Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Novelty and Impact
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(11):2756-2769. doi:10.7150/ijbs.59262 This issue Cite
Research Paper
C. tropicalis promotes chemotherapy resistance in colon cancer through increasing lactate production to regulate the mismatch repair system
Junxing Qu1,2, Zhiheng Sun1,2, Chen Peng1,2, Daoqian Li1,2, Wenyue Yan3, Zhen Xu1,2, Yayi Hou1,2, Sunan Shen1,2 ![]() , Ping Chen3
, Ping Chen3 ![]() , Tingting Wang1,2
, Tingting Wang1,2 ![]()
1. The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093.
2. Jiangsu Key Laboratory of Molecular Medicine, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093.
3. Department of Oncology, Yancheng First Hospital, Affiliated Hospital of Nanjing University Medical School, The First People's Hospital of Yancheng, Yancheng, Jiangsu, China, 224001.
Received 2021-2-9; Accepted 2021-5-22; Published 2021-7-2
Abstract

Due to chemotherapeutic drug resistance, tumor recurrence is common in patients with colorectal cancer (CRC) and chemo-resistant patients are often accompanied by defects in the mismatch repair system (MMR). Our previous study has shown that Candida tropicalis (C. tropicalis) is closely related to the occurrence and development of colorectal cancer, but whether this conditional pathogenic fungus is involved in chemotherapy needs further investigation. Here we found that C. tropicalis promoted chemotherapy resistance of colon cancer to oxaliplatin. Compared with oxaliplatin-treated group, the expression of functional MMR proteins in tumors were decreased in C.tropicalis/oxaliplatin -treated group, while the glycolysis level of tumors was up-regulated and the production of lactate was significantly increased in C.tropicalis/oxaliplatin -treated group. Inhibiting lactate production significantly alleviated the chemoresistance and rescued the decreased expression of MMR caused by C. tropicalis. Furthermore, we found that lactate down-regulated the expression of MLH1 through the GPR81-cAMP-PKA-CREB axis. This study clarified that C. tropicalis promoted chemoresistance of colon cancer via producing lactate and inhibiting the expression of MLH1, which may provide novel ideas for improving CRC chemotherapy effect.
Keywords: Candida. tropicalis, chemoresistance, glycolysis, lactate, mismatch repair
Introduction
Colorectal cancer (CRC) is the third most common cancer and the third leading cause of death in the world [1], which threatens human health and life safety. Current cancer treatments include chemotherapy, surgical treatment and immunotherapy [2, 3]. The cytotoxic drugs commonly used in the therapy of colon cancer are oxaliplatin, 5-FU and capecitabine [4]. Oxaliplatin, which is a kind of platinum, can form DNA-platinum crosslinks by covalently binding DNA to block the G2 phase, thereby suppressing tumor cell proliferation [5]. Although patients with colorectal cancer can respond to chemotherapy at the beginning, they eventually experience relapse due to drug resistance [4]. Therefore, it is very important to elucidate the mechanism of chemotherapy resistance in CRC patients.
Numerous underlying mechanisms conferring chemotherapy resistance have been elaborated in the past decades. Studies have reported that overexpression of ATP-binding cassette (ABC) transporters to enhance the efflux of anticancer medicines or escape chemotherapy-induced apoptosis can promote CRC chemotherapy resistance [6]. Furthermore, a recent study discovered that tumor cells can evade chemotherapy by down-regulating the function of the mismatch repair system (MMR) to accumulate genetic mutations [7]. MMR plays a prominent role in the correction of replicative mismatches [8]. It is thought to involve certain functional proteins hMLH1, hMSH2, and so on [9]. Studies have proved that loss of MMR could impair the ability to detect DNA damage and produce more mutations in tumor cells, which makes it more possible for tumor cells to improve adaptive variability and develop resistance to chemotherapeutics [8, 10, 11]. Clinically, loss of MMR is a significant mechanism of resistance to many important drugs.
Accumulating evidence suggests that the gut microbiota including bacteria, fungi and virus plays a vital role in the cancer context. Studies have confirmed the association of Peptostreptococcus stomatis, Parvimonas micra and Solobacterium moorei with CRC [12]. Microbial pathogens have been detected inside human tumor tissues and shown to promote cancer progression and resistance to anti-tumor therapies [13]. Studies have shown that Fusobacterium significantly increased and could promote chemotherapy resistance in the course of CRC while other studies found that Bifidobacterium enhanced the anti-tumor effects [4, 13]. Studies have found that intratumoral accumulation of Bifidobacterium facilitated CD47-based immunotherapy via STING signaling, and that the diversity and abundance of Bifidobacterium influenced the therapeutic outcome of blockade of the PD-1/PD- L1 axis [13, 14]. However, most studies of commensal microbes focused on bacteria, commensal fungi have been less reported. Our previous study proved that the burden of C. tropicalis, which is a kind of conditional pathogenic fungus, is significantly elevated in both CRC patients and colon cancer animal models. C. tropicalis aggravated CRC by inducing the differentiation of myeloid-derived suppressor cells (MDSCs) and inhibiting the function of T cells [15]. However, the potential effect of commensal fungi on chemotherapy is not examined in human literature.
In this study, we tested whether C. tropicalis had an effect on chemotherapy in the development of CRC. We have found that C. tropicalis can facilitate the glycolysis of tumor cells to produce lactate, which then inhibit the function of MMR system and promote the chemotherapy resistance of CRC to oxaliplatin.
Materials and methods
Reagents and Antibodies
The C. tropicalis strain (W4162870) was kindly provided by Dr. Sarah L. Gaffen (University of Pittsburgh, PA). Oxaliplatin(S1224), Forskolin (S2449) and Sodium Oxamate (S6871) were purchased from Selleck Chemicals (Houston, Texas, USA). Lactate was purchased from Sigma (St. Louis, MO, USA). Antibody against MLH1 (#3515), MSH2 (#2017), Cleaved caspase3 (#9664), caspase9 (#9508), p-H2AX (Ser139, #9718), PFKFB3 (#13123), LDHA (#3582), PKM2 (#4053), PGAM1 (#12098), GPI (#57893), GLUT1(#12939), ALDOA (#8060), HK2(#2867), PKAC-α(#4782) and p-CREB (Ser133, #9198)were purchased from Cell Signaling Technology (Boston, MO, USA). Antibody against β-tublin (# 66240-1-Ig) was purchased from Proteintech (Wuhan, Hubei, P.R.C). Antibody against GPR81 (YN2554) was purchased from Immunoway (Plano, TX, USA). CCK8 Kit (CK04) was purchased from Dojindo Laboratories (Japan). Annexin V FITC/PI Apoptosis Kit (A211-01) was purchased from Vazyme Biotech (Nanjing, China). F4/80-APC (#123115), CD11b-FITC (#301330), Ly6G-PE (#127607), Ly6C-APC (#128016), Gr-1-FITC (#108405) were all from Biolegend (San Diego, CA, USA). Lactate Assay Kit I (#120001100P) and Glucose Assay Kit I (#120003100P) were purchased from Eton Bioscience (San Diego, USA). Cyclic AMP Complete ELISA Kit (ab133051) was purchased from abcam (Cambridge, England).
Mice
For CRC xenograft experiments, four-week-old male BALB/c nude mice were purchased from Model Animal Research Center of Nanjing University and housed in laminar flow cabinets under specific pathogen-free conditions. Colorectal cancer cells (1 × 107 SW480 cells) were injected subcutaneously into the right axilla of each mouse. Six days after subcutaneous inoculation, mice were randomly divided into different groups (n=6 each group). C. tropicalis was given by multipoint intratumoral injection, twice per week for three weeks. Oxaliplatin (10 mg/kg) and Sodium oxamate (500 mg/kg) were administered by intraperitoneal injection, twice per week for three weeks. The length and width of the tumors were measured every three days. After three weeks, all mice were sacrificed and subcutaneous tumors were collected and weighed. All animal procedures were performed in accordance with the “National Institutes of Health Guidelines for the Care and Use of Laboratory Animals” and were approved by the Institutional Animal Care and Use Committee of Nanjing University Medical School.
Fungi Strain and Growth Conditions
The Candida tropicalis strain (W4162870) was kindly provided by Dr. Sarah L. Gaffen (University of Pittsburgh, PA). C. tropicalis was cultured overnight at 30 °C in liquid Sabouraud Dextrose Medium and shake cultivation at 220 rpm/min to expand.
TUNEL Assay
Tumor cell apoptosis in the xenograft tumor tissues were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) technology according to standard procedures.
Immunohistochemistry
Mice tumor sections were obtained from CRC xenograft models and formalin fixed, paraffin-embedded, and processed according to standard procedures. Sections were analyzed with the following antibodies: anti-MLH1 (1:100, Proteintech, Wuhan, China); anti-MSH2 (1:200, CST, USA) and PCNA (1:200, CST, USA).
RNA Extraction and Quantitative PCR
Total RNA was extracted from CRC cells using TRIzol reagent, and reverse transcriptions were performed in a 20 µL mixture with1 pg-1 µg of total RNA according to the manufacturer's instructions (Vazyme company, China). The oligonucleotide primers used for Quantitative real-time PCR amplification are listed in Table 1. PCR amplification consisted of 45 cycles of denaturation at 95 °C for 2 min, annealing at 60 °C for 30s, and extension at 72 °C for 30s on an Applied Biosystem Viia 7 quantitative PCR system (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed by using SYBR Green with an ABI Step One Plus system (Life Technologies). All reactions were run in triplicate. The gene expression levels were normalized to β-tublin.
Primer sequence
| Gene | Sense (5′-3′) | Anti-sense (3′-5′) |
|---|---|---|
| MLH1 | CAGAGCTTGGAGGGGGATA | TTTCGGGAATCATCTTCCAC |
| MSH2 | AGGCATCCAAGGAGAATGATTG | GGAATCCACATACCCAACTCCAA |
| GLUT1 | GGCCAAGAGTGTGCTAAAGAA | ACAGCGTTGATGCCAGACAG |
| HK2 | GAGCCACCACTCACCCTACT | CCAGGCATTCGGCAATGTG |
| GPI | CAAGGACCGCTTCAACCACTT | CCAGGATGGGTGTGTTTGACC |
| PFKFB3 | TTGGCGTCCCCACAAAAGT | AGTTGTAGGAGCTGTACTGCTT |
| ALDOA | ATGCCCTACCAATATCCAGCA | GCTCCCAGTGGACTCATCTG |
| PKM2 | ATGTCGAAGCCCCATAGTGAA | TGGGTGGTGAATCAATGTCCA |
| LDHA | ATGGCAACTCTAAAGGATCAGC | CCAACCCCAACAACTGTAATCT |
| PGAM1 | GTGCAGAAGAGAGCGATCCG | CGGTTAGACCCCCATAGTGC |
| GPR81 | AATTTGGCCGTGGCTGATTTC | CCGTAAGGAACACGATGCTCC |
| β-actin | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
Western Blot
The protein samples were obtained from lysis buffer-treated tumor tissues (30mg tumor section added 300ul lysis buffer and homogenized with 3 steel beads) or cells. Lysates were put on ice for 20 min and then centrifuged at14,000 × g for 10 min. Subsequently, BCA kit (Beyotime Biotechnology) was used to detect the protein concentration and 30 µg of protein per lane was separated on 10% and 15% polyacrylamide gels and transferred on to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween 20, and then the membranes were incubated with specific antibodies. The values were normalized to the β-tublin intensity levels.
Nuclear protein extraction
A nuclear protein extraction kit was purchased from Beyotime Biotechnology (Wuhan, China) and used according to the manufacturer's instructions.
Cell culture
Human colorectal cancer cell line SW480 and HCT116 were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and were cultured in RPMI-1640 medium (GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 1% (100 U/mL penicillin and 100 ug/mL streptomycin) at 37 °C in a humidified 5% CO2 atmosphere. Cells were seeded onto different types of plates for further experiments when the cell density reached ~80%.
Cell viability assay, apoptosis detection and migration experiment
To assess the effect of C. tropicalis and lactate on cell viability, a CCK-8 assay was used according to the manufacturer's instructions (Dojindo Laboratories, Japan). CRC cells were seeded onto 96-well plates at a concentration of ~5 × 104 cells/well with 100 μL culture medium. Different concentrations of C. tropicalis and lactate were used to treat cells for different length of time. The 10 μL of CCK-8 solution was added to the cells at specific time points and cells were incubated for 2 hr at 37 °C.
Apoptosis was examined by flow cytometric analysis. An Annexin V FITC/PI double stain assay (Vazyme, China) was performed following the manufacturer's protocol. Cell migration was measured by wound healing assay. CRC cells were divided into plates and the head of the gun was used to line with a ruler next day, then the cells were washed gently with PBS for 3 times. The cells were cultured in serum-free medium and photographed in certain times.
ELISA
The concentration of lactate and glucose in cell supernatant and mice serum were detected using the corresponding enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's instructions (Eton Bioscience), and cyclic AMP complete in the cell was detected using the corresponding mouse enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's instructions (abcam).
Immunofluorescence
Cultured cells were seeded on glass coverslips in six-well plates. After three PBS washes, the samples were fixed for 10 min at room temperature with 4% paraformaldehyde. Fixed cells were rinsed with PBS and then incubated for 10 min at 4 °C with 0.2%Triton X-100. Following permeabilization, non-specific binding in the cells was blocked by 5% BSA in PBS for 1 h at room temperature. Cell samples were incubated with anti-p-H2AX, anti-PKA Cα, and anti-p-CREB primary antibodies at a1:200 dilution for 2 h at room temperature. Samples were further incubated with Alexa 647-conjugated secondary antibody at a 1:400 dilution for 1.5 h in the dark. After washed with PBS, the nuclei were stained by DAPI. Slides were visualized using a confocal laser scanning microscope (FV3000, Olympus).
siRNA Transfection
siRNA was transfected according to the product instructions (Ruibo company, China). The concentration of siRNA used in the study was 50 nM. The GPR81 siRNA target sequence is CTGCTAGACTCTATTTCCT. The target sequence of non-coding (NC) siRNA was a random sequence with no biological effects.
Flow cytometric analysis
All splenocytes were flushed out from spleen by 1× PBS and then passed through a 200-mesh sieve to obtain single cell suspension. For flow cytometric detection, the following anti-mouse antibodies (all from Biolegend, San Diego, CA, US) were used: F4/80-APC, CD11b-FITC, Ly6G-PE, Ly6C-APC, Gr-1-FITC. Cells were detected using a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Data were analyzed using FlowJo software (Treestar, Inc., San Carlos, CA).
Statistical Analysis
Statistical analysis was conducted in GraphPad Prism 7. A two-tailed Student's t-test was employed to analyze statistical significance between two groups. Data are shown as means ± SD. P < 0.05 was considered to be statistically significant.
Results
C. tropicalis promotes the chemotherapy resistance to oxaliplatin in colon cancer
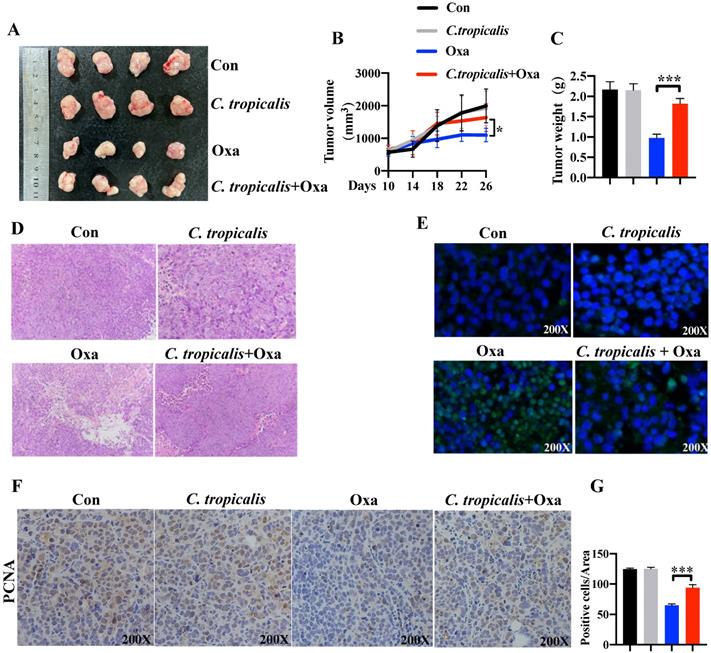
To explore the potential role of C. tropicalis on chemotherapy effect, oxaliplatin and C. tropicalis were injected into mice intraperitoneally and intratumorally in separately to establish the SW480 cells-induced xenograft CRC mouse model (Figure S1A). We found oxaliplatin treatment inhibited tumor growth, presenting as decreased tumor volume and tumor weight. C. tropicalis treatment significantly blocked the inhibitory effect of oxaliplatin (Figure 1A-C). However, C. tropicalis treated alone has no effect on tumor growth. Moreover, HE staining and analysis showed that tumors in oxaliplatin and C. tropicalis co-treatment mice had higher histological scores than those in oxaliplatin-treated mice (Figure 1D). TUNEL assay showed that C. tropicalis inhibited the tumor apoptosis caused by oxaliplatin (Figure 1E). In consistence with that, IHC staining showed more PCNA positive cells in tumors tissues from co-treatment mice than those from oxaliplatin-treated mice (Figure 1F-G). We also detected the proportion of immune cells in spleen of the tumor-bearing mice. As shown in Figure S1B, compared to oxaliplatin simply-treated mice, more proportion of macrophage, myeloid-derived suppressor cells (MDSCs) and G-MDSCs but lower frequency of M-MDSCs were found in co-treatment tumor-bearing mice.
C. tropicalis promotes the chemotherapy resistance of colon cancer to oxaliplatin in mouse xenograft tumor model. Mice were treated as described in Figure S1A. Tumors were acquired. (A) Representative data of tumors in mice under different conditions. (B-C) Statistical analysis of tumor volumes (B) and weights (C) in different groups, n=4/group. (D) Histological analysis of tumors was shown by hematoxylin and eosin staining. Tumors were microscopically analyzed. (E) TUNEL assays were performed to detect tumor cell apoptosis in tumor tissues. (F and G) Representative images of immunohistochemical staining of tumors in different groups for PCNA. Positive cells of PCNA were counted using Image-Pro Plus software 6.0. Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p<0.001 as determined by unpaired Student's t test.
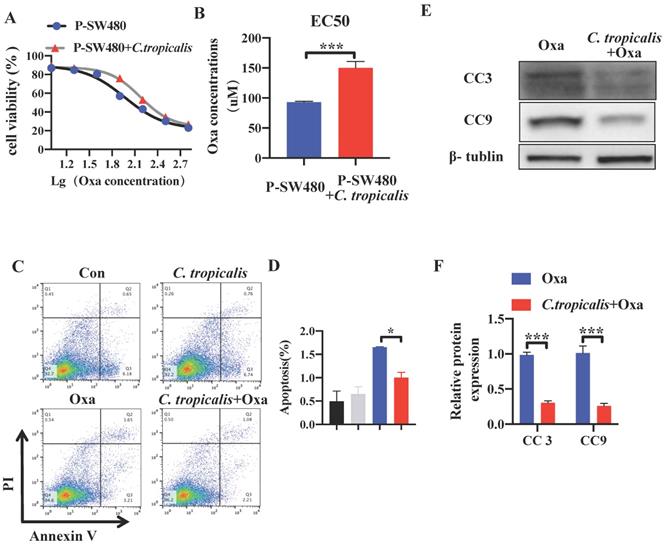
The effect of C. tropicalis on chemotherapy resistance was further confirmed in vitro. Although C. tropicalis (MOI = 1) itself had no effect on proliferation, migration and apoptosis in SW480 cells (Figures S1C-F). We compared cell viability among the parental SW480 cells and the C. tropicalis-co-cultured parental SW480 cells in the presence of different concentrations of oxaliplatin (Oxa). We found that cell viability was decreased upon oxaliplatin treated (Figure 2A). The EC50 value of oxaliplatin was higher in C. tropicalis-cultured SW480 cells, compared with parental SW480 cells (Figure 2B). Then we examined the inhibitory role of C. tropicalis in oxaliplatin-induced apoptosis, presenting as decreased proportion of apoptotic cells (Figure 2C-D) and decreased protein expression of cleaved-caspase 3 (CC3) and cleaved-caspase 9 (CC9) (Figure 2E-F) upon C. tropicalis treatment. These results suggested that C. tropicalis promotes the chemotherapy resistance to oxaliplatin in both xenograft CRC mice and SW480 cancer cells.
C. tropicalis promotes the chemotherapy resistance in SW480 cells. SW480 cells were treated with oxaliplatin in the presence or absence of C. tropicalis. (A) Cell viability was detected by using CCK8 assay. (B) The EC50 value in different SW480 cells was calculated. (C-D) Proportion of apoptotic cells was detected by flow cytometry. (E-F) Cleaved caspases 3(CC3) and 9 (CC9) expression were detected by western blot. Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p<0.001as determined by unpaired Student's t test.
MMR is inhibited in C. tropicalis-induced CRC chemotherapy resistance
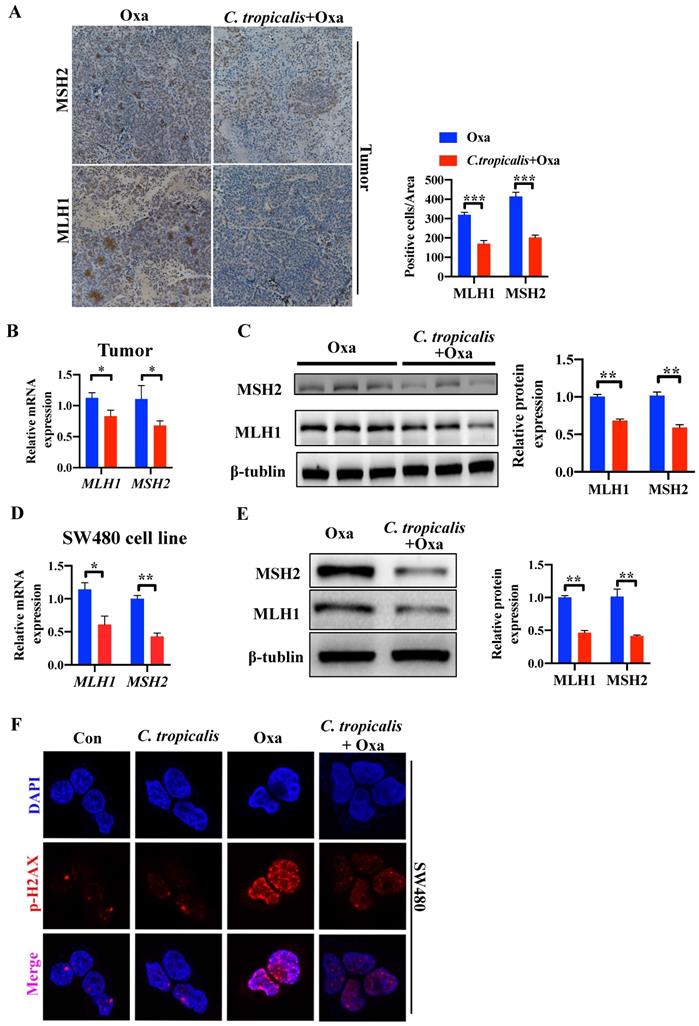
In our preliminary experiment, oxaliplatin treatment could significantly inhibit tumors in mice bearing SW480 tumor cells. However, oxaliplatin treatment had less inhibitory effect in mice bearing HCT116 tumor cells (Figure S2A-C). Several studies have shown that HCT116 is an MMR-deficient cell line while SW480 is an MMR-proficient cell line, and that loss of MMR leads to increased adaptive variability and chemoresistance in CRC [10, 16-18]. Therefore, we detected the protein and mRNA expression of functional proteins in MMR. Compared with oxaliplatin-treated mice, C. tropicalis injection led to lower expression of MLH1 and MSH2 (Figure 3A-C). Similar result was found in SW480 cells (Figure 3D-E). Furthermore, quantification of phosphorylation of H2AX at Ser139, a common marker of DNA damage [10], suggested C. tropicalis inhibited oxaliplatin-induced DNA damage in SW480 cells (Figure 3F). These results suggest the expression of MMR functional proteins was inhibited upon C. tropicalis treatment.
MMR is inhibited upon C. tropicalis treatment. (A-C) Mice were treated as described in Figure S1A. Tumors were acquired. Tumor tissues were stained for MLH1 and MSH2. The percentages of MLH1 and MSH2-positive tumor cells were quantified (A). mRNA expression of MLH1 and MSH2 in tumor tissues were detected by qPCR (B). Protein expression of MLH1 and MSH2 were detected by western blot (C). (D-F) SW480 cells were treated with oxaliplatin in the presence or absence of C. tropicalis. mRNA expression of MLH1 and MSH2 in cells were detected by qPCR (D). Protein levels of MLH1 and MSH2 were detected by western blot (E). p-H2AX expression were detected by immunofluorescence (F). Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. Magnifications are 40× (scale bar, 0.05 mm) *p < 0.05, **p < 0.01 and ***p<0.001 as determined by unpaired Student's t test.
C. tropicalis promotes glycolysis and production of lactate in colon cancer
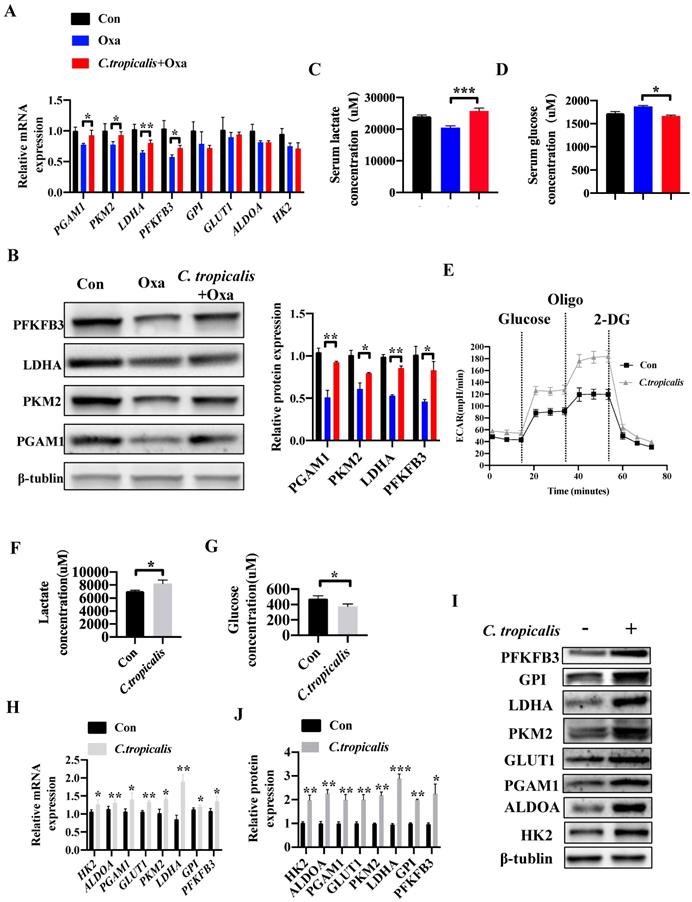
Since “Warburg effect” is emerging as a symbol of cancer and a potential cause of chemotherapy resistance [19-24]. We next explored whether C. tropicalis induced chemoresistance through regulating the level of glycolysis. Compared to oxaliplatin-treated mice, combination of C. tropicalis up-regulated the mRNA expression of pivotal glycolysis-related enzymes in tumor tissues, including Pgam1, Pkm2, Ldha and Pfkfb3 (Figure 4A). Similar results were found in the protein levels of pivotal glycolytic enzymes (Figure 4B). Compared with oxaliplatin-treated mice, production of lactate in the serum was significantly increased in oxaliplatin and C. tropicalis co-treated mice while glucose concentration in the serum was decreased in co-treated mice (Figure 4C-D). We also detected the role of C. tropicalis on glycolysis pathway in colon cancer cells. Consistently, seahorse extracellular flux analysis showed that SW480 treated with C. tropicalis exhibited significantly higher glycolytic capacity than parental SW480 (Figure 4E). Lactate production and glucose consumption were also enhanced in SW480 cells upon C. tropicalis treatment (Figure 4F-G). C. tropicalis also up-regulated expression of vital glycolysis-related enzymes in SW480 cells in both mRNA and protein levels (Figure 4H-J). Therefore, these data suggest C. tropicalis promotes glycolysis and production of lactate in colon cancer.
C. tropicalis promotes glycolysis and production of lactate in colon cancer. (A-D) Mice were treated as described in Figure S1A. Tumors were acquired. mRNA expression of glycolysis-related enzymes in tumor tissues were detected using qPCR(A). Protein levels of glycolysis-related enzymes were detected using western blot (B). Lactate concentration in serum was detected by ELISA (C). Glucose concentration in serum was measured by ELISA (D). (E-J) SW480 cells were treated with or without C. tropicalis. The ECAR of SW480 cells was measured with a seahorse analyzer (E). Lactate level in cell supernatant was detected by ELISA (F). Glucose concentration in cell supernatant was detected by ELISA (G). mRNA expressions of key glycolysis-related enzymes were detected by qPCR (H). Protein expressions of key glycolysis-related enzymes were detected by western blot (I-J). Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p<0.001 as determined by unpaired Student's t test.
Inhibiting lactate attenuates C. tropicalis-induced chemoresistance to oxaliplatin
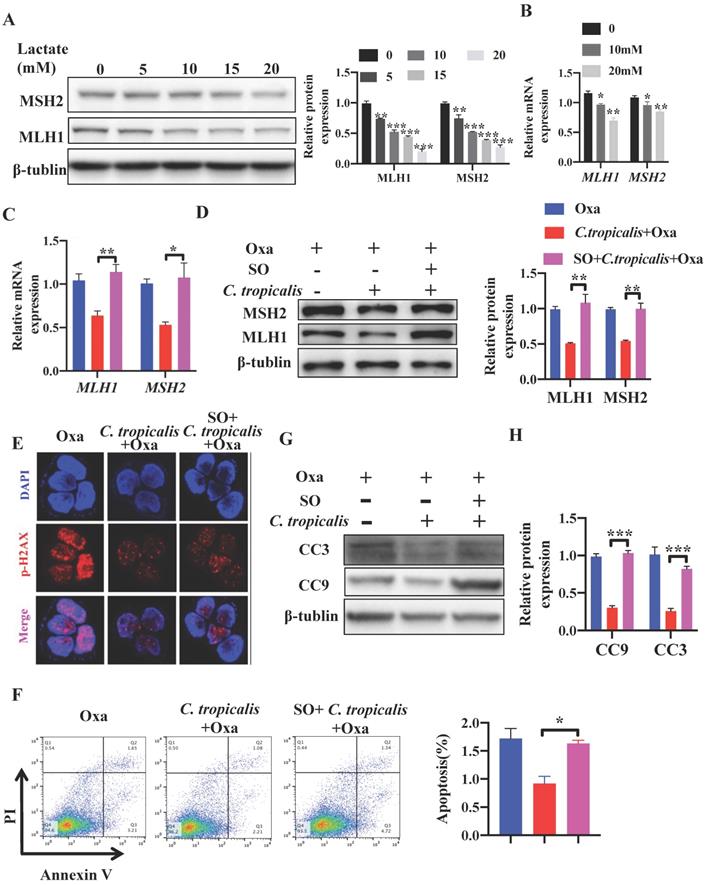
Studies have reported that lactate is no longer just a metabolic waste, but a crucial signaling molecule that refer to the regulation of metabolic pathways [20, 23, 25-30]. Next, we detected whether lactate contributed to the inhibition on MMR in C. tropicalis-induced chemoresistance. SW480 cells were treated with different concentrations of lactate. We found that 30 mM lactate significantly inhibited cell viability, therefore we used the concentration 0-20 mM in the following experiment (Figure S3A). Exogenous lactate triggered a dose-dependent decrease of MLH1 and MSH2, in both protein and mRNA levels (Figure 5A-B). To confirm whether lactate was critical in C. tropicalis-triggered chemoresistance, Sodium Oxamate (SO), a kind of LDHA inhibitor, was added in SW480 cells in the presence of oxaliplatin and C. tropicalis. We found that the decreased expression of MLH1 and MSH2 in SW480 cells treated with oxaliplatin and C. tropicalis was rescued due to the inhibition of LDHA (Figure 5C-D). Consistently, the decreased expression of p-H2AX was also rescued in the presence of SO (Figure 5E). Moreover, C. tropicalis-induced chemoresistance was also abolished by SO treatment in SW480 cells, presenting as increased proportion of apoptotic cells (Figure 5F) and increased expression of CC3 and CC9 (Figure 5G-H).
C. tropicalis promotes CRC chemotherapy resistance to oxaliplatin via producing lactate. (A-B) SW480 cells were treated with lactate (5-20mM), protein and mRNA expression of MLH1 and MSH2 were detected by western blot and qPCR, respectively. (C-H) SW480 cells were stimulated with oxaliplatin in the presence or absence of C. tropicalis and Sodium Oxamate (SO). mRNA expressions of MLH1 and MSH2 in SW480 cells were measured by qPCR(C). (D) Protein expression of MLH1 and MSH2 were measured by western blot. p-H2AX expression was detected by immunofluorescence (E). Proportion of apoptotic cells was detected by flow cytometry (F). Protein levels of CC3 and CC9 were detected by western blot (G and H). Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p<0.001 as determined by unpaired Student's t test.
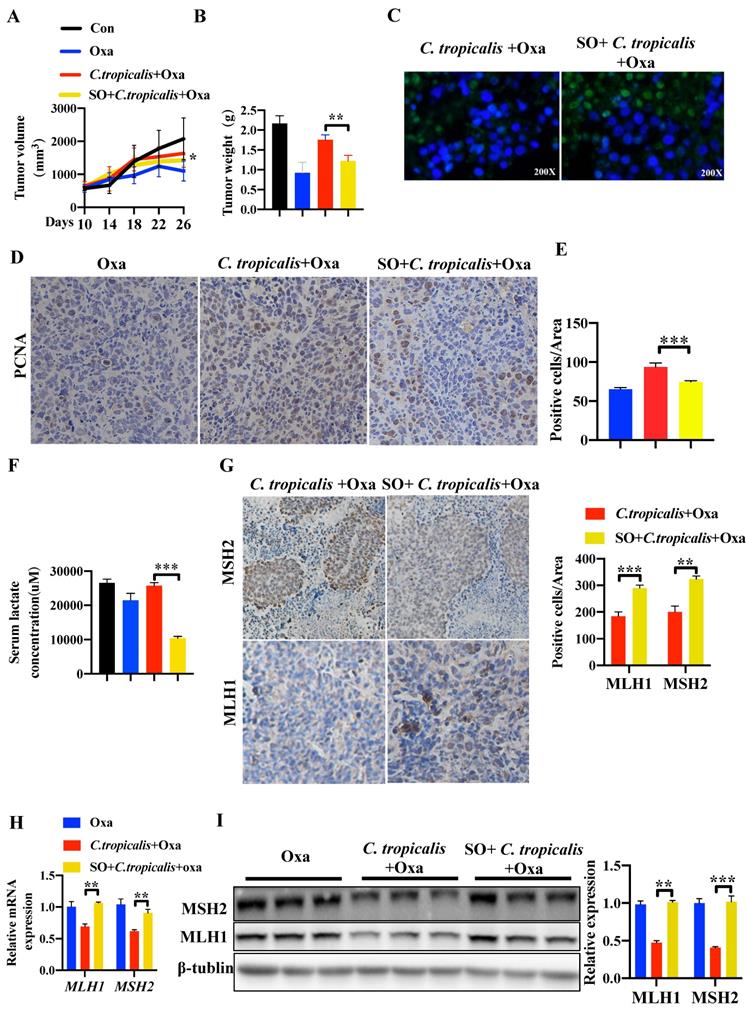
We further confirmed this critical role of lactate in tumor-bearing mouse model. SO was intraperitoneal injected into mice during oxaliplatin and C. tropicalis treatment indicated as Figure S1A. We found that SO partly recovered the effect of oxaliplatin blocked by C. tropicalis, presenting as decreased tumor volume (Figure 6A) and tumor weight (Figure 6B). TUNEL assay showed that the inhibited tumor apoptosis caused by C. tropicalis was increased in presence of SO (Figure 6C). Consistently, IHC staining showed PCNA positive cells were increased in tumors tissues due to the inhibition of lactate (Figure 6D-E). We also confirmed that lactate production was indeed inhibited in the presence of SO (Figure 6F). Furthermore, IHC showed increased staining levels in the SO co-treated group compared with the oxaliplatin and C. tropicalis-treated group in tumor tissues, suggesting that the expression of MLH1 and MSH2 was rescued by SO (Figure 6G). Similar results were found in the mRNA and protein expression of MLH1 and MSH2 (Figure 6H-I). Our results demonstrated that inhibition of lactate can restore the down-regulated MMR functional proteins and attenuate chemotherapy resistance to oxaliplatin in CRC mice caused by C. tropicalis.
Inhibiting lactate attenuates C. tropicalis-induced chemoresistance to oxaliplatin in CRC mice. Mice were treated as described in Figure S1A and were injected SO intraperitoneally additionally. Tumors were acquired. (A and B) Statistical analysis of tumor volumes and tumor weights. (C) Apoptotic cells in tumor tissues were detected by TUNEL assays. (D-E) Representative images of IHC staining of tumors in different groups for PCNA. Positive cells of PCNA were counted using Image-Pro Plus software 6.0. (F) Lactate concentration in the serum was detected by ELISA. (G) Immunohistochemical staining of tumors and statistical analysis in different groups with anti-MLH1 and anti-MSH2 antibodies. (H) mRNA expression of MLH1 and MSH2 in tumor tissues was detected by qPCR. (I) Protein expression of MLH1 and MSH2 in tumor tissues was detected by Western blot. Data with error bars are represented as mean ± SD. Magnifications are 40× (scale bar, 0.05 mm). Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p<0.001 as determined by unpaired Student's t test.
Lactate reduces the expression of MLH1 via GPR81-cAMP-PKA-CREB axis
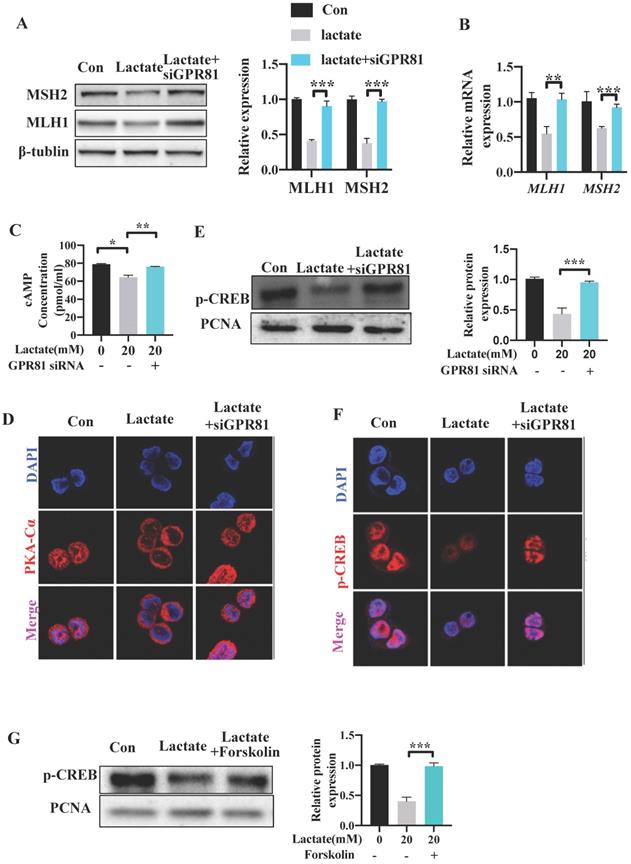
Several studies have reported that lactate can bind and activate the G-protein coupled receptor GPR81 on the cell surface in an autocrine or paracrine manner [23, 31]. To explore whether GPR81 mediates the inhibitory effect of lactate on the expression of MMR functional proteins, GPR81 was knocked down in SW480 cells by small interfering RNA siGPR81 (Figure S3B-C). Cells transfected with siGPR81 showed increased expression of MLH1 and MSH2 levels upon lactate stimulation (Figure 7A). Similar result was found in the mRNA expression of MLH1 and MSH2 (Figure 7B). Previous study has demonstrated that activation of GPR81 inhibited adenylate cyclase and results in a decreased cAMP level [32, 33]. We found a decreased level of cAMP after lactate stimulation, which was recovered after transfecting with siGPR81 (Figure 7C). The activation and nuclear translocation of protein kinase A (PKA), which is the downstream of cAMP, was also inhibited by lactate, and was recovered by siGPR81 (Figure 7D). Next, we detected phosphorylation of cAMP response element-binding protein (p-CREB), which is a direct indicator of cAMP accumulation and PKA activation [23] and is also the upstream transcriptional factor of MLH1. Indeed, as a result of attenuation of cAMP concentration, p-CREB (Ser133) was decreased in response to lactate (Figure 7E-F). Meanwhile, siGPR81 blocked the inhibitory effect of lactate on PKA and p-CREB (Figure 7D-F). Cells were further treated with forskolin, a pharmacological activator of cAMP which can induce the production of cAMP. We found that increase of cAMP can enhance the nuclear translocation of PKA and the expression of p-CREB (Figure 7G, Figure S3D-E). These data showed that lactate regulated MMR protein expression via GPR81-cAMP-PKA-CREB pathway.
Lactate reduces the expression of MLH1 via GPR81-cAMP-PKA-CREB axis. SW480 cells were transfected with GPR81 siRNA and were then treated with or without lactate. (A) Protein expression of MLH1 and MSH2 was detected by Western blot. (B) mRNA expression of MLH1 and MSH2 was detected by qPCR. (C) Intracellular cAMP concentration was determined by ELISA. (D) Immunofluorescence indicated the nucleus translocation of PKACα cells. (E-F) Protein expression of p-CREB in nuclear was detected by western blot and immunofluorescence. (G) SW480 cells were treated with lactate in the presence or absence of Forskolin. Protein expression of p-CREB in nuclear was detected by western blot. Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *p < 0.05, **p < 0.01 and ***p <0.001 as determined by unpaired Student's t test.
Discussion
In this study, we find that C. tropicalis promotes CRC chemotherapy resistance and MMR functional proteins are down-regulated in these chemo-resistant tumor cells. C. tropicalis can markedly upregulate the glycolysis flux in CRC. As a product of glycolysis, lactate secretion is therefore significantly increased. Furthermore, lactate down-regulates the expression of MLH1 and MSH2 via GPR81-cAMP-PKA-CREB axis. Of note, C. tropicalis-induced CRC chemoresistance as well as the expression of MMR system is rescued after inhibition of lactate production. Thus, our study uncovers the role of C. tropicalis in the CRC chemotherapy resistance to oxaliplatin and the mechanisms by which lactate mediates the inhibition of MLH1 and MSH2. This consequence is the first to correlate conditional fungus with CRC chemoresistance. It plays an important role in solving the problem of relapse.
Current research has shown that the intestinal flora is related to CRC. Specific types of bacteria that promote tumorigenesis have been identified [34] and metagenomic analysis of the fecal microbiome has become a biomarker for colorectal cancer [12]. Recent mouse studies have shown that the gut microbiota not only can modulate local immune responses but also affect chemotherapy [35, 36]. F. nucleatum increases in patients with CRC recurrence and can promote colon cancer chemotherapy resistance by regulating modulating autophagy [4]. Although bacteria are dominate in the microbe community, the role of fungi should not be underestimated. As for the commensal fungus, our previous studies have found C. tropicalis is specifically increased in Dectin3-/-colitis and Card9-/- CRC mice [15, 37]. However, no study has confirmed the role of commensal fungi in CRC chemoresistance until now. Here, we uncover the crucial role of C. tropicalis in the CRC chemotherapy resistance. By multipoint intratumoral injection of C. tropicalis into CRC xenograft mouse model, we provide compelling evidence that C. tropicalis activates glycolysis-related pathways to produce more lactate which down-regulates the MMR, and that C. tropicalis promotes CRC chemoresistance.
The Warburg effect is now emerging as a hallmark of cancer and an underlying cause of chemotherapy resistance [19-24]. It is mainly manifested as enhanced glucose uptake, production of lactate [21-22], and expression of rate-limiting enzymes. Studies have reported that the Warburg effect promotes drug resistance by increasing drug efflux and epigenetic alterations, mutations in drug targets, activation of survival pathways and evasion of cell death [38]. In this study, we find that C. tropicalis promotes CRC chemoresistance by activating glycolysis pathway and producing much lactate which could down-regulate MMR functional proteins for the first time. As bacteria or fungi activate TLR receptors on the cell surface, a series of cellular changes could phosphorylate downstream AMPK. Activated AMPK can elevate glycolysis by raising the expression of PFKFB3 and exert biological effects [39]. However, the specific mechanism by which C. tropicalis enhances glycolysis in CRC cells is not discussed in depth, so this needs our further investigation.
As a product of glycolysis, much researches have shown that lactate is no longer just a metabolic waste, but a crucial signaling molecule that participates in the regulation of metabolic pathways [20,23,25-30]. GPR81, as a receptor for lactate, has now been found to couple to Gi-type protein subunit and mediate lactate-induced decrease in cAMP levels. We report that lactate down-regulates intracellular cAMP levels through GPR81 receptor in an autocrine manner, thereby reducing the nucleus entry of the PKA catalytic subunit. Hence, the transcription factor CREB in the nucleus cannot be phosphorylated, leading to decreased expression of MLH1 and promotion in the chemotherapy resistance of CRC ultimately. Accordingly, biochemical inhibition of lactate, genetic GPR81 block and activation of cAMP enhance the sensitivity of C. tropicalis-treated CRC cells to oxaliplatin. In this study, lactate and GPR81 are reported to be related with MMR via cAMP-PKA-CREB axis for the first time. Because glucose metabolism is a complex network with diverse intermediate products [20], whether only lactate is involved in the regulation of MMR needs further experimental confirmation.
It is of great clinical significance to clarify whether and how C. tropicalis participates in the chemotherapy resistance in CRC patients. This reminds us that clinical detection of C. tropicalis can help advance the chemotherapy effect. For patients with higher C. tropicalis, platinum drugs can be utilized in combination with an appropriate amount of glycolysis and LDHA inhibitors or given certain anti-fungal treatment during chemotherapy. Consequently, we should quantify levels of C. tropicalis, MMR functional proteins and glycolysis components especially lactate production in chemo-resistant CRC patients in the next experiments.
Novelty and Impact
Candida tropicalis (C. tropicalis) is closely related to the occurrence and development of colorectal cancer (CRC), but whether this conditional pathogenic fungus is involved in chemoresistance remains largely unknown. Here, we find that C. tropicalis promotes CRC chemoresistance both in vivo and in vitro. C. tropicalis increases glycolysis flux and induces lactate production in colon cancer. Lactate, in turn, down-regulates the expression of MLH1 through the GPR81-cAMP-PKA-CREB axis to promote the CRC chemoresistance. These findings provide evidence that C. tropicalis promotes CRC chemoresistance, which may provide novel ideas for improving CRC chemotherapy effect.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding
This work is supported by grants from National Natural Science Foundation of China (82072648 and 81772542), the Fundamental Research Funds for the Central Universities (0214 14380500), and Medical Research Foundation of Jiangsu Health Commission (Z2018044).
Ethical approval
This study was carried out in accordance with the principles of the Basel Declaration and recommendations of the US NIH with Specific Pathogen Free conditions. The protocol was approved by the Model Animal Research Center of Nanjing University. All-time-available Standard rodent chow and water were also provided.
Availability of data and material
The datasets generated for this study are available on request to the corresponding author. This study did not generate any additional resources.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683-91
2. Cartwright TH. Treatment decisions after diagnosis of metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11(3):155-66
3. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409-13
4. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170(3):548-63 e16
5. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573-84
6. Hu T, Li Z, Gao CY, Cho CH. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterol. 2016;22(30):6876-89
7. McKenzie GJ, Rosenberg SM. Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr Opin Microbiol. 2001;4(5):586-94
8. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4(1):1-6
9. Jiricny J, Nystrom-Lahti M. Mismatch repair defects in cancer. Curr Opin Genet Dev. 2000;10(2):157-61
10. Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S. et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019;366(6472):1473-80
11. Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42(5):399-435
12. Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y. et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66(1):70-8
13. Shi Y, Zheng W, Yang K, Harris KG, Ni K, Xue L. et al. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J Exp Med. 2020 217(5)
14. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084-9
15. Wang T, Fan C, Yao A, Xu X, Zheng G, You Y. et al. The Adaptor Protein CARD9 Protects against Colon Cancer by Restricting Mycobiota-Mediated Expansion of Myeloid-Derived Suppressor Cells. Immunity. 2018;49(3):504-14 e4
16. Bouvet D, Bodo S, Munier A, Guillerm E, Bertrand R, Colas C. et al. Methylation Tolerance-Based Functional Assay to Assess Variants of Unknown Significance in the MLH1 and MSH2 Genes and Identify Patients With Lynch Syndrome. Gastroenterology. 2019;157(2):421-31
17. Yang Z, Wei D, Liu F, Liu J, Wu X, Stevens MFG. et al. Temozolomide analog PMX 465 downregulates MGMT expression in HCT116 colorectal carcinoma cells. J Cell Biochem. 2018;119(7):5350-8
18. Hassen S, Ali AA, Kilaparty SP, Al-Anbaky QA, Majeed W, Boman BM. et al. Interdependence of DNA mismatch repair proteins MLH1 and MSH2 in apoptosis in human colorectal carcinoma cell lines. Mol Cell Biochem. 2016;412(1-2):297-305
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
20. Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J. et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell. 2019;178(1):176-89 e15
21. DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132-44
22. Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13(9):611-23
23. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B. et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36(42):5829-39
24. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351-9
25. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem Sci. 2019;44(2):153-66
26. Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF. et al. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. 2020;39(16):3292-304
27. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W. et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115-8
28. Gao F, Tang Y, Liu WL, Zou MZ, Huang C, Liu CJ. et al. Intra/Extracellular Lactic Acid Exhaustion for Synergistic Metabolic Therapy and Immunotherapy of Tumors. Adv Mater. 2019;31(51):e1904639
29. Brown TP, Ganapathy V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther. 2020;206:107451
30. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71(22):6921-5
31. Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S. et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014;74(18):5301-10
32. Kuei C, Yu J, Zhu J, Wu J, Zhang L, Shih A. et al. Study of GPR81, the lactate receptor, from distant species identifies residues and motifs critical for GPR81 functions. Mol Pharmacol. 2011;80(5):848-58
33. Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J. et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. 2009;284(5):2811-22
34. Tilg H, Adolph TE, Gerner RR, Moschen AR. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell. 2018;33(6):954-64
35. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA. et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967-70
36. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D. et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971-6
37. Wang T, Pan D, Zhou Z, You Y, Jiang C, Zhao X. et al. Dectin-3 Deficiency Promotes Colitis Development due to Impaired Antifungal Innate Immune Responses in the Gut. PLoS Pathog. 2016;12(6):e1005662
38. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1-11
39. Silwal P, Kim JK, Yuk JM, Jo EK. AMP-Activated Protein Kinase and Host Defense against Infection. Int J Mol Sci. 2018 19(11)
Author contact
![]() Corresponding authors: Professor Tingting Wang, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093. E-mail: wangttedu.cn. Professor Sunan Shen, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093. E-mail: shensnedu.cn. Professor Ping Chen, Department of Oncology, Yancheng First Hospital, Affiliated Hospital of Nanjing University Medical School, The First People's Hospital of Yancheng, Yancheng, Jiangsu, China, 224001. Email: doctorchenpingnjucom.
Corresponding authors: Professor Tingting Wang, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093. E-mail: wangttedu.cn. Professor Sunan Shen, The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing, China, 210093. E-mail: shensnedu.cn. Professor Ping Chen, Department of Oncology, Yancheng First Hospital, Affiliated Hospital of Nanjing University Medical School, The First People's Hospital of Yancheng, Yancheng, Jiangsu, China, 224001. Email: doctorchenpingnjucom.