Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(11):2871-2883. doi:10.7150/ijbs.60263 This issue Cite
Research Paper
Crucial Role of Mammalian Glutaredoxin 3 in Cardiac Energy Metabolism in Diet-induced Obese Mice Revealed by Transcriptome Analysis
Ninghui Cheng1 ![]() , Qianxing Mo2, Jimmonique Donelson1, Lingfei Wang3, Ghislain Breton4, George G. Rodney5,6, Jin Wang3,7, Kendal D. Hirschi1, Xander H. T. Wehrens1,5,6, Paul A. Nakata1
, Qianxing Mo2, Jimmonique Donelson1, Lingfei Wang3, Ghislain Breton4, George G. Rodney5,6, Jin Wang3,7, Kendal D. Hirschi1, Xander H. T. Wehrens1,5,6, Paul A. Nakata1
1. USDA/ARS Children Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, Texas 77030, USA.
2. Department of Biostatistics & Bioinformatics, H. Lee Moffitt Cancer Center & Research Institute, Tampa, FL 33612, USA.
3. Department of Pharmacology and Chemical Biology, Baylor College of Medicine, Houston, TX 77030, USA.
4. Department of Integrative Biology & Pharmacology, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas 77030, USA.
5. Department of Molecular Physiology & Biophysics, Baylor College of Medicine, Houston, TX 77030, USA.
6. Cardiovascular Research Institute, and Center for Drug Discovery, Baylor College of Medicine, Houston, TX 77030, USA.
7. Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Received 2021-3-9; Accepted 2021-6-25; Published 2021-7-13
Abstract

Obesity is often associated with metabolic dysregulation and oxidative stress with the latter serving as a possible unifying link between obesity and cardiovascular complications. Glutaredoxins (Grxs) comprise one of the major antioxidant systems in the heart. Although Grx3 has been shown to act as an endogenous negative regulator of cardiac hypertrophy and heart failure, its metabolic impact on cardiac function in diet-induced obese (DIO) mice remains largely unknown. In the present study, analysis of Grx3 expression indicated that Grx3 protein levels, but not mRNA levels, were significantly increased in the hearts of DIO mice. Cardiac-specific Grx3 deletion (Grx3 CKO) mice were viable and grew indistinguishably from their littermates after being fed a high fat diet (HFD) for one month, starting at 2 months of age. After being fed with a HFD for 8 months (starting at 2 months of age); however, Grx3 CKO DIO mice displayed left ventricular systolic dysfunction with a significant decrease in ejection fraction and fractional shortening that was associated with heart failure. ROS production was significantly increased in Grx3 CKO DIO cardiomyocytes compared to control cells. Gene expression analysis revealed a significant decline in the level of transcripts corresponding to genes associated with processes such as fatty acid uptake, mitochondrial fatty acid transport and oxidation, and citrate cycle in Grx3 CKO DIO mice compared to DIO controls. In contrast, an increase in the level of transcripts corresponding to genes associated with glucose uptake and utilization were found in Grx3 CKO DIO mice compared to DIO controls. Taken together, these findings indicate that Grx3 may play a critical role in redox balance and as a metabolic switch in cardiomyocytes contributing to the development and progression of heart failure.
Keywords: Heart failure, glutaredoxin, oxidative stress, cardiac energy metabolism, transcriptome
Introduction
Overweight/obesity is an independent risk factor for cardiovascular disease (CVD) and has been associated with adverse effects on cardiac structure, hemodynamics, and function [1]. Recent epidemiological studies; however, suggests that heart failure patients with mild obesity have a better prognosis than their leaner counterparts [2]. The underlying mechanisms that explain this obesity paradox remain to be fully elucidated. Obesity is often associated with metabolic dysregulation and oxidative stress. Both are considered a unifying link between obesity and cardiovascular complications [3]. Experimental studies in rodents indicate that enhancement of antioxidant capacity in the heart plays a protective role in diabetes induced cardiac dysfunction, however, clinical investigations have failed to support the beneficial effect of antioxidants [4]. This apparent contradiction may be attributed to the complexity of the regulatory pathways controlling redox balance and cardiac energy metabolism in the heart of obese subjects.
Glutaredoxins (Grxs) are ubiquitous, small heat-stable disulfide oxidoreductases which are one of the major antioxidant systems in the heart [5]. Grxs can be categorized into dithiol Grxs, which contain two cysteine residues in their active motifs, and monothiol Grxs which contain a single cysteine residue in their putative motifs [5]. Grx3, a monothiol Grx, also termed thioredoxin-like 2 (Txnl2) or protein kinase C interacting cousin of thioredoxin (PICOT), interacts with the protein kinase C theta isoform [6]. Previous studies demonstrate that Grx3 is essential for early embryonic growth and development [7, 8]. Recent reports revealed that forced expression of Grx3 in transgenic rat heart could enhance cardiomyocyte contractility by modulating calcineurin-NFAT-mediated signaling and PKCζ activity in the progression of pressure-overload induced heart hypertrophy [9-11]. Our recent study indicated, using a cardiac-specific Grx3 deletion mouse model, that Grx3 is an important factor in regulating cardiac hypertrophy and heart failure by modulating both cellular redox homeostasis and Ca2+ handling in the heart [12]. The metabolic impact of Grx3 on cardiac energy metabolism in diet-induced obese (DIO) mice; however, remains largely unknown.
In this study, we investigated the expression of Grx3 in the heart of DIO mice compared to controls. We characterized the Grx3 CKO DIO mice and showed that disruption of Grx3 in DIO mice led to cardiac dysfunction and heart failure. In addition, we documented the transcriptional regulatory mechanism underlying Grx3 function in cardiomyocytes. Taken together, our findings suggest that the presence of Grx3 is critical for maintaining redox balance and cardiac energy metabolism.
Materials and Methods
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise. Anti-GAPDH antibody was bought from Chemicon International, Inc. Grx3 monoclonal antibody was made in-house using the full-length human Grx3 recombinant protein. This antibody was validated and used in our previous studies [8, 13, 14].
Animals
Grx3 cardiac specific knockout (Grx3 CKO) mice were generated as previously described [12]. All mouse strains were housed in temperature-controlled environment and fed a standard chow diet (LabDiet 5V5R chow; LabDiet, St. Louis, MO) ad libitum unless stated otherwise. Male mice were used in this study and Grx3 floxed mouse (Grx3flox/flox) littermates were used as controls. Mice were fed with high fat diet (D12492, rodent diet with 60 kcal% fat, Research Diets, Inc., New Brunswick, NJ, USA) for one month starting at the age of 2 months old (until 3 months of age) and for eight months starting at the age of 2 months old (until 10 months of age). All studies were performed according to protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine conforming to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Echocardiography
Lightly anesthetized mice (1.5-2.0% isoflurane) were imaged in the left lateral decubitus position with a linear 30-MHz probe (Visualsonic Vevo 2100 Imaging System, Baylor Mouse Phenotyping Core) [12]. Digital images were collected at a frame rate of 180 images/s. Two-dimensional images were recorded in parasternal long- and short-axis projections with guided M-mode recordings. All measurements were done blinded to genotypes.
ROS production assay
Dihydroergotamine (DHE) staining to measure ROS production in cardiomyocytes was done as previously described [12]. The DHE fluorescence signals were detected at 610 nm (excitation at 520 nm) using an Olympus Fluoview scanning laser confocal microscopy system. The fluorescent intensity was quantified using Image J.
NAD+ and NADH Assay
20 mg of heart (ventricle) tissues from chow-fed and HFD-fed Grx3 CKO DIO and control mice were used in the measurement of NAD+ and NADH using an NAD+/NADH assay kit (BioAssay, CA, USA) following the manufacturer's instructions. The NAD+/NADH ratio was calculated by dividing the measured amount of NAD+ by the amount of NADH.
Quantitative RT-PCR and microarray analysis
Total RNA was extracted from heart (ventricle) tissues from Grx3 CKO DIO mice and DIO controls either using Trizol reagent (Thermo Fisher Scientific, Waltham, MA) or using the Quick-RNA Miniprep Plus Kit (Zymo Research, Irvine, CA) following the manufacturer's instructions. For q-PCR analysis, purified RNA samples underwent reverse transcription to yield cDNA. qPCR was performed using the SYBR Green-based assay and the Bio-Rad CFX96™ Real-Time PCR Detection System. Differences in gene expression between control and knockout mice were quantified by comparative CT method. Gapdh was selected to serve as the internal control because its expression level was consistent between WT and KO and relatively stable across different stages. Primers used are listed in supplementary Table S7. For microarray analysis, RNA sample quality was first monitored using an Agilent 2100 bioanalyzer. Three microarrays for each genotype were performed at Baylor College of Medicine Microarray Core Facility using the GeneChip™ Mouse Genome 430 2.0 Array (Thermo Fisher Scientific, Waltham, MA). The Bioconductor limma package was used to analyze the microarray data. Briefly, the data was background corrected using the 'normexp' method with an offset of 16 added to the intensities. The background corrected data then was log2-transformed and quantile normalized. Probes without gene description were filtered out before hypothesis testing. Moderated t-statistics were used to test if genes were differentially expressed between the groups of interest and the Benjamini-Hochberg method was used to estimate false discovery rate (FDR). Probes with a p-value < 0.01 and a fold change >1.5 were considered the top differentially expressed probes (genes).
Western blot analysis
Heart lysates were prepared from flash-frozen mouse hearts and run on SDS-PAGE gels. Western blot analysis was conducted following an established procedure [12]. Antibodies against Grx3 and GAPDH were used at 1:1000 and 1:2000 dilution, respectively.
Statistical analysis
All results were shown as means ± SEM. A two-way ANOVA was used to analyze data. Student's t test was used to compare the two genotypes. * p <0.05, **p<0.01, and *** p <0.001 were used as indicators of the level of significance. For microarray data analysis, moderated t-statistics were used to test if genes were differentially expressed between the groups of interest and the Benjamini-Hochberg method was used to estimate false discovery rate (FDR). Probes with a p-value < 0.01 and a fold change >1.5 were considered the top differentially expressed probes (genes).
Results and Discussion
Grx3 expression in the heart of DIO mice
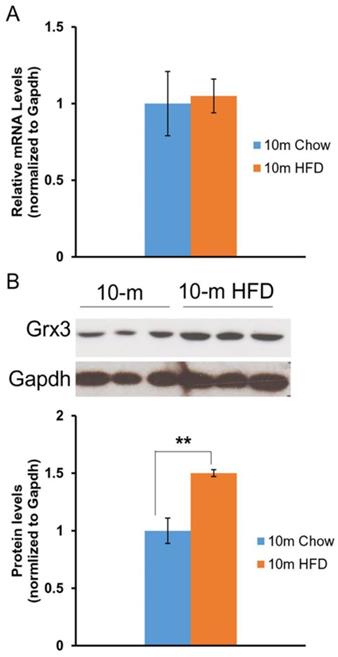
Our previous studies in mice indicated that Grx3 is ubiquitously expressed in many tissues and organs [8]. Differential expression was observed in the developing mammary glands [14] and also in muscle cells following hydrogen peroxide treatment [8]. As regards to the current investigation, Grx3 expression was also found to decline in the heart during aging [12]. Here we wanted to determine the impact of HFD feeding on Grx3 expression in the heart. Both Grx3 transcripts and proteins were measured in the hearts from 10-month-old mice fed either a chow diet- or HFD fed (starting at 2 months of age) by q-PCR and western-blot, respectively (Figure 1). The level of Grx3 transcripts remained unchanged in HFD-fed mice compared to controls (Figure 1A), while the level of Grx3 protein showed a significant increase in response to HFD feeding (Figure 1B). From our previous findings in aged mice, it appears that the HFD doesn't lead to further increase in Grx3 mRNA in 10-month-old mouse hearts as compared to controls but to an increase in the protein level. It is currently unclear what factors are responsible for the discrepancy in Grx3 mRNA and protein levels in the heart under HFD feeding. It has been shown that in mouse skeletal muscle, the high-fat diet feeding significantly down-regulates the muscle extracellular matrix structural gene expression at the mRNA level, but not the protein level [15]. The association only seen between the mRNA levels and muscle function could be due to the effect of HFD feeding on muscle structural gene expression (mRNA) and function, rather than structural changes mediating functional changes [15]. In our study, it is possible that the HFD feeding potentiates the Grx3 mRNA stability or the turnover of Grx3 proteins in cardiomyocytes. Interestingly, a recent report has shown that AtGRXS17, a homolog of Grx3 from Arabidopsis thaliana, is a target of the ubiquitin E3 ligases RGLG3/4 [16]. Whether Grx3 could be a target of the ubiquitin E3 ligase in the heart and whether turnover of Grx3 could be regulated by HFD feeding warrants future investigation. Nevertheless, the current finding suggests that the increased Grx3 protein levels may provide adaptation to oxidative and metabolic stresses under HFD feeding.
Grx3 expression in the heart of DIO mice. (A). Q-PCR analysis indicates that Grx3 mRNA levels were similar in 10-m old mice fed either a chow diet or high fat diet. (B). Grx3 protein levels were increased in response to high fat diet feeding. Student t test, n=3, * p<0.05, ** p<0.01.
Enhanced ROS production in the heart of Grx3 CKO mice independently of feeding regime
To determine whether the increase of Grx3 protein in the aged mice under HFD provides protection against oxidative stress, we compared loss of Grx3 specifically in the heart (Grx3 cardiac KO (CKO)) against wild type littermate control after being fed HFD or chow diet for 8 months. Histological analysis of left ventricular (LV) sections of 10 months old Grx3 CKO mice and wild type littermate controls fed either a chow diet- or HFD starting at 2 months of age was conducted. Sections were stained with dihydroethidium (DHE) to detect superoxide levels in the LV cardiomyocytes. First, the level of oxidative stress in the heart of 10 months old mice fed with HFD was similar to chow diet-fed mice suggesting that HFD hearts have adapted to the 8-month feeding regime and the previously detected increase in Grx3 protein did not reduce further the ROS level (Figure 2). However, Grx3 CKO hearts from mice fed a chow diet- and HFD exhibited a significant increase in DHE stained signals (Figure 2), indicating increased ROS production in comparison to wild type littermate controls and the essential role of Grx3 in ROS protection (Figure 2). This observation suggests that deletion of Grx3 induces oxidative stress in Grx3 CKO hearts under both chow diet- and HFD-feeding. The NAD+/NADH ratio were measured (Figure S1) to further examine whether the redox state were changed in the heart of Grx3 CKO DIO mice compared to wild type controls. Although the levels of ROS were increased in chow-fed Grx3 CKO heart vs wild type controls (Figure 2), the NAD+/NADH ratios were similar between the two, suggesting that the redox balance was still maintained under chow-diet feeding. However, under HFD feeding, Grx3 CKO DIO mice showed a significant increase in the NAD+/NADH ratio (Figure S1), indicating a redox imbalance in the hearts. There is growing evidence that cardiac energy metabolism and redox balance are crucial for the development and progression of heart diseases including heart failure [17-19]. Thus, these results suggest that deletion of Grx3 in the Grx3 CKO DIO hearts leads to oxidative stress and redox imbalance that are detrimental to cardiac function and eventually cause the progression of heart failure in these mice (see below).
Grx3 deletion enhanced ROS production in cardiac sections of older Grx3 CKO DIO mice. (A) Representative images showing DHE-fluorescence indicative of elevated ROS levels in cardiac sections of Grx3 CKO and Grx3 CKO DIO mice. Bars= 20µm. (B) Quantification of signal intensity per area revealing increased ROS levels in cardiac sections from Grx3 CKO and Grx3 CKO DIO mice compared to littermate controls. * p<0.05.

Disruption of Grx3 has impaired cardiac function in DIO mice
To determine if the increase of Grx3 protein in old mice fed with HFD leads to change in metabolic stress, we compared Grx3 CKO mice and the wild type littermate controls following 1 month (young) or 8 months (old) of HFD. Both feeding schedules were started at 2 months of age. In young mice fed HFD, a lack of Grx3 expression in the heart did not affect heart function or growth compared to littermate controls following one month of HFD feeding. No significant difference between Grx3 CKO and controls were measured in body weight, heart weight, heart-to-body weight ratio, and heart rate (Figure S2A-D). In addition, assessment of the LV function by echocardiography revealed no differences in cardiac contractility or dimensions (Figure S2E-H) and there was no difference detected in LV ejection fraction and fractional shortening between CKO mice and littermate controls (Figure S2I-J).
We also tested the same condition in older mice after being fed HFD for 8 months. Grx3 CKO mice and wild type littermate controls both gained weight and became obese (Figure S3A) compared to chow diet fed mice (WT: 35.1±6.3g vs CKO: 41.5±8.2g; p=0.438). As with younger mice, there was little difference between CKO and control DIO mice in body weight (51.1±1.6g vs. 53.2±1.2g; p=0.302), heart weight (223.6±9.3mg vs. 226.6±8.4mg; p=0.816), heart-to-body weight ratio (4.4±0.2 vs. 4.3±0.1; p=0.555) (Figure S3A-C). Interestingly, the heart rate of the CKO DIO mice was significantly higher than that of their littermate DIO controls (553.4±22.1 vs. 461.5±12.3; p<0.001) (Figure S3D). The in vivo cardiac function analyses using echocardiography revealed a drastic LV dysfunction was observed in Grx3 CKO DIO mice (Figures 3, S3). In detail, LV end-systolic diameter, not end-diastolic diameter was significantly increased in Grx3 CKO DIO hearts (LVESD: 3.7±0.2; LVEDD: 4.7±0.1) compared to littermate DIO controls (LVESD: 3.1±0.2; LVEDD: 4.5±0.2; p<0.01, p=0.245) (Figure 3A, B). This change was also observed in chow diet fed mice, but with lower diameter (Figure 3A, B). These changes were concomitant with drastic decline in fractional shortening and ejection fraction in Grx3 CKO (EF: 40.3±5.4; FS: 20.1±3.0) versus wild type (EF: 57.7±2.8; FS: 30.5±1.8; p<0.001) (Figure 3C, D), along with decreased cardiac output (CO) (21.3±1.7 vs. 24.6±2.1; p=0.096) and stroke volume (39.0±3.6 vs. 53.2±4.4; p<0.01) (Figure S3H, I). Similar but milder decline in fractional shortening and ejection fraction in Grx3 CKO chow diet fed mice were observed (Figure 3C, D).
Cardiac dysfunction and heart failure in Grx3 CKO DIO mice. In vivo echocardiography tracings of 10-m old chow diet- (n=6) and HFD-(n=8) fed controls and Grx3 CKO DIO mice showing left ventricular dilation and impaired contractility. Quantification of echo tracings revealed (A-B) increased left ventricular end-systolic diameters (LVESD), but not end-diastolic (LVEDD) in Grx3 CKO and Grx3 CKO DIO mice; (C-D) a significantly decline in ejection fraction (EF) and fraction shortening (FS) in Grx3 CKO and Grx3 CKO DIO mice compared to littermate controls. * p<0.5, ** p<0.01, and *** p<0.001 indicate a significance between Grx3 CKO mice and their littermate controls. # indicates a significance between chow diet- and HFD-fed Grx3 CKO mice.
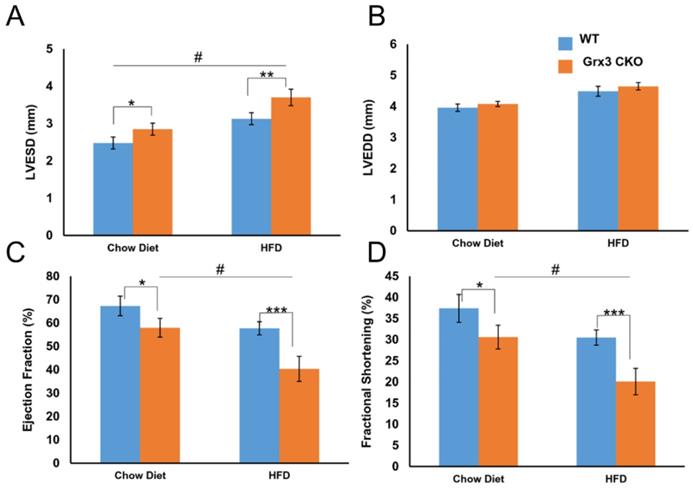
Overall, these results indicated that at a young age, high fat diet feeding did not impair cardiac function of Grx3 CKO compared to littermate controls (Figure S2), which is consistent with the our previous findings [12]. Grx3 CKO mice started to show some cardiac dysfunction at age of 10 months old even under regular chow diet feeding (Figure 3). However, DIO significantly exacerbated cardiac function in Grx3 CKO mice compared to littermate controls (Figures 3, S3), suggesting that the nutritional imbalance might potentiate cardiac energy metabolism in Grx3 CKO mice.
Identification of differential expressed genes (DEGs) in Grx3 CKO DIO mice
To gain insight into the transcriptional changes occurring in the heart of old mice lacking Grx3 under HFD, four libraries, two from Grx3 CKO hearts and two from littermate controls fed either chow diet- or HFD (3 biological replicates for each library) were constructed and subjected to DNA microarray analysis. At a 1.5-fold change cut off, a total of 1175 DEGs were identified, of which 178 (127 up- and 51 down-regulated), 796 (409 up- and 387 down-regulated), 87 (64 up- and 23 down-regulated), and 114 (75 up- and 39 down-regulated) were identified between four comparisons (Grx3 CKO vs control hearts under chow diet feeding, Grx3 CKO hearts vs controls under HFD feeding, WT mice under chow diet vs HFD diet feeding, and Grx3 CKO hearts under chow diet vs HFD feeding) (Figure S4). To validate the microarray analysis results, 9 genes having significantly differential expression in Grx3 CKO DIO mice compared to DIO littermate controls were selected to conduct q-RT-PCR analysis. As shown in Figure S5, the results indicated that 8 out of 9 genes having high correlation between the q-RT-PCR and microarray analysis data, which validated the transcriptome results.
To further explore the function of the DEGs in Grx3 CKO DIO mice compared to DIO littermate controls (Figure S6, Tables S1, S2), Gene Ontology (GO) enrichment analyses was utilized to group the DEGs according to biological processes (Tables S3, S4). As shown in Figure 4A, the top five represented groups were “regulation of metabolic process”, “single-organism process”, “establishment of localization”, “cell communication”, and “single-organism signaling” for the up-regulated DEGs, while “cellular process”, “single-organism process”, “metabolic process”, “establishment of localization”, and “single-organism transport” were the top five represented groups for the down-regulated DEGs (Figure 4B).
Identification of differentially expressed genes (DEGs) in Grx3 CKO DIO mice. (A-B) GO enrichment of identified up-regulated (A) and down-regulated (B) DEGs in Grx3 CKO DIO mice compared to littermate DIO controls. (C-D) KEGG pathway assignment of identified up-regulated (C) and down-regulated (D) DEGs in Grx3 CKO DIO mice compared to littermate controls.
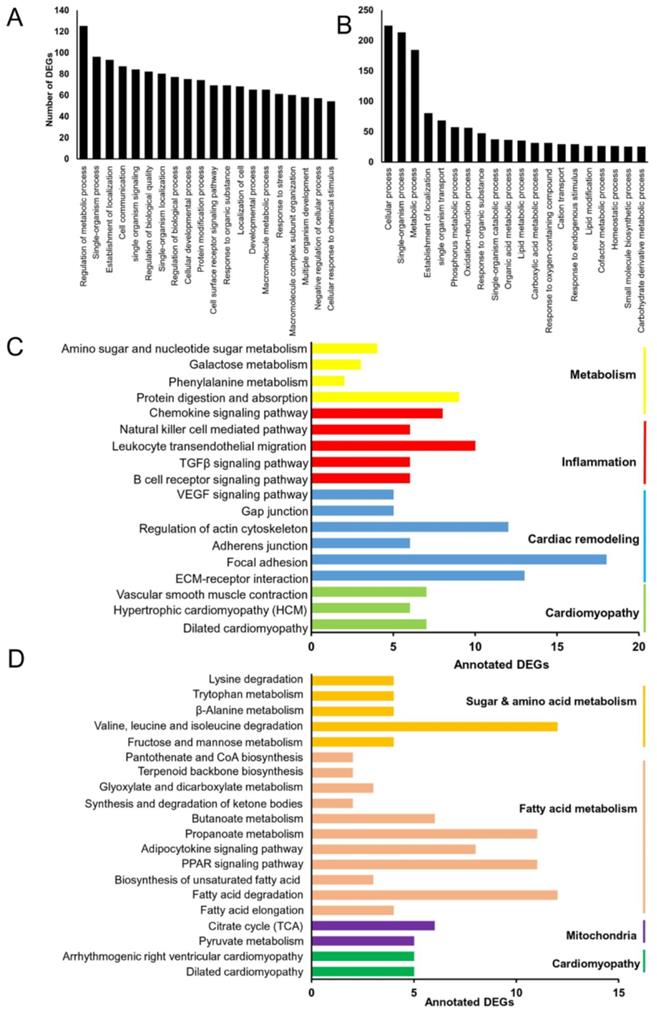
As a step toward assigning each DEG to a specific pathway, Kyoto encyclopedia of genes and genomes (KEGG) analysis was conducted. For the upregulated DEGs, a total of 133 DEGs were assigned into 18 KEGG pathways, which composed four categories: cardiomyopathy, cardiac remodeling, inflammation, and metabolism (Figure 4C). Among the four groups, the inflammation and cardiac remodeling groups have the richest numbers of pathways (Figure 4C) which include the pathway associated with focal adhesion (mmu04510, 18 genes), extracellular matrix (ECM)-receptor interaction (mmu04512, 13 genes), regulation of actin cytoskeleton (mmu04810, 12 genes), TGFβ signaling (mmu04350, 6 genes), and VEGF signaling (mmu04370, 5 genes) (Figure 4C). These results suggest that up-regulation of inflammatory responses and cardiac tissue remodeling may play an important role in developing cardiac dysfunction in Grx3 CKO DIO mice. For the down-regulated DEGs, a total of 83 DEGs were assigned into 19 KEGG pathways which could be grouped into four categories: cardiomyopathy, mitochondria, fatty acid metabolism, and sugar & amino acid metabolism (Figure 4D). In these down-regulated pathways, the ones associated with mitochondria and fatty acid metabolism groups are the most common, which include PPAR signaling (mmu03320, 11 genes), Adipocytokine signaling (mmu04920, 8 genes), propanoate metabolism (mmu00640, 11 genes), and the citrate cycle (TCA cycle, mmu00020, 6 genes) (Figure 4D). These results suggest that down-regulation of mitochondrial function and fatty acid metabolism may contribute to cardiac energy metabolism imbalance and play a crucial role in the pathogenesis of heart failure in Grx3 CKO DIO mice.
Identification of gene signatures in Grx3 CKO DIO mice
Gene clusters involved in cardiomyopathy in Grx3 CKO DIO mice
Changes in gene expression play an important role in the development of cardiac hypertrophy and heart failure [20-22]. According to the KEGG annotations, 13 DEGs involved in dilated cardiomyopathy were identified in Grx3 CKO DIO mice compared to DIO controls (Figures 4C, 4D, 5A). The expression of six genes, an ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (Atp2a2 or Serca2a), calcium voltage-gated channel subunit alpha1 S (Cacna1s), desmoplakin (Dsp), sarcoglycan gamma (Sgcg), phospholamban (Pln), and ryanodine receptor 2 (Ryr2) were significantly reduced in Grx3 CKO DIO mice compared to DIO controls (Figure 5A). All six genes encode proteins that are involved in muscle cell contraction, cardiac calcium transport and calcium handling. Previous studies demonstrated that reduction of Serca2a, Ryr2, and Pln in the heart was significantly correlated with the development of human dilated cardiomyopathy and heart failure [7, 23, 24]. DSP is the most abundant desmosomal protein, which binds intermediate filament cytoskeletons to desmosomal plaques and desmosomes are major cellular junctions that are abundant in tissues undergoing constant physical stress, like the heart [25]. Mutations in Dsp lead to the early onset severe arrhythmias, left ventricular cardiomyopathy, and heart failure [26, 27]. Although less is known about the function of Sgcg and Cacna1s in the pathogenesis of cardiomyopathy, previous reports indicated that disruption of the sarcoglycan complex and/or mutations in the cardiac voltage-dependent L-type calcium channel (LTCC) have been associated with severe cardiomyopathy and a number of inherited cardiac arrhythmia syndromes [28, 29]. In addition, Hrc (histidine rich calcium binding protein), Scn4b (sodium voltage-gated channel beta subunit 4), Kcnk3 (potassium two pore domain channel subfamily K member 3), Atp2a1 (Serca1a, ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1), Slc8a3 (solute carrier family 8 member A3) were significantly decreased in Grx3 CKO DIO mice (Figure S7). Overall, these findings imply that down-regulation in the expression of those genes may contribute to dysfunction of cardiac excitation-contraction in Grx3 CKO DIO mice (Figures 3, S3) and alteration of Ca2+ signaling and handling in hearts that lack Grx3 expression [7, 12].
Identification of gene signatures in Grx3 CKO DIO mice. (A) Heat map diagrams show gene clusters involved in cardiomyopathy in Grx3 CKO DIO mice. (B) Heat map diagrams show gene clusters involved in fatty acid metabolism and mitochondrial function. (C) Overview of fatty acid and glucose oxidation in cardiomyocytes and alteration of gene expression in Grx3 CKO DIO mice.
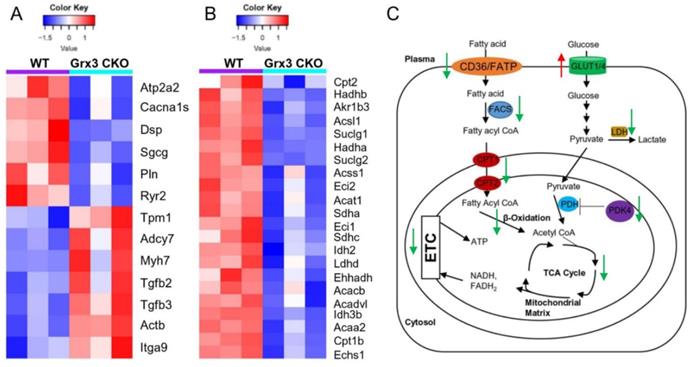
In contrast, seven genes encoding tropomyosin 1 (Tpm1), adenylate cyclase 7 (Adcy7), myosin heavy chain 7 (Myh7), actin beta (Actb), integrin subunit alpha 9 (Itga9), transforming growth factor beta 2 (Tgfb2), and transforming growth factor beta 3 (Tgfb3) were significantly up-regulated in Grx3 CKO DIO mice compared to DIO controls (Figures. 4C, 4D, 5A). Tpm1, Myh7, and Actb are known to be involved in the pathogenesis of dilated cardiomyopathy [30]. Enhanced expression of Myh7 is a hallmark in the development of hypertrophic cardiomyopathy (HCM) in humans and animal models [31]. Mutations and altered expression of Tpm1 and Actb lead to cardiac hypertrophy and heart failure [32-34]. Adcy encodes adenylate cyclase, an enzyme that can catalyze the conversion of adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP). Increased expression of Adcy7, coupled with β-adrenergic receptors and Gαs, may result in enhanced cAMP-mediated signaling which could contribute to the pathogenesis of dilated cardiomyopathy [35]. Integrins are cell surface receptors that can potentiate signals from transforming growth factor β (TGFβ) and play essential roles in the fibrotic process [36]. Increased expression of Itga9, Tgfb2, and Tgfb3 in Grx3 CKO DIO mice may promote myofibroblast proliferation and the fibrotic process that result in cardiomyocyte apoptosis and heart failure [37-39].
Gene clusters involved in cardiac tissue remodeling and inflammation
Cardiac remodeling and inflammatory responses are strongly associated with extreme obesity that may lead to the development of heart failure in obese subjects and animal models as well [40, 41]. Based on the KEGG enrichment analysis, 60 DEGs were assigned to the category of tissue remodeling and inflammation (Figure 4C, Table S5). Eleven genes, encoding collagen type III alpha 1 chain (Col3a1), collagen type V alpha 2 chain (Col5a2), collagen type VI alpha 1 chain (Col6a1), collagen type I alpha 1 chain (Col1a1), collagen type I alpha 2 chain (Col1a2), thrombospondin 3 (Thbs3), thrombospondin 4 (Thbs4), cartilage oligomeric matrix protein (Comp), fibronectin 1 (Fn1), tenascin C (Tnc), and Itag9, were significantly up-regulated in Grx3 CKO DIO mice compared to DIO controls (Figure 4C, Table S5). These genes are involved in ECM interaction and focal adhesion signaling. The ECM is a complex architectural network that play a crucial role in the pathogenesis of heart failure with a reduced ejection fraction [42] as seen in Grx3 CKO DIO mice (Figures 3, S3). The aberrant signal transductions in ECM interaction and focal adhesion have been shown to be crucial for myocardial remodeling and its progression to heart failure [43-48].
Epidermal growth factor receptor (Egfr) and fibroblast growth factor receptor 1(Fgfr1)-mediated signaling is involved in gap junctions (GJs) and adherens junctions (AJs) and critical for cardiac remodeling [49, 50]. For example, cardiac-specific over-expression of epidermal growth factor receptor (ErbB2) induced hypertrophic cardiomyopathy [51]. Both Egfr and Fgfr1 were found to be up-regulated in Grx3 CKO DIO mice (Table S5). Interestingly, EGFR-mediated signaling, like ErbB2, is down-regulated in Grx3 KD breast cancer cells [13], suggesting that Grx3 may play different roles in heart diseases and cancer.
VEGF and TGFβ signaling pathways are important in inflammation and cardiac remodeling [38, 52, 53]. Most significantly, both VEGF and TGF signaling pathways, which include genes encoding protein kinase C beta (Prkcb), Rac family small GTPase 2 (Rac2), phospholipase A2 group IVA (Pla2g4a), prostaglandin-endoperoxide synthase 2 (Ptgs2), sphingosine kinase 1 (Sphk1), cyclin dependent kinase inhibitor 2B (Cdkn2b), Comp, Tgfb2, Tgfb3, Thbs3, and Thbs4, were activated in Grx3 CKO DIO mice compared to DIO controls (Figure 4C, Table S5), which may contribute to the progression to heart failure in Grx3 CKO DIO mice [38, 54-59].
Gene clusters involved in fatty acid metabolism and mitochondrial function
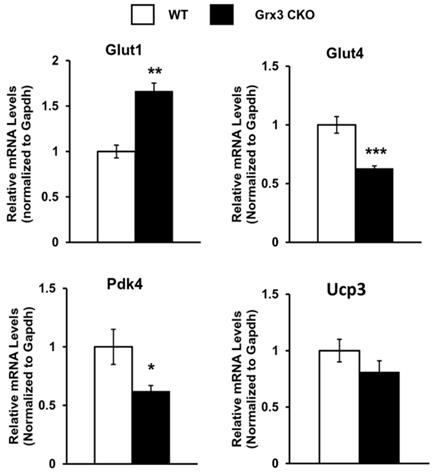
Proper energy metabolism is essential for cardiac function. In order to adapt to an altered physiological or metabolic environment, the healthy heart has the capacity to select the most efficient substrate for ATP production, however, this metabolic flexibility is diminished in the failing heart [60]. In obese subjects, in particular, alterations in cardiac fatty acid metabolism and mitochondrial function may play a causal role in the development and progression of heart failure because of elevated circulating free fatty acids, lipid accumulation (lipotoxicity), and marked shift in fuel utilization [61-63]. In Grx3 CKO DIO hearts with profound cardiac dysfunction and heart failure, expression of genes related to fatty acid oxidation (FAO) and mitochondrial function were significantly decreased in comparison to DIO control hearts (Figures 4D, 5B). A total of 35 annotated DEGs were identified and assigned to 10 KEGG pathways which included fatty acid elongation and degradation as well as PPAR and adipocytokine signaling pathways (Figure 4D). Cd36 encoding the CD36 molecule, a multifunctional immuno-metabolic receptor, and Slc27a1 encoding the solute carrier family 27 member 1 (FATP1), important in the uptake of long-chain fatty acid in the heart [64-66], were significantly reduced in Grx3 CKO DIO mice compared to DIO controls (Figure 4D, Table S6). This reduction in expression suggests a likely decrease in fatty acid uptake in cardiomyocytes of Grx3 CKO DIO hearts. Acsl1 encoding acyl-CoA synthetase long chain family member 1, Cpt1b encoding carnitine palmitoyltransferase 1B, and Cpt2 encoding carnitine palmitoyltransferase 2 are rate-limiting enzymes controlling the mitochondrial uptake of long-chain acyl-CoAs for β-oxidation and are indispensable for energy metabolism in the heart [67-69]. Down-regulation of Acsl1, Cpt1b, and Cpt2 in Grx3 CKO DIO mice (Figure 5B) implies a reduction of mitochondrial fatty acid acyl-CoA uptake. Acadvl encodes a very long chain acyl-CoA dehydrogenase and Echs1 encodes enoyl-CoA hydratase, short chain 1. These enzymes catalyzes the first and second steps, respectively, in mitochondrial fatty acid β-oxidation which is an essential part of cardiac energy metabolism [70, 71]. Acetyl-CoA acyltransferase 2 (Acaa2), hydroxyacyl-CoA dehydrogenase subunit alpha (Hadha), and hydroxyacyl-CoA dehydrogenase subunit beta (Hadhb) form a mitochondrial trifunctional protein complex that is critical for fatty acid β-oxidation [72-74]. Reduction of Acadvl and Echs1 combined with a reduction of Acaa2, Hadha, and Hadhb suggest a dysregulation of mitochondrial fatty acid oxidation in Grx3 CKO DIO hearts. In addition, succinate-CoA ligase GDP/ADP-forming subunit α/β (Suclg1/2), succinate dehydrogenase complex subunit C (Sdhc), succinate dehydrogenase complex flavorprotein subunit A (Sdha), isocitrate dehydrogenase (NAD+) 3 non-catalytic subunit beta (Idh3b), and isocitrate dehydrogenase (NADP+) 2 (Idh2) are key enzymes involved in the citrate cycle (TCA) which is essential for mitochondrial bioenergetics in cardiomyocytes [75]. Decreased expression of these genes (Suclg1/2, Sdha, Sdhc, Idh3b, and Idh2) suggested insufficient production of ATP from the mitochondrial TCA in Grx3 CKO DIO hearts (Figure 5B, Table S6). This lack of ATP may be a critical factor in the progression of heart failure [75-79]. Most interestingly, peroxisome proliferator-activated receptors (PPARs), the retinoid X receptors (RXRs), and the PPAR co-activator g (PGC-1a) play an essential role in the transcriptional regulation of genes encoding enzymes involved in cardiac fatty acid metabolism [80, 81]. In Grx3 CKO DIO hearts, the PPAR signaling pathway (Pparα, Rxrγ, Ppargc1α), especially PPARα targets in the cellular fatty acid oxidation pathway, was significantly down-regulated compared to DIO controls (Figures 4D, 5B, Table S6). Interestingly, while the fatty acid utilization as substrates was diminished in Grx3 CKO DIO mice, glucose uptake and utilization as fuel substrates was enhanced in Grx3 CKO DIO hearts compared to DIO controls as evident by increased expression of Glut1 and reduced expression of Pkd4 (Figure 6). Together, these findings suggest that dysregulation of fatty acid oxidation and altered glucose metabolism in cardiomyocytes, a fuel utilization switch, may contribute to cardiac function and heart failure in Grx3 CKO DIO mice [61, 82, 83].
Gene expression in the Grx3 CKO DIO heart. Q-PCR analysis indicates an increased Glut1, but reduced Glut4 and Pdk4 mRNA levels in the Grx3 CKO DIO heart, while Ucp3 expression was not altered. Gapdh was used as an internal control. Student t test, n=3, * p<0.05, ** p<0.01, and *** p<0.001 indicate the significance vs controls.
Grx3 is an iron-sulfur binding protein shown to regulate cellular iron homeostasis [84, 85]. Deletion of Grx3 results in iron accumulation in the cell, which could generate cellular ROS and cause redox imbalance through the Fenton chemical reaction. Grx3 has been shown to play an important role in cytosolic iron-sulfur assembly transferring [2Fe-2S] clusters to monomeric apo proteins [86-88]. Interestingly, a recent study reports that human Grx3 can bind and effectively deliver a [4Fe-4S] cluster to apo iron regulatory protein 1 (IRP1) and convert IRP1 into an aconitase [89]. Therefore, these studies suggest that the absence of Grx3 could impair Fe-S cluster assembly, which is required for many mitochondrial metabolic enzymes and nuclear proteins and may increase mitochondrial ROS production in Grx3 CKO DIO hearts. Our current findings indicate that expression of gene clusters involved in mitochondrial energy metabolism was significantly decreased (Figure 5B, Table S6), supporting the notion that mitochondrial function might be impaired in Grx3 CKO DIO heart. However, the mechanisms by which Grx3 regulates ROS and redox homeostasis in cardiomyocytes, especially under HFD feeding, remain to be fully investigated and warrant future study.
In conclusion, disruption of Grx3 increases ROS accumulation in cardiomyocytes and impairs cardiac function leading to heart failure under diet-induced obesity. Transcriptional analysis revealed a significant decrease in expression of genes related to fatty acid uptake, mitochondrial transfer, beta-oxidation, TCA, and PPAR signaling pathway and an increase in expression of genes related to glucose uptake and utilization, suggesting a metabolic switch occurred in Grx3 CKO DIO hearts (Figure 5C). Our findings indicate that Grx3 plays a critical role in cardiac energy metabolism and offers a clue to the elaborate regulatory interplay between redox homeostasis and metabolic control in the heart.
Supplementary Material
Supplementary figures.
Supplementary table 1.
Supplementary table 2.
Supplementary table 3.
Supplementary table 4.
Supplementary table 5.
Supplementary table 6.
Supplementary table 7.
Acknowledgements
This work was supported by the United States Department of Agriculture ⁄Agricultural Research Service under Cooperation Agreement 58-3092-5-001. The contents of this publication do not necessarily reflect the views or policies of the U.S. Department of Agriculture, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. NIH grant R01-GM115622 (to J.W). NIH grants R01HL147188, R01HL153358, and R01HL089598 (to X.H.T.W).
Author Contributions
Data curation, Qianxing Mo; Formal analysis, Ninghui Cheng, Qianxing Mo, Ghislain Breton, Kendal D. Hirschi, Xander H.T. Wehrens, and Paul A. Nakata; Funding acquisition, Ninghui Cheng, Xander H.T. Wehrens, and Paul A. Nakata; Investigation, Ninghui Cheng, Jimmonique Donelson and Lingfei Wang; Methodology, Qianxing Mo; Project administration, Ninghui Cheng and Paul A. Nakata; Resources, George G. Rodney, Jin Wang and Xander H.T. Wehrens; Writing - original draft, Ninghui Cheng; Writing - review & editing, Ghislain Breton, George G. Rodney, Jin Wang, Kendal D. Hirschi, Xander H.T. Wehrens, and Paul A. Nakata.
Competing Interests
J.W. is the co-founder of CoActigon Inc. and Chemical Biology Probes LLC.
References
1. Carbone S, Lavie CJ, Elagizi A, Arena R, Ventura HO. The Impact of Obesity in Heart Failure. Heart failure clinics. 2020;16:71-80
2. Lavie CJ, De Schutter A, Milani RV. Healthy obese versus unhealthy lean: the obesity paradox. Nat Rev Endocrinol. 2015;11:55-62
3. Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J Am Coll Cardiol. 2017;70:230-51
4. Xu Y-J, Tappia P, Neki N, Dhalla N. Prevention of diabetes-induced cardiovascular complications upon treatment with antioxidants. Heart Failure Reviews. 2014;19:113-21
5. Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304-17
6. Isakov N, Witte S, Altman A. PICOT-HD: a highly conserved protein domain that is often associated with thioredoxin and glutaredoxin modules. Trends in biochemical sciences. 2000;25:537-9
7. Cha H, Kim JM, Oh JG, Jeong MH, Park CS, Park J. et al. PICOT is a critical regulator of cardiac hypertrophy and cardiomyocyte contractility. J Mol Cell Cardiol. 2008;45:796-803
8. Cheng N-H, Zhang W, Chen W-Q, Jin J, Cui X, Butte NF. et al. A mammalian monothiol glutaredoxin, Grx3, is critical for cell cycle progression during embryogenesis. FEBS Journal. 2011;278:2525-39
9. Jeong D, Cha H, Kim E, Kang M, Yang DK, Kim JM. et al. PICOT inhibits cardiac hypertrophy and enhances ventricular function and cardiomyocyte contractility. Circ Res. 2006;99:307-14
10. Jeong D, Kim JM, Cha H, Oh JG, Park J, Yun SH. et al. PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circ Res. 2008;102:711-9
11. Oh JG, Jeong D, Cha H, Kim JM, Lifirsu E, Kim J. et al. PICOT increases cardiac contractility by inhibiting PKCzeta activity. J Mol Cell Cardiol. 2012;53:53-63
12. Donelson J, Wang Q, Monroe TO, Jiang X, Zhou J, Yu H. et al. Cardiac-specific ablation of glutaredoxin 3 leads to cardiac hypertrophy and heart failure. Physiological Reports. 2019;7:e14071
13. Qu Y, Wang J, Ray PS, Guo H, Huang J, Shin-Sim M. et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-κB signaling. The Journal of Clinical Investigation. 2011;121:212-25
14. Pham K, Dong J, Jiang X, Qu Y, Yu H, Yang Y. et al. Loss of glutaredoxin 3 impedes mammary lobuloalveolar development during pregnancy and lactation. American Journal of Physiology Endocrinology and Metabolism. 2017;312:E136-E49
15. Tam CS, Power JE, Markovic TP, Yee C, Morsch M, McLennan SV. et al. The effects of high-fat feeding on physical function and skeletal muscle extracellular matrix. Nutr Diabetes. 2015;5:e187
16. Nagels Durand A, Inigo S, Ritter A, Iniesto E, De Clercq R, Staes A. et al. The Arabidopsis Iron-Sulfur Protein GRXS17 is a Target of the Ubiquitin E3 Ligases RGLG3 and RGLG4. Plant & Cell Physiology. 2016;57:1801-13
17. Zuurbier CJ, Bertrand L, Beauloye CR, Andreadou I, Ruiz-Meana M, Jespersen NR. et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. Journal of Cellular and Molecular Medicine. 2020;24:5937-54
18. Xu W, Li L, Zhang L. NAD+ Metabolism as an Emerging Therapeutic Target for Cardiovascular Diseases Associated With Sudden Cardiac Death. Frontiers in Physiology. 2020;11:901
19. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA. et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883-94
20. Molkentin JD, Dorn GW 2nd. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391-426
21. Barrans JD, Allen PD, Stamatiou D, Dzau VJ, Liew C-C. Global Gene Expression Profiling of End-Stage Dilated Cardiomyopathy Using a Human Cardiovascular-Based cDNA Microarray. The American Journal of Pathology. 2002;160:2035-43
22. Barth AS, Kuner R, Buness A, Ruschhaupt M, Merk S, Zwermann L. et al. Identification of a Common Gene Expression Signature in Dilated Cardiomyopathy Across Independent Microarray Studies. Journal of the American College of Cardiology. 2006;48:1610-7
23. Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. The Journal of Clinical Investigation. 1995;95:888-94
24. Gorski Przemek A, Ceholski Delaine K, Hajjar Roger J. Altered Myocardial Calcium Cycling and Energetics in Heart Failure—A Rational Approach for Disease Treatment. Cell Metabolism. 2015;21:183-94
25. Patel DM, Green KJ. Desmosomes in the Heart: A Review of Clinical and Mechanistic Analyses. Cell Communication & Adhesion. 2014;21:109-28
26. Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H. et al. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. Journal of Medical Genetics. 2006;43:e5-e
27. Heliö K, Kangas-Kontio T, Weckström S, Vanninen SUM, Aalto-Setälä K, Alastalo T-P. et al. DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC Medical Genetics. 2020;21:19
28. Barresi R, Di Blasi C, Negri T, Brugnoni R, Vitali A, Felisari G. et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. Journal of Medical Genetics. 2000;37:102-7
29. Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circulation Research. 2011;108:607-18
30. McNally EM, Mestroni L. Dilated Cardiomyopathy. Circulation Research. 2017;121:731-48
31. Bonne G, Carrier L, Richard P, Hainque B, Schwartz K. Familial Hypertrophic Cardiomyopathy. Circulation Research. 1998;83:580-93
32. Karibe A, Tobacman LS, Strand J, Butters C, Back N, Bachinski LL. et al. Hypertrophic Cardiomyopathy Caused by a Novel α-Tropomyosin Mutation (V95A) Is Associated With Mild Cardiac Phenotype, Abnormal Calcium Binding to Troponin, Abnormal Myosin Cycling, and Poor Prognosis. Circulation. 2001;103:65-71
33. Karam CN, Warren CM, Rajan S, de Tombe PP, Wieczorek DF, Solaro RJ. Expression of tropomyosin-κ induces dilated cardiomyopathy and depresses cardiac myofilament tension by mechanisms involving cross-bridge dependent activation and altered tropomyosin phosphorylation. Journal of Muscle Research and Cell Motility. 2011;31:315-22
34. Despond EA, Dawson JF. Classifying Cardiac Actin Mutations Associated With Hypertrophic Cardiomyopathy. Frontiers in Physiology. 2018;9:405 -
35. Movsesian MA. Altered cAMP-mediated signalling and its role in the pathogenesis of dilated cardiomyopathy. Cardiovascular Research. 2004;62:450-9
36. Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. Journal of Molecular and Cellular Cardiology. 2016;93:162-74
37. Dullens HFJ, Schipper MEI, van Kuik J, Sohns W, Scheenstra M, van Wichen DF. et al. Integrin expression during reverse remodeling in the myocardium of heart failure patients. Cardiovascular Pathology. 2012;21:291-8
38. Doetschman T, Barnett JV, Runyan RB, Camenisch TD, Heimark RL, Granzier HL. et al. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell and Tissue Research. 2012;347:203-23
39. Liu G, Ma C, Yang H, Zhang P-Y. Transforming growth factor β and its role in heart disease. Exp Ther Med. 2017;13:2123-8
40. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiological Reviews. 2008;88:389-419
41. Cavalera M, Wang J, Frangogiannis NG. Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Translational research: The Journal of Laboratory and Clinical Medicine. 2014;164:323-35
42. Frangogiannis NG. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circulation Research. 2019;125:117-46
43. Terasaki F, Okamoto H, Onishi K, Sato A, Shimomura H, Tsukada B. et al. Higher Serum Tenascin-C Levels Reflect the Severity of Heart Failure, Left Ventricular Dysfunction and Remodeling in Patients With Dilated Cardiomyopathy. Circulation Journal. 2007;71:327-30
44. Frangogiannis NG. Syndecan-1: a critical mediator in cardiac fibrosis. Hypertension (Dallas, Tex: 1979). 2010;55:233-5
45. Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F. et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation. 2018;138:1236-52
46. Querejeta R, López B, González A, Sánchez E, Larman M, Ubago JLM. et al. Increased Collagen Type I Synthesis in Patients With Heart Failure of Hypertensive Origin. Circulation. 2004;110:1263-8
47. López B, Ravassa S, González A, Zubillaga E, Bonavila C, Bergés M. et al. Myocardial Collagen Cross-Linking Is Associated With Heart Failure Hospitalization in Patients With Hypertensive Heart Failure. Journal of the American College of Cardiology. 2016;67:251-60
48. Samarel AM. Focal adhesion signaling in heart failure. Pflugers Archiv: European Journal of Physiology. 2014;466:1101-11
49. Itoh N, Ohta H. Pathophysiological roles of FGF signaling in the heart. Frontiers in Physiology. 2013;4:247 -
50. Makki N, Thiel KW, Miller FJ. The Epidermal Growth Factor Receptor and Its Ligands in Cardiovascular Disease. International Journal of Molecular Sciences. 2013;14:20597-613
51. Sysa-Shah P, Xu Y, Guo X, Belmonte F, Kang B, Bedja D. et al. Cardiac-Specific Over-Expression of Epidermal Growth Factor Receptor 2 (ErbB2) Induces Pro-Survival Pathways and Hypertrophic Cardiomyopathy in Mice. PLOS ONE. 2012;7:e42805
52. Gogiraju R, Bochenek ML, Schäfer K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Frontiers in Cardiovascular Medicine. 2019;6:20
53. Hanna A, Frangogiannis NG. The Role of the TGF-β Superfamily in Myocardial Infarction. Frontiers in Cardiovascular Medicine. 2019 6
54. Martin R, Gutierrez B, Cordova C, Roman AS, Alvarez Y, Hernandez M. et al. Secreted Phospholipase A2-IIA Modulates Transdifferentiation of Cardiac Fibroblast through EGFR Transactivation: An Inflammation-Fibrosis Link. Cells. 2020 9
55. Brown JH, Re DPD, Sussman MA. The Rac and Rho Hall of Fame. Circulation Research. 2006;98:730-42
56. Singh RM, Cummings E, Pantos C, Singh J. Protein kinase C and cardiac dysfunction: a review. Heart failure reviews. 2017;22:843-59
57. Zhang F, Xia Y, Yan W, Zhang H, Zhou F, Zhao S. et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. American Journal of Physiology-Heart and Circulatory Physiology. 2016;310:H250-H61
58. Wang Y, Streicher JM. The Role of COX-2 in Heart Pathology. Cardiovascular & Hematological Agents in Medicinal Chemistry. 2008;6:69-79
59. Frolova EG, Sopko N, Blech L, Popovic ZB, Li J, Vasanji A. et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB Journal. 2012;26:2363-73
60. Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V. et al. Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circulation Research. 2016;118:1659-1701
61. Bertero E, Maack C. Metabolic remodelling in heart failure. Nature Reviews Cardiology. 2018;15:457-70
62. Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circulation Research. 2007;100:795-806
63. Lopaschuk GD, Folmes CDL, Stanley WC. Cardiac Energy Metabolism in Obesity. Circulation Research. 2007;101:335-47
64. Umbarawan Y, Syamsunarno M, Koitabashi N, Obinata H, Yamaguchi A, Hanaoka H. et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Scientific Reports. 2018;8:12035
65. Abumrad NA, Goldberg IJ. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochimica Biophysica Acta. 2016;1861:1442-9
66. Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Molecular Aspects of Medicine. 2013;34:516-28
67. He L, Kim T, Long Q, Liu J, Wang P, Zhou Y. et al. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation. 2012;126:1705-16
68. Ellis JM, Mentock SM, DePetrillo MA, Koves TR, Sen S, Watkins SM. et al. Mouse Cardiac Acyl Coenzyme A Synthetase 1 Deficiency Impairs Fatty Acid Oxidation and Induces Cardiac Hypertrophy. Molecular and Cellular Biology. 2011;31:1252-62
69. Pereyra AS, Hasek LY, Harris KL, Berman AG, Damen FW, Goergen CJ. et al. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J Biol Chem. 2017;292:18443-56
70. Sharpe AJ, McKenzie M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells. 2018;7:46
71. Strauss AW, Powell CK, Hale DE, Anderson MM, Ahuja A, Brackett JC. et al. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proceedings of the National Academy of Sciences. 1995;92:10496-500
72. Marı́n-Garcı́a J, Goldenthal MJ. Fatty acid metabolism in cardiac failure: biochemical, genetic and cellular analysis. Cardiovascular Research. 2002;54:516-27
73. Orii KE, Aoyama T, Wakui K, Fukushima Y, Miyajima H, Yamaguchi S. et al. Genomic and Mutational Analysis of the Mitochondrial Trifunctional Protein β-Subunit (HADHB) Gene in Patients with Trifunctional Protein Deficiency. Human Molecular Genetics. 1997;6:1215-24
74. Abo Alrob O, Lopaschuk G. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochemical Society Transactions. 2014;42:1043-51
75. Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovascular Research. 2011;90:202-9
76. Demirbas D, Harris D, Arn P, Huang X, Waisbren S, Anselm I. et al. Phenotypic variability in deficiency of the α subunit of succinate-CoA ligase. JIMD Reports. 2019;46:63-9
77. Levitas A, Muhammad E, Harel G, Saada A, Caspi VC, Manor E. et al. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet. 2010;18:1160-5
78. Akbay EA, Moslehi J, Christensen CL, Saha S, Tchaicha JH, Ramkissoon SH. et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes & Development. 2014;28:479-90
79. Ku HJ, Ahn Y, Lee JH, Park KM, Park J-W. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radical Biology and Medicine. 2015;80:84-92
80. Duncan JG, Finck BN. The PPARalpha-PGC-1alpha Axis Controls Cardiac Energy Metabolism in Healthy and Diseased Myocardium. PPAR Research. 2008;2008:253817
81. Oka S-i, Sabry AD, Cawley KM, Warren JS. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Frontiers in Cardiovascular Medicine. 2020 7
82. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818-28
83. Wende AR, Brahma MK, McGinnis GR, Young ME. Metabolic Origins of Heart Failure. JACC: Basic to Translational Science. 2017;2:297-310
84. Haunhorst P, Berndt C, Eitner S, Godoy JR, Lillig CH. Characterization of the human monothiol glutaredoxin 3 (PICOT) as iron-sulfur protein. Biochem Biophys Res Commun. 2010;394:372-6
85. Haunhorst P, Hanschmann EM, Brautigam L, Stehling O, Hoffmann B, Muhlenhoff U. et al. Crucial function of vertebrate glutaredoxin 3 (PICOT) in iron homeostasis and hemoglobin maturation. Molecular Biology of the Cell. 2013;24:1895-903
86. Banci L, Ciofi-Baffoni S, Gajda K, Muzzioli R, Peruzzini R, Winkelmann J. N-terminal domains mediate [2Fe-2S] cluster transfer from glutaredoxin-3 to anamorsin. Nature Chemical Biology. 2015;11:772-8
87. Frey AG, Palenchar DJ, Wildemann JD, Philpott CC. A Glutaredoxin-BolA Complex Serves as an Iron-Sulfur Cluster Chaperone for the Cytosolic Cluster Assembly Machinery. J Biol Chem. 2016
88. Camponeschi F, Prusty NR, Heider SAE, Ciofi-Baffoni S, Banci L. GLRX3 Acts as a [2Fe-2S] Cluster Chaperone in the Cytosolic Iron-Sulfur Assembly Machinery Transferring [2Fe-2S] Clusters to NUBP1. Journal of the American Chemical Society. 2020;142:10794-805
89. Xia H, Li B, Zhang Z, Wang Q, Qiao T, Li K. Human glutaredoxin 3 can bind and effectively transfer [4Fe-4S] cluster to apo-iron regulatory protein 1. Biochem Biophys Res Commun. 2015;465:620-4
Author contact
![]() Corresponding author: Ninghui Cheng, Ph.D., USDA/ARS Children Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, 1100 Bates Street, Houston, TX 77030. Tel: 7137989326; Email: nchengedu
Corresponding author: Ninghui Cheng, Ph.D., USDA/ARS Children Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, 1100 Bates Street, Houston, TX 77030. Tel: 7137989326; Email: nchengedu