Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(12):3224-3238. doi:10.7150/ijbs.62808 This issue Cite
Research Paper
BC-N102 suppress breast cancer tumorigenesis by interfering with cell cycle regulatory proteins and hormonal signaling, and induction of time-course arrest of cell cycle at G1/G0 phase
Bashir Lawal1,2*, Yu-Cheng Kuo3,4*, Alexander T. H. Wu5,6,7,8 ![]() , Hsu-Shan Huang1,2,8,9,10
, Hsu-Shan Huang1,2,8,9,10 ![]()
1. PhD Program for Cancer Molecular Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University and Academia Sinica, Taipei 11031, Taiwan
2. Graduate Institute for Cancer Biology & Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei 11031, Taiwan
3. Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan
4. School of Post-baccalaureate Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung 40402, Taiwan
5. The PhD Program of Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei 11031, Taiwan
6. Clinical Research Center, Taipei Medical University Hospital, Taipei Medical University, Taipei 11031, Taiwan
7. TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei 11031, Taiwan
8. Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei 11490, Taiwan
9. School of Pharmacy, National Defense Medical Center, Taipei 11490, Taiwan
10. PhD Program in Drug Discovery and Development Industry, College of Pharmacy, Taipei Medical University, Taipei 11031, Taiwan
*These authors contributed equally to this work.
Received 2021-5-16; Accepted 2021-7-9; Published 2021-7-25
Abstract

Mechanisms of breast cancer progression and invasion, often involve alteration of hormonal signaling, and upregulation and/or activation of signal transduction pathways that input to cell cycle regulation. Herein, we describe a rationally designed first-in-class novel small molecule inhibitor for targeting oncogenic and hormonal signaling in ER-positive breast cancer. BC-N102 treatment exhibits dose-dependent cytotoxic effects against ER+ breast cancer cell lines. BC-N102 exhibited time course- and dose-dependent cell cycle arrest via downregulation of the estrogen receptor (ER), progesterone receptor (PR), androgen receptor (AR), phosphatidylinositol 3-kinase (PI3K), phosphorylated (p)-extracellular signal-regulated kinase (ERK), p-Akt, CDK2, and CDK4 while increasing p38 mitogen-activated protein kinase (MAPK), and mineralocorticoid receptor (MR) signaling in breast cancer cell line. In addition, we found that BC-N102 suppressed breast cancer tumorigenesis in vivo and prolonged the survival of animals. Our results suggest that the proper application of BC-N102 may be a beneficial chemotherapeutic strategy for ER+ breast cancer patients.
Keywords: ER+ breast cancer, G1/G0 cell cycle arrest, cell cycle proteins, chromatin immunoprecipitation, tumor progression, hormonal signaling
Introduction
Breast cancer is an invasive breast ductal carcinoma, and the most commonly diagnosed cancer (11.7% of total cases) representing 2.3 million new cases in 2020 [1, 2]. With an estimated 685,000 deaths, breast cancer is the fifth leading cause of cancer mortality worldwide, in both men and women [3]. Among women, breast cancer respectively accounts for 25% and 16% of new cancer cases and deaths, ranking first for incidence and mortality in more than 100 countries [3]. Unfortunately, reduced access to healthcare facilities, and delays in diagnosis and treatment due to the coronavirus disease 2019 (COVID-19) pandemic may lead to a short-term drop in the cancer incidence followed by an increase in advanced-stage disease and ultimately increased mortality [4, 5]
Hormone receptors including the estrogen receptor (ER), progesterone receptor (PR), androgen receptor (AR), and mineralocorticoid receptor (MR) are steroid receptors with similar hormone binding, nuclear translocation, DNA binding, and transactivation domains [6]. The ER is a ligand-activated nuclear receptor that mediates the transcription of estrogen-responsive genes [7], which play pivotal roles in cell growth, differentiation, proliferation, and malignant transformation [8], and thus serve as important therapeutic targets [9-12]. On the basis of the ER, breast cancer is subdivided into estrogen receptor-positive (ER+) and negative (ER-) sub-types, among which ER+ accounts for ~70% of all breast cancers (expressing ER protein), while the remaining breast cancers are ER- subtypes [13], which do not express ER. PR plays an important role in normal breast development and its over-activation is detrimental for late-stage breast cancers, providing some rationale for dual targeting of ER and PR in advanced tumors [14]. AR on the other hand is present in about 80 % of breast cancers making it more commonly expressed than either ER or PR, and plays an important oncogenic role in breast cancer [15]. Significant progress has been made in understanding the role of ER as a transcription factor that directly regulates expressions of target genes [16] or indirectly by mediating other transcription factors on promoter targets [16, 17].
Cell cycle progression is tightly regulated by the collective activities of cyclins, CDKs, and CDK inhibitors (CDKIs). The p27, p21, and p53 are important CDKIs that negatively regulate cell cycle progression via inhibition of the cyclin/CDKs. p27 mediate G 1 -phase arrest of the cell cycle, via cell-cell contact and growth factors dependent mechanisms, and in response to agents that inhibit cell cycle progression [18, 19]. p21 binds to and inhibits the cyclin/CDK complexes, thereby preventing RB phosphorylation and thus inhibit cell proliferation [20]. p53 a known tumor suppressor gene is well associated with p21 expression and exhibited dependent mechanisms of regulating tumor progression. However, it has been demonstrated that p21 expression may also be activated by p53-independent pathways [21].
However, considering the heterogeneity of breast cancer with its multiple genetic alterations and resistance to multiple treatment modalities [22, 23], targeting a single pathway by inhibiting the activity of one component would unlikely yield reasonable outcomes in the long run [24, 25]. Thus, targeting multiple oncogenic signaling networks would be preferable for the discovery and development of effective anticancer drugs [26]. Consequently, human epidermal growth factor receptor 2 (HER2), mammalian target of rapamycin (mTOR), cyclin-dependent kinases (CDKs), and phosphatidylinositol 3-kinase (PI3K/p110α)/protein kinase B (PKB/Akt)/extracellular signal-regulated kinase (ERK) signaling has attracted considerable attention as potential therapeutic targets for the development of new anticancer drugs to treat breast and other human malignancies [27-29]. In this report, we demonstrate that BC-N102, a novel small molecule, exhibited anticancer activity against breast cancer cell lines via interfering with cell cycle regulatory proteins and hormonal receptors signaling in vitro, and exhibited potent antitumor activity at tolerated doses in an ER+ human xenograft breast cancer model. Our results provide preclinical justification to support phase I clinical trials with BC-N102 in ER+ breast cancer patients.
Materials and Methods
Bioinformatics analysis of breast cancer in The Cancer Genome Atlas (TCGA) pan-cancer database
We analyzed differential gene expression profiles of CDK2/CDK4 among normal breast tissues, tumor stages, metastasis, and subtypes across the TCGA pan-cancer database using the TNMplot webserver algorithm [30]. Genetic and epigenetic alteration profiles of CDK2/CDK4 and survival differences between breast cancer cohorts with differential expressions or a genetically altered status of CDK2/CDK4 across the TCGA pan-cancer database were analyzed using the cBioportal [31], GSCLite [32], and MethSurve [33] webserver algorithms, while gene expression correlations were analyzed using the TIMER2.0 resource [34].
Cell lines and culture
A normal breast cell line (MCF-10A) and a breast cancer cell line (ER+HER2-Luminal 1; MCF-7) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), while MDB-MB-231/ATC, HS 578T, T-47D, and MDB-MB-468 were sources from the US National Cancer Institute. Cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 25 units/mL of penicillin, 25 units/mL of streptomycin, and 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 and 95% humidity incubator. Culture media were replaced after 72 h, and cells were subcultured to 70%~80% confluence.
Drugs and chemicals
The clinical drug (paclitaxel) was purchased from Selleckchem (Houston, TX, USA), while BC-N102 was synthesized through established protocols in our lab [35, 36]. A 10-mM stock solution of the drugs was prepared in dimethyl sulfoxide (DMSO) and kept frozen at a temperature of -20 °C. DMEM was obtained from Gibco-Invitrogen (Grand Island, NY, USA). Other reagents and chemicals used included Cremophor EL (Sigma, St. Louis, MO, USA), dimethyl acetamide (DMA) (Sigma), estradiol cyclopentyl propionate (Estol-depot injection, Astar, Taiwan), FBS (HyClone, Logan, UT, USA), Matrigel (BD Biosciences, USA), and phosphoric acid (Wako, Japan).
In vitro anticancer assay
In vitro anticancer activities of BC-N102 were assayed using established protocols of the sulforhodamine B (SRB) reagent [37]. Approximately 5000~40,000 viable cells of the NCI's 60 human tumor cell lines representing leukemia, melanoma, non-small-cell lung cancer (NSCLC), colon, central nervous system (CNS), ovarian, renal, prostate, and breast cancer cell lines were sown in wells of 96-well plates overnight (15 h). After incubation, cells were treated with a single dose (10 μM) or multiple doses (0, 0.1, 1.0, 10, and 100 μM) and incubated for 2 days. After incubation, the medium was removed, and plates were washed thrice with phosphate-buffered saline (PBS) (1%), followed by treatment with 10% trichloroacetic acid and incubation (at -4 °C) for 1 h. The plates were washed with double-distilled (dd)H2O, and incubated with SRB (0.4%) for 1 h. The unbound SRB dye was removed using 1% acetic acid, and the plates were air-dried. The contents of the plates were re-dissolved in a 20 mM Tris-based solution for 15 min under constant agitation. Cell viability was determined with a spectrophotometer at a wavelength of 515 nm. Growth inhibition was calculated in relation to cells without drug treatment and the time-zero control. GI50 is calculated from [(Ti-Tz)/(C-Tz)] × 100 = 50, while TGI is calculated from Ti = Tz, where Tz, Ti, and C represent absorbance at time 0, after treatment and for the control respectively [38, 39].
Chromatin immunoprecipitation (ChIP) analysis
MCF-7 cells were seeded into 10-cm Petri dishes and incubated for 24 h. After 24 h of cell incubation, the medium was replaced with phenol red-free DMEM containing 5% dextran-coated charcoal (DCC)-treated FBS, and cells were incubated for another 24 h. ChIP analyses were conducted after treatment with estrogen (17β-estradiol; Sigma Aldrich, St. Louis, MO, USA), and chromatin was immunoprecipitated with 2 µg of antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) against ERα as previously described [13]. The CDK2, CDK4, and GSTM3 promoter regions were amplified by a polymerase chain reaction (PCR). PCR products were analyzed by agarose gel electrophoresis.
Flow cytometric analysis
A flow cytometric analysis was conducted according to the protocol described by Gao et al. [13] Briefly, MCF-7 cells were seeded in six-well plates and incubated for 24 h. Cells were harvested, washed with cold PBS, and then fixed in ice-cold (4 °C) 75% ethanol for 12 h. After fixation, cells were washed in PBS, incubated with propidium iodide (BD Bioscience, San Jose, CA, USA) and RNase for 30 min at room temperature. Cells were then analyzed using the BD FACSVerse flow cytometer (BD Biosciences).
Western blotting
Cell lysates from BC-N102-treated MCF-7 cell lines, were subjected to protein expression analyses using sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) with a Mini-Protean III system (Bio-Rad, Taipei, Taiwan) and transferred onto polyvinylidene difluoride membranes using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were incubated with primary antibodies for the PI3K (CAT: #E11-1224B, 1:1000), p-ERK (CAT: #4370, 1:1000), p-AKT (CAT: #4060, 1:1000), P38 MAPK (1:1000), CDK2 (CAT: #2546, 1:1000), CDK4 (CAT: #2906, 1:1000), cyclin D1 (CAT: #2926, 1:1000), p-CDC35C (1:1000), p21 (CAT: #2947, 1:1000), p27 (CAT: #2552, 1:1000), p53 (CAT:2557 #1:1000), and GAPDH (CAT: #10494-1-AP 1:1000) for 24 h at 4 °C, followed by incubation with the respective secondary antibodies at room temperature for 1 h. Protein signals were detected and visualized with the aid of an enhanced chemiluminescence (ECL) detection kit, and images were captured using the UVP BioDoc-It system (Upland, CA, USA).
Luciferase reporter gene assay
We used a mouse mammary tumor virus (MMTV)-Luc reporter to examine the effect of BC-N102 on PR, AR, and MR activity. This reporter contains a degenerate glucocorticoid response element that is responsive to androgens, progesterone, and glucocorticoids/aldosterone (through the AR, PR, and MR, respectively) but is not activated to any significant extent by estrogen [40]. Briefly, cells were co-transfected overnight with MMTV-Luc reporter and with BC-N102, and either progesterone (P, 300 nM), dihydrotestosterone (10 nM), or mineralocorticoid (100 nM) for T = 24 h and assayed for luciferase activity as described in a previous study [41]. After subtracting the blank, the luciferase values were expressed relative to negative control conditions (MMTV-Luc reporter only, no progesterone, dihydrotestosterone, or mineralocorticoid), or with the presence of the hormone.
Maximum tolerated dose (MTD) assay
To estimate the appropriate dose level for the anticancer study, the MTD of BC-N102 was determined in BALB/c nude mice (three per group) by intraperitoneal administration of BC-N102 in a 10-day toxicity study at various concentrations; 0, 5, 10, 20, 30, and 60 mg/kg body weight (BW). The MTD is defined as the highest concentration that causes no more than a 10% BW decrement compared to an appropriate control group and produces no mortality or any external signs of toxicity that would be predicted to shorten the natural lifespan of the animal [42, 43]. All animals were examined daily for signs of toxicity. At the end of 9 days, all animals were sacrificed, and their oral cavity, liver, colon, small intestine, kidneys, and stomach were examined for any abnormalities under a dissection microscope.
In vivo anticancer assay in a human breast tumor xenograft model
Female nude (nu/nu) mice aged 6~7 weeks obtained from BioLasco Taiwan (Taipei City, Taiwan; under license from Charles River Laboratories) were used. Four animals were housed in an individually ventilated cage (IVC, 26.7 x 20.7 x 14.0 cm) with controlled temperature (20~24 °C), humidity (30%~70%) and a 12-h light/dark cycle. Free access to a standard lab diet (Oriental Yeast, Tokyo, Japan) and autoclaved tap water was provided. All aspects of this work including housing, experimentation, and animal disposal were performed in general accordance with the Guide for the Care and Use of Laboratory Animals: Eighth Edition (National Academies Press, Washington, D.C., 2011) in an AAALAC-accredited laboratory animal facility. In addition, the animal care and use protocol was reviewed and approved by the IACUC at Eurofins Panlabs Taiwan (New Taipei City, Taiwan). Viable human breast tumor cells (MCF-7, ATCC HTB-22) at 1.5 x 107 cells in a 0.2-mL mixture (Matrigel: medium = 1:1) were subcutaneously implanted into the dorsal back region of the animals, and estradiol cyclopentyl propionate at 100 μg/mouse was subcutaneously injected twice a week as a supplement during the study period [44]. When the tumor had reached ≥ 5 mm in diameter (denoted as day 1), the animals were randomly divided into three groups (of six animals each). The test substance, BC-N102 (dissolved in 30% DMA/20% Cremophor EL/10 mM H3PO4) at 1 mg/kg BW was administered once daily by an intraperitoneal injection for 45 days. The reference substance, paclitaxel, at 10 mg/kg BW, was intravenously injected once weekly. The tumor size, BW, and overt signs of toxicity were observed and recorded every 4 days for 45 days. The tumor volume (mm3) was estimated according to the formula: length x (width)2 x 0.5. Tumor growth inhibition (T/C) was calculated by the following formula: T/C = (Tn - T1/Cn - C1) x 100%, where C1 (Cn) is the tumor weight measured on day 1 (day n) in the control group, and T1 (Tn) is the tumor weight measured on day 1 (day n) in the treated group.
Molecular docking of BC-N102 with CDK2 and CDK4
The Avogadro molecular builder and visualization tool vers. 1.XX (http://avogadro.cc/) was used to obtain the mol2 file of the three-dimensional (3D) structure of BC-N102, while the SDF file of palbociclib (CID: 5330286) was obtained from the PubChem database. The PDB files (3D structures) of the receptors, CDK2 (PDB; 4EK3) and CDK4 D3 (PDB; 3G33), were retrieved from the Protein Data Bank. SDF files were converted to PDB files using the PyMOL Molecular Graphics System, vers. 1.2r3pre (Schrödinger, https://pymol.org/edu/?q=educational/). All PDB files were converted into PDBQT files using AutoDock VINA (vers. 0.8) [45]. Pre-docking preparation of the ligands and receptors was conducted following the protocol described in previous studies [39]. Docking was conducted using AutoDock VINA software as described previously [46]. Docking results were visualized using the PyMOL software and Discovery studio visualizer (vers. 19.1.0.18287, BIOVIA, San Diego, CA, USA) [47].
Statistical analysis
All experiments were conducted with at least three replicates (n≥3). GraphPad Prism software (San Diego, CA, USA) was used for data analysis and visualization. Experimental data are presented as the mean ± standard deviation (SD) and were calculated using Student's t-test. Purity-adjusted Pearson's correlations were used to analyze gene expression correlations in breast cancer patients. All survival analyses are presented using Kaplan-Meier plots. A p value of <0.05 indicated a significant difference. Statistical significance was denoted as * p<0.05, ** p<0.01, and *** p<0.001.
Results
CDK2 and CDK4 are clinical biomarkers of breast cancer (BRCA) staging and therapeutic responses
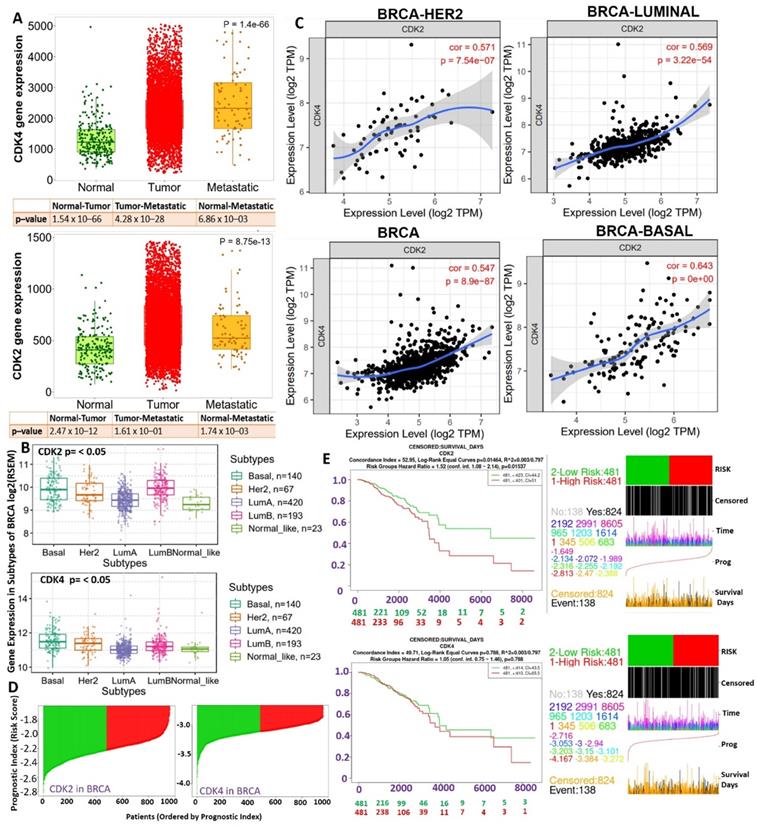
To evaluate the clinical relevance of CDK2 and CDK4 in breast cancer, we analyzed CDK2- and CDK4-related clinical information of breast cancer patients from TCGA pan-cancer database. Our differential expression analysis of CDK2 and CDK4 expression profiles revealed that both CDK2 and CDK4 are overexpressed in breast cancer tumors compared to adjacent normal tissues (Figure 1A). In addition, expression profiles of CDK2 and CDK4 were associated with tumor metastasis (Figure 1A) and breast cancer subtypes, with higher expression levels in basal and Her2 subtypes than the other subtypes (Figure 1B). The expression correlation analysis revealed strong expression correlations (r=0.54~064, all p<0.001) of CDK2 and CDK4 expressions in BRCA, basal, Her2, and luminal subtypes (Figure 1C). As expected, the higher expression profiles of CDK2 and CDK4 were associated with high risk scores and shorter survival periods of the breast cancer cohorts (Figure 1D).
Boxplot of differential expression profiles of cyclin-dependent kinase 2 (CDK2) and CDK4 between (A) breast cancer (BRCA) tumors, normal and metastatic tissues, (B) and among breast cancer subtypes. (C) Correlation plot between CDK2 and CDK4 expressions in BRCA, basal, Her2, and luminal subtypes. (D and E) Risk scores and Kaplan-Meir plot of the differential survival analysis between breast cancer cohorts with high and cohorts with low expression profiles of CDK2 and CDK4.
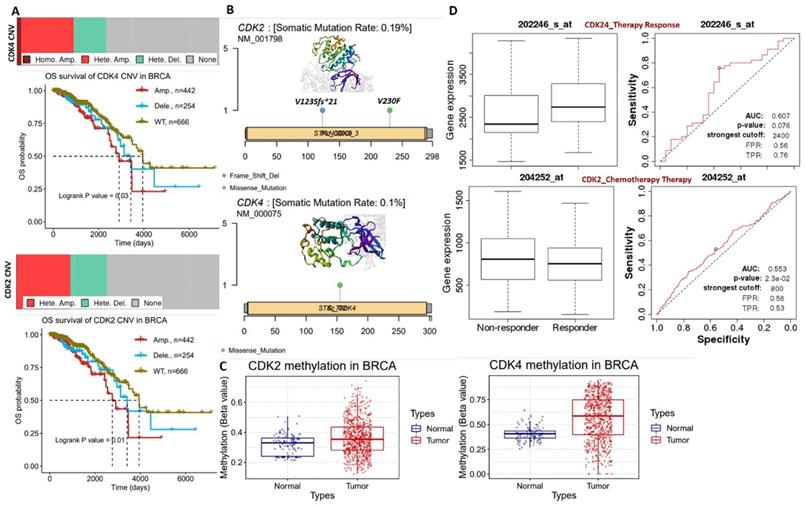
Our analysis of genetic and epigenetic alterations revealed that gene amplifications and heterozygous deletions were the most frequent copy number variations of CDK2 and CDK4 in breast cancer patients and were significantly (p<0.05) associated with poorer survival of breast cancer cohorts compared to cohorts with wild-type CDK2 and CDK4 (Figure 2A). In addition, CDK2 and CDK4 are hypomethylated but are less frequently mutated: CDK2 (2 cases 0.19%) and CDK2 (1 case 0.10%) in breast cancer (Figure 2B, C). Interestingly, we found that higher CDK2 and CDK4 expressions were associated with non-responsiveness of breast cancer patients to therapy (Figure 2D). Altogether, our bioinformatics analysis of clinical information from breast cancer patients strongly suggested that CDK2 and CDK4 are biomarkers of tumor progression, metastasis, and prognostic and therapy responses in breast cancer.
(A) Copy number variation landscape and Kaplan-Meir plot of breast cancer cohort with copy number variation (CNV) of cyclin-dependent kinase 2 (CDK2) and CDK4. (B) Somatic mutation plot of CDK2 and CDK4 in breast cancer. (C) Boxplot of differential methylation statuses of CDK2 and CDK4 between breast cancer tumors and adjacent normal tissues. (D) Gene expression and therapy response curve, showing the association of CDK2 and CDK4 expressions with non-responsiveness of breast cancer patients to therapy.
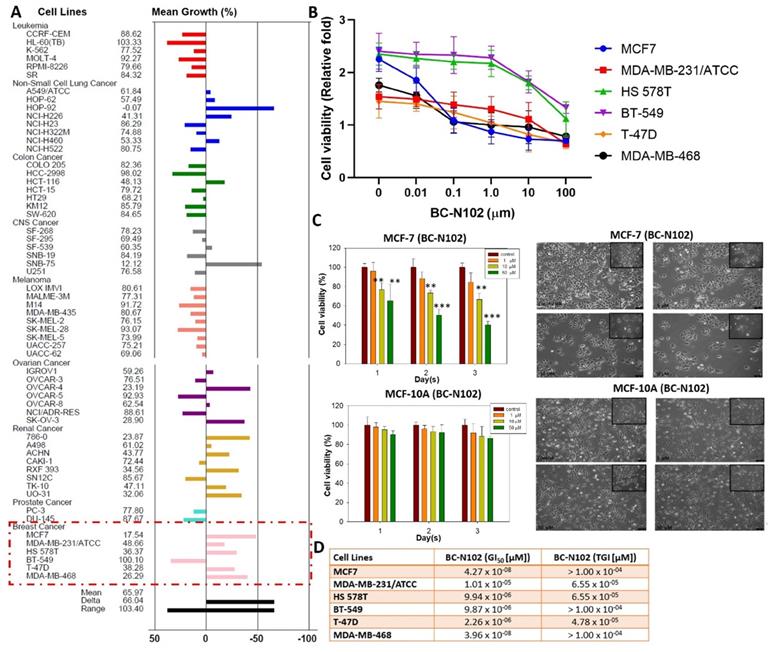
BC-N102 demonstrated dose-dependent cytotoxic effects against breast cancer cell lines but no significant activities against a normal breast cell line
We evaluated the anticancer activities of BC-N102 against the NCI60 cell line panel of breast, prostate, renal, ovarian, colon, melanoma, CNS, leukemia, and NSCL cancers. Interestingly, we found that BC-N102 at 10 μM exhibited antiproliferative effects against all 60 cell lines (Figure 3A). In addition to the antiproliferative effects, BC-N102 demonstrated cytotoxic effects against NSCLC cell lines (A549/ATCC, HOP-62, HOP-92, NCI-H226, and NCI-H460), renal cancer cell lines (786-0, A498, ACHN, RXF393, TK-10, and UO-31), and breast cancer cell lines (MCF7, MDB-MB-231/ATC, HS 578T, T-47D, and MDB-MB-468). Furthermore, we found that BC-N102 exhibited dose-dependent cytotoxic effects against breast cancer cell lines (Figure 3B, C) with GI50 values of 1.01 x 10-05~4.27 x 10-08 µM and TGI values of 4.78 x 10-05~1.00 x 10-05 (Figure 3D) but demonstrated no significant antiproliferative or cytotoxic activities against a normal breast cancer cell line (MCF-10A, Figure 3C). Taken together, these findings indicated that BC-N102 has cytotoxic effects against breast cancer cell lines but no significant activities against a normal breast cell line, and could serve as a novel candidate for treating breast cancer.
In vitro anticancer activities of BC-N102. Inhibitory activities of BC-N102 against panels of 60 human cancer cell lines. Each cell line was treated with a single dose of 10 μM of BC-N102. The zero point on the X-axis denotes the mean percentage of cell growth. The percentage growth of each cell line relative to the mean is represented by horizontal bars (Y axis). The right side bars indicate greater sensitivity, and left side bars indicate lower sensitivity. (B and C) Dose-response curve of BC-N102 against breast cancer cell lines and a normal breast cell line. (D) The 50% growth inhibition (GI50) and total growth inhibition (TGI) concentrations of BC-N102 against the breast cancer cell lines.
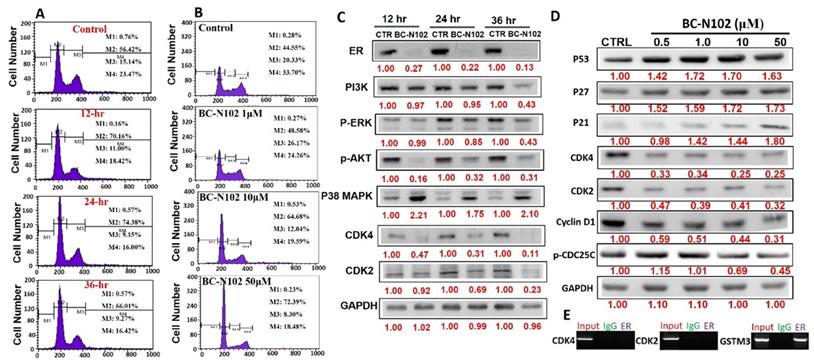
BC-N102 induced time-course arrest of the cell cycle at GO/G1 phase via interfering with the signaling of cell cycle regulatory proteins in breast cancer cell line
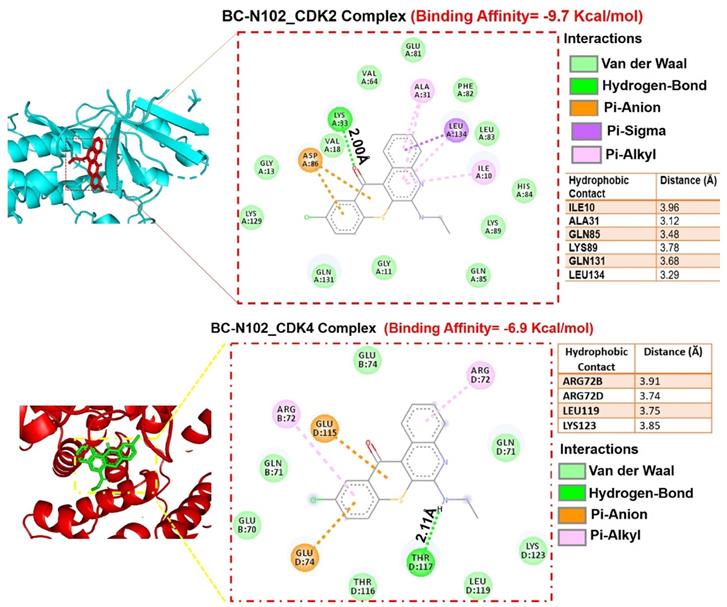
We used flow cytometry to analyze the effects of BC-N102 on different stages of MCF-7 cell cycle progression. Interestingly we found that treatment of MCF-7 cells with BC-N102 led to time-dependent arrest of the cell cycle at the G0/G1 phase with peak activities after 24 h of treatment (Figure 4A). In addition, BC-N102 treatment also led to dose-dependent arrest of the MCF-7 cell cycle at the G0/G1 phase. Cell arrest at this phase increased to 0%, 48.55%, 64.68%, and 72.39% with 0, 1, 10, and 50 μM of BC-N102, respectively (Figure 4B). The roles of the ER and cell cycle-related genes in cell cycle regulation were reported in several studies. Therefore, we wondered whether effects of BC-N102 on cell cycle progression and cell proliferation were linked to downregulation of these genes. We performed a Western blot analysis and found that treatment of MCF-7 cells with BC-N102 led to a time course-dependent downregulation of the ER, PI3K, p-ERK, p-Akt, p38 MAPK, CDK2, and CDK4 while increasing p38 MAPK expression in the MCF-7 cell line (Figure 4C). In addition, BC-N102 exhibited dose-dependent downregulation of CDK2, CDK4, cyclin D1, and phosphorylated CDC35C while increasing the expression levels of cell cycle regulatory proteins (p21, p27, and p53, Figure 4D). It is already known that the ER is a transcription factor that binds to promoters of estrogen-responsive target genes [48, 49]. To investigate whether the downregulation of CDK2 and CDK4 is a result of the effect of BC-N102, we evaluated whether the ER directly regulates these cell cycle-related genes by binding to their promoter regions. Our ChIP assay showed that the ER did not interact with the CDK2 or CDK4 promoter region, suggesting that BC-N102 downregulated the expression of CDK2 and CDK4 independent of ER positivity in breast cancer cell lines (Figure 4E). Moreover, our molecular docking study revealed robust interactions of BC-N102 with CDK2 and CDK4 binding cavities with binding affinities (ΔG) of -9.7 and -6.9 kcal/mol respectively. The BC-N102 bound with CDK2 and CDK4 by several hydrogen bonding, pi-interactions, hydrophobic contact, and several van der wall forces (Figure 5).
BC-N102 exhibited time course- and dose-dependent cell cycle arrest of the MCF-7 cell line. Flow cytometry showing the (A) time-dependent and (B) concentration-dependent effects of BC-N102 against different stages of MCF-7 cell cycle progression. (C) Western blot showing the time-dependent effect of BC-N102 treatment on the expression levels of the estrogen receptor (ER), phosphatidylinositol 3-linase (PI3K), phosphorylated (p)-extracellular signal-regulated kinase (ERK), p-Akt, p38 mitogen-activated protein kinase (MAPK), cyclin-dependent kinase 2 (CDK2), and CDK4 expressions in the MCF-7 cell line. (D). western blot showing the dose dependent effect of BC-N102 treatment on the expression levels of CDK2, CDK4, cyclin D1, phosphorylated CDC35C, p21, p27, and p53 in MCF-7 cell line. Numbers in red indicate relative expression ratios from the band quantification. (E) Chromatin Immunoprecipitation (ChIP) assay of the binding capacity of the ER for CDK2 and CDK4. The ChIP assay showed that the ER did not interact with the CDK2 or CDK4 promoter regions.
Molecular docking profiles of BC-N102 with cyclin-dependent kinase 2 (CDK2)/CDK4. (A) Two dimensional (2D) representations of ligand-receptor complexes, showing the interacting amino acid residues and types of interactions occurring between the ligand (BC-N102) and receptors (CDK2 and CDK4). The accompanying table shows the hydrophobic contacts between the ligand and receptors.
BC-N102 interfered with ligand-mediated and ligand-independent activation of hormonal receptors signaling in breast cancer
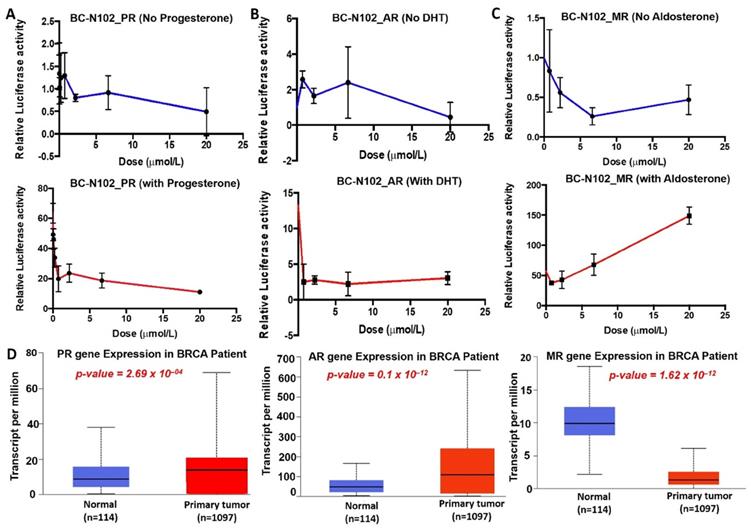
We investigated the dose dependence effects of BC-N102 on PR, AR, and MR signaling pathways using the luciferase reporter gene assay in T47D cells. These studies revealed that BC-N102 produced a dose-dependent inhibition of PR activity in the absence of progesterone. In addition, BC-N102 produced dose-dependent inhibition of the progesterone-stimulated PR activity in a dose-dependent manner (Figure 6A). Similarly, BC-N102 caused dose-dependent inhibition of AR activity in the presence or absence of DHT (Figure 6B). The maximum inhibitory activities of BC-N102 on AR and PR occurred at a concentration of 20 μM. Our results also demonstrated that in the absence of aldosterone, BC-N102 mediated increase inhibition of MR activity with increasing concentrations from 1.25, 2.5, and 7.5 μM, however, with further increase in BC-N102 concentrations, MR activity became activated. Similarly, in the presence of aldosterone, BC-N102 results in a dose-dependent induction of aldosterone-mediated MR activity in a dose-dependent manner (Figure 6C). We evaluated differential expression profiles of PR, AR, and MR genes between tumor samples from TCGA breast cancer patients and adjacent normal tissue and found that PR and AR were over-expressed while MR gene is under-expressed in breast cancer patients compared to normal tissue (Figure 6D). Altogether, our results indicate that BC-N102 exhibited its therapeutic effect via suppression of PR and AR signaling while mediating the activation of MR signaling irrespective of the presence or absence of the receptor's ligand in the breast cancer cell line.
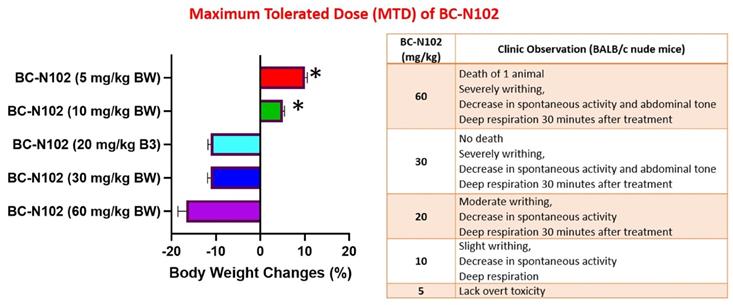
Clinical observations and body weight changes of mice treated with various concentrations of BC-N102 in the maximum tolerated dose (MTD) study.
In vivo maximum tolerated dose (MTD) study
We examined the MTD of BC-N1025, after a single i.v. injection into BALB/c nude mice. The MTD was estimated based on the threshold at which all animals survived with no more than 10% BW loss. We found that all mice treated with 5, 10, and 20, mg/kg BW tolerated the drug. Mice treated with 5 mg/kg BW of BC-N102 were completely devoid of toxicity and showed an increased percentage of BW gain after 2 weeks. During the observation period, none of the mice exhibited weight loss of >10%, and no deterioration in health was observed in mice treated with BC-N102 at up to 20 mg/kg BW. However, decreases in spontaneous activity, writhing, and deep respiration of the animals were observed at 30 min after administration. In contrast, high loss of BW (>10%) in mice treated with 30 and 60 mg/kg BW and death of one animal treated with 60 mg/kg BW were observed (Figure 7).
BC-N102 interfered with the ligand-mediated and ligand-independent activation of hormonal receptors signaling in breast cancer. Dose dependence plot of the effects of BC-N102 on (A) progesterone receptor (PR), (B) androgen receptor (AR), and (C) mineralocorticoid receptor (MR) signaling pathways in the absence (upper panel) and presence of progesterone, dihydrotestosterone, and aldosterone respectively (lower panel) respectively using the luciferase reporter gene assay in T47D cells. (D) Differential expression profile of PR, AR, and MR genes between tumor samples from TCGA breast cancer patients and adjacent normal tissue.
BC-N102 suppressed breast cancer tumorigenesis and prolonged survival of animals
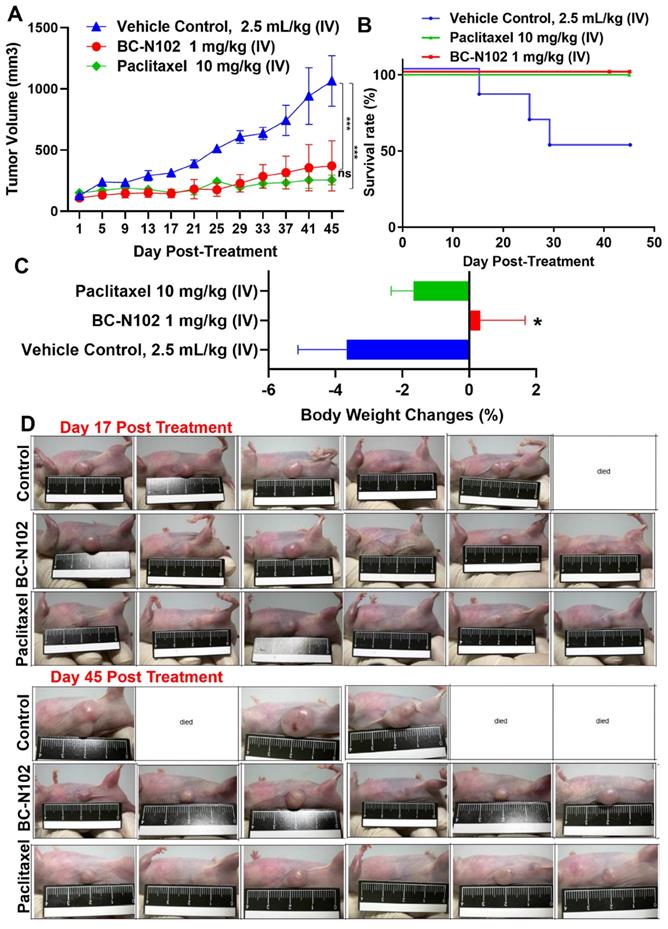
Following our in vitro studies, we evaluated the in vivo antitumor activities of BC-N102 against the MCF-7 cell line. Treatments with BC-N102 significantly (p<0.001) reduced MCF-7 tumor growth (Figure 8A) and prolonged the survival rate of animals (Figure 8B) compared to the control counterparts. In addition, BC-N102 enhanced the percentage BW gain of animals (Figure 8C). The in vivo therapeutic efficacy of BC-N102 was comparable to the observed therapeutic effect of paclitaxel (Figure 8A, D). However, treatment with paclitaxel could not prevent weight loss in the animals.
BC-N102 suppressed MCF-7 tumorigenesis in vivo and prolonged the survival of animals. (A) An average tumor volume versus time curve shows that treatments with BC-N102 significantly delayed MCF-7 tumor growth and the tumor burden. (B) The Kaplan-Meier survival curve shows a higher overall survival ratio of animals treated with BC-N102 compared to control counterparts. (C) Graph of percentage body weight changes of mice; animals treated with BC-N102 showed a significant increase in the percentage body weight gain compared to the control counterparts. * p<0.05, *** p<0.001.
Discussion
The development of targeted therapies for breast cancer is impeded by the heterogeneity of cancer cells which enhances their proliferation and invasive phenotypes and regulates their responsiveness to therapies [50, 51]. In addition, the molecular targets of current therapies, such as ER/PI3K/Akt, human epidermal growth factor receptor 2 (HER2), mammalian target of rapamycin (mTOR), and CDK4/CDK6, exhibit considerable intra- and inter-patient variations in their expression levels [28, 29]. In line with previous studies, our analysis of clinical data revealed that CDK2/CDK4 are overexpressed in breast cancer patients and contribute to tumor progression, metastasis, invasive phenotypes and non-responsiveness to chemotherapy. Moreover, we found that our novel tetracyclic derivative, BC-N102 suppressed tumorigenesis via inhibition of ER/Akt/PI3K/ERK/CDK2/4, and induced time-course G0/G1 cell cycle arrest of human breast cancer cells.
Our results demonstrated that BC-N102 exhibited dose-dependent suppression of breast cancer cell lines in vivo and in vitro. Our mechanistic study further revealed that BC-N102 treatment led to time-course and dose-dependent arrest of the cell cycle at G0/G1 phase with concomitant downregulation of ER, PI3K, p-ERK, p-AKT, cyclin D1, CDK2, and CDK4. Cyclin-dependent kinases play an important role in cell cycle progression and tumorigenesis in various cancers [28]. In line with our observation, a number of studies have associated the inhibition of CDKs to cell cycle arrest [52, 53]. Zhou et al. reported that treatment of human breast cancer cells with staurosporine causes a reduction in the expression level of CDK that accompanies a G1 cell cycle arrest [54], while Ishida et al. reported that treatment of breast cancer cell lines with herbimycin A caused cell cycle arrest-related reduction of the CDK4 and CDK6 expression levels [55]. In another study, it was reported that treatment of MCF-7 breast cancer cell line with 1,25(OH)2D3 induces cell cycle arrest at G0/G1phase via inhibition of CDK2 and CDK4 [56-60].
The cell cycle regulatory proteins; P53, P21, P27, respond to intrinsic or extracellular anti-mitogenic signals by binding to and inhibiting cyclin/Cdks and thereby blocking cell cycle progression [61, 62]. Interestingly, our results demonstrated that treatment of MCF-7 cells with BC-N102 significantly increases the activities of these cell cycle regulatory proteins and P38 MAPK. Altogether, these findings strongly suggest that BC-N102 suppresses breast cancer proliferation via cell cycle arrest by enhancing the activities of cell cycle regulatory protein which in turn inhibit the activities of cell cycle proteins including the CDK2, CDK4 and cyclin D1, and consequently led to cell cycle arrest.
ER, AR, PR, and MR are oncogenic steroid hormones that are activated by hormone binding [63]. Once activated they act as transcription factors by binding to the promoter's region of the target genes [6]. However, in the absence of the ligands (hormones), these hormone receptors are activated through phosphorylation by kinases such as ERK, MAPK, PI3K, and AKT in human malignancies [64]. In addition, activated receptors could also act as coactivators that mediate the activities of other transcription factors [65]. Therefore, inhibition of these steroid hormone signaling would be an effective strategy of cancer therapy. Interestingly, our results revealed that BC-N102 treatment-induced dose-dependent inhibition of AR and PR in the present as well as in the absence of hormone ligands. in addition, we found that the activities of AKT, PI3K, and ERK which are known alternative activator of these hormone receptors [64, 66] were significantly downregulated by TCN-02 treatment, suggesting inhibition of both canonical (classical; ligand-mediated) and non-canonical (alternative; ligand-independent) pathways of ER, AR, and PR activation (Figure 9).
Schematic representation of the therapeutic effect of BC-N102 in breast cancer. BC-N102 attenuated breast cancer growth via downregulation of the estrogen receptor (ER), progesterone receptor (PR), androgen receptor (AR), phosphatidylinositol 3-kinase (PI3K), phosphorylated (p)-extracellular signal-regulated kinase (ERK), p-Akt, CDK2, and CDK4 while increasing p38 mitogen-activated protein kinase (MAPK), and mineralocorticoid receptor (MR) signaling in breast cancer cell line.
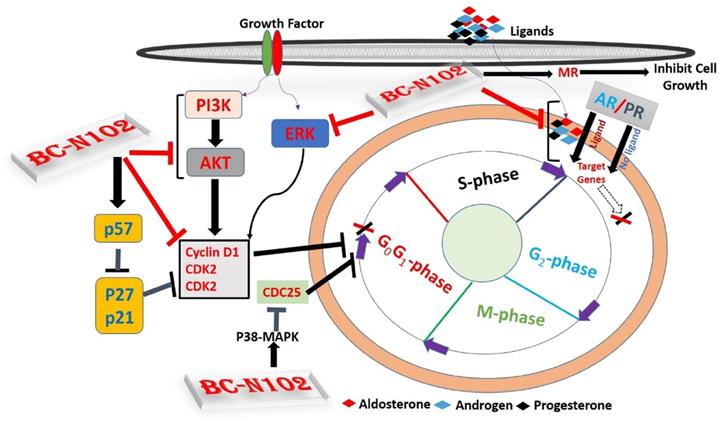
However, our observed effect of BC-N102 on mineralocorticoid receptor suggest a distinct role of mineralocorticoid in cancer development and response to therapeutic intervention. In order to understand the translational relevance of MR inhibition by BC-N102, we evaluated the differential expression profile of PR, AR, and MR genes and found that PR and AR were over-expressed while MR gene is under-expressed in breast cancer patients compared to normal tissue. Our findings are supported by a clinical study that reported that MR expression is lower in breast cancer compared to normal breast tissues and that MR is an independent predictor of metastasis-free survival in tamoxifen-treated breast cancer patients [67]. In addition, studies have reported that activation of mineralocorticoid receptor induces cell adhesion and growth inhibition in breast cancer [68, 69] and another cancer type [70]. This mineralocorticoid receptor-mediated tumor inhibition has been linked to inhibition of the Warburg effect [69, 70].
Previous studies revealed that ER downregulation attenuated the mitogenic effects of estrogen and induction of target genes in the MCF-7 cell line [71, 72]. To investigate whether the downregulation of CDK2 and CDK4 is a result of the effects of BC-N102, we evaluated whether the ER directly regulates these cell cycle-related genes by binding to their promoter regions. Disappointedly, our ChIP assay showed that the ER did not interact with the CDK2 or CDK4 promoter regions, suggesting that BC-N102 downregulates the expression of CDK2 and CDK4 independent of the ER in breast cancer cell lines.
Patient-derived xenograft (PDX) models have translational relevance in preclinical studies of anticancer drug discovery, due to their high stability and similarity to human tumors [73]. To evaluate the inhibitory effects of BC-N102 against breast cancer, a PDX model of MCF-7 was established for our in vivo study. Results showed that BC-N102 significantly suppressed tumor cell proliferation and prolonged the survival of animals in the PDX mice model. In contrast to the non-treated control, mice treated with BC-N102 exhibited increase body weight gain, suggesting that in addition to the therapeutic effect on tumor growth, BC-N102 ameliorated the tumor induce loss of body weight and does not mediate obvious adverse effect in the animal.
Conclusions
Our results demonstrated that BC-N102 suppresses human breast cancer tumorigenesis via time-course and dose-dependent cell cycle arrest at G0/G1 phase, and regulation of cell cycle proteins. In addition, BC-N102 suppresses both ligands mediated (classical) and non-ligand mediated (alternative) pathways of ER, AR, and PR activation while mediating the activation of MR signaling in breast cancer. Our results suggest that the proper application of BC-N102 may be a beneficial chemo-preventive strategy for breast cancer patient.
Abbreviations
ER: estrogen receptor; PR: progesterone receptor; AR: androgen receptor; PI3K: phosphatidylinositol 3-kinase; p-ERK: phosphorylated extracellular signal-regulated kinase; MAPK: mitogen-activated protein kinase; CDK2: cyclin dependent kinase 2; CDK4: cyclin dependent kinase 4; HER2: human epidermal growth factor receptor 2; mTOR: mammalian target of rapamycin; AKT: protein kinase B; TCGA: The Cancer Genome Atlas; SRB: sulforhodamine B; NSCLC: non-small-cell lung cancer; CNS: central nervous system; TCA: trichloroacetic acid; ChIP: Chromatin immunoprecipitation; MTD: Maximum tolerated dose; PDB: protein data bank.
Acknowledgements
We acknowledged the NCI Developmental Therapeutics Program (DTP) of the National Cancer Institute, National Institutes of Health (NIH‐NCI) for the 60‐cancer‐cell line screening of BC-N102.
Author Contributions
B.L. wrote the manuscript; Y-C.K. helped in data collection A.T.H.W. and H.-S.H. designed and oversaw the study. All authors have read and agreed to the final version of the manuscript.
Funding
Hsu-Shan Huang was funded by the Ministry of Science and Technology (MOST 109-2113-M-038-003 and MOST 110-2314-B-038-120).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Clèries R, Rooney R, Vilardell M, Espinàs JA, Dyba T, Borras J. Assessing predicted age-specific breast cancer mortality rates in 27 European countries by 2020. Clinical and Translational Oncology. 2018;20:313-21
2. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A. et al. Cancer statistics for the year 2020: An overview. International Journal of Cancer. 2021
3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. n/a.
4. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians. 2021;71:7-33
5. Arduino PG, Conrotto D, Broccoletti R. The outbreak of Novel Coronavirus disease (COVID-19) caused a worrying delay in the diagnosis of oral cancer in north-west Italy: The Turin Metropolitan Area experience. Oral Diseases. 2021;27:742-3
6. Piasecka D, Braun M, Kitowska K, Mieczkowski K, Kordek R, Sadej R. et al. FGFs/FGFRs-dependent signalling in regulation of steroid hormone receptors - implications for therapy of luminal breast cancer. Journal of Experimental & Clinical Cancer Research. 2019;38:230
7. Lin Z, Reierstad S, Huang C-C, Bulun SE. Novel Estrogen Receptor-α Binding Sites and Estradiol Target Genes Identified by Chromatin Immunoprecipitation Cloning in Breast Cancer. Cancer Research. 2007;67:5017-24
8. Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annual review of physiology. 2001;63:165-92
9. Kun Y, How LC, Hoon TP, Bajic VB, Lam TS, Aggarwal A. et al. Classifying the estrogen receptor status of breast cancers by expression profiles reveals a poor prognosis subpopulation exhibiting high expression of the ERBB2 receptor. Human molecular genetics. 2003;12:3245-58
10. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K. et al. Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492-502
11. Santos-Martínez N, Díaz L, Ordaz-Rosado D, García-Quiroz J, Barrera D, Avila E. et al. Calcitriol restores antiestrogen responsiveness in estrogen receptor negative breast cancer cells: a potential new therapeutic approach. BMC Cancer. 2014;14:230 -
12. Osborne CK, Yochmowitz MG, Knight III WA, McGuire WL. The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer. 1980;46:2884-8
13. Gao S, Ge A, Xu S, You Z, Ning S, Zhao Y. et al. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3β/β-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J Exp Clin Cancer Res. 2017;36:179 -
14. Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Research and Treatment. 1998;51:227-38
15. Osborne CK, Elledge RM, Fuqua SA. Estrogen receptors in breast cancer therapy. Sci Med. 1996;3:32-41
16. O'Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Molecular endocrinology (Baltimore, Md). 2004;18:1859-75
17. Göttlicher M, Heck S, Herrlich P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. Journal of molecular medicine (Berlin, Germany). 1998;76:480-9
18. Sánchez-Beato M, Sáez AI, Martínez-Montero JC, Sol Mateo M, Sánchez-Verde L, Villuendas R. et al. Cyclin-dependent kinase inhibitor p27KIP1 in lymphoid tissue: p27KIP1 expression is inversely proportional to the proliferative index. Am J Pathol. 1997;151:151-60
19. Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC. et al. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154:313-23
20. Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer research. 2000;60:3689-95
21. Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K. et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes & development. 1995;9:935-44
22. Polyak K. Heterogeneity in breast cancer. The Journal of clinical investigation. 2011;121:3786-8
23. Turashvili G, Brogi E. Tumor heterogeneity in breast cancer. Frontiers in medicine. 2017;4:227
24. Yan X, Xie Y, Yang F, Hua Y, Zeng T, Sun C. et al. Comprehensive description of the current breast cancer microenvironment advancements via single-cell analysis. Journal of Experimental & Clinical Cancer Research. 2021;40:142
25. Videira M, Reis RL, Brito MA. Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2014;1846:312-25
26. Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J. et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Research. 2008;10:R33
27. Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Annals of the New York Academy of Sciences. 2007;1113:202-16
28. Lawal B, Lin L-C, Lee J-C, Chen J-H, Bekaii-Saab TS, Wu ATH. et al. Multi-Omics Data Analysis of Gene Expressions and Alterations, Cancer-Associated Fibroblast and Immune Infiltrations, Reveals the Onco-Immune Prognostic Relevance of STAT3/CDK2/4/6 in Human Malignancies. Cancers (Basel). 2021;13:954
29. Lawal B, Liu Y-L, Mokgautsi N, Khedkar H, Sumitra MR, Wu ATH. et al. Pharmacoinformatics and Preclinical Studies of NSC765690 and NSC765599, Potential STAT3/CDK2/4/6 Inhibitors with Antitumor Activities against NCI60 Human Tumor Cell Lines. Biomedicines. 2021;9:92
30. Bartha Á, Győrffy B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. International Journal of Molecular Sciences. 2021;22:2622
31. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer discovery. 2012;2:401-4
32. Liu C-J, Hu F-F, Xia M-X, Han L, Zhang Q, Guo A-Y. GSCALite: a web server for gene set cancer analysis. Bioinformatics (Oxford, England). 2018;34:3771-2
33. Modhukur V, Iljasenko T, Metsalu T, Lokk K, Laisk-Podar T, Vilo J. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10:277-88
34. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509-w14
35. Chen T-C, Wu C-L, Lee C-C, Chen C-L, Yu D-S, Huang H-S. Structure-based hybridization, synthesis and biological evaluation of novel tetracyclic heterocyclic azathioxanthone analogues as potential antitumor agents. European journal of medicinal chemistry. 2015;103:615-27
36. Huang H-s, Yu D-s, Chen T-C. Thiochromeno [2, 3-c] quinolin-12-one derivatives, preparation method and application thereof. Google Patents. 2015
37. Orellana EA, Kasinski AL. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016;6:e1984
38. Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Development Research. 1995;34:91-109
39. Lawal B, Lee C-Y, Mokgautsi N, Sumitra MR, Khedkar H, Wu ATH. et al. mTOR/EGFR/iNOS/MAP2K1/FGFR/TGFB1 Are Druggable Candidates for N-(2,4-Difluorophenyl)-2′,4′-Difluoro-4-Hydroxybiphenyl-3-Carboxamide (NSC765598), With Consequent Anticancer Implications. Frontiers in Oncology. 2021 11
40. Scheidereit C, von der Ahe D, Cato AC, Wenz M, Suske G, Carlson C. et al. Protein-DNA interactions at steroid hormone regulated genes. Endocrine research. 1989;15:417-40
41. Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA. et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Molecular endocrinology (Baltimore, Md). 2006;20:14-34
42. Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proceedings of the National Academy of Sciences. 2011;108:1850-5
43. Rao CV, Reddy BS, Steele VE, Wang C-X, Liu X, Ouyang N. et al. Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Molecular cancer therapeutics. 2006;5:1530-8
44. Kang JS, Kang MR, Han SB, Yoon WK, Kim JH, Lee TC. et al. Low dose estrogen supplementation reduces mortality of mice in estrogen-dependent human tumor xenograft model. Biological & pharmaceutical bulletin. 2009;32:150-2
45. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-61
46. Lee J-C, Wu ATH, Chen J-H, Huang W-Y, Lawal B, Mokgautsi N. et al. HNC0014, a Multi-Targeted Small-Molecule, Inhibits Head and Neck Squamous Cell Carcinoma by Suppressing c-Met/STAT3/CD44/PD-L1 Oncoimmune Signature and Eliciting Antitumor Immune Responses. Cancers (Basel). 2020;12:3759
47. Visualizer DS. BIOVIA, Dassault Systèmes, BIOVIA Workbook, Release 2020; BIOVIA Pipeline Pilot, Release 2020, San Diego: Dassault Systèmes. 2020.
48. Weisz A, Rosales R. Identification of an estrogen response element upstream of the human c-fos gene that binds the estrogen receptor and the AP-1 transcription factor. Nucleic acids research. 1990;18:5097-106
49. Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends in Endocrinology & Metabolism. 2005;16:46-52
50. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clinical therapeutics. 2016;38:1551-66
51. Sledge GW, Mamounas EP, Hortobagyi GN, Burstein HJ, Goodwin PJ, Wolff AC. Past, present, and future challenges in breast cancer treatment. Journal of Clinical Oncology. 2014;32:1979
52. Morgan DO. Principles of CDK regulation. Nature. 1995;374:131-4
53. Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27 Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325-31
54. Zhou W, Lin Y, Wersto R, Chrest J, Gabrielson E. Staurosporine-induced G1 arrest in cancer cells depends on an intact pRB but is independent of p16 status. Cancer Letters. 2002;183:103-7
55. Ishida T, Kobayashi N, Tojo T, Ishida S, Yamamoto T, Inoue J-i. CD40 signaling-mediated induction of Bcl-XL, Cdk4, and Cdk6. Implication of their cooperation in selective B cell growth. The Journal of Immunology. 1995;155:5527-35
56. James SY, Mackay AG, Colston KW. Effects of 1, 25 dihydroxyvitamin D3 and its analogues on induction of apoptosis in breast cancer cells. The Journal of steroid biochemistry and molecular biology. 1996;58:395-401
57. Hansen CM, Rohde L, Madsen MW, Hansen D, Colston KW, Pirianov G. et al. MCF-7/VDR: A new vitamin D resistant cell line. Journal of cellular biochemistry. 2001;82:422-36
58. Flanagan L, Packman K, Juba B, O'Neill S, Tenniswood M, Welsh J. Efficacy of Vitamin D compounds to modulate estrogen receptor negative breast cancer growth and invasion. The Journal of steroid biochemistry and molecular biology. 2003;84:181-92
59. Wu G, Fan RS, Li W, Ko TC, Brattain MG. Modulation of cell cycle control by vitamin D 3 and its analogue, EB1089, in human breast cancer cells. Oncogene. 1997;15:1555-63
60. Verlinden L, Verstuyf A, Convents R, Marcelis S, Van Camp M, Bouillon R. Action of 1, 25 (OH) 2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7 cells. Molecular and cellular endocrinology. 1998;142:57-65
61. Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH. et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570-3
62. Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P. et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59-66
63. Rosen EM, Fan S. Inhibition of estrogen receptor signaling. Breast Cancer Online. 2005;8:e14
64. Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. The EMBO journal. 1996;15:2174-83
65. Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Seminars in cancer biology: Elsevier. 2001 p. 339-52
66. Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M. et al. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocrine-related cancer. 2005;12(Suppl 1):S99-s111
67. Muscat GE, Eriksson NA, Byth K, Loi S, Graham D, Jindal S. et al. Research resource: nuclear receptors as transcriptome: discriminant and prognostic value in breast cancer. Molecular endocrinology (Baltimore, Md). 2013;27:350-65
68. Leo JC, Guo C, Woon CT, Aw SE, Lin VC. Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology. 2004;145:1314-21
69. Doan TB, Cheung V, Clyne CD, Hilton HN, Eriksson N, Young MJ. et al. A tumour suppressive relationship between mineralocorticoid and retinoic acid receptors activates a transcriptional program consistent with a reverse Warburg effect in breast cancer. Breast Cancer Research. 2020;22:122
70. Nie H, Li J, Yang XM, Cao QZ, Feng MX, Xue F. et al. Mineralocorticoid receptor suppresses cancer progression and the Warburg effect by modulating the miR-338-3p-PKLR axis in hepatocellular carcinoma. Hepatology. 2015;62:1145-59
71. James S, Mackay A, Binderup L, Colston K. Effects of a new synthetic vitamin D analogue, EB1089, on the oestrogen-responsive growth of human breast cancer cells. Journal of Endocrinology. 1994;141:555-63
72. Colston K. Mechanisms implicated in the growth regulatory effects of vitamin D in breast cancer. Endocrine-related cancer. 2002;9:45-59
73. Sia D, Moeini A, Labgaa I, Villanueva A. The future of patient-derived tumor xenografts in cancer treatment. Pharmacogenomics. 2015;16:1671-83
Author contact
![]() Corresponding authors: A.T.H. and H-S.H.
Corresponding authors: A.T.H. and H-S.H.