Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Classes of Histone deacetylases...
Preclinical studies of HDACs and...
Conclusion and Perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(13):3381-3400. doi:10.7150/ijbs.62001 This issue Cite
Review
Preclinical and clinical progress for HDAC as a putative target for epigenetic remodeling and functionality of immune cells
Sijia Zhang1#, Lingjun Zhan4#, Xue Li1, Zhenhong Yang1, Yumin Luo1,2,3 ![]() , Haiping Zhao1,2
, Haiping Zhao1,2 ![]()
1. Institute of Cerebrovascular Disease Research, Xuanwu Hospital of Capital Medical University, Beijing, China.
2. Beijing Geriatric Medical Research Center and National Clinical Research Center for Geriatric Disorders, Beijing, China.
3. Beijing Institute for Brain Disorders, Capital Medical University, Beijing, China.
4. Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences, Beijing, China.
#Co-first authors.
Received 2021-4-24; Accepted 2021-7-26; Published 2021-8-3
Abstract

Genetic changes are difficult to reverse; thus, epigenetic aberrations, including changes in DNA methylation, histone modifications, and noncoding RNAs, with potential reversibility, have attracted attention as pharmaceutical targets. The current paradigm is that histone deacetylases (HDACs) regulate gene expression via deacetylation of histone and nonhistone proteins or by forming corepressor complexes with transcription factors. The emergence of epigenetic tools related to HDACs can be used as diagnostic and therapeutic markers. HDAC inhibitors that block specific or a series of HDACs have proven to be a powerful therapeutic treatment for immune-related diseases. Here, we summarize the various roles of HDACs and HDAC inhibitors in the development and function of innate and adaptive immune cells and their implications for various diseases and therapies.
Keywords: Epigenetics, HDAC, neutrophils, mast cells, macrophage, dendritic cells, lymphocyte, microglia, astrocyte
Introduction
Although the immune system is generally defined as a defense system against pathogens, it is increasingly recognized as playing a crucial role in tissue repair after injury. The plasticity of immune cells has extensive implications in the pathogenesis and resolution of cancers, autoimmune diseases, chronic inflammatory disorders, and metabolic disorders. With highly coordinated and dynamic alterations in chromatin structure and gene expression programs, immune cells can exquisitely identify, respond, and produce a rapid and augmented response to pathogens or tissue injury in immune-related diseases. Increasing evidence has shown that immune cell plasticity can be altered by epigenetic modulation.
Epigenetics is typically defined as heritable changes in genome function due to DNA and histone modifications, nucleosome remodeling, and expression of noncoding RNAs, as opposed to the genetic code, which distinctly influence several aspects of cell physiology and pathology [1, 2]. The high plasticity of epigenetic modifications due to environmental variability provides a novel area for therapeutic intervention. The epigenetic regulation of gene expression requires a balance in the acetylation/deacetylation of histone and nonhistone proteins controlled by histone deacetylases (HDACs) and histone acetyltransferases (HATs) [3, 4]. Recently, several HDAC-based diagnostic biomarkers and drugs have received regulatory approval or entered clinical practice or both. For example, HDAC inhibitors (HDACi) are being evaluated for the treatment of hematological malignancies and solid tumors. Here, we review recent advances in our understanding of classical HDACs as potential targets for epigenetic regulation of distinct immune cell types in both preclinical studies and clinical drug trials in inflammatory and autoimmune diseases and discuss the potential translational implications and challenges.
Classes of Histone deacetylases (HDACs) and HDAC inhibitors (HDACi)
Classes of Histone deacetylases (HDACs)
To date, 18 types of mammalian HDAC isoforms have been identified. Based on their structure, function, and homology of accessory domains to histone deacetylases, HDACs are categorized into four classes: Class I (HDAC 1, 2, 3, and 8); Class II (HDAC 4, 5, 6, 7, 9, and 10); Class III (SIRT 1, 2, 3, 4, 5, 6, and 7), and Class IV (HDAC11). Classes I, II, and IV HDACs are zinc-dependent and also termed classical HDACs, while class III HDACs, termed non-classical HDACs, are NAD(+)-dependent [5]. Whether these HDAC isoforms are redundant or specific for controlling gene expression programs in eukaryotic cells remains unclear. Pharmacological studies using selective HDAC inhibitors (HDACIs) and HDAC-knockout mice have revealed the specificity of individual HDACs in the development of various diseases [6].
Regulation of deacetylation by HDACs
Acetylation is a major post-translational protein modification that introduces an acetyl functional group into amino acids, such as lysine (the residue), to yield an acetate ester bond, neutralizing its positive charge. Presently, the acetylome encompasses nearly 40,000 unique protein acetylation sites, as classified in the PhosphoSitePlus database [7]. Regulation of protein functions by acetylation occurs in multiple ways: by affecting protein stability, activity, localization, and interaction with other proteins or DNA [8]. HATs and HDACs are the enzymes involved in this modification and are thought to act as critical gene silencers or activators. HATs increase the acetylation of histones, presenting a less condensed DNA segment, which is prone to transcription. HDACs are evolutionarily conserved enzymes that remove acetyl groups from histones and nonhistone proteins such as hormone receptors, signaling proteins, chaperones, transcription factors, and DNA damage response proteins, with functional consequences on chromatin remodeling and gene expression profiles [9]. HDACs regulate target genes via ε-amino group deacetylation of histone lysine residues, thus promoting DNA condensation, and therefore, transcriptional silencing. Moreover, certain HDACs are not lysine deacetylases, but rather lysine fatty-acid deacylases [10]; others process non-protein substrates by acting as polyamine deacetylases [11].
Classes of HDAC inhibitors (HDACi)
HDACi modulate gene expression by interacting with HDACs. These processes are target-specific reactions among various types of HDACis and HDACs [12]. HDACis are classified according to their chemical structures as follows: short-chain fatty acids (sodium butyrate [NaBu] and valproic acid [VPA]); benzamides (entinostat [MS-275] and mocetinostat [MGCD0103]); hydroxamic acids (panobinostat [LBH-589], belinostat (PXD101), vorinostat (SAHA), scriptaid (ST), and trichostatin-A [TSA]); cyclic peptides (romidepsin [FK228]), and miscellaneous compounds (depudecin) [13] (Table 1).
The chemical structures and targets of HDAC inhibitors
| Structure | Chemical Name (Abbreviation) | Targets |
|---|---|---|
| Short-chain fatty acids | Valproic acid [VPA] | pan-HDACi |
| Sodium butyrate [NaBu] | class I and II HDACi | |
| Benzamides | DMA-PB | pan-HDACi |
| Entinostat [MS-275] | class I HDACi | |
| Mocetinostat [MGCD0103] | class I HDACi | |
| MI192 | selective HDAC2/3 inhibitor | |
| M344 | selective HDAC6-inhibitor | |
| Hydroxamic acids | Panobinostat [LBH-589] | pan-HDACi |
| Belinostat [PXD101] | pan-HDACi | |
| Vorinostat [SAHA] | pan-HDACi | |
| Trichostatin-A [TSA] | pan-HDACi | |
| Dacinostat [LAQ824] | pan-HDACi | |
| AR-42 | pan-HDACi | |
| Panobinostat [LBH-589] | pan-HDACi | |
| Ky-2 | pan-HDACi | |
| Scriptaid [ST] | class I HDACi | |
| Givinostat [ITF2357] | class I and II HDACi | |
| MC-1568 | class IIa HDACi | |
| 17a | selective HDAC6-inhibitor | |
| Santacruzamate A [CAY10683] | selective HDAC2-inhibitor | |
| Tubacin | selective HDAC6-inhibitor | |
| PTG-0861 | selective HDAC6-inhibitor | |
| KA2507 | selective HDAC6-inhibitor | |
| Citarinostat [ACY-241] | selective HDAC6-inhibitor | |
| ACY-738 | selective HDAC6-inhibitor | |
| ACY-1215 (ricolinostat) | selective HDAC6-inhibitor | |
| ACY-241 (citarinostat) | selective HDAC6-inhibitor | |
| Ricolinostat [ACY-1215] | selective HDAC6-inhibitor | |
| CKD-L | selective HDAC6-inhibitor | |
| Cyclic peptides | Apicidin | pan-HDACi |
| KBH-A42 | pan-HDACi | |
| Romidepsin [FK228] | selective HDAC1/2-inhibitor | |
| Miscellaneous compounds | Depudecin | pan-HDACi |
| RGFP966 | selective HDAC3-inhibitor |
Pharmacokinetic and pharmacodynamic evaluation of HDACi treatment
To improve the clinical potential of HDACIs, a prodrug strategy has been utilized to improve the in vivo pharmacokinetic and pharmacodynamic performances of HDACIs, such as hydroxamic acid, carboxylic acid, thiol, benzamide, anticancer drug, and carrier-conjugated prodrugs [14]. Early research focused on developing the pharmacokinetic/pharmacodynamic tools necessary for novel HDACi studies in canine cancer clinical setting [15]. A positive correlation was found between VPA dose and histone hyperacetylation in peripheral blood mononuclear cells [15]. In examining compound stability and cellular permeability, PTG-0861, a novel HDAC6-selective inhibitor, revealed a promising in vitro pharmacokinetic profile [16]. Besides, structure-activity relationship studies have been performed to verify the influence of the linker on the biological profile of the compounds [17]. Recently, a novel photo-activated HDACi was identified to cage metacept-3 15, with pharmacokinetic activation characteristics and biological properties that may make it suitable for evaluation as a novel coating for targeted drug eluting balloon catheter angioplasty interventions [18]. These studies could help enhance the stability, water solubility, lipophilicity, oral bioavailability, tumor cell selectivity, and further structural modification of HDACIs with high potential as drug candidates.
Clinical investigation of HDACi
HDACs are considered potential next-generation therapeutics, given the promising potential of their broad HDAC inhibition or HDAC isoform-selective inhibition for multiple diseases. An emblematic example is the prospect of HDACi in treating cancer, which is attracting extensive attention. HDACIs have exhibited potency against various cancer types, and four of them: SAHA, FK228, PXD101, and LBH-589, have been approved by the US FDA (Foods and Drugs Authority) for cancer treatment [14]. SAHA and FK228 have proven to be effective in treating cutaneous T-cell lymphoma (CTCL) and have already been applied clinically, while chidamide has been approved as the first oral subtype-selective HDACi in China. SAHA and FK228 were both demonstrated to have significant and sustainable single-agent activity with acceptable safety profiles in patients with CTCL in phase IIb multicenter trials [19-21]. PXD101, a non-specific HDAC inhibitor (pan-HDACi), was proven to be effective and well-tolerated in patients with peripheral T-cell lymphoma (PTCL) or cutaneous T-cell lymphoma (CTCL) in a multicenter study [22]. The safety and activity of LBH-589, a nonselective HDACi, in relapsed/refractory Hodgkin's lymphoma were also proven in a multicenter, phase II trial, and showed a significant reduction in tumor size [23]. Some specific HDACi are currently undergoing clinical trials. Chidamide is the first oral subtype-selective HDACI (HDAC1,2,3,10) approved in China and the first HDACI of the benzamide class approved for treating relapsed and refractory PTCL [24]. In a phase I study, KA2507, a potent and selective inhibitor of HDAC6, showed selective target engagement, no significant toxicities, and prolonged disease stabilization in a subset of patients with refractory solid tumors [25].
HDACis have also been trialed in other diseases. VPA is a widely prescribed antiepileptic drug, and a longitudinal observational study showed that VPA efficacy for focal or generalized seizures is superior to that of mixed-type seizures [26]. VPA has been used in treating bipolar disorder since the 1990s [27], and has shown positive results in spinal muscular atrophy in cohort studies [28]. The successful trial of HDACis, which are currently available for clinical use in the treatment of cancers, hints at the existence of the cures for various human diseases such as epilepsy through epigenetic regulation.
Preclinical studies of HDACs and HDACi in different types of immune cells
Immune cells in the circulatory system can be divided into innate immune cells (neutrophils, eosinophils, basophils, mast cells, macrophages, dendritic cells, and natural killer cells) and adaptive immune cells (T and B lymphocytes). The immune system of the central nervous system, which is relatively independent, primarily consists of two types of glial cells, microglia and astrocytes. These glial cells intimately participate in immune responses in multiple CNS diseases.
HDAC and neutrophils
Neutrophils, eosinophils, basophils, and mast cells are crucial in host defense against pathogens. Neutrophils, which are key in the first line of defense, extensively exist in the circulatory system and are deemed indispensable for HDACs. Neutrophils act as effector and regulatory cells to defend against pathogens, and perform functions such as phagocytosis, generating antimicrobial enzymes and toxic factors, releasing inflammatory cytokines, and discharging nuclear DNA and chromatin proteins which form neutrophil extracellular traps (NETs) [29]. The mechanism of NET generation (NETosis) is based on the special cell death process of neutrophils. After liberation, NETs may block invaders and facilitate the restoration of homeostasis. Neutrophils are classically defined as terminally differentiated, short-lived cells; however, they can be long-lived with phenotypic plasticity. As regards their high plasticity, neutrophils can acquire an anti-inflammatory N1 or proinflammatory N2 phenotype. Besides, a subset of neutrophils transdifferentiate into neutrophil-dendritic cell hybrids (PMN-DCs), a group of cells with both neutrophil and dendritic cell properties during inflammation.
Emerging evidence has supported the essential role of HDACs in regulating the proliferation, differentiation, and apoptosis of neutrophils. Knockdown of HDAC1 or HDACi (SAHA and MS-275) reduced cell expansion, increased apoptosis of neutrophil progenitor cells, and decreased the percentage of mature neutrophils [30]. HDAC11 is a recently discovered member of the HDAC family. Among myeloid cells, neutrophils are considered the largest producers of HDAC11. The HDAC11 yield increases throughout the differentiation and maturation of neutrophils. However, the migratory and phagocytic capacities of inflammatory cytokines were elevated in neutrophils from mice lacking HDAC11, which were found to be more vulnerable to lipopolysaccharide (LPS)-induced sepsis, granulocytic expansion, and splenomegaly due to increased extramedullary hematopoiesis [31]. This implies that HDAC11 is a vital factor that influences neutrophil function. However, the links between HDAC11 and some noteworthy matters, such as neutrophil NETosis, neutrophil activation, and antagonistic neutrophil populations, remain largely unknown. Recently, researchers found that histone acetylation has prompted NETosis in human neutrophils via induction by NOX-dependent or NOX-independent pathway agonists. HDACi belinostat and panobinostat, which cause neutropenia in clinical practice, could shift neutrophil NETosis to apoptosis [32, 33]. Another study showed that NETs may influence the autoimmune response in systemic lupus erythematosus (SLE) by providing extra autoantigens [34]. The paper argued that histones in NETs produced by SLE-derived neutrophils showed higher levels of acetylation and methylation. Furthermore, pan-HDACi TSA treatment performed before NETosis induction was found to stimulate hyperacetylation of histones in NETs, thus enhancing their ability to activate macrophages, eventually prompting a shorter exposure period to NET antigens [35].
In contrast, HDACi can inhibit disease progression by depressing neutrophil activity and migration. SAHA, HDAC6-shRNAs treatment, or Tubacin (selective HDAC6-inhibitor) could reduce neutrophil activities, thus regressing cystic fibrosis-lung disease [36]. HDACi NaBu was demonstrated to mediate the therapeutic effect on the LPS-induced endometritis mouse model by reducing neutrophil granulocyte concentration and TNF-α and IL-1β levels [37]. The HDAC1-3 inhibitor MS-275 attenuated neutrophil infiltration into the lungs in a model of cigarette smoke-exposed mice [38]. A class I HDACi, CI-994, suppressed the accumulation of neutrophils and expression of inflammatory cytokines in a mouse model of spinal cord injury (SCI), thus contributing to promoting functional recovery, especially during the early stages of SCI [39]. Our group demonstrated HDAC2-mediated neutrophil infiltration and N1/N2 phenotypic shift following ischemic stroke [40]. In summary, HDAC and HDACi play necessary roles in the differentiation, NETosis, immune responses, and migration of neutrophils under normal and pathological conditions. However, the effect of HDAC on different diseases is inconsistent, which is related to the pathological mechanism by which neutrophils influence the disease pathogenesis.
HDAC and eosinophils
Eosinophils exhibit intense proinflammatory effects in some pathological situations. Besides, eosinophils can contribute to immunoregulation and antiviral activity. Eosinophils lineage-restricted progenitors (EoP), the progenitors of eosinophils, are reasoned to be downstream of the granulocyte/macrophage progenitors (GMP) in mice and the common myeloid progenitor (CMP) or the upstream pre-CMP progenitor in humans [41, 42]. Various environmentally adaptive phenotypes of mature eosinophils are located in the mucosae of multiple organs, such as the gastrointestinal tract and lungs. Functional heterogeneity is discovered not only among eosinophils that reside in different tissues, but also between residential eosinophils under homeostasis and eosinophils recruited after immune responses [43, 44].
The epigenetic regulation by HDAC and HDACi has been shown to be involved in the control of eosinophil function under normal homeostatic and pathogenic conditions in various tissues. Lung eosinophilia is a hallmark of asthma, and eosinophils are reasoned to affect the pathogenesis of allergic inflammatory diseases significantly. RGFP966 was recently shown to reduce HDAC3 expression and HDAC3 activities; eosinophil and mast cell recruitment, goblet cell proliferation, and inflammatory cytokine levels were decreased, triggering the alleviation of allergic and inflammatory responses in an OVA-induced allergic rhinitis mouse model [45]. A previous study showed that short chain fatty acids (SCFAs) attenuated human eosinophils at several functional levels, including (1) adhesion to the endothelium, (2) migration, and (3) survival [46]. In vivo, butyrate ameliorated allergen-induced airway and lung eosinophilia, reduced type 2 cytokine levels in bronchial fluid, and improved airway hyperresponsiveness in mice. In vitro and in vivo findings highlight the importance of SCFAs, especially butyrate, as a promising therapeutic agent for allergic inflammatory diseases [46]. In a mouse asthma model, TSA reduced allergen-induced airway inflammation and airway constriction partly by reducing the eosinophils numbers in bronchoalveolar lavage fluid [47-49]. Moreover, TSA promoted the apoptosis of human eosinophils, indicating the potential therapeutic effect of HDACi on eosinophil-related diseases such as chronic obstructive pulmonary disease (COPD) and asthma (Figure 1) [50]. HDAC1 could be a molecularly targeted treatment for specific granule deficiency with homozygous mutation for two amino acids, arginine (R247) and serine (S248), which were deleted in the basic leucine zipper domain of C/EBPε (ΔRS) [51]. In summary, the present research mainly demonstrated the abrogation of HDACi on eosinophil function, suggesting a promising therapeutic effect of HDACi on eosinophil-related inflammatory disorders.
Schematic diagram of effect of HDAC inhibitors on immune cells during type 1 allergy. The upward arrows represent the promotion effect by HDAC inhibitors and the downward arrows represent the suppression effect by HDAC inhibitors.
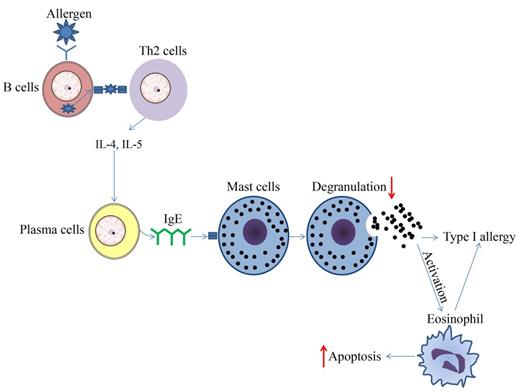
HDAC and mast cells
Mast cells are multifunctional. They perform different tasks when encountering different immune triggers, circumstances, and cytokine stimulation. Mast cells (MCs) are enduring pluripotent leukocytes that mainly reside in the mucosa, airways, and skin, while a small number are found in the perivascular tissue of large blood vessels and between cardiomyocytes [52]. They are components of the innate immune system, characterized by their granules that contain inflammatory mediators such as histamine and tryptase, and can produce many more inflammatory cytokines, such as leukotrienes and prostaglandins [53]. Until now, two subtypes of MCs have been recognized: MCTCs, which are typically found in the mucosa and can produce tryptase and chymase, and MCTs, which contain tryptase but not chymase. MCTs are mainly present in connective tissue [53].
The role of HDACi in the inhibition of MC-mediated inflammatory responses has been demonstrated in previous studies; for example, SAHA and MGCD0103 were able to inhibit antigen-induced histamine release in rat peritoneal mast cells (Figure 1) [54]. When binding to IgE, bone marrow-derived mast cells (BMMCs) activate and produce higher levels of IL-4, IL-6, TNF-α, and IL-13. This process could be reversed to some extent by TSA pretreatment. The results showed that with TSA pretreatment, cytokines secreted by mast cells were altered, while both FcεRI amount and mast cell degranulation declined (Figure 1). After TSA treatment, I-κBα transcription increased while phospho-RelA levels and NF-κB activation were decreased. TSA treatment may play far more roles than previously thought. It could also impact MC experienced non-IgE stimuli, and continuous TSA exposure could accelerate mast cell apoptosis (Figure 1). Finally, an ovalbumin-induced food allergy mice model induced showed that TSA treatment remarkably relieved allergic diarrhea and decreased mast cell activation [55]. In summary, these findings demonstrate that histone acetylation may be an important factor affecting epigenetic modification during mast cell activation, and exposure to dietary substances may affect the epigenetic regulation of mast cells. Current studies have demonstrated the anti-allergy effect on mast cells, indicating the potential future application of HDACi in related fields.
HDAC and macrophages
Macrophages and neutrophils mount early immune protection against intracellular pathogens; however, neutrophils are short-lived cells, and removal of apoptotic cells by resident macrophages is a key event in resolving inflammation and tissue repair. Monocytes and macrophages, the most plastic cells of the hematopoietic system, are derived from bone marrow progenitors that continually proliferate and shed their progeny in the bloodstream as promonocytes. Circulating monocytes then migrate into almost all tissues, where they differentiate into macrophages under local growth factors, proinflammatory cytokines, and microbial products, and display great functional diversity and affect development, homeostasis, tissue repair, and immunity [56]. Although macrophages are activated in various microenvironments, they are recognized to exist in two distinct phenotypes: 1) the proinflammatory M1 macrophages, which are polarized by microbicidal stimuli including LPS or Th1 cytokines such as IFN-γ, and could respond to the stimuli by triggering the expression of a series of proinflammatory cytokines and chemokines such as IL-1β, IL-6, IL-12, IL-23, and TNF-α; 2) anti-inflammatory M2 macrophages, which are polarized by Th2 cytokines such as IL-4 and IL-13 and dampen inflammation by expressing and releasing anti-inflammatory cytokines such as IL-10 and TGF-β [57]. M1 and M2 macrophages have different transcriptional profiles and differentially affect health and disease.
Recently, macrophage epigenome profiling has revealed thousands of chromosomal loci that are differentially active in macrophages, revealing chromosome elements that drive macrophage gene expression. Nucleosomal histone acetylation is one of the most dynamic epigenomic marker. This marker is found at gene promoters and enhancers and correlates adequately with gene expression changes. Imbalance of acetylation and deacetylation in macrophages could result in numerous pathological conditions, such as the development of chronic obstructive pulmonary disease (COPD) [58-60], asthma [61], smoking-induced pro-tumor effect [62] or inflammation [63, 64], sterile inflammation and diabetes comorbidities [65], viral immune evasion [66], disruption of intestinal immune homeostasis [67], and chronic inflammatory responses during atherogenesis [68]. HDACi are promising therapeutic agents for these macrophage-related diseases.
Compared to infection with the histone deacetylase-like enzyme (Gc-HDAC)-deficient mutant, higher H3K9ac enrichment at the promoters of proinflammatory mediator genes, various TLRs, adaptor proteins, and transcription factors were detected after infection with Neisseria gonorrhoeae [69]. The transcription factor ATF-2 correlates with p38, which is associated with stress-induced apoptosis and Toll-like receptor (TLR)-mediated signaling. ATF-2 and LPS synergistically improved the transcription of the Socs-3 promoter. However, HDAC1 could barely interact with ATF-2 after LPS treatment. Treating RAW264.7 cells with trichostatin A, an HDACi, triggered a decrease in LPS-induced Socs-3 expression. This result indicates that HDAC1 promotes Socs-3 transcription after LPS induction in TLR-mediated transcriptional control in macrophage cells [70]. In another study, HDACi increased mice vulnerability to bacterial and fungal infections while reinforcing defense against toxic and septic shock, identifying an essential role for HDACi in regulating the expression of innate immune genes and host defenses against microbial pathogens [71]. Although prolonged HDACi treatment might injure host defense, selective HDACi may contribute to treating acute bacterial infections [72]. TSA could essentially affect anti-apoptosis in LPS-treated macrophages, which was supposed to contribute to the protective effect of TSA in a mouse model of cecal ligation and puncture (CLP)-induced lethal sepsis [73]. During polymicrobial sepsis, TSA can adjust the phenotypes of macrophages by facilitating autophagy and reducing inflammation [74]. HDACi and DNA methyl transferase inhibitor DNMTi synergistically ablated macrophage death mediated by endotoxemia via STAT3-JMJD3 signaling [75]. Similarly, in acute lung injury, combination treatment with HDACi and DNMTi reduces inflammation, while M2 macrophages exhibited an evident increase in quantity, which could be a potential treatment for sepsis [76].
The motility of macrophages, which can infiltrate almost all tissues and exert non-specific immunoregulation and other effects, is an irreplaceable element during self-protective inflammation. Butyrate, a noncompetitive, reversible inhibitor of HDACs, was able to reduce LPS-mediated macrophage migration by suppressing Src and focal adhesion kinase activity [77]. TSA was demonstrated to downregulate MMP-9 the expression of in RAW264.7 murine macrophages [78] and reduce macrophage infiltration in tubulointerstitial injury induced by ureteral obstruction [79]. Phenylthiobutanoic acids (PTBAs), a new class of HDACi, were able to reduce peritubular macrophage infiltration, thus contributing to recovery and postinjury fibrosis recovery in a progressive mouse model of acute kidney injury [80]. Treatment with selective class I HDACi MS-275 could impede macrophage activation and decrease the macrophage infiltration, thus improving the outcome of the cerulein-induced pancreatitis mouse model [81]. MGCD0103, a novel orally available and isotype-selective HDACi, could ameliorate macrophage infiltration in a streptozotocin-induced type I diabetes model and thus protects the pancreas from oxidative stress and cell death [82]. Administration of class I HDAC and HDAC3 inhibitors MS-275 and RGFP966 in Ang II-induced hypertensive mice could reduce systolic blood pressure and thickness of the aortic wall partly by reducing macrophage infiltration [83]. Treatment of Ang II-infused ApoE-/- mice abdominal aortic aneurysm (AAA) models with a class I HDACi (MS-275) or a class IIa HDACi (MC-1568) blocked macrophage infiltration and decreased proinflammatory cytokines [84]. Notably, a considerable amount of evidence on HDACi-mediated reduction of macrophage infiltration has been elucidated, and this outstanding discovery provides a promising therapeutic target for multiple human diseases.
In vitro, HDACi exert both pro- and anti-inflammatory effects on macrophages. The pro- and anti-inflammatory effects of TSA were separable within a particular concentration range, which means that each HDAC may differentially affect macrophages. MS-275 exhibits proinflammatory effects while downregulating certain inflammatory reactions. In contrast, an HDAC6-selective inhibitor, 17a, which possesses anti-inflammatory functions but no proinflammatory properties. Besides, no significant difference in HDAC-dependent gene expression was detected between HDAC6(-/-) macrophages and normal macrophages, implying that HDAC6 is not the only intermediary for the anti-inflammatory effects of 17a [85]. TLR or IFN-γ treatment reduced HDAC5 transcriptional levels in several macrophage cell lines, and blocking HDAC5 expression by specific siRNA significantly decreased the production of proinflammatory cytokines [86]. TSA can also block LPS-induced expression of NO synthase II in macrophages by reducing C/EBPβ phosphorylation without affecting DNA binding, which facilitates the production of NO and the cytostatic or cytotoxic activity of macrophages [87]. VPA can also induce a phenotype switch towards the M2 subtype [88]. The anti-rheumatic activities of the class I/IIb and HDAC1-3 inhibitors, SAHA and MS-275, respectively, such as growth arrested in RA synovial fibroblasts and decreased proinflammatory cytokines and NO, were mediated by p21 expression induction and suppressed NF-κB nuclear accumulation [89]. Ky-2, a hybrid-compound HDACi, downregulated the expression of proinflammatory cytokines in LPS-driven human monocyte-like THP-1 cells, thus suppressing M1 macrophage polarization [90]. KBH-A42, a new HDACi, plays anti-inflammatory roles by suppressing the transcriptional levels of TNF-α and NO in LPS-stimulated RAW264.7 cells and peritoneal macrophages [91].
The confrontation and balance between M1 and M2 macrophages are necessary to appropriately control the inflammatory response and restore organ function. The anti-inflammatory and antioxidant effects of HDACi VPA on lung macrophages were demonstrated in a nitrogen mustard-induced lung injury model, which was accompanied by an increased M2 phenotype and decreased M1 phenotype of macrophages [92]. Inhibition of class I/II HDACs or class III HDACs (Sirtuin) potently influences the activation of macrophages derived from the inflamed joints of patients with rheumatoid arthritis by inhibiting the production of proinflammatory factors, such as IL-6 and TNF-α, and inducing macrophage apoptosis [93]. TSA can disrupt inflammatory cytokines by reducing the stability of IL-6 mRNA in IL-1β, TNFα, or Toll-like receptor ligands stimulated macrophages, thus suppressing inflammation in rheumatoid arthritis [94]. Recent in vivo studies have found that HDACi, specifically, deletion of HDAC3, could promote M2 polarization while dampening the activation of M1 macrophages, thus helping treat atherosclerosis [95, 96]. One attractive approach for atherosclerosis therapy is to target macrophage-mediated inflammation. EM-conjugated HDACi, which specifically targets cells expressing carboxylesterase-1, such as monocytes and macrophages, limited peritoneal macrophage maturation but had no inhibitory effect on bone marrow-derived macrophages from WT mice; however, it showed limited efficacy in an atherosclerosis model, [97]. In the acute myocardial infarction mouse model, SAHA upregulated M2 markers, thereby facilitating ventricular remodeling and functional recovery [98]. In another model of skin wounds, TSA significantly induced an improvement in wound closure by increasing the number of CD11blow/Ly6Clow subsets of macrophages [99]. It has been shown that HDAC11 is a negative regulator of LPS-induced IL-10 production in mouse and human macrophages [100]. Furthermore, the suppression of IL-10 in imipramine-treated L. donovani-infected macrophages [101] was reported to depend on enhanced HDAC11 [102]. HDAC11 binds to the distal region of the IL-10 promoter, generating a chromatin configuration with less binding of transcription factors, including SP1 and STAT3 [100]. Interestingly, miR-145 was shown to target the 3' UTR of HDAC11, positively affecting IL-10 transcription, and IFN-γ stimulation was shown to reduce mouse miR-145 levels through the JAK/STAT signaling pathway [103]. Additionally, treating murine macrophages with a pan-HDACi called LAQ824 inhibits IL-10 expression and recruits two transcriptional repressors, including HDAC11, to the distal site of the IL-10 promoter [104]. This observation may partially explain why, after an LAQ824 treatment, an increase in a more inflammatory macrophage phenotype, such as production of higher levels of proinflammatory cytokines and capacity to restore anergic CD4+ T cells [104], is observed. The generation of APCs with elevated inflammatory characteristics after HDACi treatment has also been observed in other settings. For example, treatment of murine and human DCs and macrophages, and colitis-induced mice with certain HDACIs, was shown to induce secretion of IL-1β from these cells via a previously unknown caspase-1 independent mechanism [105]. Although further verification is required, a pharmacological exclusion approach suggests the involvement of HDAC11 in this process [105].
Another subset of innate immune cells consists of myeloid-derived suppressor cells (MDSCs). They are a mixed population of cells with T cell response suppressing ability, and they consist of precursors and various immature stages of DCs, macrophages, and granulocytes such as neutrophils [106]. Consistent with its regulatory role in the expression of the anti-inflammatory cytokine IL-10 in LPS-stimulated macrophages, HDAC11 negatively regulated MDSC expansion and function [107].
Besides, HDACi may also contribute essentially to other functional aspects of macrophages. The pan-HDACi TSA has been demonstrated to decrease CD9 expression in murine peritoneal macrophages, which is implicated in macrophage activation, indicating that HDACi could regulate the expression of membrane proteins involved in matrix interaction; this process may partly contribute to macrophage function modification [108].
In conclusion, HDACs are therapeutic targets in many inflammatory disease models. HDACi may exert their regulatory effects on macrophages in various pathological models via multiple mechanisms, including suppression of macrophage activation, promotion of M2 phenotype affinity, decrease release of proinflammatory cytokine release, inhibition or exacerbation of cell apoptosis, and regulation of phagocytosis or recognition capacity during inflammation.
HDAC and dendritic cells
Dendritic cells (DCs) originate from hematopoietic stem cells and are distinct from other leukocytes. The main function of DCs is to present antigens to T cells and initiate immune responses. Generally, human DCs can be classified into plasmacytoid DCs, which mainly accumulate in the blood and lymphoid tissues and enter the lymph nodes through the blood circulation, and conventional DCs (cDCs), which refer to all other DCs and comprise cDC1 and cDC2 [109, 110]. Plasmacytoid DCs express low levels of MHC-II, costimulatory molecules, and integrin CD11c in the steady state and a smaller number of pattern recognition receptors, including TLR-7 and 9. Once TLRs on plasmacytoid DCs are activated, they can produce numerous type I IFN, chemokines, and cytokines and acquire the ability to present foreign cells. cDCs are found in most lymphoid and nonlymphoid tissues and have a strong capacity to sense and capture antigens, which are then processed and presented to T lymphocytes [111].
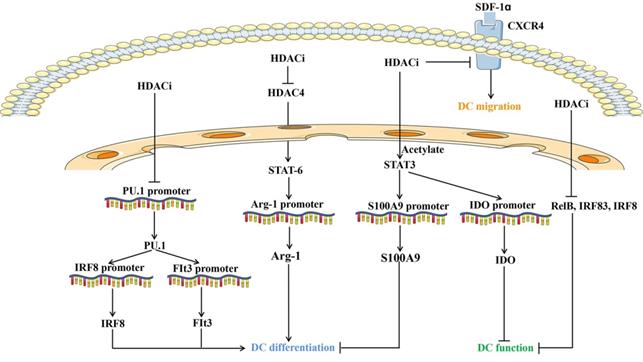
Interestingly, DC functions are modulated by HDACs (Figure 2). Inhibition of HDAC represses the hallmark gene CD1a and adhesion molecule expression, thus skewing DC differentiation. Defective cytokine secretion, impaired ligand 19-induced migration, and decreased immunostimulatory capacity of DCs upon HDACi exposure appears to prevent signaling through NF-κB, IRF-3, and IRF-8 [112, 113]. The transcription of RelB, the NF-κB component required for DC differentiation and maturation, is regulated by chromatin modifiers, including HDAC3 [114]. HDAC4 cross-talks with STAT6 to regulate the transcription of arginase 1, which is essential during mouse DC differentiation [115]. Inhibiting HDAC activity prevents mouse DC differentiation by upregulating STAT-3 and indoleamine 2,3-dioxygenase expression [116]. Gene expression profiling revealed that HDACs are required for the induction of PU.1 and IRF8 expression and DC development, which is further supported by HDACi studies using TSA, VPA, and MS-275 [117]. Emerging studies have suggested that liver X receptors are involved in DC-dependent immune responses. Liver X receptor activation and HDAC inhibition balanced the recruitment of STAT3 to the S100A9 promoter and distinctly modified STAT3 [118]. Moreover, histone acetylation suppresses the chemotactic activity of DCs via the CXCR4-dependent pathway and inhibition of MAP kinases [119]. VPA-treated DCs reduced the capacity to activate Th17 cells under LPS stimulation, which is critical for promoting inflammatory responses [120]. Collectively, HDAC mainly regulates the differentiation, antigen presenting capacity, and migration of DCs (Figure 2).
Overview of molecules and pathways implicated in the effect of HDAC inhibitors on dendritic cells development, function and migration. HDACi: HDAC inhibitors; DC: Dendritic cells; Arg1: Arginase 1; SDF-1α: Stromal cell derived factor-1α; IRF: IFN regulatory factor; Flt3: Fms-like tyrosine kinase 3; CXCR4: CXC chemokine receptor 4; IDO: Indoleamine 2,3-dioxygenase.
HDAC and lymphocytes
Lymphocytes include natural killer (NK) cells, T and B cells, lymphoid tissue inducing (LTi) cells, and natural helper (NH) cells, among which T and B cells constitute the major components of the adaptive immune system; NK cells, LTi cells, and NH cells are defined as innate lymphoid cells (ILCs). T lymphocytes are subdivided into 1) CD4+ T cells, helper T cells (Th cells), 2) CD4+ regulatory T cells (Tregs), and 3) CD8+ T cells, which are cytotoxic T cells. These naïve cells are exported to the periphery, circulate through the lymph nodes, and find antigens, leading to the activation and differentiation of T cells into effector T cells and memory T cells. CD4+ Th cells further differentiate into several functionally distinct Th effector subsets: Th1 cells can stimulate cell-mediated immune responses against intracellular bacteria by producing and secreting IFN-γ. Th2 cells generate Th2 cytokines, such as IL-4, IL-5, and IL-13, for helminth infection. Th17 cells produce interleukins such as IL-17A, IL-21, and IL-22 and initiate immune responses in response to extracellular bacterial and fungal infections. Other subsets, such as T follicular helper cells, Th9, Th22, and TGM-CSF, also play essential roles in regulating immune responses [121]. CD8+ T lymphocytes mediate immediate and long-term protection against intracellular pathogens NK cells and consist of two main subsets: the cytotoxic CD56dim/CD16pos (CD56dim) population, which accounts for 90% of peripheral NK cells, and regulatory CD56bright/CD16neg (CD56br) NK cell subset, which generates pro-inflammatory cytokines [122-124]. LTi cells can secrete the pro-inflammatory cytokines IL-17 and IL-22, whereas NH cells can generate large numbers of Th2-type cytokines, such as IL-5, IL-13, IL-6, and IL-9 [125]. As described above, T and B cells constitute a major part of the adaptive immune system, and ILCs represent another branch of innate immune cells, aside from myeloid cells. Activated B cells, generated from mature B cells, are also known as plasma cells and specialize in the production and secretion of immunoglobulins. Interactions between the innate and adaptive immune systems are crucial for an appropriate immune response against invading pathogens, which requires precise genetic and epigenetic regulation.
HDACi has been demonstrated to regulate the differentiation and function of multiple subsets of CD4 and CD8 T cells [126]. The RORγT gene encodes a Th17 lymphocyte signature transcription factor. The differentiation stage-specific effect of HDACi-mediated H4 acetylation by butyrate and apicidin is involved in epigenetic regulation of RORγT expression in human Th17 cells, suggesting the therapeutic potential of HDACi for diseases in which Th17 cells play vital roles [127]. TSA was shown to improve allogeneic islet transplantation in diabetic C57BL/6 mice by enhancing Treg cells while suppressing Th17 cells [128]. Moreover, in a unilateral ureteral obstruction mouse model, TSA decreased the conversion of Tregs into inflammation-associated IL-17+ Tregs, thus attenuating the degree of fibrosis [129]. Similarly, a specific class I and II HDAC inhibitor (ITF2357) decreased the proportion of Th17 cells while increasing Tregs in a dose-dependent manner and acetylating Foxp3 in an NZB/W mice model, indicating its potential to relieve systemic lupus erythematosus (SLE) [130]. The studies described above demonstrated the crucial effect of HDACi in reducing the Th17/Treg ratio, thus releasing immune responses in T cell-associated diseases. HDACs mediate the suppressive effect of delphinidin on the proliferation, differentiation, and activation of T lymphocytes towards Th1, Th17, and Treg in patients with metabolic syndrome showing cardiovascular risks [131].
A recent functional study showed that the expression level of HDAC2 correlates linearly with muscle power improvement, indicating that the baseline level of HDAC2 in peripheral blood mononuclear cells is a good biomarker for the steroid response in patients with Duchenne muscular dystrophy [132]. The HDAC2-STAT4 pathway can also reverse Th1 shift-mediated neuronal and sensorimotor functional damage, as revealed in an ischemic stroke mouse model treated with antagomir-494 [133]. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice by increasing the Treg phenotype and altering the early stage of B cell development, thus decreasing the overproduction of autoimmune antibodies [134]. Another study confirmed that a novel HDAC6 inhibitor, CKD-L, exerts therapeutic effects on collagen-induced arthritis in vivo and rheumatoid arthritis in vitro by increasing the immunosuppressive function and inducing the expression of cytotoxic T-lymphocyte associated protein-4 in Treg cells [135]. Other HDAC6-selective inhibitors such as ACY-1215 (ricolinostat) and ACY-241 (citarinostat) limited melanoma progression by reducing the production of Th2 cytokines and augmenting T-cell immune properties, providing a promising rationale for translational medicine [136]. In addition, ACY-1215 suppressed effector T cell maturation and regulated the functions of CD8+ T cells in murine CD8+ T cell-related skin inflammation models [137]. One approach for the management of chronic obstructive pulmonary disease (COPD) is combining theophylline with low-dose cyclosporine A to reduce inflammation. Silencing of lymphocytes in COPD is correlated with the loss of HDAC2 in CD28nullCD8+ T and NKT-like cells [138]. CD8+ T lymphocytes can inhibit viral replication by suppressing HIV-1 gene expression in HIV-1-infected individuals However, this capability is impaired after blocking of HDACs, demonstrating that HDACs control the expression of some suppressive factors [139]. An effective adaptive immune response against microbial infection requires activation of heterogeneous T lymphocytes. Increased activity of HDAC7, 9, and 11 and decreased HAT of memory T lymphocytes were involved in the protective effect of You-Gui pills in asthma, which is airway inflammation mediated by eosinophilic cells [140]. HDAC11 has been shown to be involved in the regulation of different aspects of T cell biology. A recent study showed that HDAC11 is responsible for controlling the acetylation status of T-bet and Eomes gene promoters in naïve T cells and that it is rapidly removed from these promoters after T cell activation [141]. Furthermore, HDAC11-deficient effector memory mouse T cells exhibited a more inflammatory phenotype and were less responsive to suppression by Tregs [141]. An in vivo study revealed more rapid and potent development of graft versus host disease (GVHD) after allograft transplantation of T cells lacking HDAC11 [141]. These findings indicate that similar to its role in neutrophils, HDAC11 is a negative regulator of inflammatory T cell responses. Furthermore, exacerbation of GVHD upon transfer of T cells lacking HDAC11 may partially explain why not all HDACi have beneficial effects on allograft models. Although vorinostat, an inhibitor of class I and class II HDACs, is effective both in animal models and patients [142-144], treatment with an HDACi with potency against HDAC11, including trichostatin A or panobinostat, was reported to accelerate GVHD by increasing proinflammatory cytokine production [145, 146]. HDAC11-deficient T cells not only accelerate GVHD, but also may provide a better anti-tumor response when considering their elevated inflammatory capabilities. In line with this prediction, adoptive transfer of HDAC11-deficient T cells into a syngeneic lymphoma tumor model generated a better anti-tumor response than WT T cells [141]. Furthermore, silencing of HDAC11 in Hodgkin lymphoma cells induces expression of OX40 ligand, which is known to be important for the development of an efficient anti-tumor response [147-149], and proinflammatory cytokines, including TNF-ɑ and IL-17, which generate an overall favorable anti-tumor response with more effector and fewer regulatory T cells [150].
Currently, surgery is the only effective clinical management for abdominal aortic aneurysms (AAAs). Gene expression analysis showed that HDAC is deregulated in human AAA, and treatment of an AAA mouse model with MS-275 (class I HDACi) or MC-1568 (class IIa HDACi), which decreases lymphocyte and macrophage infiltration, limited aneurysm expansion [151]. HDACi is a promising pharmacotherapy for overcoming human aneurysm progression [84]. Activation of HDAC7 was demonstrated to mediate Flagellin-triggered IgE expression in B cells, which initiates food allergy [152].
Based on the above studies, the regulatory effects of HDAC and HDACi on lymphocytes include altering their differentiation and conversion, modulating the intensity of immune responses, and reducing cell infiltration.
HDAC and innate lymphoid cells
Innate lymphoid cells (ILCs) play key roles in host defense, barrier integrity, and homeostasis, and mirror adaptive CD4+ Th cell subtypes in both the usage of effector molecules and transcription factors. Chromatin in proximity to effector genes is selectively accessible in ILCs prior to high-level transcription upon activation. Accessibility of these regions is acquired during development and shows only minor changes after activation. In contrast, dramatic chromatin remodeling occurred in naïve CD4+ T cells during Th cell differentiation in a type-2-infection model. This alteration resulted in a substantial convergence of Th2 cells toward ILC2 regulomes. These data indicate extensive sharing of regulatory circuitry across the innate and adaptive compartments of the immune system, despite their divergent development pathways [153].
A recent study demonstrated that the HDACi butyrate suppresses type 2 cytokine production in mouse and human ILC2s and inhibits the proliferation and GATA3 expression of mouse ILC2s. Moreover, butyrate suppresses IL-33- and Alternaria-induced lung inflammation [154]. This suppressive effect depends on the HDAC inhibitory effect, rather than on GPR41 or GPR43 [154]. Similarly, TSA suppressed ILC2-mediated lung inflammation induced by IL-33 and Alternaria [49].
HDAC and microglia
Microglia, the major resident immune cells in the CNS, are recognized as the brain-specific macrophage population and genetically differ from circulating myeloid cells. Like macrophages in the periphery, they are the most multifunctional and plastic CNS inhabitants and can dynamically adapt to their ever-changing environments. They can also be divided into M1 pro-inflammatory and M2 anti-inflammatory subtypes. M1 microglia may exacerbate neuronal damage by releasing inflammatory cytokines such as TNF-α and nitric oxide and impairing axon regeneration after injury. M2 microglia are healthier cells with enhanced phagocytic activity and a reduced production of inflammatory mediators, and can promote the survival of cortical neurons under both normal and ischemic/hypoxic conditions.
The regulatory effect of HDACi on microglial activation has been revealed in multiple previous studies. LPS-activated microglia exhibited histone hyperacetylation [155], and HDAC2 protein levels were significantly upregulated in microglia in the substantia nigra of Parkinson's disease compared to normal controls [156], indicating the regulatory effect of HDACs on microglia in neuroinflammation. In vivo studies demonstrated that VPA or NaBu treatment decreased the number of microglia in the ischemic brain, which contributed to reduced brain infarct volume and neurological deficits in a rat model of permanent middle cerebral artery occlusion [157, 158]. NaBu administration in a similar model significantly downregulated the expression of TNF-α and NOS2 and upregulated IL-10 expression in activated microglia, thus alleviating neuroinflammation [159]. In a traumatic brain injury model, administration of a novel HDAC inhibitor, DMA-PB, reduced the density of phagocytic microglia [160]. In a spinal cord injury (SCI) model, injection of VPA attenuated the accumulation of microglia in the injured spinal cord and thus contributed to reducing gliosis after SCI [161]. VPA decreased the number of activated microglia and mRNA levels of pro-inflammatory marker genes in an LRRK2 R1441G transgenic Parkinson's disease mouse model [162]. In an APP/PS1 transgenic mouse model of Alzheimer's disease, inhibition of HDAC3 in the hippocampus alleviated microglial activation, which showed therapeutic potential for Alzheimer's disease [163]. MS-275 alleviates postoperative cognitive dysfunction by reducing the number of activated microglia and neuroinflammation in the hippocampus of rats via HDAC inhibition pretreatment with MS-275 [164]. The anti-neuroinflammatory effect of HDACi has been demonstrated in various animal disease models, providing a basis for related clinical transformation.
However, in vitro studies have shown contradictory results. The HDAC inhibitors TSA, CAY10683, apicidin, MS-275, and MI192 reduced the expression of inflammatory mediators such as IL-6, IL-1β, and TNF-α in LPS-treated BV-2 microglial cells [165-167]. VPA, TSA, and NaBu can induce apoptosis of microglia cells, and pretreatment with sodium butylate or TSA in mesencephalic neuron-glia cultures led to a decrease in LPS-induced pro-inflammatory responses and damage to dopaminergic neurons [168]. TSA administration in BV-2 and primary microglia reversed the suppressive effect of LPS on Rgs10 transcription, which was able to regulate the production of pro-inflammatory cytokines in microglia [169]. In a rat model of SCI, VPA treatment also promoted the differentiation of microglia into the anti-inflammatory M2 subtype by inhibiting HDAC3 and subsequent STAT1-mediated acetylation of the NF-κB pathway [170]. SAHA treatment in LPS-stimulated glial cultures was found to strengthen the immunosuppressive effects of dexamethasone [171]. In addition, Scriptaid, a novel HDACi, stimulated microglia to switch to the M2 phenotype. This phenotypic switch relieved the inflammatory response in a mouse model of traumatic brain injury [172]. In a neonatal rat model of hypoxia-ischemia, NaBu treatment exerted a similar effect as Scriptaid by promoting neurogenesis in the subventricular zone [173]. The transition effect of sodium butylate also reversed the proliferative and neurotoxic effects of microglia mediated by Runt-related transcription factor 1, translocated to 1, which was reported to interact with HDACs and is involved in epigenetic modification of Cdk4 and LAT2 [174].
After LPS treatment, however, the expression level of HDACs was increased in both primary microglia cultures and acutely isolated microglia from LPS-treated mice, and HDACi TSA and SAHA strongly suppressed LPS-induced cytokine release by microglia. Furthermore, the expression of M1- and M2-associated activation markers was suppressed and the migratory behavior of microglia was attenuated [175]. Similarly, TSA and SAHA treatment in LPS-activated murine N9 and primary microglia increased the expression of pro-inflammatory cytokines such as IL-6, TNF-α, and nitric oxide [176], suggesting a pro-inflammatory effect for HDACi. VPA and NaBut increased the release of pro-inflammatory cytokines in activated microglia [177]. The variations among different studies may be related to differences in the doses of administration and between models used in vivo and in vitro.
Thus, although there are some contradictory results, the anti-inflammatory effect of HDAC on microglia has been demonstrated in most related studies and HDAC may function by reducing the number of microglia, generating cytokines, and promoting the phenotype switch towards M2.
HDAC and astrocytes
As the largest group of glial cells in the CNS, astrocytes play major roles in various manners, such as by maintaining blood-brain barrier integrity, wiring neural circuitry, regulating metabolism, and neuroinflammation, and when pathologically activated, further undergoing a series of morphological and functional adaptations, including proliferation, to form a barrier that separates the injury site and by releasing cytokines and chemokines [178]. These reactive astrocytes can be classified into the A1 and A2 anti-inflammatory subtypes [179, 180]. A1-reactive astrocytes are thought to be harmful to neurons and oligodendroglia in vivo. In contrast, A2-reactive astrocytes promote neuronal survival and tissue repair by upregulating neurotrophic factors.
Epigenetic regulation by HDAC can coordinate astrocyte function by affecting activation and cytokine production. It has been demonstrated that reduced HDAC levels in astrocytes by dimethyl fumarate treatment contribute to the suppression of inflammatory responses [181]. In addition, the mood-stabilizing and anticonvulsant drug VPA modulated the mRNA levels of neuroligin-1 and neuregulin-1, which function in post-synaptic cell adhesion, and two extracellular matrix proteins (neuronal pentraxin-1 and thrombospondin-3) in astrocytes to balance excitatory/inhibitory synapse formation in neuro-glial mixed cultures [182]. In LPS-or 1-methyl-4-phenylpyridinium-treated neuron-glia cultures, VPA, TSA, and NaBu protected dopaminergic neurons by upregulating glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor expression in astrocytes [183, 184]. Moreover, VPA, sodium butylate, TSA, MS-275, and apicidin recruit the transcription co-activator p300 and upregulate H3K4me2 modification at the HSP70 promoter, enhancing neuroprotective HSP70 expression in rat astrocytes [185]. M344, a specific HDAC 1/6 inhibitor, and VPA increased acetylated glutamate carboxypeptidase II (GCPII) protein and decreased the GCPII ubiquitination level. In contrast, C646, a histone acetyltransferase inhibitor of p300/CBP, significantly reduced the level of GCPII protein. Therefore, the increase in GCPII induced by VPA does not occur via a classical epigenetic mechanism, but rather by enhanced acetylation of lysine residues in GCPII [186]. A highly selective HDAC3 inhibitor, RGFP966, downregulates astrocyte activation, showing protective effects against huntingtin-induced neuronal toxicity and cell death in an N171-82Q transgenic mouse model of huntingtin [187]. These results demonstrate that HDACi-treated astrocytes exert neuroprotective effects in diverse manner. Moreover, although there is emerging evidence that astrocytes exhibit regional heterogeneity, whether HDACs control this diversity remains unknown.
HDAC and hematological malignant cells
In addition to regulating the biological activities of immune cells, HDACs play a role in several hematological pathologies (Table 2). Therefore, HDACi have been gradually applied in the clinical treatment of bone marrow failure and hematopoietic malignancies. For example, combining immunotherapy with HDACi helps overcome immunotherapy resistance in B-cell lymphoma [188].
Inhibition of HDAC6 inhibitors on immune cells function in different disease models
| Inhibitor Name | Disease Models | Immune cells | Effect of HDAC6 inhibitors | References |
|---|---|---|---|---|
| HDAC6(-/-) | LPS | Macrophages | Showed normal LPS-induced expression of HDAC-dependent inflammatory genes | [85] |
| HDAC6-shRNAs, or Tubacin | Cystic fibrosis-lung disease | Neutrophils | Reduce the release of IL-6 and MPO | [36] |
| ACY-738 | SLE pathogenesis | T-cell; B-cell | Increase the Treg phenotype and alter the early stage of B cell development thus decrease the overproduction of autoimmune antibody | [134] |
| CKD-L | Collagen-induced arthritis | T-cell | Increase the suppressive function of Treg cells | [135] |
| ACY-1215, ACY-241 | Melanoma | T-cell | Decreased Th2 cytokine production, augmented T-cell immune properties | [136] |
| ACY-1215 | Skin inflammation models | T-cell | Suppress the generation of effector T cells from naive CD8+T cells and regulating the activation and functions of CD8+T cells | [137] |
Expression of HDAC1, HDAC2, and HDAC6 was significantly upregulated in PTCL and diffuse large B-cell lymphoma, and HDAC6 levels indicate a favorable prognosis [189]. In Hodgkin's lymphoma, HDAC1, 2, and 3 are highly expressed, whereas low levels of HDAC1 may be a marker of worse outcomes [190]. HDAC activity in CD19+ B cells purified from patients with chronic lymphocytic leukemia was found to be negatively correlated with treatment-free survival and overall survival [191]. The expression of HDACs is enhanced in NK cells from both aggressive and chronic NK leukemia [192]. The transcription of HDAC3 was repressed by the chimeric transcription factor PML-RARalphain, which leads to the development of acute promyelocytic leukemia [193].
HDAC inhibitors and their anti-tumor effect in hematological malignant cells
| HDAC inhibitors | Hematological malignant cells | References |
|---|---|---|
| TSA | B lymphoma cell line | [194] |
| Lymphoblastoid cell line | [195] | |
| Myeloid leukemia (AML) cell line | [196] | |
| Acute lymphoblastic leukemia cell line | [196] | |
| Eosinophilic leukemia EoL-1 cell line | [197] | |
| MS-275 | Leukemia and lymphoma cell lines | [199] |
| Vorinostat (SAHA) | Myeloid leukaemia (AML) cell line | [198] |
| Myelodysplastic syndromes cell line | [198] | |
| Leukaemic NK cells | [192] | |
| AR-42 | Malignant mast cell line | [201] |
| Butyrate | Mastocytoma P815 cell line | [200] |
| LBH-589 | multiple myeloma cell | [202] |
HDACi can target hematological malignant cells. Histone deacetylation has been shown to downregulate Bcl-6 activity, and TSA treatment can accumulate inactive acetylated Bcl-6, thus leading to cell cycle arrest and apoptosis of B-cell lymphoma cells [194]. Similarly, TSA exerted anti-cancer effects on Epstein-Barr virus-transformed lymphoblastoid cell lines by inducing cell cycle arrest, apoptosis, and the Epstein-Barr virus lytic cycle [195]. Addition of TSA to a cytokine cocktail containing TNF-α, GM-CSF, and ckit ligand significantly enhanced the treatment effects to promote the differentiation of acute myeloid leukemia and acute lymphoblastic leukemia cell lines towards DCs [196]. In another study, HDACi TSA, n-butyrate, and apicidin induced the differentiation of the eosinophilic leukemia EoL-1 cell line into eosinophils [197]. SAHA arrests the cell cycle, decreases cell survival, and induces differentiation of acute myeloid leukemia and myelodysplastic syndrome cell lines [198]. SAHA also strengthened the cytotoxic effects of cladribine towards leukemic NK cells through intrinsic mitochondrial and extrinsic death receptor pathways [192]. MS-275 arrests the cell cycle at low concentrations while inducing apoptosis at higher concentrations in several human leukemia and lymphoma cell lines [199]. Similarly, butyrate likely inhibits the proliferation and increases the apoptosis of mastocytoma P815 cells [200], indicating the anti-cancer capacity of HDACi and providing insight into their clinical transformation. AR-42, a novel HDACi, induced growth inhibition, cell cycle arrest, apoptosis, and activation of caspases-3/7 in malignant mast cell lines [201]. Additionally, LBH-589 inhibits multiple myeloma cell growth by degrading the catalytic subunit of calcineurin, PPP3CA. This degradation may be mediated by suppression of the chaperone function of HSP90 due to HDAC6 inhibition [202].
Conclusion and Perspective
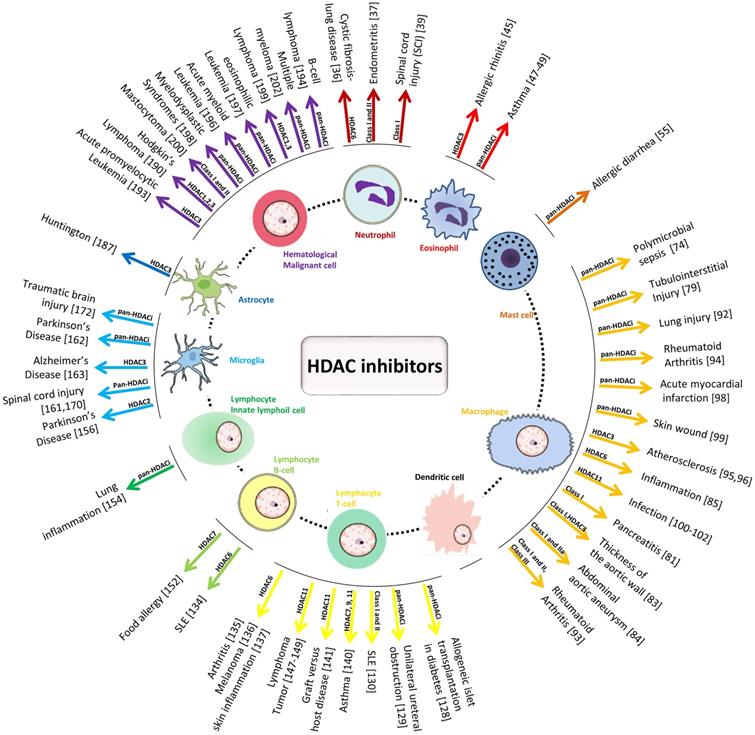
In this review, we summarized the results of preclinical investigations of the regulatory effects of HDACs and HDACi on immune cells in related diseases (Figure 3). As illustrated, there are several ways by which HDAC and HDACi modulate the development and functions of immune cells, affect the proliferation and differentiation of hematopoietic stem and precursor cells, and regulate inflammatory responses by adapting the phenotype switch of mature immune cells, inducing or inhibiting the apoptosis of effective immune cells and their infiltration into inflammatory sites.
Schematic diagram of the effect of HDAC inhibitors on immune cells and the detailed HDACs involved in different inflammation-related diseases.
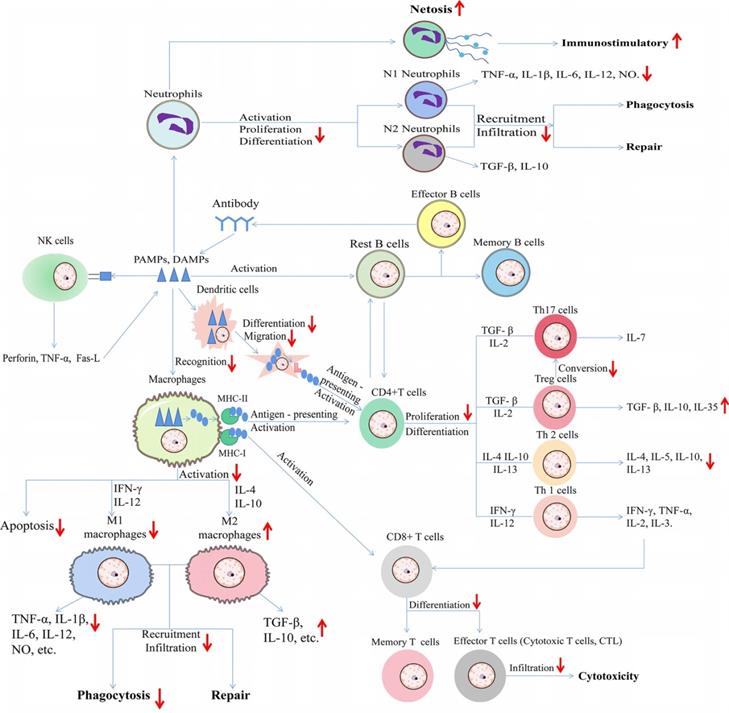
In particular, under certain pathological conditions, different immune cells interact with each other. In one hand, we summed up the effect of HDACi on immune cells during type 1 allergy (Figure 1). In another hand, we summerize the network diagram of cell-cell interaction, as well as the effect of HDACi on immune cells during immune responses to diverse pathogen-associated molecular patterns (PAMPs) from microorganisms as well as damage-associated molecular patterns (DAMPs) from dead or dying host cells (Figure 4).
Schematic diagram of effect of HDAC inhibitors on immune cells during immune responses to diverse pathogen-associated molecular patterns (PAMPs) from microorganisms as well as damage-associated molecular patterns (DAMPs) from dead or dying host cells. The upward arrows represent the promotion effect by HDAC inhibitors and the downward arrows represent the suppression effect by HDAC inhibitors. TNF-α: Tumor necrosis factor-α; IL: Interleukin; NO: Nitric oxide; TGF-β: Transforming growth factor; IFN-γ: Interferon-γ; MHC: major histocompatibility complex; Fas-L: Fas ligand.
HDACi treatment decreases inflammatory responses in most cases and can alleviate symptoms in animal models of autoimmune and inflammatory diseases. Notably, HDACi show bidirectional regulatory effects on immune responses; for example, pretreatment with TSA relieved the inflammatory responses of microglia in the mouse brain after LPS injection [165], whereas TSA aggravated the inflammatory response when added at the same time as LPS in vitro [176]. HDACi have been shown to suppress overactivated autoimmune responses [135, 137], although in other studies, HDACi enhanced the cytotoxic effect of lymphocytes in malignant tumors [136] and HIV-1 [139]. These results suggest that the regulatory role of HDACi in immune cells is phase-and context-dependent. Selective inhibition of HDAC6 exerted different functions in different immune cells. The mechanism underlying this biphasic role remains unclear, and further fundamental research is needed to improve the use of HDACi in immune-related diseases.
Moreover, there are concerns regarding the effectiveness and safety of treating other diseases, although the therapeutic benefit of HDACi on hematological malignancies is unclear. For example, most currently used HDACi are non-selective pan-HDACi, and their relatively low specificity may alter the expression of thousands of vital genes, thus leading to undesirable consequences and impeding the widespread clinical application of HDACi. Therefore, the specificity of these molecular compounds should be further improved and toxicity should be carefully controlled. VPA, a widely prescribed antiepileptic drug, is known to induce hepatotoxicity. To date, selective HDACi have been modified in several regions [203-205]. However, because of the similarity among the crystal structures of different HDACs, their selectivity remains unclear, as most current 'selective' inhibitors may actually bind to multiple HDAC isoforms [206]. Thus, more accurate knowledge of the crystal structures of HDACs and HDACi is required to develop more sophisticated and better modified HDACi to match the subtle differences among different HDAC isozymes.
Researchers currently have a general understanding of how epigenetic modulation by HDAC and HDACi regulates the development and function of immune cells. HDACi may serve as epigenetic modifiers, which have been explored as single agents and in combination with other established therapies and can reverse some pathological conditions. In future research, more efforts should be focused on the issues of specificity to broaden the field of clinical application, which will open new pathways for pharmacological interventions in multiple related diseases.
Acknowledgements
Funding
This project was supported by the National Natural Science Foundation of China (82071314, 81771412 and 81971222) and Beijing Natural Science Foundation Program, and the Scientific Research Key Program of the Beijing Municipal Commission of Education (KZ201810025041).
Authors' contributions
ZSJ and ZLJ contributed to the literature review and manuscript writing. YZH and LX contributed to the literature review. ZHP and LYM have revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bird A. Perceptions of epigenetics. Nature. 2007;447:396-8
2. Li H, Ye Q, Lin Y, Yang X, Zou X, Yang H. et al. CpG oligodeoxynucleotides attenuate RORgammat-mediated Th17 response by restoring histone deacetylase-2 in cigarette smoke-exposure asthma. Cell Biosci. 2021;11:92
3. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693-705
4. Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016;32:42-56
5. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-49
6. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32-42
7. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512-20
8. Dang F, Wei W. Targeting the acetylation signaling pathway in cancer therapy. Semin Cancer Biol. 2021
9. Benedetti R, Conte M, Altucci L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid Redox Signal. 2015;23:99-126
10. Moreno-Yruela C, Galleano I, Madsen AS, Olsen CA. Histone Deacetylase 11 Is an epsilon-N-Myristoyllysine Hydrolase. Cell Chem Biol. 2018;25:849-56 e8
11. Hai Y, Shinsky SA, Porter NJ, Christianson DW. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat Commun. 2017;8:15368
12. Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G. et al. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes (Basel). 2020 11
13. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-84
14. Fan W, Zhang L, Jiang Q, Song W, Yan F, Zhang L. Histone deacetylase inhibitor based prodrugs. Eur J Med Chem. 2020;203:112628
15. Wittenburg LA, Gustafson DL, Thamm DH. Phase I pharmacokinetic and pharmacodynamic evaluation of combined valproic acid/doxorubicin treatment in dogs with spontaneous cancer. Clin Cancer Res. 2010;16:4832-42
16. Gawel JM, Shouksmith AE, Raouf YS, Nawar N, Toutah K, Bukhari S. et al. PTG-0861: A novel HDAC6-selective inhibitor as a therapeutic strategy in acute myeloid leukaemia. Eur J Med Chem. 2020;201:112411
17. Tavares MT, de Almeida LC, Kronenberger T, Monteiro Ferreira G, Fujii de Divitiis T, Franco Zannini Junqueira Toledo M. et al. Structure-activity relationship and mechanistic studies for a series of cinnamyl hydroxamate histone deacetylase inhibitors. Bioorg Med Chem. 2021;35:116085
18. Sama GR, Liu H, Mountford S, Thompson P, Robinson A, Dear AE. Synthesis and biological evaluation of a novel photo-activated histone deacetylase inhibitor. Bioorg Med Chem Lett. 2020;30:127291
19. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S. et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109-15
20. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M. et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485-91
21. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH. et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410-7
22. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A. et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br J Haematol. 2015;168:811-9
23. Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong TC, Prince HM. et al. Panobinostat in patients with relapsed/refractory Hodgkin's lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30:2197-203
24. Xu Y, Zhang P, Liu Y. Chidamide tablets: HDAC inhibition to treat lymphoma. Drugs Today (Barc). 2017;53:167-76
25. Tsimberidou AM, Beer PA, Cartwright CA, Haymaker C, Vo HH, Kiany S. et al. Preclinical Development and First-in-Human Study of KA2507, a Selective and Potent Inhibitor of Histone Deacetylase 6, for Patients with Refractory Solid Tumors. Clin Cancer Res. 2021
26. Peng Q, Ma M, Gu X, Hu Y, Zhou B. Evaluation of Factors Impacting the Efficacy of Single or Combination Therapies of Valproic Acid, Carbamazepine, and Oxcarbazepine: A Longitudinal Observation Study. Front Pharmacol. 2021;12:641512
27. Yokoyama S, Yasui-Furukori N, Nakagami T, Miyazaki K, Ishioka M, Tarakita N. et al. Association Between the Serum Carnitine Level and Ammonia and Valproic Acid Levels in Patients with Bipolar Disorder. Ther Drug Monit. 2020;42:766-70
28. Elshafay A, Hieu TH, Doheim MF, Kassem MAM, MF EL, Holloway SK. et al. Efficacy and Safety of Valproic Acid for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis. CNS Drugs. 2019;33:239-50
29. Ostuni R, Natoli G, Cassatella MA, Tamassia N. Epigenetic regulation of neutrophil development and function. Semin Immunol. 2016;28:83-93
30. Govers A, Wiggers CRM, van Boxtel R, Mokry M, Nieuwenhuis EES, Creyghton MP. et al. Transcriptomic and Epigenomic Profiling of Histone Deacetylase Inhibitor Treatment Reveals Distinct Gene Regulation Profiles Leading to Impaired Neutrophil Development. Hemasphere. 2019;3:e270
31. Sahakian E, Chen J, Powers JJ, Chen X, Maharaj K, Deng SL. et al. Essential role for histone deacetylase 11 (HDAC11) in neutrophil biology. J Leukoc Biol. 2017;102:475-86
32. Hamam HJ, Khan MA, Palaniyar N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules. 2019 9
33. Hamam HJ, Palaniyar N. Histone Deacetylase Inhibitors Dose-Dependently Switch Neutrophil Death from NETosis to Apoptosis. Biomolecules. 2019 9
34. Tokuyama M, Gunn BM, Venkataraman A, Kong Y, Kang I, Rakib T. et al. Antibodies against human endogenous retrovirus K102 envelope activate neutrophils in systemic lupus erythematosus. J Exp Med. 2021 218
35. Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol. 2015;179:68-74
36. Bodas M, Mazur S, Min T, Vij N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir Res. 2018;19:2
37. Guo J, Wang Y, Jiang P, Yao H, Zhao C, Hu X. et al. Sodium butyrate alleviates lipopolysaccharide-induced endometritis in mice through inhibiting inflammatory response. Microb Pathog. 2019;137:103792
38. Leus NG, van den Bosch T, van der Wouden PE, Krist K, Ourailidou ME, Eleftheriadis N. et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci Rep. 2017;7:45047
39. Shapiro E. Market forces and vulnerable elderly people: who cares? CMAJ. 1998;159:151-2
40. Li F, Zhao H, Li G, Zhang S, Wang R, Tao Z. et al. Intravenous antagomiR-494 lessens brain-infiltrating neutrophils by increasing HDAC2-mediated repression of multiple MMPs in experimental stroke. FASEB J. 2020;34:6934-49
41. Iwasaki H, Mizuno S, Mayfield R, Shigematsu H, Arinobu Y, Seed B. et al. Identification of eosinophil lineage-committed progenitors in the murine bone marrow. J Exp Med. 2005;201:1891-7
42. Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A. et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med. 2009;206:183-93
43. Abdala Valencia H, Loffredo LF, Misharin AV, Berdnikovs S. Phenotypic plasticity and targeting of Siglec-F(high) CD11c(low) eosinophils to the airway in a murine model of asthma. Allergy. 2016;71:267-71
44. Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D. et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest. 2016;126:3279-95
45. Zhang W, Sun X, Ba G, Tang R, Lin H. RGFP966, a selective HDAC3 inhibitor, ameliorates allergic and inflammatory responses in an OVA-induced allergic rhinitis mouse model. Int Immunopharmacol. 2021;93:107400
46. Theiler A, Barnthaler T, Platzer W, Richtig G, Peinhaupt M, Rittchen S. et al. Butyrate ameliorates allergic airway inflammation by limiting eosinophil trafficking and survival. J Allergy Clin Immunol. 2019;144:764-76
47. Choi JH, Oh SW, Kang MS, Kwon HJ, Oh GT, Kim DY. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin Exp Allergy. 2005;35:89-96
48. Banerjee A, Trivedi CM, Damera G, Jiang M, Jester W, Hoshi T. et al. Trichostatin A abrogates airway constriction, but not inflammation, in murine and human asthma models. Am J Respir Cell Mol Biol. 2012;46:132-8
49. Toki S, Goleniewska K, Reiss S, Zhou W, Newcomb DC, Bloodworth MH. et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax. 2016;71:633-45
50. Kankaanranta H, Janka-Junttila M, Ilmarinen-Salo P, Ito K, Jalonen U, Ito M. et al. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J Inflamm (Lond). 2010;7:9
51. Muraoka M, Akagi T, Ueda A, Wada T, Koeffler HP, Yokota T. et al. C/EBPepsilon DeltaRS derived from a neutrophil-specific granule deficiency patient interacts with HDAC1 and its dysfunction is restored by trichostatin A. Biochem Biophys Res Commun. 2019;516:293-9
52. Huber M, Cato ACB, Ainooson GK, Freichel M, Tsvilovskyy V, Jessberger R. et al. Regulation of the pleiotropic effects of tissue-resident mast cells. J Allergy Clin Immunol. 2019;144:S31-S45
53. Hermans M, Lennep JRV, van Daele P, Bot I. Mast Cells in Cardiovascular Disease: From Bench to Bedside. Int J Mol Sci. 2019 20
54. Assem el SK, Peh KH, Wan BY, Middleton BJ, Dines J, Marson CM. Effects of a selection of histone deacetylase inhibitors on mast cell activation and airway and colonic smooth muscle contraction. Int Immunopharmacol. 2008;8:1793-801
55. Krajewski D, Kaczenski E, Rovatti J, Polukort S, Thompson C, Dollard C. et al. Epigenetic Regulation via Altered Histone Acetylation Results in Suppression of Mast Cell Function and Mast Cell-Mediated Food Allergic Responses. Front Immunol. 2018;9:2414
56. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445-55
57. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012;53:11-24
58. Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451-64
59. Milara J, Navarro A, Almudever P, Lluch J, Morcillo EJ, Cortijo J. Oxidative stress-induced glucocorticoid resistance is prevented by dual PDE3/PDE4 inhibition in human alveolar macrophages. Clin Exp Allergy. 2011;41:535-46
60. Cosio BG, Jahn A, Iglesias A, Shafiek H, Busquets X, Agusti A. Haemophilus influenzae induces steroid-resistant inflammatory responses in COPD. BMC Pulm Med. 2015;15:157
61. Cosio BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF. et al. Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med. 2004;170:141-7
62. Edderkaoui M, Xu S, Chheda C, Morvaridi S, Hu RW, Grippo PJ. et al. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget. 2016;7:7747-60
63. Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm Pharmacol Ther. 2012;25:286-92
64. Li M, Zhong X, He Z, Wen M, Li J, Peng X. et al. Effect of erythromycin on cigarette-induced histone deacetylase protein expression and nuclear factor-kappaB activity in human macrophages in vitro. Int Immunopharmacol. 2012;12:643-50
65. Filgueiras LR, Brandt SL, Ramalho TR, Jancar S, Serezani CH. Imbalance between HDAC and HAT activities drives aberrant STAT1/MyD88 expression in macrophages from type 1 diabetic mice. J Diabetes Complications. 2017;31:334-9
66. Adhya D, Dutta K, Kundu K, Basu A. Histone deacetylase inhibition by Japanese encephalitis virus in monocyte/macrophages: a novel viral immune evasion strategy. Immunobiology. 2013;218:1235-47
67. Kobayashi T, Matsuoka K, Sheikh SZ, Russo SM, Mishima Y, Collins C. et al. IL-10 regulates Il12b expression via histone deacetylation: implications for intestinal macrophage homeostasis. J Immunol. 2012;189:1792-9
68. Kong X, Fang M, Li P, Fang F, Xu Y. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol. 2009;46:292-9
69. Zughaier SM, Rouquette-Loughlin CE, Shafer WM. Identification of a Neisseria gonorrhoeae Histone Deacetylase: Epigenetic Impact on Host Gene Expression. Pathogens. 2020 9
70. Hirose N, Maekawa T, Shinagawa T, Ishii S. ATF-2 regulates lipopolysaccharide-induced transcription in macrophage cells. Biochem Biophys Res Commun. 2009;385:72-7
71. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T. et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117:1205-17
72. Ariffin JK, das Gupta K, Kapetanovic R, Iyer A, Reid RC, Fairlie DP. et al. Histone Deacetylase Inhibitors Promote Mitochondrial Reactive Oxygen Species Production and Bacterial Clearance by Human Macrophages. Antimicrob Agents Chemother. 2015;60:1521-9
73. Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X. et al. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg. 2015;78:378-85
74. Cui SN, Chen ZY, Yang XB, Chen L, Yang YY, Pan SW. et al. Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis. Int Immunopharmacol. 2019;77:105973
75. Samanta S, Zhou Z, Rajasingh S, Panda A, Sampath V, Rajasingh J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int J Biochem Cell Biol. 2018;102:117-27
76. Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan YT, Dawn B. et al. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J Cell Sci. 2015;128:3094-105
77. Maa MC, Chang MY, Hsieh MY, Chen YJ, Yang CJ, Chen ZC. et al. Butyrate reduced lipopolysaccharide-mediated macrophage migration by suppression of Src enhancement and focal adhesion kinase activity. J Nutr Biochem. 2010;21:1186-92
78. Kim S, Bong N, Kim OS, Jin J, Kim DE, Lee DK. Lithium chloride suppresses LPS-mediated matrix metalloproteinase-9 expression in macrophages through phosphorylation of GSK-3beta. Cell Biol Int. 2015;39:177-84
79. Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol. 2010;298:F133-41
80. Novitskaya T, McDermott L, Zhang KX, Chiba T, Paueksakon P, Hukriede NA. et al. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am J Physiol Renal Physiol. 2014;306:F496-504
81. Bombardo M, Saponara E, Malagola E, Chen R, Seleznik GM, Haumaitre C. et al. Class I histone deacetylase inhibition improves pancreatitis outcome by limiting leukocyte recruitment and acinar-to-ductal metaplasia. Br J Pharmacol. 2017;174:3865-80
82. Lee HA, Lee E, Do GY, Moon EK, Quan FS, Kim I. Histone deacetylase inhibitor MGCD0103 protects the pancreas from streptozotocin-induced oxidative stress and beta-cell death. Biomed Pharmacother. 2019;109:921-9
83. Ryu Y, Kee HJ, Sun S, Seok YM, Choi SY, Kim GR. et al. Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension. PLoS One. 2019;14:e0213186
84. Galan M, Varona S, Orriols M, Rodriguez JA, Aguilo S, Dilme J. et al. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: therapeutic potential of HDAC inhibitors. Dis Model Mech. 2016;9:541-52
85. Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C. et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol. 2010;87:1103-14
86. Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B. et al. Histone deacetylase 5 regulates the inflammatory response of macrophages. J Cell Mol Med. 2015;19:2162-71
87. Serrat N, Sebastian C, Pereira-Lopes S, Valverde-Estrella L, Lloberas J, Celada A. The response of secondary genes to lipopolysaccharides in macrophages depends on histone deacetylase and phosphorylation of C/EBPbeta. J Immunol. 2014;192:418-26
88. Wu C, Li A, Leng Y, Li Y, Kang J. Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol. 2012;31:592-9
89. Choo QY, Ho PC, Tanaka Y, Lin HS. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (Oxford). 2010;49:1447-60
90. Kaneko J, Okinaga T, Ariyoshi W, Hikiji H, Fujii S, Iwanaga K. et al. Ky-2, a hybrid compound histone deacetylase inhibitor, regulated inflammatory response in LPS-driven human macrophages. Cell Biol Int. 2018;42:1622-31
91. Choi Y, Park SK, Kim HM, Kang JS, Yoon YD, Han SB. et al. Histone deacetylase inhibitor KBH-A42 inhibits cytokine production in RAW 264.7 macrophage cells and in vivo endotoxemia model. Exp Mol Med. 2008;40:574-81
92. Venosa A, Gow JG, Hall L, Malaviya R, Gow AJ, Laskin JD. et al. Regulation of Nitrogen Mustard-Induced Lung Macrophage Activation by Valproic Acid, a Histone Deacetylase Inhibitor. Toxicol Sci. 2017;157:222-34
93. Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME. et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J Immunol. 2010;184:2718-28
94. Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis. 2012;71:424-31
95. Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MC, van der Velden S. et al. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455:396-402
96. Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE. et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6:1124-32
97. Luque-Martin R, Van den Bossche J, Furze RC, Neele AE, van der Velden S, Gijbels MJJ. et al. Targeting Histone Deacetylases in Myeloid Cells Inhibits Their Maturation and Inflammatory Function With Limited Effects on Atherosclerosis. Front Pharmacol. 2019;10:1242
98. Kimbrough D, Wang SH, Wright LH, Mani SK, Kasiganesan H, LaRue AC. et al. HDAC inhibition helps post-MI healing by modulating macrophage polarization. J Mol Cell Cardiol. 2018;119:51-63
99. Cabanel M, da Costa TP, El-Cheikh MC, Carneiro K. The epigenome as a putative target for skin repair: the HDAC inhibitor Trichostatin A modulates myeloid progenitor plasticity and behavior and improves wound healing. J Transl Med. 2019;17:247
100. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M. et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92-100
101. Mukherjee S, Mukherjee B, Mukhopadhyay R, Naskar K, Sundar S, Dujardin JC. et al. Imipramine is an orally active drug against both antimony sensitive and resistant Leishmania donovani clinical isolates in experimental infection. PLoS Negl Trop Dis. 2012;6:e1987
102. Mukherjee S, Mukherjee B, Mukhopadhyay R, Naskar K, Sundar S, Dujardin JC. et al. Imipramine exploits histone deacetylase 11 to increase the IL-12/IL-10 ratio in macrophages infected with antimony-resistant Leishmania donovani and clears organ parasites in experimental infection. J Immunol. 2014;193:4083-94
103. Lin L, Hou J, Ma F, Wang P, Liu X, Li N. et al. Type I IFN inhibits innate IL-10 production in macrophages through histone deacetylase 11 by downregulating microRNA-145. J Immunol. 2013;191:3896-904
104. Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J. et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011;186:3986-96
105. Stammler D, Eigenbrod T, Menz S, Frick JS, Sweet MJ, Shakespear MR. et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J Immunol. 2015;195:5421-31
106. Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53-65
107. Sahakian E, Powers JJ, Chen J, Deng SL, Cheng F, Distler A. et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63:579-85
108. Wang XQ, Alfaro ML, Evans GF, Zuckerman SH. Histone deacetylase inhibition results in decreased macrophage CD9 expression. Biochem Biophys Res Commun. 2002;294:660-6
109. Amon L, Lehmann CHK, Heger L, Heidkamp GF, Dudziak D. The ontogenetic path of human dendritic cells. Mol Immunol. 2020;120:122-9
110. Wang A, Bai Y. Dendritic cells: The driver of psoriasis. J Dermatol. 2020;47:104-13
111. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563-604
112. Nencioni A, Beck J, Werth D, Grunebach F, Patrone F, Ballestrero A. et al. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007;13:3933-41
113. Nascimento CR, Freire-de-Lima CG, da Silva de Oliveira A, Rumjanek FD, Rumjanek VM. The short chain fatty acid sodium butyrate regulates the induction of CD1a in developing dendritic cells. Immunobiology. 2011;216:275-84
114. Dong X, Lutz W, Schroeder TM, Bachman LA, Westendorf JJ, Kumar R. et al. Regulation of relB in dendritic cells by means of modulated association of vitamin D receptor and histone deacetylase 3 with the promoter. Proc Natl Acad Sci U S A. 2005;102:16007-12
115. Yang Q, Wei J, Zhong L, Shi M, Zhou P, Zuo S. et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol Cell Biol. 2015;35:63-75
116. Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T. et al. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182:5899-903
117. Chauvistre H, Kustermann C, Rehage N, Klisch T, Mitzka S, Felker P. et al. Dendritic cell development requires histone deacetylase activity. Eur J Immunol. 2014;44:2478-88
118. Zhong L, Yang Q, Xie W, Zhou J. Liver X receptor regulates mouse GM-CSF-derived dendritic cell differentiation in vitro. Mol Immunol. 2014;60:32-43
119. Kim YH, Han SB, Lee JK. Histone deacetylase inhibitors suppress CXCR4-mediated dendritic cell migration by regulation of maturation process. Cell Immunol. 2013;284:139-45
120. Leu SJ, Yang YY, Liu HC, Cheng CY, Wu YC, Huang MC. et al. Valproic Acid and Lithium Meditate Anti-Inflammatory Effects by Differentially Modulating Dendritic Cell Differentiation and Function. J Cell Physiol. 2017;232:1176-86
121. Loo TT, Gao Y, Lazarevic V. Transcriptional regulation of CD4(+) TH cells that mediate tissue inflammation. J Leukoc Biol. 2018;104:1069-85
122. Thapa P, Farber DL. The Role of the Thymus in the Immune Response. Thorac Surg Clin. 2019;29:123-31
123. Ikawa T. Genetic and epigenetic control of early lymphocyte development. Curr Top Microbiol Immunol. 2014;381:1-20
124. Di Vito C, Mikulak J, Mavilio D. On the Way to Become a Natural Killer Cell. Front Immunol. 2019;10:1812
125. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G. et al. Innate lymphoid cells-a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145-9
126. Sheng Y, Wu T, Dai Y, Ji K, Zhong Y, Xue Y. The effect of 6-gingerol on inflammatory response and Th17/Treg balance in DSS-induced ulcerative colitis mice. Ann Transl Med. 2020;8:442
127. Salkowska A, Karas K, Walczak-Drzewiecka A, Dastych J, Ratajewski M. Differentiation stage-specific effect of histone deacetylase inhibitors on the expression of RORgammaT in human lymphocytes. J Leukoc Biol. 2017;102:1487-95
128. Sugimoto K, Itoh T, Takita M, Shimoda M, Chujo D, SoRelle JA. et al. Improving allogeneic islet transplantation by suppressing Th17 and enhancing Treg with histone deacetylase inhibitors. Transpl Int. 2014;27:408-15
129. Wu WP, Tsai YG, Lin TY, Wu MJ, Lin CY. The attenuation of renal fibrosis by histone deacetylase inhibitors is associated with the plasticity of FOXP3(+)IL-17(+) T cells. BMC Nephrol. 2017;18:225
130. Regna NL, Chafin CB, Hammond SE, Puthiyaveetil AG, Caudell DL, Reilly CM. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin Immunol. 2014;151:29-42
131. Dayoub O, Le Lay S, Soleti R, Clere N, Hilairet G, Dubois S. et al. Estrogen receptor alpha/HDAC/NFAT axis for delphinidin effects on proliferation and differentiation of T lymphocytes from patients with cardiovascular risks. Sci Rep. 2017;7:9378
132. Pradhan S, Das A, Singh H, Chaturvedi S, Singh K, Mishra R. et al. Steroid Responsiveness in Duchenne Muscular Dystrophy - Predictive Value of Epigenetic Regulator Histone Deacetylase 2. Indian J Pediatr. 2020
133. Zhao H, Li G, Wang R, Tao Z, Ma Q, Zhang S. et al. Silencing of microRNA-494 inhibits the neurotoxic Th1 shift via regulating HDAC2-STAT4 cascade in ischaemic stroke. Br J Pharmacol. 2020;177:128-44
134. Regna NL, Vieson MD, Luo XM, Chafin CB, Puthiyaveetil AG, Hammond SE. et al. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin Immunol. 2016;162:58-73
135. Oh BR, Suh DH, Bae D, Ha N, Choi YI, Yoo HJ. et al. Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res Ther. 2017;19:154
136. Laino AS, Betts BC, Veerapathran A, Dolgalev I, Sarnaik A, Quayle SN. et al. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J Immunother Cancer. 2019;7:33
137. Tsuji G, Okiyama N, Villarroel VA, Katz SI. Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. J Allergy Clin Immunol. 2015;135:1228-39
138. Hodge G, Jersmann H, Tran HB, Roscioli E, Holmes M, Reynolds PN. et al. Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes. Respir Res. 2015;16:130
139. Saunders KO, Freel SA, Overman RG, Cunningham CK, Tomaras GD. Epigenetic regulation of CD8(+) T-lymphocyte mediated suppression of HIV-1 replication. Virology. 2010;405:234-42
140. Zhang HP, Fu JJ, Fan T, Zhang WB, Wang ZL, Wang L. et al. Histone deacetylation of memory T lymphocytes by You-Gui-Wan alleviates allergen-induced eosinophilic airway inflammation in asthma. Chin Med. 2015;10:9
141. Woods DM, Woan KV, Cheng F, Sodre AL, Wang D, Wu Y. et al. T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood. 2017;130:146-55
142. Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA. et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101:3921-6
143. Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S. et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34:776-87
144. Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y. et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood. 2015;125:815-9
145. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL. et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695-704
146. Wang D, Iclozan C, Liu C, Xia C, Anasetti C, Yu XZ. LBH589 enhances T cell activation in vivo and accelerates graft-versus-host disease in mice. Biol Blood Marrow Transplant. 2012;18:1182-90 e1
147. Zaini J, Andarini S, Tahara M, Saijo Y, Ishii N, Kawakami K. et al. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J Clin Invest. 2007;117:3330-8
148. Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206-15
149. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825-39
150. Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, Younes A. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood. 2011;117:2910-7
151. Rughani G, Robertson L, Clarke M. Medical treatment for small abdominal aortic aneurysms. Cochrane Database Syst Rev. 2012: CD009536.
152. Li LJ, Ma N, Zeng L, Mo LH, Li XX, Xu LZ. et al. Flagellin modulates IgE expression in B cells to initiate food allergy in mice. Am J Transl Res. 2016;8:2748-57
153. Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR. et al. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell. 2016;165:1120-33
154. Thio CL, Chi PY, Lai AC, Chang YJ. Regulation of type 2 innate lymphoid cell-dependent airway hyperreactivity by butyrate. J Allergy Clin Immunol. 2018;142:1867-83 e12
155. Harrison IF, Smith AD, Dexter DT. Pathological histone acetylation in Parkinson's disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci Lett. 2018;666:48-57
156. Tan Y, Delvaux E, Nolz J, Coleman PD, Chen S, Mastroeni D. Upregulation of histone deacetylase 2 in laser capture nigral microglia in Parkinson's disease. Neurobiol Aging. 2018;68:134-41
157. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892-901
158. Xuan A, Long D, Li J, Ji W, Hong L, Zhang M. et al. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012;90:463-8
159. Patnala R, Arumugam TV, Gupta N, Dheen ST. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol Neurobiol. 2017;54:6391-411
160. Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM. et al. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008;1226:181-91
161. Lu WH, Wang CY, Chen PS, Wang JW, Chuang DM, Yang CS. et al. Valproic acid attenuates microgliosis in injured spinal cord and purinergic P2X4 receptor expression in activated microglia. J Neurosci Res. 2013;91:694-705
162. Kim T, Song S, Park Y, Kang S, Seo H. HDAC Inhibition by Valproic Acid Induces Neuroprotection and Improvement of PD-like Behaviors in LRRK2 R1441G Transgenic Mice. Exp Neurobiol. 2019;28:504-15
163. Zhu X, Wang S, Yu L, Jin J, Ye X, Liu Y. et al. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer's disease. Aging Cell. 2017;16:1073-82
164. Wu Y, Dou J, Wan X, Leng Y, Liu X, Chen L. et al. Histone Deacetylase Inhibitor MS-275 Alleviates Postoperative Cognitive Dysfunction in Rats by Inhibiting Hippocampal Neuroinflammation. Neuroscience. 2019;417:70-80
165. Hsing CH, Hung SK, Chen YC, Wei TS, Sun DP, Wang JJ. et al. Histone Deacetylase Inhibitor Trichostatin A Ameliorated Endotoxin-Induced Neuroinflammation and Cognitive Dysfunction. Mediators Inflamm. 2015;2015:163140
166. Durham BS, Grigg R, Wood IC. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem. 2017;143:214-24
167. Jiao FZ, Wang Y, Zhang HY, Zhang WB, Wang LW, Gong ZJ. Histone Deacetylase 2 Inhibitor CAY10683 Alleviates Lipopolysaccharide Induced Neuroinflammation Through Attenuating TLR4/NF-kappaB Signaling Pathway. Neurochem Res. 2018;43:1161-70
168. Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H. et al. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 2007;149:203-12
169. Alqinyah M, Maganti N, Ali MW, Yadav R, Gao M, Cacan E. et al. Regulator of G Protein Signaling 10 (Rgs10) Expression Is Transcriptionally Silenced in Activated Microglia by Histone Deacetylase Activity. Mol Pharmacol. 2017;91:197-207
170. Chen S, Ye J, Chen X, Shi J, Wu W, Lin W. et al. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-kappaB pathway dependent of HDAC3. J Neuroinflammation. 2018;15:150
171. Faraco G, Pittelli M, Cavone L, Fossati S, Porcu M, Mascagni P. et al. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol Dis. 2009;36:269-79
172. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y. et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A. 2015;112:2853-8
173. Jaworska J, Zalewska T, Sypecka J, Ziemka-Nalecz M. Effect of the HDAC Inhibitor, Sodium Butyrate, on Neurogenesis in a Rat Model of Neonatal Hypoxia-Ischemia: Potential Mechanism of Action. Mol Neurobiol. 2019;56:6341-70
174. Baby N, Li Y, Ling EA, Lu J, Dheen ST. Runx1t1 (Runt-related transcription factor 1; translocated to, 1) epigenetically regulates the proliferation and nitric oxide production of microglia. PLoS One. 2014;9:e89326
175. Kannan V, Brouwer N, Hanisch UK, Regen T, Eggen BJ, Boddeke HW. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J Neurosci Res. 2013;91:1133-42
176. Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem. 2003;87:407-16
177. Singh V, Bhatia HS, Kumar A, de Oliveira AC, Fiebich BL. Histone deacetylase inhibitors valproic acid and sodium butyrate enhance prostaglandins release in lipopolysaccharide-activated primary microglia. Neuroscience. 2014;265:147-57
178. Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V. et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 2016;131:323-45
179. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481-7
180. Hartmann K, Sepulveda-Falla D, Rose IVL, Madore C, Muth C, Matschke J. et al. Complement 3(+)-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol Commun. 2019;7:83
181. Kalinin S, Polak PE, Lin SX, Braun D, Guizzetti M, Zhang X. et al. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J Neuroimmunol. 2013;263:13-9
182. Wang CC, Chen PS, Hsu CW, Wu SJ, Lin CT, Gean PW. Valproic acid mediates the synaptic excitatory/inhibitory balance through astrocytes-a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry. 2012;37:111-20
183. Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC. et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116-25
184. Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC. et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123-34
185. Marinova Z, Leng Y, Leeds P, Chuang DM. Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology. 2011;60:1109-15
186. Choi JY, Kim JH, Jo SA. Acetylation regulates the stability of glutamate carboxypeptidase II protein in human astrocytes. Biochem Biophys Res Commun. 2014;450:372-7
187. Jia H, Wang Y, Morris CD, Jacques V, Gottesfeld JM, Rusche JR. et al. The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington's Disease Mice. PLoS One. 2016;11:e0152498
188. Wang X, Waschke BC, Woolaver RA, Chen SMY, Chen Z, Wang JH. HDAC inhibitors overcome immunotherapy resistance in B-cell lymphoma. Protein Cell. 2020
189. Marquard L, Poulsen CB, Gjerdrum LM, de Nully Brown P, Christensen IJ, Jensen PB. et al. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology. 2009;54:688-98
190. Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin's lymphoma. Expert Opin Ther Targets. 2010;14:577-84
191. Van Damme M, Crompot E, Meuleman N, Mineur P, Dessars B, El Housni H. et al. Global histone deacetylase enzymatic activity is an independent prognostic marker associated with a shorter overall survival in chronic lymphocytic leukemia patients. Epigenetics. 2014;9:1374-81
192. Sun X, Hasanali ZS, Chen A, Zhang D, Liu X, Wang HG. et al. Suberoylanilide hydroxamic acid (SAHA) and cladribine synergistically induce apoptosis in NK-LGL leukaemia. Br J Haematol. 2015;168:371-83
193. Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun. 2006;345:1471-80
194. Pasqualucci L, Bereshchenko O, Niu H, Klein U, Basso K, Guglielmino R. et al. Molecular pathogenesis of non-Hodgkin's lymphoma: the role of Bcl-6. Leuk Lymphoma. 2003;44(Suppl 3):S5-12
195. Seo JS, Cho NY, Kim HR, Tsurumi T, Jang YS, Lee WK. et al. Cell cycle arrest and lytic induction of EBV-transformed B lymphoblastoid cells by a histone deacetylase inhibitor, Trichostatin A. Oncol Rep. 2008;19:93-8
196. Moldenhauer A, Frank RC, Pinilla-Ibarz J, Holland G, Boccuni P, Scheinberg DA. et al. Histone deacetylase inhibition improves dendritic cell differentiation of leukemic blasts with AML1-containing fusion proteins. J Leukoc Biol. 2004;76:623-33
197. Kaneko M, Ishihara K, Takahashi A, Hong J, Hirasawa N, Zee O. et al. Mechanism for the differentiation of EoL-1 cells into eosinophils by histone deacetylase inhibitors. Int Arch Allergy Immunol. 2007;143(Suppl 1):28-32
198. Silva G, Cardoso BA, Belo H, Almeida AM. Vorinostat induces apoptosis and differentiation in myeloid malignancies: genetic and molecular mechanisms. PLoS One. 2013;8:e53766
199. Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63:3637-45
200. Zhang H, Du M, Yang Q, Zhu MJ. Butyrate suppresses murine mast cell proliferation and cytokine production through inhibiting histone deacetylase. J Nutr Biochem. 2016;27:299-306
201. Lin TY, Fenger J, Murahari S, Bear MD, Kulp SK, Wang D. et al. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood. 2010;115:4217-25
202. Imai Y, Hirano M, Kobayashi M, Futami M, Tojo A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers (Basel). 2019 11
203. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188-93
204. Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D. et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064-9
205. Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF. et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720-8
206. Gupta P, Reid RC, Iyer A, Sweet MJ, Fairlie DP. Towards isozyme-selective HDAC inhibitors for interrogating disease. Curr Top Med Chem. 2012;12:1479-99
Author contact
![]() Corresponding authors: Dr. Haiping Zhao, Institute of Cerebrovascular Diseases Research, Xuanwu Hospital of Capital Medical University, 45 Changchun Street, Beijing 100053, China. Tel: +86 10 83198129. E-mail: zhaohaipingorg; or to Dr. Yumin Luo, E-mail: yumin111edu.cn.
Corresponding authors: Dr. Haiping Zhao, Institute of Cerebrovascular Diseases Research, Xuanwu Hospital of Capital Medical University, 45 Changchun Street, Beijing 100053, China. Tel: +86 10 83198129. E-mail: zhaohaipingorg; or to Dr. Yumin Luo, E-mail: yumin111edu.cn.