Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The roles of m6A modification in...
The biogenesis and functions of...
The roles of circRNA in HCC
Cross-talk and interplay between...
Introduction to common databases...
m6A modifiers and circRNAs as...
Conclusions and perspectives
Abbreviation
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(12):3059-3079. doi:10.7150/ijbs.62767 This issue Cite
Review
The functional roles, cross-talk and clinical implications of m6A modification and circRNA in hepatocellular carcinoma
Sha Qin1#, Yitao Mao2,3#, Xue Chen4, Juxiong Xiao2, Yan Qin2, Luqing Zhao1,3 ![]()
1. Department of Pathology, Xiangya Hospital, Central South University, Changsha, Hunan, China; and Department of Pathology, School of Basic Medical Science, Xiangya School of Medicine, Central South University, Changsha, Hunan, China.
2. Department of Radiology, Xiangya Hospital, Central South University, Changsha, Hunan, China.
3. National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan, China.
4. Early Clinical Trial Center, Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, China.
# These authors contributed equally to this work.
Received 2021-5-15; Accepted 2021-7-6; Published 2021-7-22
Abstract

Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide. HCC has high rates of death and recurrence, as well as very low survival rates. N6-methyladenosine (m6A) is the most abundant modification in eukaryotic RNAs, and circRNAs are a class of circular noncoding RNAs that are generated by back-splicing and they modulate multiple functions in a variety of cellular processes. Although the carcinogenesis of HCC is complex, emerging evidence has indicated that m6A modification and circRNA play vital roles in HCC development and progression. However, the underlying mechanisms governing HCC, their cross-talk, and clinical implications have not been fully elucidated. Therefore, in this paper, we elucidated the biological functions and molecular mechanisms of m6A modification in the carcinogenesis of HCC by illustrating three different regulatory factors ("writer", "eraser", and "reader") of the m6A modification process. Additionally, we dissected the functional roles of circRNAs in various malignant behaviors of HCC, thereby contributing to HCC initiation, progression and relapse. Furthermore, we demonstrated the cross-talk and interplay between m6A modification and circRNA by revealing the effects of the collaboration of circRNA and m6A modification on HCC progression. Finally, we proposed the clinical potential and implications of m6A modifiers and circRNAs as diagnostic biomarkers and therapeutic targets for HCC diagnosis, treatment and prognosis evaluation.
Keywords: N6-methyladenosine (m6A) modification, circRNA, Hepatocellular carcinoma (HCC), Biomarkers, Therapeutic targets
Introduction
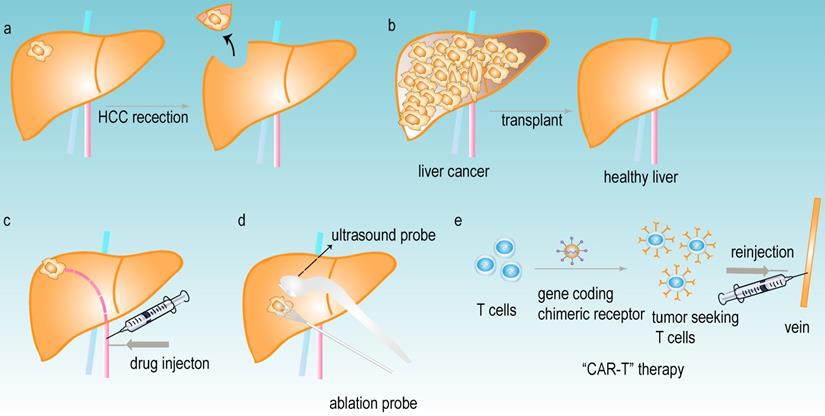
Primary liver cancer is one of the most common malignancies, which has a low survival rate. It is currently the sixth most commonly diagnosed cancer and the third leading cause of cancer mortality worldwide, and the hepatocellular carcinoma (HCC) comprises 75%-85% of primary liver cancer [1]. With recognition of the severity and danger of HCC, pharmacological and nonpharmacological treatments have achieved great progress in the therapeutic field of HCC. The nonpharmacological treatments mainly include HCC resection (Fig 1a) [2], liver transplantation (Fig 1b) [3], trans-arterial chemoembolization (TACE) (Fig 1c) [4], percutaneous ablation (Fig 1d) [5] and chimeric antigen receptor engineered T-cell immunotherapy (CAR-T) (Fig 1e) [6]. The pharmacological treatments largely consist of small molecule targeted drug therapies, such as sorafenib [7], lenvatinib, regrofenib, and monoclonal antibodies such as nivolumab [8]. These drugs are used mainly for the systemic treatment of advanced HCC. However, most patients are diagnosed at advanced stages due to the lack of obvious early symptoms and limitations in early detection or screening. Despite the variety of treatments for HCC, the overall survival rate of patients is still far from satisfactory; therefore, developing more effective early diagnosis and treatment methods for HCC patients is critical.
The common treatments of HCC. (a) HCC resection. (b) liver transplant. (c) trans-arterial chemoembolization (TACE). (d) percutaneous ablation. (e) chimeric antigen receptor-engineered T-cell therapy (CAR-T).
N6-methyladenosine (m6A) has been shown to be the most abundant modification in eukaryotic RNAs and plays an essential role in a variety of biological processes, including RNA stability, translation, splicing, degradation, and export [9, 10]. An increasing number of studies have proven that aberrant regulation of m6A modification is associated with a wide variety of human malignancies, including breast cancer, lung cancer, colorectal cancer, and HCC [11, 12].
circRNA is a circular noncoding RNA formed by back-splicing. Due to its covalent closed-loop structure, circRNA can prevent degradation from RNA exonuclease; thus, circRNA is more stable than linear mRNA. Accumulating evidence has indicated that the functions of circRNAs in gene regulation are closely related to the development of HCC and that circRNAs might be promising potential biomarkers and therapeutic targets for the diagnosis and treatment of HCC [13].
In this paper, we elucidated the functional roles, cross-talk, and underlying interplay mechanisms between m6A modification and circRNA in HCC carcinogenesis and development. And we demonstrated the clinical implications of m6A modifiers and circRNAs in the diagnosis and treatment of HCC as promising diagnostic biomarkers and therapeutic targets. In particular, we also briefly identified common databases and research methods for investigating the roles and interactions of m6A and circRNA, which could provide novel in-depth research insights and avenues for further studies on the functions and mechanisms of m6A and circRNAs in HCC.
The roles of m6A modification in HCC
The components of m6A modification
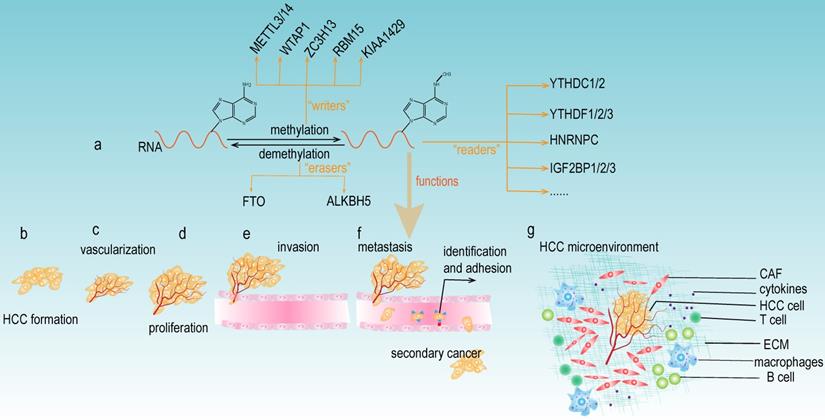
m6A modification consists of the following three main components: m6A "writers", "erasers", and "readers" (Fig 2a). The "writers" are methyltransferases that function to promote the methylation modification of RNA. They mainly include METTL3, METTL14, WTAP1, RBM15, and KIAA1429 [14]. The "erasers" are the enzymes that promote the demethylation of m6A modification. Currently, the "erasers" mainly include FTO and ALKBH5. The presence of these enzymes indicates that m6A modification is a dynamic and reversible process. Notably, m6A modification relies on “readers” to perform its biological functions. The "readers" selectively recognize and bind the m6A modification site on the RNA and subsequently change the RNA structures, thus leading to different functions in RNA biology. The YTH protein domain families are the major m6A “readers”, which consist of YTHDC1 (in the nucleus), YTHDC2, YTHDF1, YTHDF2 and YTHDF3 (in the cytoplasm). In addition, the heterogeneous nuclear ribonucleoprotein (HNRNP) families (including HNRNPA2B1, HNRNPC and HNRNPG), NF-κβ-associated protein (NKAP) and IGF2BP families are also m6A "readers" that exert important roles in various biological functions of m6A modification.
The functions of m6A modification in HCC. (a) The components of m6A modification. m6A “writers” catalyze the m6A modification of adenosine on RNA and mainly include METTL3/14, WTAP, ZC3H13, RBM15, and KIAA1492. Removing the methylation of RNA needs the functions of m6A “erasers” that mainly consist of FTO and ALKBH5. m6A “readers” (such as YTHDC1/2, YTHDF1/2/3, HNRNPC, and IGF2BP1/2/3) recognize m6A modification sites and exert various biological functions. (b-g) Multiple functions of m6A modification in HCC development. m6A modifiers could affect the tumor formation, vascularization, proliferation, invasion, metastasis, and the microenvironment of HCC.
The m6A “writers” and HCC
METTL3 and HCC
METTL3 has a catalytic subunit that can bind S-adenosylmethionine (SAM) (which provides the methyl group) and catalyze methylation [15], and METTL3 was the first identified methyltransferase catalyzing m6A modifications of mRNA and noncoding RNA [16]. Transcriptome sequencing revealed that METTL3 was significantly upregulated in HCC tissues. Knockdown of METTL3 significantly reduced the proliferation, migration and colony formation capacities of HCC cells. Overexpression of METTL3 was associated with poor prognosis in HCC patients [17]. Additionally, a gene set enrichment analysis of HCC specimens demonstrated that METTL3 expression was closely related to glycolysis and the mTOR signaling pathway. Overexpression of METTL3 could activate the mTOR complex 1 (mTORC1) and facilitate glycolysis in HCC cells through mTORC1 pathway [18]. Moreover, another study discovered that the Hippo pathway was involved in the METTL3-mediated formation of vasculogenic mimicry and this m6A modification could affect the translation of YAP1 mRNA, thus promoting the angiogenesis of HCC [19]. Knockdown of METTL3 expression under hypoxic conditions was also reported to be able to promote the expression of angiogenic genes and activate autophagy-related pathways in HCC cells. Furthermore, METTL3 deletion abolished METTL3-mediated regulation of FOXO3 mRNA stability and markedly enhanced resistance to sorafenib treatment in HCC, therefore uncovering the critical functions of METTL3-mediated m6A modification in the hypoxic microenvironment of HCC [20].
METTL14 and HCC
METTL14 and METTL3 share 43% homology in their molecular structure [21]. As a homolog of METTL3, the downregulation of METTL14 was closely related to tumor metastasis and it acted as an adverse prognostic factor of recurrence-free survival in HCC patients. METTL14 could interact with the microprocessor protein DGCR8 and positively regulated pri-miR-126 biogenesis in an m6A-dependent manner [22]. METTL14 could also inhibit the metastasis of HCC by upregulating the m6A methylation of CSAD, GOT2, and SOCS2 [23]. Another report indicated that METTL14 modulated the EGFR/PI3K/AKT signaling pathway to impair the migration, invasion and epithelial-mesenchymal transition (EMT) of HCC cells [24]. Additionally, hepatocyte nuclear factor 3γ (HNF3γ) expression was significantly downregulated in HCC patients and was negatively correlated with HCC progression and survival rate. The reduction in HNF3γ expression was mediated by METTL14-dependent m6A methylation of HNF3γ mRNA. In particular, overexpression of HNF3γ could reduce the drug resistance of HCC by upregulating the expression of OATP1B1 and OATP1B3, which are the two major membrane transport proteins affecting sorafenib absorption, thus sensitizing HCC cells to sorafenib-induced growth repression and apoptosis [25].
Other “writers” and HCC
In addition to two major m6A "writers", METTL3 and METTL14, several other "writers" have also been discovered to govern HCC development and progression. For example, KIAA1429 was remarkably upregulated in HCC tissues and was strongly associated with the proliferation and metastasis of HCC cells. KIAA1429 induced m6A methylation on the 3'-untranslated region (3'-UTR) of GATA3 mRNA and led to the disassociation of the RNA-binding protein (RBP) HuR, thus resulting in the degradation of GATA3 mRNA and promoting the formation of HCC [26]. WTAP was also highly expressed in HCC and was an independent predictor of HCC patient survival. WTAP boosted HCC cell proliferation and accelerated HCC progression via the HuR-ETS1-p21/p27 axis [27]. Accumulating evidence has demonstrated that multiple growth factors can stimulate HCC cell proliferation by activating the Ras/Raf-1/ERK pathway, which is essential for the tumorigenesis of HCC [28]. Recently, bioinformatics analysis revealed that ZC3H13 and KIAA1429 could be practical and reliable prognostic indicators of HCC [29]; however, the molecular mechanisms of ZC3H13 in the RAS-ERK pathway and their interactions in HCC have not been fully elucidated, therefore further researches are needed to be conducted in the future.
The m6A “erasers” and HCC
FTO and HCC
Fat mass and obesity-associated protein (FTO) is well known for its pivotal roles in the regulation of fat mass, adipogenesis and body weight. FTO mRNA levels were reported to be significantly lower in HCC tissues. The expression of FTO was reduced in the HCC cells and was closely associated with AFP levels, tumor size, metastasis and vascular invasion [30, 31]. Another study demonstrated that overexpression of FTO was associated with poor prognosis in HCC patients. Knockdown of FTO suppressed the proliferation of HCC and induced G0/G1 phase arrest. Mechanistically, FTO resulted in the demethylation of PKM2 mRNA and accelerated its translation process, thus facilitating HCC malignant progression [32]. Moreover, SIRT1 exerted an oncogenic role by downregulating the expression of FTO. Guanine nucleotide-binding protein G (o) subunit alpha (GNAO1) was identified as the downstream target of FTO and a tumor suppressor in HCC. Depletion of FTO by SIRT1 could improve the m6A modification of GNAO1 and downregulated its mRNA expression level, therefore contributing to HCC metastasis [33]. Additionally; FTO is indispensable for the regulation of energy homeostasis and glucose metabolism in HCC. Furthermore, FTO acted as a protector in HCC carcinogenesis, and FTO-dependent dynamic mRNA demethylation of CUL4A exerted critical functions in the initiation and development of HCC [34].
ALKBH5 and HCC
ALKBH5 expression in HCC tissues was significantly lower than that in normal tissues, and the absence of ALKBH5 predicted poor overall survival and disease-free survival in HCC patients [30, 35]. ALKBH5-mediated m6A demethylation resulted in the posttranscriptional repression of LY6/PLAUR domain 1 (LYPD1), which could be stabilized by the m6A “reader” IGF2BP1. In addition, LYPD1 was identified to induce the oncogenic behaviors of HCC. These results suggested that the dysregulation of the ALKBH5/LYPD1 axis could accelerate the progression and invasiveness of HCC.
The m6A “readers” and HCC
Many m6A “readers” such as YTHDC1/2, YTHDF1/2/3, HNRNPA2B1, HNRNPC, HNRNPG, NKAP and IGF2BP1/2/3 have been reported to play essential roles in HCC development. For example, Liu et al. revealed that YTHDC2 could act as a promising target for specific therapeutic strategy in unresectable HCC patients [36]. Li et al. found that the IGF2BP1/IGF2BP3 could stabilize LINC01138 transcript and was associated with the malignant features and poor prognosis of HCC patients [37]. In addition, the overexpression of IGF2BP2 enhanced the stability of FEN1 mRNA, so as to promote HCC proliferation [38]. HNRNPA2B1 was also involved in HCC metastasis. Wang et al. showed that HNRNPA2B1decreased the stability of p52 and p65 mRNA. Besides, miR503HG could promote the degradation of HNRNPA2B1 and inhibit HCC migration through suppressing the NF-κB signaling pathway [39]. NKAP was highly expressed in HCC tissues. And NKAP promoted the proliferation and invasion in HCC cell through AKT signaling pathway [40]. Zhao et al. discovered that YTHDF1 was significantly upregulated in HCC and positively related to pathological stage. Kaplan-Meier analysis suggested that lower YTHDF1 expression levels were associated with better survival in HCC patients. Moreover, YTHDF1 acted as a key regulator of cell cycle progression and metabolism in HCC, and it became a potential novel therapeutic and prognostic target for HCC [41]. Additionally, bioinformatics analysis predicted that YTHDF1 and YTHDF2 were significantly positively correlated with overall survival, WHO stage, and pathological grade [42, 43]. Zhang et al. demonstrated that YTHDF2 expression was negatively associated with survival time in HCC. In addition, YTHDF2 expression was positively related to OCT4 expression and m6A levels in the 5'-UTR of OCT4 mRNA in HCC clinical specimens. Therefore, YTHDF2 facilitated the HCC stemness phenotype and tumor metastasis by regulating m6A methylation of OCT4 [44, 45]. Another study identified that the downregulation of YTHDF2 was specifically induced by hypoxia in HCC cells, and overexpression of YTHDF2 could inhibit cell proliferation and tumor growth in HCC cells. Mechanistically, YTHDF2 directly bound the m6A modification site in the EGFR 3'-UTR to accelerate the degradation of EGFR mRNA. This was the first report indicating that YTHDF2 might act as a tumor suppressor to suppress cell proliferation and growth by destabilizing EGFR mRNA in HCC development [46].
As mentioned above, the "writers", "erasers" and "readers" of m6A modification are significantly involved in the development of HCC (Table 1), and this modification can either facilitate or suppress the progression of HCC. These paradoxical results caused by m6A modification may be attributable to different downstream targets of m6A, which could impact diverse signaling pathways and trigger different biological functions. Further studies on the specific regulatory mechanisms are still needed. m6A regulators have been reported to be involved in various aspects of HCC carcinogenesis, including tumor formation, vascularization, proliferation, invasion, metastasis, and the tumor microenvironment (Fig 2b-g). Meanwhile, most current research investigating m6A regulators focused on the downstream molecular mechanisms of specific genes; however, there are relatively insufficient studies exploring the upstream modulatory mechanisms, thus providing new avenues and directions to conduct in-depth research concerning m6A modification and HCC development.
The relationship between m6A modification and HCC
| Category | m6A regulators | Up/down | Functions | Mechanisms | Refs |
|---|---|---|---|---|---|
| “writers” | METTL3 | up | promote proliferation, migration and colony formation of HCC | NA | [17] |
| “writers” | METTL3 | up | facilitate glycolysis | activate the mTORC1 pathway | [18] |
| “writers” | METTL3 | up | promote the formation of vasculogenic mimicry | affect the translation of YAP1 mRNA | [19] |
| “writers” | METTL3 | down | enhance the resistance to sorafenib | abolish METTL3-mediated FOXO3 mRNA stability | [20] |
| “writers” | METTL14 | up | promote metastasis | upregulate m6A methylation of CSAD, GOT2, and SOCS2 | [23] |
| “writers” | METTL14 | down | inhibit migration, invasion and EMT | regulate EGFR/PI3K/AKT signaling pathway | [24] |
| “writers” | METTL14 | up | enhance the resistance to sorafenib | reduce the expression of HNF3γ | [25] |
| “writers” | KIAA1429 | up | promote proliferation and metastasis | Induce degradation of GATA3 precursor mRNA | [26] |
| “writers” | WTAP | up | boost HCC cell proliferation | the HuR-ETS1-p21/p27 axis | [27] |
| “writers” | ZC3H13 | up | prognostic indicators for HCC | may associated with RAS-ERK pathway | [28,29] |
| “erasers” | FTO | up | poor prognosis | demethylation of PKM2 mRNA and speed up the translation | [32] |
| “erasers” | FTO | down | induce metastasis | downregulate m6A-modified GNAO1 and upregulated its mRNA expression | [33] |
| “erasers” | FTO | up | promote glucose metabolism | reduce CUL4A protein abundance | [34] |
| “erasers” | ALKBH5 | down | promote proliferation and invasive | repress LYPD1 | [30,35] |
| “readers” | YTHDC2 | up | poor prognosis | small nucleotide polymorphisms | [36] |
| “readers” | IGF2BP1/IGF2BP3 | up | poor prognosis | stabilize LINC01138 transcript | [37] |
| “readers” | IGF2BP2 | up | promote HCC proliferation | enhance the stability of FEN1 mRNA | [38] |
| “readers” | HNRNPA2B1 | down | inhibit HCC migration | decreased the stability of p52 and p65 mRNA | [39] |
| “readers” | NKAP | up | promote the proliferation and invasion | AKT signaling pathway | [40] |
| “readers” | YTHDF1 | up | influence pathological stage | regulate cell cycle progression and metabolism | [41] |
| “readers” | YTHDF2 | up | facilitate HCC stem cell phenotype and metastasis | increase OCT4 protein expression | [42,43] |
| “readers” | YTHDF2 | down | inhibit the occurrence of HCC | accelerate the degradation of EGFR mRNA | [44] |
The biogenesis and functions of circRNA
Accumulating evidence has demonstrated that a large number of circRNAs are aberrantly expressed in HCC tissues, suggesting that these circRNAs may play a vital role in the development of HCC. Based on their different origins, circRNAs can be divided into the following three groups: exonic circRNAs (ecRNAs), exon-intron circRNAs (eIciRNAs) and intronic circRNAs (ciRNAs). Exonic circRNAs are generated mainly from the exons of protein-coding genes [47]. When exons are cyclized by introns and exons, they generated eIciRNAs. eIciRNAs are largely localized in the nucleus and interact with U1 snRNP to promote the transcription of their parental genes [48]. Due to the debranching failure, ciRNA can accumulate in large quantities at their transcription sites. ciRNAs are associated with the elongation of Pol II and act as positive regulators of Pol II transcription, thus exerting cis-regulatory effects on their parental coding genes [49].
The biogenesis of circRNA
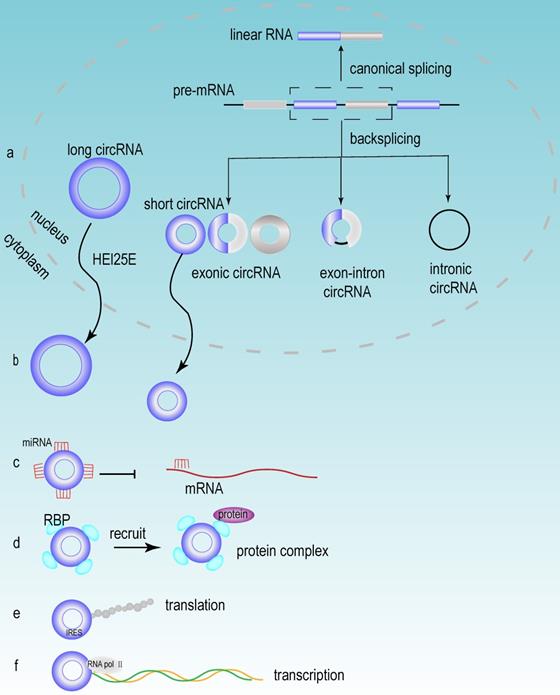
circRNA is an abundant, stable, conserved, nonrandom product generated by RNA splicing that may participate in the regulation of gene expression [50]. Typically, eukaryotic pre-mRNA splicing is catalyzed by a spliceosomal mechanism that removes introns and attaches exons, resulting in the formation of linear RNA transcripts. The generation of circRNA relies on a back-splicing mechanism that backlinks the downstream splice donor site to the upstream splice acceptor site and forms a covalently closed circular RNA transcript and a selectively spliced linear RNA that skips exons (Fig 3a) [51]. Competition exists between the back splicing of circRNA and typical splicing of linear RNA, and this competition is modulated by cis- and trans- elements [52]. Base pairing between highly supplementary transposable elements can boost back splicing [53]. The loop formation of intron sequences on the flank of the downstream splice donor site and the upstream splice acceptor site keeps these sites in close proximity, which may be the main foundation for the back splicing of circRNA. This loop formation can be achieved by base pairing between reverse repeat elements (e.g. Alu elements) located in the upstream and downstream introns [54]. The efficiency of exon loop formation can be regulated by competition between RNA pairs. The alternate formation of reverse repeat Alu elements and the competition between them results in multiple circRNA transcripts from a single gene [55]. The biogenesis of circRNA is regulated by various factors, including intron sequences, enzymes and protein factors [56]. Adenosine deaminase ADAR1 is a type of RNA editing enzyme. ADAR1 has been reported to negatively regulate circRNA expression, and the knockdown of ADAR1 induces the elevation of circRNA expression [57]. RBPs may play an activating or inhibiting role in circRNA formation by regulating variable splicing factor Quaking (QKI) levels. And QKI binding motifs of introns exert an essential impact on circRNA abundance [58].
The biogenesis and functions of circRNA. (a) The mechanisms of circRNA biogenesis. The formation of circRNA was owing to back-splicing. There were three types of circRNA named exon circRNA, exon-intronic circRNA and intronic circRNA, which were generated through different splicing methods. (b) Nuclear export of circRNA. Most circRNAs executed their first functions through exporting the nucleus circRNA to the cytoplasm, and this process mainly depended on the length of circRNA. (c) circRNA acted as miRNA sponge. (d) circRNA interacted with RBP. (e) circRNA could be translated in a cap-independent manner via the internal ribosome entry site (IRES). (f) circRNA regulated transcription.
The general functions of circRNA
Nucleus export process of circRNA
After the formation of circRNAs, most of circRNAs (except for ciRNAs) are exported to the cytoplasm to perform different biological functions [59]. Research has revealed that the process of circRNA exporting the nucleus is closely related to its own length [60, 61]. circRNA is transported from the nucleus to the cytoplasm via the ATP-dependent RNA helicase DDX39A (also known as URH49) and the spliceosomal RNA helicase DDX39B (also known as UAP56). And this mode of transport is closely related to the length of circRNA. When the circRNA length is longer than 800 bases, the nuclear export process relies on Hel25E. With the transcript length increasing, the export of circRNA becomes increasingly dependent on Hel25E (Fig 3b) [62].
circRNA acts as a miRNA sponge
miRNA is an endogenous short-stranded noncoding RNA that consists of approximately 20-25 nucleotides. Current studies have shown that miRNAs can specifically bind to mRNA and regulate gene expression through posttranscriptional regulation, thus contributing to mRNA silencing or degradation [63]. circRNAs can function as miRNA sponges and can compete for mRNA-miRNA binding sites, thereby inhibiting mRNA suppression (Fig 3c). When circRNA functions as a miRNA sponge, the stoichiometric relationship between the miRNA binding site of circRNA and the mRNA binding site of miRNA is an important factor to influence the efficiency of circRNA function [64]. Notably, circRNAs can act as sponges for multiple different miRNAs, therefore, the same circRNA can affect multiple different miRNAs and perform different biological functions [65]. Different circRNAs have different functions, but some similar biological functions can be performed by different circRNAs. For example, circRNA_0016624 could act as a molecular sponge for miR-98 and enhanced the expression of BMP2; therefore, circRNA_0016624 could prevent osteoporosis [66]. circRNA_33186 acted as a molecular sponge to directly bind and repress miR-127-5p, thereby increasing the expression of MMP-13, which is involved in the pathogenesis of osteoarthritis [67]. In glioblastoma, circNT5E acted as a sponge for miR-422a and could inhibit the activity of miR-422a, thereby promoting tumor cell proliferation, migration and invasion [68].
The translation capacity of circRNA
Although circRNA is a circular noncoding RNA, several studies in recent years have shown that it also has translation capacity [69]. In recent years, several circRNAs have been discovered to have translational functions, which have biological roles in human diseases. The discovery of these circRNAs and their encoded peptide-enriched genomes helped us to study the etiology of diseases and facilitated the development of biotechnology [70]. circMYBL2 has been proven to be translated, and it could regulate FLT3 kinase levels so as to specifically affect acute leukemia that caused by internal tandem repeat mutations of FLT3. The mechanism of this function is that circMYBL2 enhances the translation efficiency of FLT3 kinase by increasing the binding of polypyrimidine binding protein 1 (PTBP1) to the mRNA of FMS-like tyrosine kinase-3 (FLT3) [71]. Furthermore, although circRNAs lack essential components such as the 5' cap and poly(A) tail for cap-dependent translation, circRNAs can still be translated in a cap-independent manner via the internal ribosome entry site (IRES) (Fig 3e) [72].
Other functions of circRNA
circRNAs can also regulate gene expression at the transcriptional level (Fig 3f). For example, eIciRNAs, which are mainly localized in the nucleus, interact with U1 snRNP to promote the transcription of their parental genes [48]. circRNA also participates in regulating the variable splicing of transcripts, and researchers have proposed a mechanism by which circRNA can regulate transcription through cyclization, i.e., the formation of circRNA can compete with splicing of linear RNAs for common pre-mRNAs, thereby affecting the abundance of linear RNAs [52]. In addition, circRNAs can also interact with RBPs (Fig 3d) [73]. In detail, circRNAs participate in epigenetic regulation and posttranslational regulation of proteins through the following methods. They could act as protein sponges and enhance protein function. They could also act as scaffolds to mediate some complex formation between specific enzymes and substrates. Additionally, they recruit proteins to specific sites to regulate their activity or influence their expression [54]. The interaction of circRNA with RBP is considered to be an important function of circRNA and is the basic mechanism for circRNA to exert functions, including circRNA generation, translation, transcriptional regulation of target genes and extracellular transport [74].
The roles of circRNA in HCC
circRNA and HCC proliferation and apoptosis
circRNA_0001955 increased TRAF6 and MAPK11 expression through adsorption of miR-516a-5p and further promoted the tumorigenesis of HCC [75]. circRHOT1 promoted HCC cell growth of HCC by increasing the level of NR2F6 and worsened the prognosis of HCC patients [76]. Knockdown of circRNA_0067934 not only significantly inhibited the proliferation of Hep3B and HuH7 cells, but also induced the apoptosis of HCC cells though the FZD5/Wnt/β-catenin signaling pathway [77]. circRNA_0000502 promoted the malignant progression of HCC by downregulating the expression of miR-124. Knockdown of circRNA_0000502 significantly reduced the proliferation ability of HCC cells and increased cell apoptosis [78]. hsa_circ_0051443 was delivered from normal cells to HCC cells via exosomes, and it can facilitate apoptosis and block the cell cycle to repress malignant behaviors of HCC by regulating extracellular communication in the tumor microenvironment (TME) [79].
circRNA and HCC invasion and metastasis
Downregulation of circRNA cSMARCA5 (hsa_circ_0001445) in HCC was significantly associated with invasion and was an independent risk factor for overall survival and recurrence-free survival after hepatectomy. cSMARCA5 was identified to promote the expression of the tumor suppressor TIMP3 by acting as a miR-17-3p and miR-181b-5p sponge, thereby inhibiting the migration of HCC cells [80]. circASAP1 (a circRNA from exons 2 and 3 of the ASAP1 gene, hsa_circ_0085616) was identified by circRNA sequencing to be associated with metastasis in the lungs of HCC patients after radical resection. circASAP1 was overexpressed in HCC with high metastatic potential. circASAP1 promotes HCC invasion through the modulation of the miR-326/miR-532-5p-MAPK1 signaling pathway and mediation of HCC-associated macrophage infiltration through the regulation of the miR-326/miR-532-5p-CSF-1 pathway [81]. circ-ADD3 negatively correlated with vascular invasion, intrahepatic metastasis and distant metastasis. circ-ADD3 (hsa_circ_0020007) downregulated the stability of EZH2 and increased the expression of antimetastatic genes to suppress metastasis in HCC [82]. In addition, circMTO1 (hsa_circRNA_0007874/hsa_circRNA_104135) was a circRNA that was downregulated in HCC tissues and promoted HCC invasion by acting as a sponge of oncogenic miR-9 to promote p21 expression [83]. circRNA_0003998 contributed to EMT in HCC via the circRNA_0003998/miR-143-3p/FOSL2 axis and circ_0003998/PCBP1/CD44V6 axis. circRNA_0003998 might act as a ceRNA that alleviated the suppressive effect of miR-143-3p on FOSL2 (a stimulator of EMT), while circRNA_0003998 could bind to PCBP1 and increase the expression level of CD44V6 (a gene associated with EMT) [84]. In addition, overexpression of circRNA Cdr1 (hsa_circ_0001946) accelerated the migration of HCC cells. Cdr1as could promote the expression of AFP through adsorption of miR-1270. Additionally, circRNA Cdr1as exosomes from HCC cells accelerated the migration of surrounding normal cells [85]. Another survey concluded that the expression of circRNA_104718 accelerated cell migration and invasion, while inhibited the apoptosis of HCC cells through the microRNA-218-5p/TXNDC5 signaling pathway [86]. In contrast, overexpression of exosomal circRNA_100338 significantly enhanced the invasive ability of HCC cells, so overexpression of exosomal circRNA_100338 could promote metastasis by enhancing invasiveness and angiogenesis, and persistent high expression of exosomal circRNA_100338 may be a risk indicator for metastasis and poor survival of HCC [87]. In addition, circRNA_5692 expression was demonstrated to be downregulated in HCC tissues, and overexpression of circRNA_5692 attenuated malignant behavior by reducing miR-328-5p expression and enhancing DAB2IP expression. This study proved that circRNA inhibited the invasion and metastasis of HCC by regulating the circRNA-5692-miR-328-5p-DAB2IP pathway. These findings may provide potential new targets for the diagnosis and treatment of HCC [88].
circRNA and HCC angiogenesis
Angiogenesis is an extremely vital mechanism for tumors to eliminate carbon dioxide and metabolic waste and to acquire adequate nutritional support [89]. Vasculogenesis mimicry (VM), a novel pattern of vascularization, is involved in HCC progression and metastasis. Androgen receptor (AR) suppressed the formation of HCC VM by downregulating circRNA7/miRNA7-5p/VE-cadherin/Notch4 signaling axis in HCC, which may be novel therapies against HCC [90]. circGFRA1 was aberrantly highly expressed in HCC tissues and cells, which may lead to poor prognosis. Knockdown of circGFRA1 suppressed angiogenesis and caused inhibition of migration in HCC cells by binding to miR-149 [91]. hsa_circ_0000092 competitively bound to miR-338-3p and thereby upregulating the expression of HN1. And the circ_0000092 deletion or miR-338-3p elevation repressed HCC cell proliferation, migration, invasion and angiogenesis [92]. As a critical component of the TME, endothelial cells were involved in angiogenesis process and could affect the development of HCC. circ_4911 and circ_4302 were downregulated in HCC, and overexpression of circ_4911 and circ_4302 inhibited the proliferation and migration of human umbilical vein endothelial cells (HUVECs), thereby restraining the angiogenesis of tumors [93].
circRNA and HCC microenvironment
circRNAs are well established to recruit and reconstitute key components in the tumor microenvironment (TME) and to mediate various signaling pathways that involving tumorigenesis, angiogenesis, immune response, metabolism reprogramming and metastatic progression. circRNA interacts with critical cellular components of the TME (such as cancer-associated endothelial cells (CAEs), immune cells, cancer-associated fibroblasts (CAFs) and cancer stem cells) and noncellular components (cytokines, growth factors, metabolites, and extracellular matrix (ECM)) [94]. Combining targeting circRNA and coactivating components in the TME may achieve superior therapeutic efficacy and become a new pattern of future tumor treatment [95]. circRNA affects the TME from multiple perspectives, including immune monitoring, angiogenesis, hypoxia, stromal remodeling and the resistance of radiation treatments [96]. In HCC, circMET (hsa_circ_0082002) promoted the EMT process, and it enhanced the immunosuppression of HCC through the Snail/DPP4/CXCL10 axis [97]. Additionally, circUHRF1 caused immunosuppression through inducing NK cell dysfunction in HCC. circUHRF1 inhibited the secretion of NK cell-derived IFN-γ and TNF-α and it was associated with a decreased NK cell infiltration in TME [98]. circMAT2B (hsa_circ_0074854) expression was upregulated in HCC, which shortened the overall survival of patients. Mechanistically, circMAT2B promoted glycolysis and accelerated HCC progression by acting as a miR-338-3p sponge and upregulating the expression level of the miR-338-3p target gene PKM2, which encoded a key enzyme in the glycolysis process [99].
circRNA and HCC drug resistance
circRNA_101505 was diminished in HCC cisplatin-resistant tissues and cell lines, which indicated poorer survival outcomes. circRNA_101505 could sensitize HCC cells to cisplatin by acting as a sponge for miR-103, thereby stimulating the expression of oxidored-nitro domain-containing protein 1 (NOR1), which suppressed the growth of cancer cells and enhanced cisplatin toxicity [100]. In addition, the expression of circRNA-SORE (circRNA_104797) was upregulated in sorafenib-resistant HCC cells. circRNA-SORE bound to the oncogenic protein YBX1 in the cytoplasm and prevented the interaction of YBX1 with the E3 ubiquitin ligase PRP19, thereby blocking PRP19-mediated degradation of YBX1. Therefore, the deletion of circRNA-SORE could significantly enhance the cell killing ability of sorafenib [59]. In HCC, circRNA_102272 expression was upregulated, which facilitated cisplatin resistance in HCC through regulation of the miR-326/RUNX2 axis [101]. The expression of circRNA_101237 may increase DNA repair, increase drug efflux, and alter cell accumulation so as to promote cisplatin resistance in HCC, which is also associated with tumor size, lymph node metastasis and TNM stage. Therefore circRNA_101237 was an independent predictor of prognosis in HCC patients [102]. circFN1 (hsa_circ_0058124) was found to be able to promote the expression of E2F1 by acting as a miR-1205 sponge; thus, inhibition of circFN1 could enhance the sensitivity of HCC cells to sorafenib [103]. circARNT2 (hsa_circ_0104670) regulated cisplatin resistance through the miR-155-5p/PDK1 axis, while downregulation of circARNT2 inhibited cell proliferation and enhanced the sensitivity of HCC cells to cisplatin [104]. Knockdown of circ_0003998 inhibited drug-resistant cell viability, migration, invasion and EMT, and also enhanced the toxicity of doxorubicin to HCC cells. circ_0003998 acted as a ceRNA for miR-218-5p to regulate the expression of EIF5A2, and reduced sensitivity to doxorubicin in HCC cells by regulating the miR-218-5p/EIF5A2 axis [105]. Knockout of circ_0003418 stimulated the proliferation, migration and invasion of HCC cells. circ_0003418 enhanced the sensitivity of HCC cells to cisplatin by inhibiting the Wnt/β-catenin signaling pathway [106].
In conclusion, circRNA is generated by back splicing and lacks a cap and tail structure. It is also less susceptible to degradation by RNA enzymes and is more stable than linear RNA. The process of its generation is regulated by multiple factors, including intron sequences, enzymes and protein factors. Most circRNAs function in the cytoplasm, and the process of circRNAs export from the nucleus depend on their own length. circRNAs usually act as miRNA sponges to exert their biological functions. Certain circRNAs have cap-independent translation potential. circRNAs play significant roles in HCC carcinogenesis (Table 2). Most circRNAs act as miRNA sponges and inhibit the suppression of target mRNAs to affect the malignant behaviors of HCC, including cell growth, proliferation, cell cycle, apoptosis, invasion, metastasis, angiogenesis, tumor microenvironment remodeling, immune imbalance, metabolism reprogramming, and drug resistance. In addition, some circRNAs can directly affect signaling pathways to regulate the progression of HCC. The detailed regulatory mechanisms and interactions are worthwhile to conduct further investigations.
The relationship between circRNA and HCC
| CircRNA | CircBase ID/common name | Gene symbol | Up/ down | Functions | Mechanisms | Refs |
|---|---|---|---|---|---|---|
| circRNA-SORE | circRNA_104797 | NA | up | decrease cell killing ability of sorafenib | bound and stabilize YBX1 | [59] |
| circRNA_0001955 | circRNA_0001955 | NA | up | promote tumorigenesis of HCC | adsorption of miR-516a-5p | [75] |
| circRHOT1 | hsa_circRNA_102034 | NA | up | promote HCC growth and metastasis. | increase the level of NR2F6 | [76] |
| circRNA_0067934 | circRNA_0067934 | NA | up | boost proliferation, metastasis and invasion | activate FZD5/Wnt/β-catenin signaling pathway | [77] |
| circ_0000502 | circ_0000502 | NA | up | promote proliferation and invasion | mediate the expression of miR-124 | [78] |
| hsa_circ_0051443 | hsa_circ_0051443 | TRAPPC6A | up | facilitate apoptosis and block the cell cycle | regulate extracellular communication in the tumor microenvironment | [79] |
| cSMARCA5 | hsa_circ_0001445 | SMARCA5 | down | promote invasion | suppress the expression of tumor suppressor TIMP3 | [80] |
| circASAP1 | hsa_circ_0085616 | ASAP1 | up | promote proliferation and invasion | modulate miR-326/miR-532-5p-MAPK1 signaling pathway | [81] |
| circRNA-ADD3 | hsa_circ_0020007 | LOC100505933 | up | suppress metastasis | downregulate the stability of EZH2 | [82] |
| circMTO1 | hsa_circRNA_0007874/ hsa_circRNA_104135 | NA | down | promote proliferation and invasion | act as the sponge of oncogenic miR-9 | [83] |
| circRNA_0003998 | circRNA_0003998 | NA | up | promote EMT | act as ceRNA for miR-143-3p and bind to PCBP1 | [84] |
| circRNA Cdr1 | hsa_circ_0001946 | CDR1 | up | promote proliferation and migration | adsorption of miR-1270 | [85] |
| circRNA_104718 | circRNA_104718 | NA | up | promote proliferation, migration, invasion | regulate miR-218-5p/TXNDC5 signaling pathway | [86] |
| circRNA_100338 | circRNA_100338 | NA | up | promote metastasis | enhance invasiveness and angiogenesis | [87] |
| circRNA_5692 | circRNA_5692 | NA | down | promote invasion and metastasis | regulate the circRNA-5692-miR-328-5p-DAB2IP pathway | [88] |
| circGFRA1 | NA | NA | up | promote angiogenesis | binding to miR-149 | [91] |
| hsa_circ_0000092 | hsa_circ_0000092 | RPL5 | up | promote invasion and angiogenesis | binding to miR-338-3p to upregulate the expression of HN1 | [92] |
| circ_4911, circ_4302 | circ_4911, circ_4302 | NA | down | promote angiogenesis | promote the proliferation and migration of HUVECs | [93] |
| circMET | hsa_circ_0082002 | MET | up | enhance immuno-suppression effect | modulate Snail/DPP4/CXCL10 axis | [97] |
| circUHRF1 | NA | NA | up | immunosuppression | induce NK cell dysfunction | [98] |
| circMAT2B | hsa_circ_0074854 | MAT2B | up | promote glycolysis | act as a miR-338-3p sponge to upregulate PKM2 | [99] |
| circRNA_101505 | circRNA_101505 | NA | down | sensitize HCC cells to cisplatin | act as a sponge for miR-103 and stimulate the expression of NOR1 | [100] |
| circRNA_102272 | circRNA_102272 | NA | up | facilitate cisplatin resistance | regulate the miR-326/RUNX2 axis | [101] |
| circRNA_101237 | circRNA_101237 | NA | up | promote cisplatin resistance | may increase DNA repair to alter cell accumulation | [102] |
| circFN1 | hsa_circ_0058124 | FN1 | up | promote cisplatin resistance | act as miR-1205 sponge | [103] |
| circARNT2 | hsa_circ_0104670 | ARNT2 | up | promote cisplatin resistance | regulate miR-155-5p/PDK1 pathway | [104] |
| circ_0003998 | circ_0003998 | NA | up | reduce sensitivity to doxorubicin of HCC cells | act as a ceRNA for miR-218-5p to regulate the expression of EIF5A2 | [105] |
| circ_0003418 | circ_0003418 | NA | up | enhance the sensitivity of HCC cells to cisplatin | inhibit the Wnt/β-catenin pathway | [106] |
Cross-talk and interplay between m6A modification and circRNA in HCC
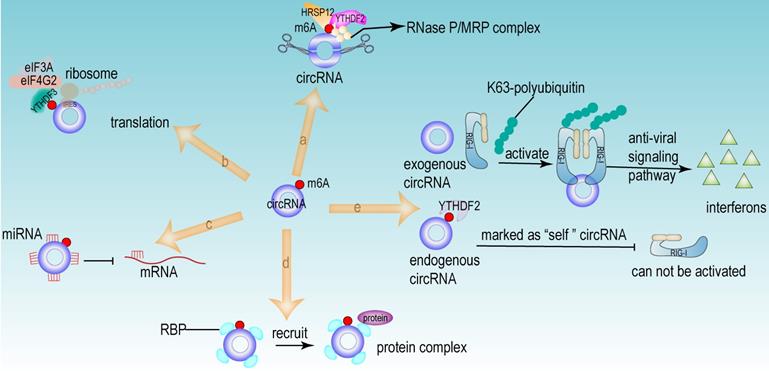
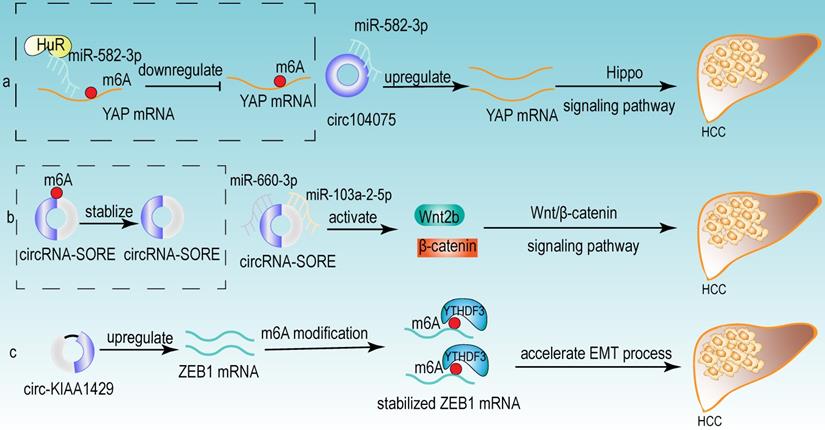
From the above studies, we can conclude that both m6A modification and circRNA are closely associated with the development of HCC. Further researches revealed that circRNA can be modified by m6A modification and that m6A modification has an important function in maintaining the biological activities of circRNAs in the following perspectives: (1) m6A modification regulates the stability of circRNA (Fig 4a). m6A modification could be recognized by YTHDF2. HRSP12 linked YTHDF2 to RNase P/MRP, and regulated circRNA degradation by the YTHDF2-HRSP12-RNase P/MRP pathway [107, 108]. (2) m6A modification promotes the translation of circRNA (Fig 4b). m6A modification promoted the translation of circRNA in a cap-independent manner. m6A modification could be recognized by “reader” YTHDF3, which further recruited translation initiation factor (such as eIF4G2 and eIF3A) and ribosome to IRES for m6A-initiated circRNA translation process [109]. (3) m6A modification influences tumor progression by regulating circRNA-miRNA interactions (Fig 4c) [110]. (4) m6A modification impacts the interactions between circRNA and RBPs (Fig 4d) [111]. (5) m6A modification labels endogenous RNA to distinguish it from exogenous RNA, so as to avoid recognition and attack by the autoimmune system (Fig 4e). Exogenous RNA (without m6A modification) activated RIG-I under the circumstance of K63-polyubiquitin, and then led to the activation of interferon production through anti-viral signaling pathway. While endogenous circRNA modified by m6A could be recognized by YTHDF2 and marked as “self” circRNA, thus blocking the activation of RIG-I [112], and serving as a novel mechanism for immune escape of exogenous RNA viruses [113]. In addition, the proportion of m6A modifications is closely related to the functions of circRNAs [114, 115]. Therefore, the cross-talk of circRNA and m6A modification may play an even more important role in HCC development. For example, the m6A modification of YAP promoted the binding between miR-582-3p and the YAP mRNA. In addition, it promoted HuR binding and stabilizing miR-582-3p, so as to downregulate the expression of YAP. circ104075 could upregulate YAP expression by absorbing miR-582-3p, thus contributing to HCC progression through Hippo signaling pathway (Fig 5a) [116]. circRNA-SORE promoted the development of HCC by acting as a miRNA sponge to segregate miR-103a-2-5p and miR-660-3p, then competitively activating Wnt/β-catenin signaling. Elevated m6A levels stabilized circRNA-SORE (Fig 5b) [117]. ZEB1 (a critical transcription factor of EMT process) was a target gene of circ-KIAA1429. YTHDF3 could enhance the stability of ZEB1 mRNA in an m6A-dependent manner in order to promote the EMT process and facilitate the development of HCC (Fig 5c) [118].
The functions of circRNA modified by m6A. (a) m6A regulated the stability of circRNA. The m6A modification site of circRNA could be recognized by “reader” YTHDF2. HRSP12 linked YTHDF2 to RNase P/MRP complex, therefore regulating circRNA degradation by YTHDF2-HRSP12-RNase P/MRP pathway. (b) m6A promoted the translation of circRNA in a cap-independent manner. m6A modification could be recognized by “reader” YTHDF3, which further recruited translation initiation factor (such as eIF4G2 and eIF3A) and ribosome to IRES for m6A-initiated circRNA translation process. (c) m6A facilitated circRNA to act as miRNA sponge. (d) m6A regulated the interaction between RBP and circRNA. (e) m6A recognized and participated the immunoreaction mediated by circRNA. Exogenous RNA (without m6A modification) activated RIG-I under the circumstance of K63-polyubiquitin, and then led to the activation of interferon production through anti-viral signaling pathway. While the endogenous circRNA (modified by m6A) could be recognized by YTHDF2 and then this endogenous circRNA was marked as “self” circRNA, thus blocking the activation of RIG-I.
Some examples of the cross-talk of circRNA and m6A modification in HCC. (a) The m6A modification of YAP promoted the binding between miR-582-3p and the YAP mRNA. In addition, it promoted HuR binding and stabilizing miR-582-3p, so as to downregulate the expression of YAP. circ104075 could upregulate YAP expression by absorbing miR-582-3p, thus contributing to HCC progression through Hippo signaling pathway. (b) m6A modification stabilized circRNA-SORE, and circRNA-SORE acted as miR-103a-2-5p and miR-660-3p sponge so as to activate Wnt/β-catenin signaling and promote the progression of HCC. (c) ZEB1 acted as a target of circ-KIAA1429 and could be recognized by m6A “reader” YTHDF3, which could enhance the stability of ZEB1 mRNA. ZEB1 was also a critical transcription factor of EMT process. Therefore, upregulating the expression of ZEB1 by circ-KIAA1429 could accelerate the EMT process, and further facilitate the development of HCC.
m6A modification has an extremely important role in circRNAs, but few studies have combined the analysis of m6A modification and circRNAs or m6A-modified circRNAs in HCC. More extensive studies on the relationship among m6A modification, circRNA, and HCC carcinogenesis can be excavated to conduct further investigations and might become a new hot spot for HCC studies.
Introduction to common databases for m6A modification and circRNA studies
m6A modification databases
Databases based on MeRIP-seq
m6A regulation based on nucleotide sequences and with the development of secondary generation sequencing, the technique of MeRIP-seq for m6A sequencing has been widely used. For example, there are downstream analysis databases based on MeRIP-seq data, such as RNAmod, which provides information about the distribution of RNA modifications, modification coverage for different gene features, and functional annotation of modified mRNAs to annotate diverse kinds of mRNA modifications in different species [119].
RMBase v2.0 expands on the RMBase database with 600 datasets from 47 studies involving 13 species and 1,397,000 modification sites. The RMBase v2.0 database integrates epigenomic transcriptome sequencing data to explore RNA posttranscriptional modifications and their association with miRNA binding, disease-related single nucleotide polymorphisms (SNPs) and relationships with RBPs. In addition, the database includes predictions for m5c, m1A and others [120, 121].
The Met-DB v2.0 database predicts significantly more m6A peaks and single base loci in a specific context compared to the Met-DB database from 185 samples of 7 species from 26 independent studies. In addition, the Met-DB v2.0 database integrates a database of m6A "writers", "erasers" and "readers” and is a good tool for understanding m6A function [122, 123].
The WHISTLE database is a transcriptome-wide database for m6A RNA methylation site prediction. In addition to conventional sequence features, WHISTLE integrates 35 other genomic features for more accurate m6A site prediction. The WHISTLE database is a network-based approach to explore biological processes affected by changes in individual m6A sites by integrating RNA methylation profiles, gene expression profiles and protein-protein interaction data [124].
The REPIC database records approximately 10 million peaks from publicly available m6A-seq and MeRIP-seq data from 672 samples from 49 studies, covering 61 cell lines or tissues from 11 organisms. REPIC allows users to query m6A modification sites by specific cell lines or tissue types, which can present a comprehensive map of m6A modification sites, histone modification sites and related chromatin [125].
Sequence analysis databases
The BERMP database, a novel cross-species identification tool, is based on deep learning, which can predict m6A regulatory sites with high confidence. The prediction accuracy is closely related to data size and compensated by the integration of a random forest classifier with a novel encoding of enhanced nucleic acid content. Excellent performance was achieved in both cross-validation tests and independent tests. The BERMP database performed well on the mammalian dataset but poorly on the yeast dataset [126].
The SRAMP database is a sequence-based m6A prediction database that combines three random forest classifiers that exploit their positional nucleotide sequence patterns, k-nearest neighbor information, and position-independent nucleotide pairing spectra features to depict the sequence context around m6A sites. SRAMP uses either genomic sequences or cDNA sequences as the input sequence and can achieve competitive performance in cross-validation tests and rigorous independent benchmark tests [127].
iMRM can simultaneously predict multiple loci for m6A, m5C, m1A, ψ and A-to-I modifications in Homo sapiens, Mus musculus and Saccharomyces cerevisiae. iMRM extracts optimally characterized modifications using feature selection techniques and is a good method for simultaneously identifying modifications occurring on different nucleotides [128].
Other m6A modification databases
Most prediction models are based only on sequence features and use SVM or random forest as classification methods, while the HSM6AP database creatively weights the HSM6AP samples during the training process and is an m6A high-precision prediction database based on multiple weights and features. The HSM6AP database contributes to the effect of the fusion sequence and gene-level feature extraction on the accuracy of methylation site recognition [129].
m6Avar, which was derived from miCLIP/PA-m6A-seq experiments (high confidence), MeRIP-seq experiments (medium confidence) and transcriptome-wide predictions (low confidence), integrated the RBP-binding regions, miRNA targets and splicing sites associated with variants [130].
In addition, m6Acorr corrects the existing laboratory bias in RNA m6A methylation profiles and performs profile comparisons on the corrected datasets [131]; M6A2Target provides a database of validated m6A targets and potential m6A targets [132], and the WITMSG database predicts m6A modification sites of introns [133] (Table 3).
The summary of m6A modification databases
| Database | Websites | Mechanisms | Functions | Refs |
|---|---|---|---|---|
| RNAmod | https://bioinformatics.sc.cn/RNAmod | based on MeRIP-seq | annotate diverse kinds of mRNA modifications in different species | [119] |
| RMBase v2.0 | http://rna.sysu.edu.cn/rmbase | based on MeRIP-seq | explore RNA post-transcriptional modifications and their association with miRNA binding, disease-related SNPs and relationships with RBPs. | [120, 121] |
| Met-DB v2.0 | http://compgenomics.utsa.edu/MeTDB/ | based on MeRIP-seq | integrate a database of m6A "writers", "erasers" and "readers” | [122, 123] |
| WHISTLE | http://whistle-epitranscriptome.com | based on MeRIP-seq | explore biological processes affected by changes in individual m6A sites by integrating RNA methylation profiles, gene expression profiles and protein-protein interaction data | [124] |
| REPIC | https://repicmod.uchicago.edu/repic | based on MeRIP-seq | query m6A modification sites by specific cell lines or tissue types | [125] |
| BERMP | http://www.bioinfogo.org/bermp | based on sequence analysis | a cross-species identification tool | [126] |
| SRAMP | http://www.cuilab.cn/sramp/ | based on sequence analysis | depict the sequence context around m6A sites | [127] |
| iMRM | http://www.bioml.cn/XG_iRNA/home | based on sequence analysis | simultaneously predict multiple loci for m6A, m5C, m1A, ψ and A-to-I modifications in Homo sapiens, Mus musculus and Saccharomyces cerevisiae | [128] |
| HSM6AP | http://lab.malab.cn/~lijing/HSM6AP.html | multiple weights and features | explore the effect of the fusion sequence and gene-level feature extraction on the accuracy of methylation site recognition | [129] |
| m6AVar | http://m6avar.renlab.org | based on miCLIP/PA-m6A-seq, MeRIP-seq and transcriptome-wide predictions | integrate the RBP-binding regions, miRNA-targets and splicing sites associated with variants | [130] |
| m6Acorr | http://www.rnanut.net/m6Acorr | based on normalizing RNA-seq and microarray data | correct laboratory bias in RNA m6A methylation profiles and perform profile comparisons on the corrected datasets | [131] |
| M6A2Target | http://m6a2target.canceromics.org | collect all reported m6A WERs | provide database of validated m6A targets and potential m6A targets | [132] |
| WITMSG | http://rnamd.com/intron/ | based on sequence features and a variety of genomic characteristics | predict all possible intronic m6A sites | [133] |
| Database | Websites | Mechanisms | Functions | Refs |
| RNAmod | https://bioinformatics.sc.cn/RNAmod | based on MeRIP-seq | annotate diverse kinds of mRNA modifications in different species | [119] |
| RMBase v2.0 | http://rna.sysu.edu.cn/rmbase | based on MeRIP-seq | explore RNA post-transcriptional modifications and their association with miRNA binding, disease-related SNPs and relationships with RBPs. | [120, 121] |
| Met-DB v2.0 | http://compgenomics.utsa.edu/MeTDB/ | based on MeRIP-seq | integrate a database of m6A "writers", "erasers" and "readers” | [122, 123] |
| WHISTLE | http://whistle-epitranscriptome.com | based on MeRIP-seq | explore biological processes affected by changes in individual m6A sites by integrating RNA methylation profiles, gene expression profiles and protein-protein interaction data | [124] |
| REPIC | https://repicmod.uchicago.edu/repic | based on MeRIP-seq | query m6A modification sites by specific cell lines or tissue types | [125] |
| BERMP | http://www.bioinfogo.org/bermp | based on sequence analysis | a cross-species identification tool | [126] |
| SRAMP | http://www.cuilab.cn/sramp/ | based on sequence analysis | depict the sequence context around m6A sites | [127] |
| iMRM | http://www.bioml.cn/XG_iRNA/home | based on sequence analysis | simultaneously predict multiple loci for m6A, m5C, m1A, ψ and A-to-I modifications in Homo sapiens, Mus musculus and Saccharomyces cerevisiae | [128] |
| HSM6AP | http://lab.malab.cn/~lijing/HSM6AP.html | multiple weights and features | explore the effect of the fusion sequence and gene-level feature extraction on the accuracy of methylation site recognition | [129] |
| m6AVar | http://m6avar.renlab.org | based on miCLIP/PA-m6A-seq, MeRIP-seq and transcriptome-wide predictions | integrate the RBP-binding regions, miRNA-targets and splicing sites associated with variants | [130] |
| m6Acorr | http://www.rnanut.net/m6Acorr | based on normalizing RNA-seq and microarray data | correct laboratory bias in RNA m6A methylation profiles and perform profile comparisons on the corrected datasets | [131] |
| M6A2Target | http://m6a2target.canceromics.org | collect all reported m6A WERs | provide database of validated m6A targets and potential m6A targets | [132] |
| WITMSG | http://rnamd.com/intron/ | based on sequence features and a variety of genomic characteristics | predict all possible intronic m6A sites | [133] |
circRNA databases
circRNA databases are essential for transcriptomics studies. The CircAtlas database was developed based on 1070 RNA-seq samples collected from 19 normal tissues of 6 vertebrate species. It contains the expression patterns, conservation and functional annotation information of 1007087 highly reliable circRNAs [134].
The circRNADisease database is based on more than 800 papers reporting on circRNAs and diseases, with a collection of 354 pairs of interconnections between 330 circRNAs and 48 diseases to provide the circRNA expression pattern, experimental detection techniques, and circRNA-associated partners [135].
The MiOncoCirc database is the first circRNA database based on clinical tumor samples, containing circRNA expression information from more than 2000 clinical tumor samples, collecting the circRNA expression data of in situ and metastatic cancers, etc. [136].
exoRBase is a repository of circRNA, lncRNA and mRNA from human blood exosome RNA sequence analysis. exoRBase is characterized by the integration and visualization of RNA expression profiles of normal individuals and patients with different diseases based on standardized RNA-seq data and provides annotation, expression level and possible original tissues [137].
The CSCD database collected 228 RNA-seq samples from cancer and normal cell lines and identified 272152 cancer-specific circRNAs. The CSCD database can predict the miRNA response element sites and RBP sites of each circRNA and predict the potential open reading frames to facilitate the exploration of translatable circRNAs. Additionally, the CSCD database predicts the splicing events of each linear transcript and can also predict the splicing events of each circRNA, which can help us better understanding the connection between linear splicing and back-splicing [138].
circBase collects and integrates information on well-known circRNAs and newly discovered circRNAs from sequencing data, including human, mouse and other species, and is a fundamental database in circRNA research field [139].
The circInteractome database serves as the circRNA, miRNA and RBP public database and provides bioinformatics analysis of circRNA binding sites. It also predicts RBP and miRNA binding sites on human circRNAs and identifies potential circRNAs that can act as RBP sponges. Moreover, it could design primers to specifically detect circRNAs, design siRNAs for circRNA silencing, and identify potential IRESs [140] (Table 4).
The summary of circRNA databases
| Database | Websites | Features | Functions | Refs |
|---|---|---|---|---|
| circAtlas | http://circatlas.biols.ac.cn/ | contain 1007087 highly reliable circRNA | provide the expression patterns, conservation and functional annotation information | [134] |
| circRNADisease | http://cgga.org.cn:9091/circRNADisease/ | based on the collection of 354 pairs of interconnections between 330 circRNAs and 48 diseases | provide the circRNA expression pattern, experimental detection techniques, circRNA-associated partners, a brief description of circRNA biological function | [135] |
| MiOncoCirc | https://mioncocirc.github.io/ | based on more than 2000 clinical tumor samples | provide the circRNA expression data of in-situ and metastatic cancers | [136] |
| exoRBase | http://www.exoRBase.org | a repository of circRNA, lncRNA and mRNA from human blood exosome RNA sequence analysis | integrate and visualize RNA expression profiles of normal individuals and patients based on RNA-seq data | [137] |
| CSCD | http://gb.whu.edu.cn/CSCD | collect 228 RNA-seq samples and identify 272152 cancer-specific circRNAs | predict the miRNA response element sites and RBP sites and the potential open reading frames to facilitate the exploration of translatable circRNAs | [138] |
| circBase | http://www.circbase.org/ | merge circRNAs and downloaded their expression profile | provide scripts to identify known and novel circRNAs in sequencing data | [139] |
| CircInteractome | http://circinteractome.nia.nih.gov | search the circRNA, miRNA and RBP public database | predict circRNA binding sites, RBP and miRNA binding sites | [140] |
In conclusion, several databases for m6A modification and circRNA studies have been developed and each of them contains different functions. We could reasonably select, integrate and apply the information from these databases according to the specific purpose of the research.
m6A modifiers and circRNAs as biomarkers and therapeutic targets for HCC
The application of biomarkers is crucial in all stages of cancer and has become one of the main avenues for cancer diagnosis, prognosis and progression monitoring [141]. A qualified biomarker should be sensitive, specific, reproducible, stable and clinically useful.
m6A modifiers as HCC biomarkers
m6A modification is the most abundant modification in eukaryotes and is closely associated with HCC development. The expression levels of m6A modifiers could be tested in liver tissue biopsy; thus, m6A modifiers are considered as potential biomarkers and prognostic indicators for HCC (Fig 6a). Qu et al. found that YTHDF1, YTHDF2, and KIAA1429 were significantly associated with the WHO stage of HCC and acted as independent prognostic markers for survival of HCC [42]. In addition, METTL3 was identified as an independent prognostic factor for recurrence-free survival (RFS) of HCC [142]. Chen et al. demonstrated that WTAP was highly expressed in HCC and its expression was an independent predictor of HCC patients' survival [27]. YTHDF1 was significantly upregulated, exerted an important role in regulating cell cycle progression and metabolism in HCC. It also positively correlated with pathological stage. Therefore, it might be a potential new therapeutic and prognostic target for HCC [41]. Higher expression of and IGF2BP2 in HCC tissues was associated with poor prognosis, which indicated another potential biomarker to predict HCC prognosis [38].
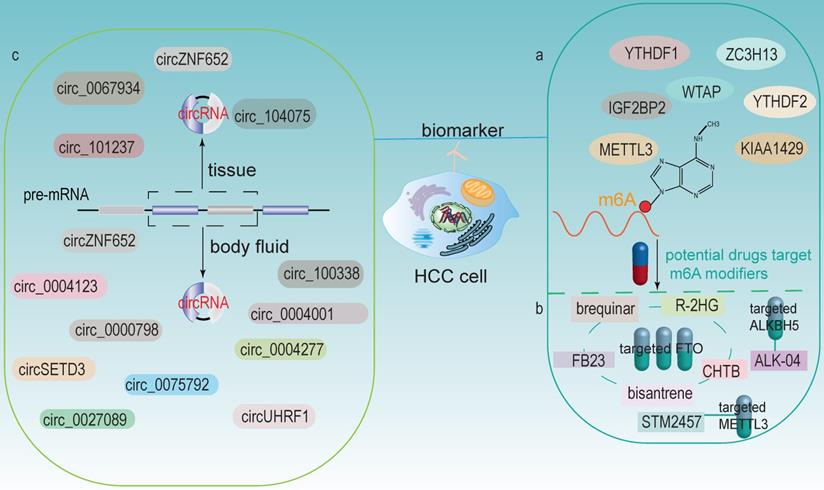
m6A modifiers and circRNAs as potential biomarkers and therapeutic targets for HCC. (a) m6A modifiers act as biomarkers in HCC. (b) Some drugs targets on m6A modifiers (including FTO, ALKBH5 and METTL3). (c) circRNAs in HCC tissues and body fluids could both be potential biomarkers.
m6A modifiers as potential therapeutic targets in cancers
In recent years, several small molecule inhibitors of m6A modifiers have been identified or developed, implying that m6A modifiers can be used as therapeutic drug targets and applied in the clinic (Fig 6b). Currently, the majority of m6A inhibitors discovered are targeting at FTO and ALKBH5. For example, Qiao et al. found that CHTB is an inhibitor of FTO, and this novel molecule provided a direction for further development of more selective and effective FTO inhibitors [143]. Huang et al. identified that targeting FTO with two potential inhibitors, FB23 and FB23-2, have the potential to treat acute myelocytic leukemia (AML). These two inhibitors could directly bind to FTO and selectively inhibit the activity of FTO, thereby significantly inhibiting cell proliferation [144]. Su et al. showed that bisantrene and brequinar (are also inhibitors of FTO), significantly attenuated the initiation of cellular self-renewal and reprogrammed leukemic stem cells by inhibiting the expression of the immune checkpoint gene LILRB4 [145]. Su et al. revealed that the tumor metabolite R-2HG could act as an inhibitor of FTO, leading to the increased methylation and decreased expression of c-MYC and CEBPA, thereby blocking proliferation, cell cycle and inducing apoptosis in AML and glioma cells [146]. ALK-04, an inhibitor of ALKBH5, might serve as a potential therapeutic target to enhance immunotherapy outcomes in melanoma, colorectal cancer and other cancers [147]. In addition, a recent research has showed that a small inhibitor STM2457 could target m6A “writer” METTL3 to reduce AML cells growth and increase its apoptosis [148].
The discovery of new m6A inhibitors has paved the way for m6A-based therapies; however, little is known about their roles in HCC. Moreover, the specificity and side effects of these drugs should be clarified to better promoting their application to HCC. In addition, determining whether these inhibitors affect other RNA modifications such as m1A is also of importance, as it could lead to uncertain consequences. Therefore, more in-depth studies of these inhibitors in vitro and in vivo are urgently requested [149]. Besides, some m6A inhibitors can affect the immunotherapy of tumors, so combining appropriate m6A inhibitors with immune checkpoint blockade might achieve more efficient therapeutic effects.
circRNAs as biomarkers for HCC
circRNA can be detected in tissue samples, plasma, serum, saliva, urine, latex, etc. And its expression profile appears to be cell-specific or stage-specific [150]. Memczak et al. sequenced RNA in human peripheral blood and identified circRNA as an easily accessible potential biomarker in body fluids [151]. Li et al. identified a large amount of exosomal RNAs in serum, which can be used as biomarkers for cancer diagnosis [152]. These features make circRNAs be possible prospective biomarkers (Fig 6c).
circRNAs in tissues
Numerous studies have now identified circRNAs in HCC tissues can be used as potential biomarkers. For example, patients with highly expressed circ-ZNF652 in HCC were more likely to suffer from vascular invasion, intrahepatic metastasis and distant metastasis, which were closely associated with a poor prognosis [153]. circRHOT1 was a potential biomarker that repressed the progression of HCC by recruiting TIP60 to initiate NR2F6 expression [76]. circ_0067934 stimulated invasion and metastasis of HCC, while circMTO1 negatively regulated the progression of HCC [154]. Zhang et al. discovered that circ_104075 has potential as a new diagnostic biomarker for HCC, and using circ_104075 as a therapeutic target may provide a new strategy for the diagnosis and treatment of HCC [116]. Moreover, the expression of circRNA_101237 was associated with tumor size, lymph node metastasis, and TNM stage. The serum circRNA_101237 level was an independent predictor of prognosis in HCC patients [102].
circRNAs in body fluids
The role of circRNAs in body fluids as biomarkers of HCC has also been identified. For example, circUHRF1 was proven in plasma exosomes, and its high expression was associated with a decreased NK cell ratio. Therefore, it diminished NK cell infiltration and led to resistance of anti-PD1 immunotherapy, thus resulting in poor prognosis in HCC patients [98]. circPTGR1 (circRNAs, hsa_circ_0008043, hsa_circ_0003731, and hsa_circ_0088030), a kind of circRNA with three subtypes that was upregulated in serum exosomes of HCC patients with metastasis-free or low metastatic capacities, they were closely related to clinical staging and prognosis [155]. Besides, Sun et al. revealed that hsa_circ_0004001, hsa_circ_0004123, and hsa_circ_0075792 were upregulated in HCC blood samples and positively correlated with TNM stage and tumor size. Additionally, the combination use of these three biomarkers had much higher sensitivity and specificity in the diagnosis of HCC [156]. High expression of circ_0000798 in peripheral blood was related to poor overall survival in HCC patients, and circ_0000798 had the potential as a diagnostic or predictive biomarker for HCC [157]. Furthermore, circulating circSETD3 showed predictive ability for HCC microvascular invasion, and the expression level of circSETD3 was negatively correlated with HCC invasion and metastasis [158]. Zhu et al. identified plasma hsa_circ_0027089 as a possible new biomarker for the diagnosis of hepatitis B-associated HCC via a clinical investigation [159]. circ_0004277 was significantly upregulated in HCC cells, tissues and plasma exosomes. Exosomal circ_0004277 from HCC cells enhanced the circ_0004277 expression in surrounding normal cells through cellular communication, thus stimulating EMT process and promoting the invasion of HCC cells [160]. In addition, the expression of exosomal circZNF652 was upregulated in the serum of HCC patients and was closely associated with the proliferation, migration, invasion and glycolysis of HCC cells, suggesting that it might become a novel biomarker for predicting the malignant characteristics of HCC [161]. circ_100338 was highly expressed in metastatic HCC cells as well as exosomes, which could significantly affect tumor angiogenesis and might be a useful indicator of pulmonary metastasis and poor survival [87].
Conclusions and perspectives
Many studies have shown that alterations of m6A modification and circRNA affect the development and progression of HCC, and they have important implications in the diagnosis, treatment and prognosis of HCC. In addition, circRNAs, which are highly abundant and stable, exist in a variety of extracellular fluids, including saliva, blood and urine, and can become ideal biomarkers and therapeutic targets for HCC [152, 162]. circRNAs can be modified by m6A, which makes them more stable, so m6A modification might serves as an essential regulatory mechanism for the circRNAs to exert their functions. It is worthwhile to pay more attention to the interplay among m6A modification, circRNA, and HCC, because m6A modification as well as circRNA could significantly modulate the development of HCC, and the cross-talk of circRNA and m6A modification in HCC has been rarely studied nowadays.
Most of the current studies focused on the downstream targets of m6A modification, and further studies are needed to investigate the upstream mechanism of m6A modification. In terms of m6A modifiers as potential drug targets, some inhibitors have been investigated; however, most of these small molecule inhibitors target at FTO and ALKBH5, and their effects on HCC have not been extensively observed nowadays. Additionally, the inhibitors of other m6A regulators and their applications in HCC are needed to be investigated further. Moreover, the roles of m6A motif-associated SNPs in HCC have not been well studied, and these SNPs may alter the fate of target mRNAs, therefore leading to different consequences. Besides, the specificity of m6A modification detection has yet to be addressed; for example, antibodies used to recognize m6A modifications exhibit nonspecific binding and can also bind to another kind of RNA modification m6Am (N(6),2'-O-dimethyladenosine) [163]. Although antibody-free assays have been introduced to detect m6A sites such as DART-SEQ (deamidation near RNA modification targets), they still have limitations such as low reproducibility, and need further improvement [164].
Stable expressions of circRNAs in HCC tissues, cells, exosomes and plasma provide new avenues for cancer diagnosis and treatment. However, the translational functions of circRNAs have been rarely studied and the current tools used to predict and identify their encoded proteins or peptides are still limited. In addition, Ma et al. pointed out that most circRNAs are named according to their host genes or functions, and when multiple circRNAs originate from the same host gene and have different functional roles, the naming situation is rather confused. Therefore, a novel standard circRNA naming rule is needed to construct in the related circRNA databases [95]. Taken together, with the development of bioinformatics technology, the interaction networks of circRNA, m6A modification and HCC carcinogenesis will be more extensively studied. And more regulatory mechanisms and therapeutic targets that contribute to HCC development will be exploited.
Abbreviation
3'-UTR: 3'-untranslated region; AML: acute myelocytic leukemia; AR: androgen receptor; CAEs: cancer-associated endothelial cells; CAFs: cancer-associated fibroblasts; CAR-T: chimeric antigen receptor engineered T-cell immunotherapy; ciRNAs: intronic circRNAs; ECM: extracellular matrix; ecRNAs: exonic circRNAs; eIciRNAs: exon-intron circRNAs; EMT: epithelial-mesenchymal transition; FLT3: FMS-like tyrosine kinase-3; FTO: fat mass and obesity-associated protein; GNAO1: guanine nucleotide-binding protein G (o) subunit alpha; HCC: hepatocellular carcinoma; HNF3γ: hepatocyte nuclear factor 3γ; HNRNP: heterogeneous nuclear ribonucleoprotein; HUVECs: human umbilical vein endothelial cells; IRES: internal ribosome entry site; LYPD1: LY6/PLAUR domain 1; m6A: N6-methyladenosine; mTORC1: mTOR complex 1; NKAP: NF-κβ-associated protein; NOR1: oxidored-nitro domain-containing protein 1; PTBP1: polypyrimidine binding protein 1; QKI: Quaking; RBP: RNA-binding protein; RFS: recurrence-free survival; SAM: S-adenosylmethionine; SNPs: single nucleotide polymorphisms; TACE: trans-arterial chemoembolization; TME: tumor microenvironment; VM: vasculogenesis mimicry.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81602575, 81701847, 61572200), Natural Science Foundation of Hunan Province (2017JJ3430, 2017JJ3497), Xiang Jiang Scholars Program/Hong Kong Scholars Program (XJ2016054).
Authors' contributions
LZ designed the study. SQ and LZ drafted the manuscript. SQ, YM, XC, JX, YQ and LZ collected the data and conducted the picture processing. LZ and YM revised the manuscript. All authors have read and approved the final version of manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Pinna AD, Yang T, Mazzaferro V, De Carlis L, Zhou J, Roayaie S. et al. Liver Transplantation and Hepatic Resection can Achieve Cure for Hepatocellular Carcinoma. Ann Surg. 2018;268:868-75
3. Rich NE, Parikh ND, Singal AG. Hepatocellular Carcinoma and Liver Transplantation: Changing Patterns and Practices. Curr Treat Options Gastroenterol. 2017;15:296-304
4. Tsurusaki M, Murakami T. Surgical and Locoregional Therapy of HCC: TACE. Liver Cancer. 2015;4:165-75
5. Shiina S, Sato K, Tateishi R, Shimizu M, Ohama H, Hatanaka T. et al. Percutaneous Ablation for Hepatocellular Carcinoma: Comparison of Various Ablation Techniques and Surgery. Can J Gastroenterol Hepatol. 2018;2018:4756147
6. Chen Y, E CY, Gong ZW, Liu S, Wang ZX, Yang YS. et al. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat Dis Int. 2018;17:301-9
7. Sun W, He B, Yang B, Hu W, Cheng S, Xiao H. et al. Genome-wide CRISPR screen reveals SGOL1 as a druggable target of sorafenib-treated hepatocellular carcinoma. Lab Invest. 2018;98:734-44
8. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492-502
9. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507-17
10. Meyer KD, Jaffrey SR. Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol. 2017;33:319-42
11. Huang H, Bai Y, Lu X, Xu Y, Zhao H, Sang X. N6-methyladenosine associated prognostic model in hepatocellular carcinoma. Ann Transl Med. 2020;8:633
12. Liu ZX, Li LM, Sun HL, Liu SM. Link Between m6A Modification and Cancers. Front Bioeng Biotechnol. 2018;6:89
13. Qiu L, Xu H, Ji M, Shang D, Lu Z, Wu Y. et al. Circular RNAs in hepatocellular carcinoma: Biomarkers, functions and mechanisms. Life Sci. 2019;231:116660
14. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z. et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575-8
15. Huang H, Weng H, Chen J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37:270-88
16. Zhao W, Qi X, Liu L, Ma S, Liu J, Wu J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol Ther Nucleic Acids. 2020;19:405-12
17. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254-70
18. Lin Y, Wei X, Jian Z, Zhang X. METTL3 expression is associated with glycolysis metabolism and sensitivity to glycolytic stress in hepatocellular carcinoma. Cancer Med. 2020;9:2859-67
19. Qiao K, Liu Y, Xu Z, Zhang H, Zhang H, Zhang C. et al. RNA m6A methylation promotes the formation of vasculogenic mimicry in hepatocellular carcinoma via Hippo pathway. Angiogenesis. 2020
20. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X. et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. Embo j. 2020;39:e103181
21. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol. 2002;55:431-44
22. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529-43
23. Li Z, Li F, Peng Y, Fang J, Zhou J. Identification of three m6A-related mRNAs signature and risk score for the prognostication of hepatocellular carcinoma. Cancer Med. 2020;9:1877-89
24. Shi Y, Zhuang Y, Zhang J, Chen M, Wu S. METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner. Cancer Manag Res. 2020;12:13173-84
25. Zhou T, Li S, Xiang D, Liu J, Sun W, Cui X. et al. m6A RNA methylation-mediated HNF3γ reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Signal Transduct Target Ther. 2020;5:296
26. Lan T, Li H, Zhang D, Xu L, Liu H, Hao X. et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18:186
27. Chen Y, Peng C, Chen J, Chen D, Yang B, He B. et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18:127
28. Tsuboi Y, Ichida T, Sugitani S, Genda T, Inayoshi J, Takamura M. et al. Overexpression of extracellular signal-regulated protein kinase and its correlation with proliferation in human hepatocellular carcinoma. Liver Int. 2004;24:432-6
29. Wu X, Zhang X, Tao L, Dai X, Chen P. Prognostic Value of an m6A RNA Methylation Regulator-Based Signature in Patients with Hepatocellular Carcinoma. Biomed Res Int. 2020;2020:2053902
30. Nakagawa N, Tanaka K, Sonohara F, Kandimalla R, Sunagawa Y, Inokawa Y. et al. Novel Prognostic Implications of Methylated RNA and Demethylases in Resected HCC and Background Liver Tissue. Anticancer Res. 2020;40:6665-76
31. Zhao Y, You S, Yu YQ, Zhang S, Li PT, Ye YH. et al. Decreased nuclear expression of FTO in human primary hepatocellular carcinoma is associated with poor prognosis. Int J Clin Exp Pathol. 2019;12:3376-83
32. Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am J Transl Res. 2019;11:6084-92
33. Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y. et al. SIRT1 Regulates N(6) -Methyladenosine RNA Modification in Hepatocarcinogenesis by Inducing RANBP2-Dependent FTO SUMOylation. Hepatology. 2020
34. Mittenbühler MJ, Saedler K, Nolte H, Kern L, Zhou J, Qian SB. et al. Hepatic FTO is dispensable for the regulation of metabolism but counteracts HCC development in vivo. Mol Metab. 2020;42:101085
35. Wang P, Wang X, Zheng L, Zhuang C. Gene Signatures and Prognostic Values of m6A Regulators in Hepatocellular Carcinoma. Front Genet. 2020;11:540186
36. Liu J, Wang D, Zhou J, Wang L, Zhang N, Zhou L. et al. N6-methyladenosine reader YTHDC2 and eraser FTO may determine hepatocellular carcinoma prognoses after transarterial chemoembolization. Arch Toxicol. 2021;95:1621-9
37. Li Z, Zhang J, Liu X, Li S, Wang Q, Di C. et al. The LINC01138 drives malignancies via activating arginine methyltransferase 5 in hepatocellular carcinoma. Nat Commun. 2018;9:1572
38. Pu J, Wang J, Qin Z, Wang A, Zhang Y, Wu X. et al. IGF2BP2 Promotes Liver Cancer Growth Through an m6A-FEN1-Dependent Mechanism. Front Oncol. 2020;10:578816
39. Wang H, Liang L, Dong Q, Huan L, He J, Li B. et al. Long noncoding RNA miR503HG, a prognostic indicator, inhibits tumor metastasis by regulating the HNRNPA2B1/NF-κB pathway in hepatocellular carcinoma. Theranostics. 2018;8:2814-29
40. Song T, Liu JY, Yang JJ. NKAP plays an oncogenic function partly through AKT signaling pathway in hepatocellular carcinoma. Neoplasma. 2019;66:792-800
41. Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J. et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21:859-68
42. Qu N, Qin S, Zhang X, Bo X, Liu Z, Tan C. et al. Multiple m(6)A RNA methylation modulators promote the malignant progression of hepatocellular carcinoma and affect its clinical prognosis. BMC Cancer. 2020;20:165
43. Li W, Chen QF, Huang T, Shen L, Huang ZL, Wu P. Profiles of m(6)A RNA methylation regulators for the prognosis of hepatocellular carcinoma. Oncol Lett. 2020;19:3296-306
44. Zhang C, Huang S, Zhuang H, Ruan S, Zhou Z, Huang K. et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39:4507-18
45. Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S. et al. MicroRNA-145 Modulates N(6)-Methyladenosine Levels by Targeting the 3'-Untranslated mRNA Region of the N(6)-Methyladenosine Binding YTH Domain Family 2 Protein. J Biol Chem. 2017;292:3614-23
46. Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R. et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252-61
47. Patop IL, Kadener S. circRNAs in Cancer. Curr Opin Genet Dev. 2018;48:121-7
48. Li Z, Huang C, Bao C, Chen L, Lin M, Wang X. et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256-64
49. Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH. et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792-806
50. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J. et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19:141-57
51. Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12:381-8
52. Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M. et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55-66
53. Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S. et al. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015;29:2168-82
54. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675-91
55. Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134-47
56. Shan C, Zhang Y, Hao X, Gao J, Chen X, Wang K. Biogenesis, functions and clinical significance of circRNAs in gastric cancer. Mol Cancer. 2019;18:136
57. Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S. et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell. 2015;58:870-85
58. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA. et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125-34
59. Xu J, Ji L, Liang Y, Wan Z, Zheng W, Song X. et al. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduct Target Ther. 2020;5:298
60. Huang C, Liang D, Tatomer DC, Wilusz JE. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev. 2018;32:639-44
61. Li Z, Kearse MG, Huang C. The nuclear export of circular RNAs is primarily defined by their length. RNA Biol. 2019;16:1-4
62. Huang C, Liang D, Tatomer DC, Wilusz JE. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes & development. 2018;32:639-44
63. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202-7
64. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK. et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384-8
65. Hsiao K-Y, Lin Y-C, Gupta SK, Chang N, Yen L, Sun HS. et al. Noncoding Effects of Circular RNA CCDC66 Promote Colon Cancer Growth and Metastasis. Cancer Res. 2017;77:2339-50
66. Yu L, Liu Y. circRNA_0016624 could sponge miR-98 to regulate BMP2 expression in postmenopausal osteoporosis. Biochem Biophys Res Commun. 2019;516:546-50
67. Zhou ZB, Huang GX, Fu Q, Han B, Lu JJ, Chen AM. et al. circRNA.33186 Contributes to the Pathogenesis of Osteoarthritis by Sponging miR-127-5p. Mol Ther. 2019;27:531-41
68. Wang R, Zhang S, Chen X, Li N, Li J, Jia R. et al. CircNT5E Acts as a Sponge of miR-422a to Promote Glioblastoma Tumorigenesis. Cancer Res. 2018;78:4812-25
69. Abe N, Matsumoto K, Nishihara M, Nakano Y, Shibata A, Maruyama H. et al. Rolling Circle Translation of Circular RNA in Living Human Cells. Sci Rep. 2015;5:16435
70. Shi Y, Jia X, Xu J. The new function of circRNA: translation. Clin Transl Oncol. 2020;22:2162-9
71. Sun YM, Wang WT, Zeng ZC, Chen TQ, Han C, Pan Q. et al. circMYBL2, a circRNA from MYBL2, regulates FLT3 translation by recruiting PTBP1 to promote FLT3-ITD AML progression. Blood. 2019;134:1533-46
72. Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L. et al. Translation of CircRNAs. Mol Cell. 2017;66:9-21.e7
73. Du WW, Zhang C, Yang W, Yong T, Awan FM, Yang BB. Identifying and Characterizing circRNA-Protein Interaction. Theranostics. 2017;7:4183-91
74. Zang J, Lu D, Xu A. The interaction of circRNAs and RNA binding proteins: An important part of circRNA maintenance and function. J Neurosci Res. 2020;98:87-97
75. Yao Z, Xu R, Yuan L, Xu M, Zhuang H, Li Y. et al. Circ_0001955 facilitates hepatocellular carcinoma (HCC) tumorigenesis by sponging miR-516a-5p to release TRAF6 and MAPK11. Cell Death Dis. 2019;10:945
76. Wang L, Long H, Zheng Q, Bo X, Xiao X, Li B. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol Cancer. 2019;18:119
77. Zhu Q, Lu G, Luo Z, Gui F, Wu J, Zhang D. et al. CircRNA circ_0067934 promotes tumor growth and metastasis in hepatocellular carcinoma through regulation of miR-1324/FZD5/Wnt/β-catenin axis. Biochem Biophys Res Commun. 2018;497:626-32
78. Zhang S, Liu Y, Liu Z, Zhang Y, Chen G, Li K. et al. CircRNA_0000502 promotes hepatocellular carcinoma metastasis and inhibits apoptosis through targeting microRNA-124. J buon. 2019;24:2402-10
79. Chen W, Quan Y, Fan S, Wang H, Liang J, Huang L. et al. Exosome-transmitted circular RNA hsa_circ_0051443 suppresses hepatocellular carcinoma progression. Cancer Lett. 2020;475:119-28
80. Yu J, Xu QG, Wang ZG, Yang Y, Zhang L, Ma JZ. et al. Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J Hepatol. 2018;68:1214-27
81. Hu ZQ, Zhou SL, Li J, Zhou ZJ, Wang PC, Xin HY. et al. Circular RNA Sequencing Identifies CircASAP1 as a Key Regulator in Hepatocellular Carcinoma Metastasis. Hepatology. 2020;72:906-22
82. Sun S, Wang W, Luo X, Li Y, Liu B, Li X. et al. Circular RNA circ-ADD3 inhibits hepatocellular carcinoma metastasis through facilitating EZH2 degradation via CDK1-mediated ubiquitination. Am J Cancer Res. 2019;9:1695-707
83. Han D, Li J, Wang H, Su X, Hou J, Gu Y. et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;66:1151-64
84. Song LN, Qiao GL, Yu J, Yang CM, Chen Y, Deng ZF. et al. Hsa_circ_0003998 promotes epithelial to mesenchymal transition of hepatocellular carcinoma by sponging miR-143-3p and PCBP1. J Exp Clin Cancer Res. 2020;39:114
85. Su Y, Lv X, Yin W, Zhou L, Hu Y, Zhou A. et al. CircRNA Cdr1as functions as a competitive endogenous RNA to promote hepatocellular carcinoma progression. Aging (Albany NY). 2019;11:8182-203
86. Yu J, Yang M, Zhou B, Luo J, Zhang Z, Zhang W. et al. CircRNA-104718 acts as competing endogenous RNA and promotes hepatocellular carcinoma progression through microRNA-218-5p/TXNDC5 signaling pathway. Clin Sci (Lond). 2019;133:1487-503
87. Huang XY, Huang ZL, Huang J, Xu B, Huang XY, Xu YH. et al. Exosomal circRNA-100338 promotes hepatocellular carcinoma metastasis via enhancing invasiveness and angiogenesis. J Exp Clin Cancer Res. 2020;39:20
88. Liu Z, Yu Y, Huang Z, Kong Y, Hu X, Xiao W. et al. CircRNA-5692 inhibits the progression of hepatocellular carcinoma by sponging miR-328-5p to enhance DAB2IP expression. Cell Death Dis. 2019;10:900
89. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
90. Bao S, Jin S, Wang C, Tu P, Hu K, Lu J. Androgen receptor suppresses vasculogenic mimicry in hepatocellular carcinoma via circRNA7/miRNA7-5p/VE-cadherin/Notch4 signalling. J Cell Mol Med. 2020;24:14110-20
91. Yu YX, Ge TW, Zhang P. Circular RNA circGFRA1 promotes angiogenesis, cell proliferation and migration of hepatocellular carcinoma by combining with miR-149. Eur Rev Med Pharmacol Sci. 2020;24:11058-64
92. Pu J, Wang J, Li W, Lu Y, Wu X, Long X. et al. hsa_circ_0000092 promotes hepatocellular carcinoma progression through up-regulating HN1 expression by binding to microRNA-338-3p. J Cell Mol Med. 2020
93. Yan K, Cheng W, Xu X, Cao G, Ji Z, Li Y. Circulating RNAs, circ_4911 and circ_4302, are novel regulators of endothelial cell function under a hepatocellular carcinoma microenvironment. Oncol Rep. 2020;44:1727-35
94. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-22
95. Ma Z, Shuai Y, Gao X, Wen X, Ji J. Circular RNAs in the tumour microenvironment. Mol Cancer. 2020;19:8
96. Zhang Q, Wang W, Zhou Q, Chen C, Yuan W, Liu J. et al. Roles of circRNAs in the tumour microenvironment. Mol Cancer. 2020;19:14
97. Huang XY, Zhang PF, Wei CY, Peng R, Lu JC, Gao C. et al. Circular RNA circMET drives immunosuppression and anti-PD1 therapy resistance in hepatocellular carcinoma via the miR-30-5p/snail/DPP4 axis. Mol Cancer. 2020;19:92
98. Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM. et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110
99. Li Q, Pan X, Zhu D, Deng Z, Jiang R, Wang X. Circular RNA MAT2B Promotes Glycolysis and Malignancy of Hepatocellular Carcinoma Through the miR-338-3p/PKM2 Axis Under Hypoxic Stress. Hepatology. 2019;70:1298-316
100. Luo Y, Fu Y, Huang R, Gao M, Liu F, Gui R. et al. CircRNA_101505 sensitizes hepatocellular carcinoma cells to cisplatin by sponging miR-103 and promotes oxidored-nitro domain-containing protein 1 expression. Cell Death Discov. 2019;5:121
101. Guan Y, Zhang Y, Hao L, Nie Z. CircRNA_102272 Promotes Cisplatin-Resistance in Hepatocellular Carcinoma by Decreasing MiR-326 Targeting of RUNX2. Cancer Manag Res. 2020;12:12527-34
102. Zhou S, Wei J, Wang Y, Liu X. Cisplatin resistance-associated circRNA_101237 serves as a prognostic biomarker in hepatocellular carcinoma. Exp Ther Med. 2020;19:2733-40
103. Yang C, Dong Z, Hong H, Dai B, Song F, Geng L. et al. circFN1 Mediates Sorafenib Resistance of Hepatocellular Carcinoma Cells by Sponging miR-1205 and Regulating E2F1 Expression. Mol Ther Nucleic Acids. 2020;22:421-33
104. Li Y, Zhang Y, Zhang S, Huang D, Li B, Liang G. et al. circRNA circARNT2 Suppressed the Sensitivity of Hepatocellular Carcinoma Cells to Cisplatin by Targeting the miR-155-5p/PDK1 Axis. Mol Ther Nucleic Acids. 2021;23:244-54
105. Li X, He J, Ren X, Zhao H, Zhao H. Circ_0003998 enhances doxorubicin resistance in hepatocellular carcinoma by regulating miR-218-5p/EIF5A2 pathway. Diagn Pathol. 2020;15:141
106. Chen H, Liu S, Li M, Huang P, Li X. circ_0003418 Inhibits Tumorigenesis And Cisplatin Chemoresistance Through Wnt/β-Catenin Pathway In Hepatocellular Carcinoma. Onco Targets Ther. 2019;12:9539-49
107. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N. et al. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep. 2017;20:2262-76
108. Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK. et al. Endoribonucleolytic Cleavage of m(6)A-Containing RNAs by RNase P/MRP Complex. Mol Cell. 2019;74:494-507.e8
109. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y. et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27:626-41
110. Huang A, Zheng H, Wu Z, Chen M, Huang Y. Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics. 2020;10:3503-17
111. Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD. et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695
112. Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L. et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell. 2019;76:96-109.e9
113. Zhao J, Lee EE, Kim J, Yang R, Chamseddin B, Ni C. et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun. 2019;10:2300
114. Lewis CJ, Pan T, Kalsotra A. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat Rev Mol Cell Biol. 2017;18:202-10
115. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science. 2016;352:1408-12
116. Zhang X, Xu Y, Qian Z, Zheng W, Wu Q, Chen Y. et al. circRNA_104075 stimulates YAP-dependent tumorigenesis through the regulation of HNF4a and may serve as a diagnostic marker in hepatocellular carcinoma. Cell Death Dis. 2018;9:1091
117. Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L. et al. N(6)-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol Cancer. 2020;19:163
118. Wang M, Yang Y, Yang J, Yang J, Han S. circ_KIAA1429 accelerates hepatocellular carcinoma advancement through the mechanism of m(6)A-YTHDF3-Zeb1. Life Sci. 2020;257:118082
119. Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019;47:W548-w55
120. Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL. et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327-d34
121. Sun WJ, Li JH, Liu S, Wu J, Zhou H, Qu LH. et al. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44:D259-65
122. Liu H, Wang H, Wei Z, Zhang S, Hua G, Zhang SW. et al. MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018;46:D281-d7
123. Liu H, Flores MA, Meng J, Zhang L, Zhao X, Rao MK. et al. MeT-DB: a database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 2015;43:D197-203
124. Chen K, Wei Z, Zhang Q, Wu X, Rong R, Lu Z. et al. WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019;47:e41
125. Liu S, Zhu A, He C, Chen M. REPIC: a database for exploring the N(6)-methyladenosine methylome. Genome Biol. 2020;21:100
126. Huang Y, He N, Chen Y, Chen Z, Li L. BERMP: a cross-species classifier for predicting m(6)A sites by integrating a deep learning algorithm and a random forest approach. Int J Biol Sci. 2018;14:1669-77
127. Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91
128. Liu K, Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics. 2020;36:3336-42
129. Li J, He S, Guo F, Zou Q. HSM6AP: a high-precision predictor for the Homo sapiens N6-methyladenosine (m^6 A) based on multiple weights and feature stitching. RNA Biol. 2021:1-11
130. Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y. et al. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2018;46:D139-d45
131. Li J, Huang Y, Cui Q, Zhou Y. m6Acorr: an online tool for the correction and comparison of m(6)A methylation profiles. BMC Bioinformatics. 2020;21:31
132. Deng S, Zhang H, Zhu K, Li X, Ye Y, Li R. et al. M6A2Target: a comprehensive database for targets of m6A writers, erasers and readers. Brief Bioinform. 2020
133. Liu L, Lei X, Meng J, Wei Z. WITMSG: Large-scale Prediction of Human Intronic m(6)A RNA Methylation Sites from Sequence and Genomic Features. Curr Genomics. 2020;21:67-76
134. Wu W, Ji P, Zhao F. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020;21:101
135. Zhao Z, Wang K, Wu F, Wang W, Zhang K, Hu H. et al. circRNA disease: a manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis. 2018;9:475
136. Zhao S, Li S, Liu W, Wang Y, Li X, Zhu S. et al. Circular RNA Signature in Lung Adenocarcinoma: A MiOncoCirc Database-Based Study and Literature Review. Front Oncol. 2020;10:523342
137. Li S, Li Y, Chen B, Zhao J, Yu S, Tang Y. et al. exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018;46:D106-d12
138. Xia S, Feng J, Chen K, Ma Y, Gong J, Cai F. et al. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2018;46:D925-d9
139. Glažar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. Rna. 2014;20:1666-70
140. Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13:34-42
141. Henry NL, Hayes DF. Cancer biomarkers. Mol Oncol. 2012;6:140-6
142. Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of METTL3 as an Adverse Prognostic Biomarker in Hepatocellular Carcinoma. Dig Dis Sci. 2020
143. Qiao Y, Zhou B, Zhang M, Liu W, Han Z, Song C. et al. A Novel Inhibitor of the Obesity-Related Protein FTO. Biochemistry. 2016;55:1516-22
144. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H. et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35:677-91.e10
145. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M. et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell. 2020;38:79-96.e11
146. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y. et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell. 2018;172:90-105.e23
147. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H. et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159-70
148. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597-601
149. Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X. et al. Epitranscriptomics in liver disease: Basic concepts and therapeutic potential. J Hepatol. 2020;73:664-79
150. Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y. et al. The Landscape of Circular RNA in Cancer. Cell. 2019;176:869-81.e13
151. Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and Characterization of Circular RNAs As a New Class of Putative Biomarkers in Human Blood. PLoS One. 2015;10:e0141214
152. Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J. et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981-4
153. Guo J, Duan H, Li Y, Yang L, Yuan L. A novel circular RNA circ-ZNF652 promotes hepatocellular carcinoma metastasis through inducing snail-mediated epithelial-mesenchymal transition by sponging miR-203/miR-502-5p. Biochem Biophys Res Commun. 2019;513:812-9
154. Chien Y, Tsai PH, Lai YH, Lu KH, Liu CY, Lin HF. et al. CircularRNA as novel biomarkers in liver diseases. J Chin Med Assoc. 2020;83:15-7
155. Wang G, Liu W, Zou Y, Wang G, Deng Y, Luo J. et al. Three isoforms of exosomal circPTGR1 promote hepatocellular carcinoma metastasis via the miR449a-MET pathway. EBioMedicine. 2019;40:432-45
156. Sun XH, Wang YT, Li GF, Zhang N, Fan L. Serum-derived three-circRNA signature as a diagnostic biomarker for hepatocellular carcinoma. Cancer Cell Int. 2020;20:226
157. Lei B, Zhou J, Xuan X, Tian Z, Zhang M, Gao W. et al. Circular RNA expression profiles of peripheral blood mononuclear cells in hepatocellular carcinoma patients by sequence analysis. Cancer Med. 2019;8:1423-33
158. Wang P, Xu LL, Zheng XB, Hu YT, Zhang JF, Ren SS. et al. Correlation between the expressions of circular RNAs in peripheral venous blood and clinicopathological features in hepatocellular carcinoma. Ann Transl Med. 2020;8:338
159. Zhu K, Zhan H, Peng Y, Yang L, Gao Q, Jia H. et al. Plasma hsa_circ_0027089 is a diagnostic biomarker for hepatitis B virus-related hepatocellular carcinoma. Carcinogenesis. 2020;41:296-302
160. Zhu C, Su Y, Liu L, Wang S, Liu Y, Wu J. Circular RNA hsa_circ_0004277 Stimulates Malignant Phenotype of Hepatocellular Carcinoma and Epithelial-Mesenchymal Transition of Peripheral Cells. Front Cell Dev Biol. 2020;8:585565
161. Li Y, Zang H, Zhang X, Huang G. Exosomal Circ-ZNF652 Promotes Cell Proliferation, Migration, Invasion and Glycolysis in Hepatocellular Carcinoma via miR-29a-3p/GUCD1 Axis. Cancer Manag Res. 2020;12:7739-51
162. Hu BB, Wang XY, Gu XY, Zou C, Gao ZJ, Zhang H. et al. N(6)-methyladenosine (m(6)A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Mol Cancer. 2019;18:178
163. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767-72
164. Meyer KD. DART-seq: an antibody-free method for global m(6)A detection. Nat Methods. 2019;16:1275-80
Author contact
![]() Corresponding author: Dr. Luqing Zhao, Department of Pathology, Xiangya Hospital, Central South University, Changsha, Hunan, China, E-mail: luqingzhaoedu.cn
Corresponding author: Dr. Luqing Zhao, Department of Pathology, Xiangya Hospital, Central South University, Changsha, Hunan, China, E-mail: luqingzhaoedu.cn