-5
34. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022-3
35. Mondot S, de Wouters T, Doré J, Lepage P. The human gut microbiome and its dysfunctions. Dig Dis. 2013;31:278-85
36. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG. et al. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102-11
37. Zhou CH, Meng YT, Xu JJ, Fang X, Zhao JL, Zhou W. et al. Altered diversity and composition of gut microbiota in Chinese patients with chronic pancreatitis. Pancreatology. 2019;20:16-24
38. Rowan-Nash AD, Korry BJ, Mylonakis E, Belenky P. Cross-domain and viral interactions in the microbiome. Microbiol Mol Biol Rev. 2019;83:e00044-18
39. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264-7
40. Diaz PI, Hong BY, Dupuy AK, Strausbaugh LD. Mining the oral mycobiome: Methods, components, and meaning. Virulence. 2017;8:313-23
41. Kolev M, Markiewski MM. Targeting complement-mediated immunoregulation for cancer immunotherapy. Semin Immunol. 2018;37:85-97
42. Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A. et al. A strategy to estimate unknown viral diversity in mammals. mBio. 2013;4:e00598-13
43. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M. et al. An Adenovirus Mutant That Replicates Selectively in p53- Deficient Human Tumor Cells. Science. 1996;274:373-6
44. Martin NT, Bell JC. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol Ther. 2018;26:1414-22
45. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6:140
46. Taguchi S, Fukuhara H, Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn J Clin Oncol. 2019;49:201-9
47. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373-9
48. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642-62
49. Liu GB, Zhao L, Zhang L, Zhao KN. Virus, Oncolytic Virus and Human Prostate Cancer. Curr Cancer Drug Targets. 2017;17:522-33
50. Wei D, Xu J, Liu XY, Chen ZN, Bian H. Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Hum Gene Ther. 2018;29:151-9
51. Gaglia MM, Munger K. More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr Opin Virol. 2018;32:48-59
52. Akram N, Imran M, Noreen M, Ahmed F, Atif M, Fatima Z. et al. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017;30:20-7
53. Niemann J, Kuhnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53:700-6
54. Liang M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr Cancer Drug Targets. 2018;18:171-6
55. Hopcraft SE, Damania B. Tumour viruses and innate immunity. Philos Trans R Soc Lond B Biol Sci. 2017;372:20160267
56. Kvansakul M. Viral Infection and Apoptosis. Viruses. 2017;9:356
57. Russell WC. Adenoviruses: update on structure and function. J Gen Virol. 2009;90:1-20
58. Mangel WF, San Martín C. Structure, function and dynamics in adenovirus maturation. Viruses. 2014;6:4536-70
59. Gonçalves MA, de Vries AA. Adenovirus: from foe to friend. Rev Med Virol. 2006;16:167-86
60. Yamamoto M, Curiel DT. Current issues and future directions of oncolytic adenoviruses. Mol Ther. 2010;18:243-50
61. Lasaro MO, Ertl HC. New Insights on Adenovirus as Vaccine Vectors. Mol Ther. 2009;17:1333-9
62. Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL. et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A. 2008;105:5483-8
63. Cherubini G, Kallin C, Mozetic A, Hammaren-Busch K, Muller H, Lemoine NR. et al. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011;18:1157-65
64. Hecht JR, Bedford R, Abbruzzese JL, Lahoti S, Reid TR, Soetikno RM. et al. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res. 2003;9:555-61
65. Mulvihill S, Warren R, Venook A, Adler A, Randlev B. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: a phase I trial. Gene Ther. 2001;8:308-15
66. O'shea CC, Choi S, Mccormick F, Stokoe D. Adenovirus Overrides Cellular Checkpoints for Protein Translation. Cell Cycle. 2005;4:883-8
67. O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A. et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611-23
68. Leitner S, Sweeney K, Oberg D, Davies D, Miranda E, Lemoine NR. et al. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin Cancer Res. 2009;15:1730-40
69. Bhattacharyya M, Francis J, Eddouadi A, Lemoine NR, Halldén G. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 2011;18:734-43
70. Lockley M, Fernandez M, Wang Y, Li NF, Conroy S, Lemoine N. et al. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: comparison with E1A wild-type viruses, bioluminescence monitoring, and intraperitoneal delivery in icodextrin. Cancer Res. 2006;66:989-98
71. Heise C, Hermiston T, Johnson L, Brooks G, Sampson-johannes A, Williams A. et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134-9
72. Fueyo J, Gomez-manzano C, Alemany R, Lee PS, Mcdonnell TJ, Mitlianga P. et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2-12
73. Raki M, Särkioja M, Desmond RA, Chen D-T, Bützow R, Hemminki A. et al. Oncolytic adenovirus Ad5/3-Δ24 and chemotherapy for treatment of orthotopic ovarian cancer. Gynecol Oncol. 2008;108:166-72
74. Oberg D, Yanover E, Adam V, Sweeney K, Costas C, Lemoine NR. et al. Improved Potency and Selectivity of an Oncolytic E1ACR2 and E1B19K Deleted Adenoviral Mutant in Prostate and Pancreatic Cancers. Clin Cancer Res. 2010;16:541-53
75. Carlisle RC, Di Y, Cerny AM, Sonnen AF, Sim RB, Green NK. et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood. 2009;113:1909-18
76. Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol. 2005;79:7478-91
77. Man YKS, Davies JA, Coughlan L, Pantelidou C, Blazquez-Moreno A, Marshall JF. et al. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol Cancer Ther. 2018;17:575-87
78. Eriksson E, Moreno R, Milenova I, Liljenfeldt L, Dieterich LC, Christiansson L. et al. Activation of myeloid and endothelial cells by CD40L gene therapy supports T-cell expansion and migration into the tumor microenvironment. Gene Ther. 2017;24:92-103
79. Eriksson E, Milenova I, Wenthe J, Stahle M, Leja-Jarblad J, Ullenhag G. et al. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin Cancer Res. 2017;23:5846-57
80. Benedict CA, Norris PS, Prigozy TI, Bodmer J-L, Mahr JA, Garnett CT. et al. Three Adenovirus E3 Proteins Cooperate to Evade Apoptosis by Tumor Necrosis Factor-related Apoptosis-inducing Ligand Receptor-1 and -2. J Biol Chem. 2001;276:3270-8
81. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W. et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science. 2011;331:1612-6
82. Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152-72
83. Rojas JJ, Guedan S, Searle PF, Martinez-Quintanilla J, Gil-Hoyos R, Alcayaga-Miranda F. et al. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol Ther. 2010;18:1960-71
84. Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascallo M. et al. Safety and Efficacy of VCN-01, an Oncolytic Adenovirus Combining Fiber HSG-Binding Domain Replacement with RGD and Hyaluronidase Expression. Clin Cancer Res. 2015;21:1406-18
85. Nicol CG, Graham D, Miller WH, White SJ, Smith TA, Nicklin SA. et al. Effect of adenovirus serotype 5 fiber and penton modifications on in vivo tropism in rats. Mol Ther. 2004;10:344-54
86. Gaianigo N, Melisi D, Carbone C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers. 2017;9:122
87. Rojas JJ, Gimenez-Alejandre M, Gil-Hoyos R, Cascallo M, Alemany R. Improved systemic antitumor therapy with oncolytic adenoviruses by replacing the fiber shaft HSG-binding domain with RGD. Gene Ther. 2012;19:453-7
88. Smith JS, Xu Z, Tian J, Stevenson SC, Byrnes AP. Interaction of systemically delivered adenovirus vectors with Kupffer cells in mouse liver. Hum Gene Ther. 2008;19:547-54
89. Bayo-puxan N, Gimenez-Alejandre M, Lavilla-Alonso S, Gros A, Cascallo M, Hemminki A. et al. Replacement of Adenovirus Type 5 Fiber Shaft Heparan Sulfate Proteoglycan-Binding Domain with RGD for Improved Tumor Infectivity and Targeting. Hum Gene Ther. 2009;20:1214-21
90. Aghi MK, Chiocca EA. Genetically Engineered Herpes Simplex Viral Vectors in the Treatment of Brain Tumors: A Review. Cancer Invest. 2003;21:278-92
91. Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H. et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521-31
92. Agarwalla PK, Aghi MK. Oncolytic herpes simplex virus engineering and preparation. Methods Mol Biol. 2012;797:1-19
93. McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D. et al. The Complete DNA Sequence of the Long Unique Region in the Genome of Herpes Simplex Virus Type 1. J Gen Virol. 1988;69:1531-74
94. McGeoch DJ, Dolan A, Donald S, Brauer DH. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 1986;14:1727-45
95. Nakao A, Takeda S, Shimoyama S, Kasuya H, Kimata H, Teshigahara O. et al. Clinical Experiment of Mutant Herpes Simplex Virus HF10 Therapy for Cancer. Curr Cancer Drug Targets. 2007;7:169-74
96. Mineta T, Rabkin SD, Yazaki T, Hunter W D, Martuza RL. Attenuated multi-mutated herpes simplex virus- 1 for the treatment of malignant gliomas. Nat Med. 1995;1:938-43
97. Yazaki T, Manz HJ, Rabkin SD, Martuza RL. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 1995;55:4752-6
98. Lee JH, Federoff HJ, Schoeniger LO. G207, modified herpes simplex virus type 1, kills human pancreatic cancer cells in vitro. J Gastrointest Surg. 1999;3:127-33
99. Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber K. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant ( HSV- 1716 ) in human non - small cell lung cancer. Hum Gene Ther. 1999;10:3013-29
100. Chung RY, Saeki Y, Chiocca EA. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cell. J Virol. 1999;73:7556-64
101. Gayral M, Lulka H, Hanoun N, Biollay C, Sèlves J, Vignolle-Vidoni A. et al. Targeted Oncolytic Herpes Simplex Virus Type 1 Eradicates Experimental Pancreatic Tumors. Hum Gene Ther. 2015;26:104-13
102. Eissa IR, Naoe Y, Bustos-Villalobos I, Ichinose T, Tanaka M, Zhiwen W. et al. Genomic Signature of the Natural Oncolytic Herpes Simplex Virus HF10 and Its Therapeutic Role in Preclinical and Clinical Trials. Front Oncol. 2017;7:149
103. Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F. et al. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol. 2006;13:1078-84
104. Hirooka Y, Kasuya H, Ishikawa T, Kawashima H, Ohno E, Villalobos IB. et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. 2018;18:596
105. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ. et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99-102
106. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M. et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329-36
107. Xie Y, Sheng W, Miao J, Xiang J, Yang J. Enhanced antitumor activity by combining an adenovirus harboring ING4 with cisplatin for hepatocarcinoma cells. Cancer Gene Ther. 2011;18:176-88
108. Zhang X, Xu LS, Wang ZQ, Wang KS, Li N, Cheng ZH. et al. ING4 induces G2/M cell cycle arrest and enhances the chemosensitivity to DNA-damage agents in HepG2 cells. FEBS Letters. 2004;570:7-12
109. Wu Y, Mou X, Wang S, Liu XE, Sun X. ING4 expressing oncolytic vaccinia virus promotes anti-tumor efficiency and synergizes with gemcitabine in pancreatic cancer. Oncotarget. 2017;8:82728-39
110. Ishizaki H, Manuel ER, Song GY, Srivastava T, Sun S, Diamond DJ. et al. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunology, Immunotherapy. 2010;60:99-109
111. Yu YA, Galanis C, Woo Y, Chen N, Zhang Q, Fong Y. et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol Cancer Ther. 2009;8:141-51
112. Binz E, Berchtold S, Beil J, Schell M, Geisler C, Smirnow I. et al. Chemovirotherapy of Pancreatic Adenocarcinoma by Combining Oncolytic Vaccinia Virus GLV-1h68 with nab -Paclitaxel Plus Gemcitabine. Mol Ther Oncolytics. 2017;6:10-21
113. de Leeuw OS, Koch G, Hartog L, Ravenshorst N, Peeters BPH. Virulence of Newcastle disease virus is determined by the cleavage site of the fusion protein and by both the stem region and globular head of the haemagglutinin-neuraminidase protein. J Gen Virol. 2005;86:1759-69
114. Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16:1-15
115. Hotte SJ, Lorence RM, Hirte HW, Polawski SR, Bamat MK, Oneil JD. et al. An Optimized Clinical Regimen for the Oncolytic Virus PV701. Clin Cancer Res. 2007;13:977-85
116. Buijs PR, van Eijck CH, Hofland LJ, Fouchier RA, van den Hoogen BG. Different responses of human pancreatic adenocarcinoma cell lines to oncolytic Newcastle disease virus infection. Cancer Gene Ther. 2014;21:24-30
117. Buijs P, van Nieuwkoop S, Vaes V, Fouchier R, van Eijck C, van den Hoogen B. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses. 2015;7:2980-98
118. May V, Berchtold S, Berger A, Venturelli S, Burkard M, Leischner C. et al. Chemovirotherapy for pancreatic cancer: Gemcitabine plus oncolytic measles vaccine virus. Oncol Lett. 2019;18:5534-42
119. Bossow S, Grossardt C, Temme A, Leber MF, Sawall S, Rieber EP. et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18:598-608
120. Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3'-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol Cancer Ther. 2002;1:989-97
121. Woo Y, Kelly KJ, Stanford MM, Galanis C, Chun YS, Fong Y. et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15:2329-35
122. Chan WM, Rahman MM, Mcfadden G. Oncolytic myxoma virus: the path to clinic. Vaccine. 2013;31:4252-8
123. Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma Virus Sensitizes Cancer Cells to Gemcitabine and Is an Effective Oncolytic Virotherapeutic in Models of Disseminated Pancreatic Cancer. Mol Ther. 2012;20:759-68
124. Hastie E, Grdzelishvili VZ. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol. 2012;93:2529-45
125. Wollmann G, Rogulin V, Simon I, Rose JK, Van Den Pol AN. Some Attenuated Variants of Vesicular Stomatitis Virus Show Enhanced Oncolytic Activity against Human Glioblastoma Cells relative to Normal Brain Cells. J Virol. 2010;84:1563-73
126. Hastie E, Besmer DM, Shah NR, Murphy AM, Moerdyk-Schauwecker M, Molestina C. et al. Oncolytic Vesicular Stomatitis Virus in an Immunocompetent Model of MUC1-Positive or MUC1-Null Pancreatic Ductal Adenocarcinoma. J Virol. 2013;87:10283-94
127. Rahal A, Musher B. Oncolytic viral therapy for pancreatic cancer. J Surg Oncol. 2017;116:94-103
128. Pantelidou C, Cherubini G, Lemoine NR, Halldén G. The E1B19K-deleted oncolytic adenovirus mutant Ad Delta 19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget. 2016;7:15703-24
129. Goldstein D, Carroll S, Apte M, Keogh G. Modern management of pancreatic carcinoma. Intern Med J. 2004;34:475-81
130. Gujar SA, Clements D, Lee PW. Two is better than one: Complementing oncolytic virotherapy with gemcitabine to potentiate antitumor immune responses. Oncoimmunology. 2014;3:e27622
131. Davola ME, Mossman KL. Oncolytic viruses: how "lytic" must they be for therapeutic efficacy? Oncoimmunology. 2019;8:e1581528
132. Colunga AG, Laing JM, Aurelian L. The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther. 2010;17:315-27
133. Furukawa Y, Takasu A, Yura Y. Role of autophagy in oncolytic herpes simplex virus type 1-induced cell death in squamous cell carcinoma cells. Cancer Gene Ther. 2017;24:393-400
134. Saga K, Kaneda Y. Oncolytic Sendai virus-based virotherapy for cancer: recent advances. Oncolytic Virother. 2015;4:141-7
135. Liao Y, Wang HX, Mao X, Fang H, Wang H, Li Y. et al. RIP1 is a central signaling protein in regulation of TNF-α/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection. Oncotarget. 2017;8:43201-17
136. Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L. et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887-92
137. Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T. et al. Tumor Infection by Oncolytic Reovirus Primes Adaptive Antitumor Immunity. Clin Cancer Res. 2008;14:7358-66
138. Ljunggren H, Kärre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med. 1985;162:1745-59
139. Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675-8
140. Haanen JBAG. Converting Cold into Hot Tumors by Combining Immunotherapies. Cell. 2017;170:1055-6
141. Kaufman HL, Kim DW, Deraffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and Distant Immunity Induced by Intralesional Vaccination with an Oncolytic Herpes Virus Encoding GM-CSF in Patients with Stage IIIc and IV Melanoma. Ann Surg Oncol. 2010;17:718-30
142. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707-23
143. Shi X, Liu S, Kleeff J, Friess H, Büchler MW. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology. 2002;62:354-62
144. Wu J, Zhu Y, Xu C, Xu H, Zhou X, Yang J. et al. Adenovirus-mediated p53 and ING4 gene co-transfer elicits synergistic antitumor effects through enhancement of p53 acetylation in breast cancer. Oncol Rep. 2016;35:243-52
145. Nagahama Y, Ishimaru M, Osaki M, Inoue T, Maeda A, Nakada C. et al. Apoptotic pathway induced by transduction of RUNX3 in the human gastric carcinoma cell line MKN-1. Cancer Sci. 2007;99:23-30
146. Li M, Zhu Y, Zhang H, Li L, He P, Xia H. et al. Delivery of inhibitor of growth 4 (ING4) gene significantly inhibits proliferation and invasion and promotes apoptosis of human osteosarcoma cells. Sci Rep. 2014;4:7380
147. Di Piazza M, Mader C, Geletneky K, Herrero YCM, Weber E, Schlehofer J. et al. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J Virol. 2007;81:4186-98
148. Zhang X, Liu Q, Liao Q, Zhao Y. Pancreatic Cancer, Gut Microbiota, and Therapeutic Efficacy. J Cancer. 2020;11:2749-58
149. Thomas H. Pancreatic cancer: Intra-tumour bacteria promote gemcitabine resistance in pancreatic adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2017;14:632
150. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156-60
151. Lehouritis P, Cummins J, Stanton M, Murphy CT, McCarthy FO, Reid G. et al. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci Rep. 2015;5:14554
152. Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018;6:92
153. Vande Voorde J, Sabuncuoglu S, Noppen S, Hofer A, Ranjbarian F, Fieuws S. et al. Nucleoside-catabolizing enzymes in mycoplasma-infected tumor cell cultures compromise the cytostatic activity of the anticancer drug gemcitabine. J Biol Chem. 2014;289:13054-65
154. Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjonneland A. et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut. 2013;62:1764-70
155. Del Castillo E, Meier R, Chung M, Koestler DC, Chen T, Paster BJ. et al. The Microbiomes of Pancreatic and Duodenum Tissue Overlap and Are Highly Subject Specific but Differ between Pancreatic Cancer and Noncancer Subjects. Cancer Epidemiol Biomarkers Prev. 2019;28:370-83
156. Delitto D, Delitto AE, Divita BB, Pham K, Han S, Hartlage ER. et al. Human Pancreatic Cancer Cells Induce a MyD88-Dependent Stromal Response to Promote a Tumor-Tolerant Immune Microenvironment. Cancer Res. 2017;77:672-83
157. Kelly MG, Alvero AB, Chen R, Silasi D, Abrahams VM, Chan S. et al. TLR-4 Signaling Promotes Tumor Growth and Paclitaxel Chemoresistance in Ovarian Cancer. Cancer Res. 2006;66:3859-68
158. Hiroshima Y, Zhang Y, Murakami T, Maawy A, Miwa S, Yamamoto M. Efficacy of tumor-targeting Salmonella typhimurium A1-R in combination with anti-angiogenesis therapy on a pancreatic cancer patient-derived orthotopic xenograft (PDOX) and cellline mouse models. Oncotarget. 2014;5:12346-57
159. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W. et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178:795-806.e12
160. Chen SM, Chieng WW, Huang SW, Hsu LJ, Jan MS. The synergistic tumor growth-inhibitory effect of probiotic Lactobacillus on transgenic mouse model of pancreatic cancer treated with gemcitabine. Sci Rep. 2020;10:20319
161. Wörmann SM, Diakopoulos KN, Lesina M, Algül H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33:2956-67
162. Kita A, Fujiya M, Konishi H, Tanaka H, Kashima S, Iwama T. et al. Probioticderived ferrichrome inhibits the growth of refractory pancreatic cancer cells. Int J Oncol. 2020;57:721-32
163. Nomura T, Tsuchiya Y, Nashimoto A, Yabusaki H, Takii Y, Nakagawa S. et al. Probiotics reduce infectious complications after pancreaticoduodenectomy. Hepato-gastroenterology. 2007;54:661-3
164. Panebianco C, Adamberg K, Adamberg S, Saracino C, Jaagura M, Kolk K. et al. Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In vitro and In vivo Pancreatic Cancer Models. Nutrients. 2017;9:331
165. Gori S, Inno A, Belluomini L, Bocus P, Bisoffi Z, Russo A. et al. Gut microbiota and cancer: How gut microbiota modulates activity, efficacy and toxicity of antitumoral therapy. Crit Rev Oncol Hematol. 2019;143:139-47
166. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14:356-65
167. Panebianco C, Adamberg K, Jaagura M, Copetti M, Fontana A, Adamberg S. et al. Influence of gemcitabine chemotherapy on the microbiota of pancreatic cancer xenografted mice. Cancer Chemother Pharmacol. 2018;81:773-82
168. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M. et al. Diversity of the Human Intestinal Microbial Flora. Science. 2005;308:1635-8
169. Ley RE, Backhed F, Turnbaugh PJ, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070-5
170. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027-31
171. Arthur JC, Perezchanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120-3
172. Shin N, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496-503
173. da Rocha Lapa F, da Silva MD, de Almeida Cabrini D, Santos AR. Anti-inflammatory effects of purine nucleosides, adenosine and inosine, in a mouse model of pleurisy: evidence for the role of adenosine A2 receptors. Purinergic Signal. 2012;8:693-704
174. Haskó G, Kuhel D G, Németh Z H, Mabley J G, Stachlewitz R F, Virág L. et al. Inosine inhibits inflammatory cytokine production by a posttran-scriptional mechanism and protects against endotoxin-induced shock. J Immunol. 2000;164:1013-9
175. Imai H, Saijo K, Komine K, Otsuki Y, Ohuchi K, Sato Y. et al. Antibiotic therapy augments the efficacy of gemcitabine-containing regimens for advanced cancer: a retrospective study. Cancer Manag Res. 2019;11:7953-65
176. Corty RW, Langworthy BW, Fine JP, Buse JB, Sanoff HK, Lund JL. Antibacterial Use Is Associated with an Increased Risk of Hematologic and Gastrointestinal Adverse Events in Patients Treated with Gemcitabine for Stage IV Pancreatic Cancer. Oncologist. 2020;25:579-84
177. Vétizou M, Pitt J M, Daillère R, Lepage P, Waldschmitt N, Flament C. et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079-84
178. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D. et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science. 2013;342:971-6
179. Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106:14728-33
Author contact
![]() Corresponding authors: Xian-Yun Yu, Shanghai Pancreatic Cancer Institute, No. 270 Dong'An Road, Shanghai 200032, China; Tel.: +86-021-6417-5590; E-mail: yuxianjunorg; Si Shi, Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, No. 270 Dong'An Road, Shanghai 200032, China; Tel.: +86-021-6403-1446; E-mail: shisiorg.
Corresponding authors: Xian-Yun Yu, Shanghai Pancreatic Cancer Institute, No. 270 Dong'An Road, Shanghai 200032, China; Tel.: +86-021-6417-5590; E-mail: yuxianjunorg; Si Shi, Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, No. 270 Dong'An Road, Shanghai 200032, China; Tel.: +86-021-6403-1446; E-mail: shisiorg.