Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
MMR system
MMR and telomere attrition
MMR and mitochondrial dysfunction
MMR and epigenetic alterations
MMR and nutrient sensing
MMR and cell senescence
Conclusions and perspectives
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(14):3923-3935. doi:10.7150/ijbs.64953 This issue Cite
Review
Role of mismatch repair in aging
Jie Wen1,2,3,4, Yangyang Wang1,2, Minghao Yuan1,2, Zhenting Huang1,2, Qian Zou1,2, Yinshuang Pu1,2, Bin Zhao3,4 ![]() , Zhiyou Cai1,2
, Zhiyou Cai1,2 ![]()
1. Chongqing Key Laboratory of Neurodegenerative Diseases, Chongqing, 400013, China.
2. Department of Neurology, Chongqing General Hospital, University of Chinese Academy of Sciences, Chongqing, 400013, China.
3. Department and Institute of Neurology, Guangdong Medical University, Guangdong, 524001, China.
4. Guangdong Key Laboratory of aging related cardio cerebral diseases, Guangdong, 524001, China.
Received 2021-7-14; Accepted 2021-9-7; Published 2021-9-21
Abstract

A common feature of aging is the accumulation of genetic damage throughout life. DNA damage can lead to genomic instability. Many diseases associated with premature aging are a result of increased accumulation of DNA damage. In order to minimize these damages, organisms have evolved a complex network of DNA repair mechanisms, including mismatch repair (MMR). In this review, we detail the effects of MMR on genomic instability and its role in aging emphasizing on the association between MMR and the other hallmarks of aging, serving to drive or amplify these mechanisms. These hallmarks include telomere attrition, epigenetic alterations, mitochondrial dysfunction, altered nutrient sensing and cell senescence. The close relationship between MMR and these markers may provide prevention and treatment strategies, to reduce the incidence of age-related diseases and promote the healthy aging of human beings.
Keywords: aging, mismatch repair, genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction
Introduction
The proportion of people over 65 is rapidly increasing in most industrialized countries [1]. This demographic milestone will be accompanied by a significant increase in age-related illnesses such as Alzheimer's disease, cardiovascular disease, and cancer [2]. Aging is a progressive degenerative state, accompanied by tissue stem cell depletion, tissue inflammation, matrix changes, cell senescence, and metabolic dysfunction. The current status of aging research has many similarities with cancer research. Cancer and aging share a common origin and can be thought of as two different manifestations of the same underlying process, which is the accumulation of cell damage [3]. In addition, many diseases of premature aging, such as Werner syndrome and Bloom syndrome, are the result of increased accumulation of DNA damage [4].
The integrity and stability of DNA are constantly challenged by exogenous physical, chemical, and biological media, as well as endogenous threats, such as DNA replication errors, spontaneous hydrolysis and reactive oxygen species (ROS). Meanwhile, the completeness of genetic information depends to a large extent on the accuracy of the DNA replication process [5]. In order to maintain the integrity of the genome, cells have a variety of mechanisms for repairing DNA damage, including MMR, base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), non-homologous terminal connection (NHEJ) [6, 7]. This article reviews the close relationship between MMR and aging mechanism from several aspects, in order to provide some new insights into how to fight aging and prevent aging-related diseases.
MMR system
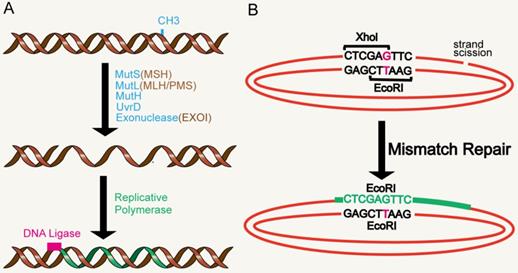
The DNA MMR system is ubiquitous among organisms, from prokaryotes to eukaryotes. In 1964, MMR was conceived simultaneously by Robin Holliday explaining genetic transformation in yeast and by Evelyn Witkin explaining brominated nucleotide processing in bacteria [8, 9]. MMR, an evolutionarily conserved process, corrects the mismatches that occur during DNA replication and escape proofread [10]. Its primary function includes excision-repair, through which a region containing the wrong base in newly synthesized DNA chain is removed and resynthesized [11] (Figure 1A). The main proteins involved in the MMR system are MLH1, MutS protein homology 2 (MSH2), MutS homology 6 (MS6) and PMS1 homology 2 (PMS2), which together interact in the form of heterodimers, whereby MSH2 binds to MSH6 or MSH3 to form MutSα and MutSβ complexes respectively, while MLH1 binds to PMS2 or MLH3 [12-14]. The basic MMR reaction is most easily understood in Escherichia coli. MMR is initiated by MutS homodimer upon recognition of mismatches, which then recruits MutL (also a homodimer complex) to the mismatch sites. MutL interacts with the third MMR component called MutH, resulting in the activation of the potential endonuclease activity of MutH and formation of a gap in the newly replicated chain. Chain recognition is a basic feature of MMR, which limits the use of amphiphilic chains as templates for DNA re-synthesis, thus eliminating misincorporation errors [11, 15, 16]. Eukaryotic MMR is similar to bacterial MMR in mechanism of action. However, its mismatch recognition uses two MutS homologous complexes, MSH2-MSH6 and MSH2-MSH3, which have partially overlapping mismatch recognition specificity, rather than a single MutS homodimer as in prokaryotes. The third MSH complex (MSH4-MSH5) does not have MMR-related functions, yet is very important in the process of meiosis and recombination [17, 18]. Reconstruction of the MMR reaction in E. coli began in 1983 using DNA substrates containing two overlapping restriction enzyme sites with central mismatches [19] (Figure 1B). The most significant MutL activity in human cells is provided by MutLα heterodimer MLH1/PMS2. In addition, human MLH1 also exists as heterodimers containing PMS1 and MLH3, called MutLβ and MutLγ [20, 21]. Studies have found that MMR plays an important role in the process of aging.
MMR reaction. A. MMR excision-resynthesis process. The reaction degrades one strand and uses the complementary strand as a repair template to eliminate the mismatch. The γ-proteobacteria components directing the specific excission are shown in blue. Bacterial (besides γ-proteobacteria), archaeal, and eukaryotic components are shown in brown. Resynthesis at the exonuclease gap is accomplished by replicating polymerase, and the remaining strand breaks are blocked by DNA ligase. B. Illustration of a simple MMR DNA substrate that contains overlapping restriction sites containing mismatches leading to restriction resistance to endonuclease.
Genomic instability, one of the key hallmarks of aging, leads to stem cell exhaustion and triggers inflammation. Genomic stability is supported by complex repair mechanisms, damage tolerance, and checkpoint pathways that counteract DNA damage. DNA damage is responsible for the development of cancer and many age-related diseases [22]. Causal evidence for a link between lifelong increase in genomic damage and aging comes from studies on mice and humans, showing that defects in DNA repair mechanisms cause accelerated aging in mice and are the basis for several human genetic syndromes, including Werner syndrome, Bloom syndrome, xeroderma pigmentosa, and trichothiodystrophy [22-24]. Several mutation patterns in somatic cells are either associated with exposure to substances that damage DNA or with genomic instability due to DNA repair failure [25]. Loss of MMR function induces a hypermutant phenotype clinically identified as microsatellite instability (MSI). dMMR (MMR deficiency)/MSI tumors exhibit proteome-wide protein instability associated with numerous unstable mutations [26]. Summarizing the specific relationship between MMR and the aging process may provide some strategies for humans to delay aging.
MMR and telomere attrition
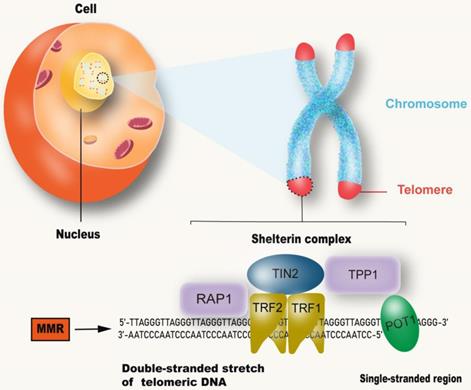
Although the process and potential molecular mechanisms of aging have not been fully understood [27], more evidence points to telomeres, which are believed to be the initiators or amplifiers of the molecular circuits that drive the aging process and related diseases [28] (Figure 2). Telomere shortening appears to be one of the biological manifestations of aging [3]. Short telomeres, rather than average-length telomeres, determine cell viability and chromosome stability [29]. Reaching the critical telomere length can lead to replicative senescence or programmed cell death [30]. Telomeres are made up of repeated nucleotide sequences to form a “cap structure” which function to maintain the integrity of chromosomes. Human telomere maintenance related gene defects are associated with reproductive and somatic degenerative diseases, such as congenital keratosis, idiopathic pulmonary fibrosis, ulcerative colitis, etc. [31]. Telomeres are strictly regulated by telomerase. In most eukaryotes, telomerase uses a complete RNA template consiting of telomerase RNA and telomerase reverse transcriptase (TERT), to synthesize telomere repeat sequences (TTAGGG) at the end of chromosomes [32]. These repeat sequences compensate for telomere loss caused by incomplete replication at the end of the genome [33]. In humans, telomerase expression is up-regulated during embryonic development and cancer, and mutations that damage telomerase function can lead to disease [34]. Furthermore, telomeres are covered by special proteins called protegerin complexes, which are polymers composed of six protein subunits: TRF1, TRF2, TPP1, POT1, TIN2 and RAP1 [35]. Protegerin subunits organize telomeres into spherical structures through complex interactions with telomere DNA [36, 37]. These advanced structures of telomeres suppresses DNA damage signals from telomere terminals, prevent DNA repair mechanisms from fusing the terminals through recombination or classical/alternative NHEJ, and regulate telomerase pathway and activity. Accordingly, mutations in the above components can destroy the protegerin-telomere complex, leading to terminal fusion and premature senescence [38, 39].
MMR and telomere attrition. Telomeres are located at the end of linear chromosomes in eukaryotic cells and are protected by polymers composed of six protein subunits: TRF1, TRF2, TPP1, POT1, TIN2 and RAP1. The absence of MMR system can accelerate the shortening of telomeres.
For example, defects in the MMR system were suggested to lead to HR of telomere ends resulting in telomerase-independent telomeres in gastric cancer [40]. The DNA MMR system maintains the stability of the genome not only by repairing DNA replication errors, but also by preventing homologous chromosome recombination [41]. Indeed, the absence of MMR helps cells overcome the cellular crisis caused by telomere dysfunction through telomere recombination in telomerase-deficient yeast or mammalian cells. The loss of MMR function promoted the proliferation of telomerase deficient yeast cells [42], inhibited telomerase activity and accelerated the ALT-like telomere elongation of MMR-deficient human colon cancer cells [43]. DNA MMR is essential for genomic stability and inheritance of MMR genes. Mutated MMR genes are most commonly caused by MSH2 or MLH1, leading to cancer susceptibilities such as in Lynch syndrome and hereditary nonpolyposis colorectal cancer (HNPCC). The average telomere shortening rate of MSH2 deletion clones was significantly higher than that of control clones. This was the first evidence that MSH2 deficiency alone can lead to accelerated telomere shortening in normal human cells [44].
Pathological telomere shortening can lead to genomic instability and lymphatic transformation. At least two main pathways have been proposed to lead to colorectal cancer (CRC): loss of heterozygosity caused by chromosome instability and DNA MMR defects caused by microsatellite instability [45-47]. Severe genomic instability in telomere crisis accelerates secondary genetic changes that lead to carcinogenesis and thus emphasize the significance of pathological telomere length changes in cancer pathways, including colorectal cancer [48-50]. Relative telomere length (RTL) is closely related to clinicopathological predictive markers of sporadic colorectal cancer. The expression of MMR proteins including MLH1, MSH2, PMS2, MSH6 and p53 protein is normal in sporadic colorectal cancer [51]. In the late stage of tumorigenesis, telomere loss leads to genomic instability and telomerase activation promotes immortalization [52].
MMR and mitochondrial dysfunction
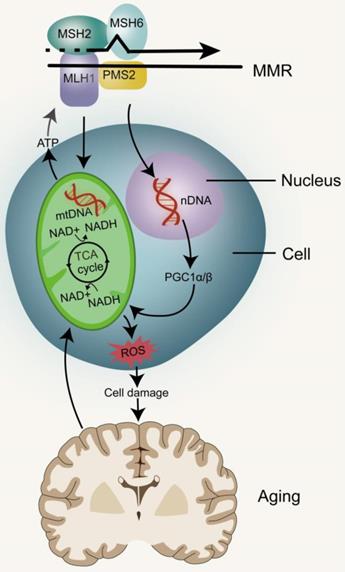
Mitochondria are important organelles for executing and coordinating various metabolic processes in cells and participating in ion homeostasis, cell growth, redox state, and cell signaling (Figure 3). They are also main source of intracellular ATP and play prominent roles in cell life and death [53, 54]. Mitochondria are unique because they contain their own DNA (mtDNA), which encodes many proteins that are essential for the assembly and activity of mitochondrial respiratory complexes, promote mitochondrial replication, transcription and repair of mtDNA. With a few exceptions, animal mitochondrial DNA encodes 13 key proteins, and about 1500 nuclear DNA (nDNA)-encoded proteins are destined for the mitochondria [55]. Defects or mutations in mitochondrial DNA can lead to several diseases, including cancer, and mitochondrial diseases, while damaged mtDNA can be eliminated by mitosis [56-58]. The mutation rate of mtDNA is significantly higher than that of nDNA, because it is close to high concentrations of toxic metabolites and the repair mechanism is relatively inefficient [59]. DNA deletion and point substitution can lead to serious disorders of electron transport chain (ETC) function, mitochondrial genome replication and mitochondrial gene expression [7]. ATP is produced through the Krebs cycle in the mitochondrial matrix and oxidative phosphorylation (OXPHOS) in the mitochondrial inner membrane, with ROS and heat as byproducts [55]. Mitochondrial dysfunction leads to reduction of ATP produced oxidative phosphorylation, inability to regulate excessive ROS and nitrogen production, calcium regulation disorders, opening of permeability transition pores and initiation of apoptosis [60]. Studies in the free-living transparent nematode, Caenorhabditis elegans, have shown that reducing the activity of mitochondrial ETC in the early stage of life leads to extensive chromatin recombination. This is often required to activate mitochondrial unfolded proteins in response to stress, a process that promotes the restoration of mitochondrial protein homeostasis and regulate stress-induced lifespan [61, 62]. Therefore, partial inhibition of mitochondrial activity has been shown to prolong the lifespan of worms, flies and mice, but complete or nearly complete loss of mitochondrial function is harmful to the organism [63-66]. The causes of mitochondrial dysfunction include changes in mitochondrial dynamics caused by the imbalance between fission and fusion events, quality control of defects caused by mitosis, a form of bulk autophagy peculiar to organelles and protein degradation targeted at defective mitochondria [67]. mtDNA mutations, deletions or impaired DNA replication are the most common causes of mitochondrial dysfunction [68].
MMR and mitochondrial dysfunction. The stable inheritance of mtDNA and nDNA requires the participation of MMR. mtDNA mutations directly promote mitochondrial dysfunction. nDNA damage can lead to mitochondrial dysfunction by inhibiting PGC1α and PGC1β, and eventually lead to aging. Impairment of mitochondrial function can affect the production of ATP thereby reducing the energy needed by MMR, as well as produce metabolic toxins such as ROS to promote aging.
The general mechanism of MMR includes locating the mismatch and repairing the newly synthesized chain through excision and DNA re-synthesis, followed by reconnecting to complete the repair process [69]. Spontaneous errors in the mechanism of mtDNA replication can lead to point mutations and deletions. The MMR system corrects post-replication errors and maintains genetic stability. Efficient MMR systems also identify and cut sequence variants in mtDNA [70]. For a long time, research work was focused on the identification of MMR proteins located in the nucleus, because few key factors involved in MMR had been identified in mitochondria, and only relative recognition complexes MutSα and MutSβ had been detected in mitochondrial MMR activity. In fact, the mtDNA repair depends to a large extent on the mechanism of nDNA repair [69]. YB-1 protein is another nuclear MMR factor, which is partially located in mitochondria. When small interference RNA (siRNA) was used to inhibit the expression of Yb-1, the activity of MMR in mitochondrial extract decreased by more than 3 times [71]. So far, no other participants of nuclear MMR have been detected in mitochondria [7]. mtDNA has a large non-coding sequence, the displacement-loop (D-loop), which contains the essential elements for transcription and replication [72], and studies have shown that this region suffers more damage than other regions of the mtDNA [73]. As the MMR system was damaged, sequence mutations in the D-loop region were not repaired, resulting in impaired mtDNA transcription. It has been shown that in diabetes, mitochondrial dysfunction in the retina and capillary cells cause initiation of apoptosis, mtDNA damage, electron transport chain damage, mtDNA biogenesis, and copy number reduction [74, 75]. Mitochondria become dysfunctional, and even after hyperglycemic condition is reversed to normal, they remain dysfunctional, leading to the development and progression of diabetic retinopathy. Therefore, strategies that target mtMMR mechanisms can help maintain mitochondrial homeostasis [76]. MLH1, one of the key proteins involved in the MMR pathway, suppresses a few mitochondrial genes, including POLG and PINK1, that induce synthetic death in MLH1-deficient cells [76-78]. Recent studies have shown that the loss of MLH1 is related to mitochondrial metabolism disorders, with basal oxygen consumption rate reduced and spare respiratory capacity reduced. In addition, loss of MLH1 resulted in decreased OCR, decreased complexⅠactivity, and increased reactive oxygen species [79]. The relationship between mtDNA and base mismatch has been doubted for a long time, and better understanding of its intricacies is still an important challenge in aging research.
Elevated levels of DNA damage activate p53, and the increase in p53 levels results in mitochondrial dysfunction by inhibiting PPARγ coactivator1α (PGC1α) and PGC1β (which promotes mitochondrial biogenesis). The p53-mediated mitochondrial dysfunction triggers the DNA damage cycle by affecting the production of ROS, Fe-S clusters and NADH/NAD, which leads to further activation of p53 and mitochondrial damage [80]. The deficiency of ATP production and the increase of ROS level can promote cell senescence. With the aging of cells and organisms, mitochondria gradually become inefficient and potentially toxic; the efficacy of the respiratory chain tends to weaken, thereby increasing electron leakage and reducing the production of ATP [81]. The initiation of MMR in eukaryotes depends on the recognition of DNA mispairs by partially redundant MSH2-MSH6 and MSH2-MSH3 heterodimers which are homologues of bacterial MutS homodimers [82, 83]. MSH2-MSH6, MSH2-MSH3 and MutS belong to the ABC family of ATPases, and there are two nucleotide binding sites at the interface of their dimers. When the MSH2-MSH6 complex binds to mispairs, it also binds to ATP at its two nucleotide binding sites, resulting in a conformational change. This ATP-activated MutS state promotes its interaction with one or more MutL proteins. ATP binding, hydrolysis and ADP release are intrinsically related to the conformational changes that play a key role in MMR [17, 84-86]. MMR of DNA can lead to mtDNA mutation and nDNA damage, which in turn leads to mitochondrial dysfunction and accelerates the aging process. The key potential factor of cell change in the process of aging is metabolic disorder and mitochondrial dysfunction in turn hinders metabolic processes, resulting in a vicious circle [87].
MMR and epigenetic alterations
Epigenetics, which includes DNA methylation, histone modification, chromatin remodeling and non-coding RNA, represents a reversible genetic mechanism that occurs without changing the underlying DNA sequence [3, 88, 89]. Although all cells have the same DNA, the epigenetic mechanism determines the fate of cells, such as whether they become hepatocytes or neurons, as well as the maturation of different types of cells. Importantly, epigenetics can be influenced by environmental factors. Thus epigenetic alterations can be spontaneous or driven by external or internal factors. For example, identical twins share the same DNA, and epigenetic markers are similar among young twins. Studies on human longevity have shown that genetic factors can explain a small portion of the observed differences in the lifespan of identical twins (20% to 30%), while the bulk of remaining variations are thought to be caused by epigenetic drift over their lifetime [90-93]. Epigenetic alterations are thus related to aging, however, how this affects aging is not clear. Studies have suggested increased histone H4K16 acetylation, H4K20 trimethylation or H3K4 trimethylation, as well as decreased H3K9 methylation or H3K27 trimethylation, as age-related epigenetic markers [94, 95]. The MMR pathway initiates epigenetic alterations during inflammation-induced tumorigenesis [96].
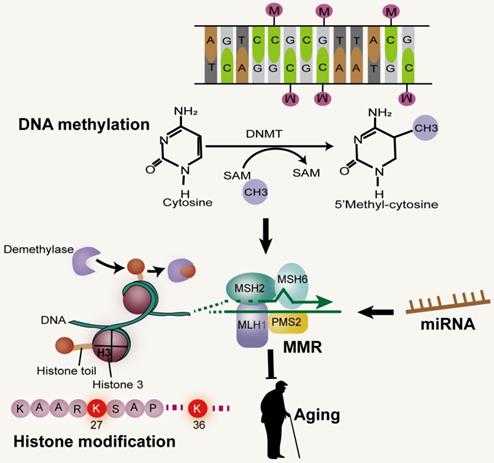
DNA methylation at C of CpG dyads (CpG) in vertebrate genomes is vital for gene regulation, genome stability and development [97]. In the early stages of development, CpG methylation increases in the brain. Although there is no significant change in the global level during aging, the CpG methylation pattern seems to be changed. In some tissues, reduced CpG methylation was observed in repetitive sequences, including transposable elements [98]. The high fidelity of DNA methylation patterns in mammals after each cell division is regulated by DNA methyltransferase (Dnmt) [94]. Studies have shown that the normal function of post-replicative DNA MMR in mammalian cells depends on the existence of genomic CpG and the maintenance of Dnmt1, independent of its catalytic activity. Furthermore, the efficient monitoring of mammalian MMR is achieved through the hemi-CpG-Np95 (Uhrf1)-Dnmt1 axis, through which the MMR monitoring surveillance complex is recruited by Dnmt1 to the replicated DNA, a process that requires it to bind to MutSα and Np95 on the hemi-methylated CpG site. Therefore, the efficiency of MMR monitoring of mammalian genome in vivo is improved at the epigenetic level [97] (Figure 4). In addition, MSH2 and MSH6 recruits epigenetic silencing proteins Dnmt1 and EZH2 to oxidative damage sites through a protein-protein interaction-dependent mechanism. It has been proven that the heterodimer of MMR protein, MSH2-MSH6, is involved in the recruitment of Dnmt1 to chromatin impaired by oxidative damage. The role of Dnmt1 in the oxidative damage site is to reduce transcription and potentially block the repair process by transcriptional interference [99].
MMR and epigenetic alterations. Mammalian MMR is related to Dnmt1 and CpG. Reductions of H3K36me3 levels can impair the MMR pathway. miRNA, a non-coding RNA, has been shown to be involved in the DNA MMR.
Studies have shown that the level of H3K27me3 in wild-type fruit flies increases with age. One possible reason that aging promotes the drift of H3K27me3 modification is that age-related DNA damage reduces the fidelity of epigenetic markers. Methylation of histone H3K9 and H3K27, acetylation of histone H3 and citrullination of histone H2A1R3 were up-regulated in patients with colorectal cancer [100]. UV-C radiation can induce the increase of H3K27me3 mediated by polycomb repressive complex2 (PRC2) in the silkworm, Bombyx mori [101]. Meanwhile, in Neurospora crassa, the increase and redistribution of H3K27me3 can be caused by loss of the H3K9 methyltransferase complex [102]. H3K36me3 is an important regulator of MMR pathway as earlier reported, and its imbalance may be due to the resultant interaction between damaged DNA and alterations in the level of epigenetic markers during senescence [103]. The epigenetic inhibition of DNA damage repair genes caused by hypermethylation of MLH1 and MSH2 promoters, and the inefficient recruitment of MMR complex at DNA damage site caused by the decrease of H3K36me3 level can impair the MMR pathway [104]. In addition, many studies have shown that histone deacetylases (HDACs) and histone acetyltransferases (HATs) play an important role in DNA repair and DNA damage response(DDR), by identifying DNA double strand breaks [105] and promoting non-homologous terminal connections [106]. HDAC1 is also associated with other proteins involved in DDR, including proliferating cell nuclear antigens (PCNA), breast cancer 1 (BRCA1), ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3 related) [107]. There is evidence that HDAC6 deacetylates and ubiquitinates MSH2, and regulates DDR and MMR activity. However, it is not known whether MSH2 is regulated by other HDAC (except HDAC6) and HATS [108].
Epigenetic alterations in histone modification represent a prominent marker of aging [109]. Research on Drosophila found that aging leads to the loss of epigenetic marker distortion and the drift of H3K27me3, which in turn decrease expression of glycolysis genes, resulting in a negative effect on energy production and the redox state [110]. The epigenetic regulation of chromatin remodeling on gene expression is crucial to the function of adult stem cells. The decline of stem cell function can be observed during aging, accompanied by currently unexplainable changes in chromatin structure [111]. There is coordination between histone modifiers and chromatin remodeling resulting in a coordinated response to prevent DNA damage [112]. Epigenetic proteins participate in DDR. “Active” chromatin modifiers such as HATS and chromatin remodeling complex are first recruited to the DNA damage site to enable DNA repair proteins to enter the “open” local chromatin structure. Once DNA repair proteins enter DNA, they recruit “inhibitory” chromatin modifiers such as HDAC to “turn off” chromatin and inhibit transcriptional activity. Finally, when the repair is completed, the chromatin returns to its original state [113]. It has been proven that JAK2 is recruited into chromatin and JAK2 interacts with MSH2 and MSH6 in the nucleus. Inhibition of JAK2 changes the chromatin interaction between MSH2, MSH6, Dnmt1 and PRC2 members [114].
MicroRNAs (miRNAs or miRs), short (~22s nucleotide) single-stranded RNAs, act as gene expression regulators by binding to target mRNAs and disrupting their stability or inhibiting their translation [115-117]. miRNAs have been shown to be involved in the DNA MMR pathway, a major genomic maintenance system. Evidence suggests that overexpression of miR-155 remarkably down-regulates core MMR proteins hMSH2, hMSH6, and hMLH1, inducing mutant phenotypes and MSI. The expression of miR-155 was found to be negatively correlated with the expression of MLH1 or MSH2 protein in human colorectal cancer [118]. In mammalian cells, primary miRNA transcripts (pri-miRNAs) are initially processed into precursor miRNAs (pre-miRNAs) in the nucleus and further processed in the cytoplasm to produce mature miRNAs [115-117]. In vitro and in vivo studies have demonstrated that MLH1 and miR-422a are involved in regulating feedback loops at both molecular levels. MutLα stimulated the transformation of pri-miR-422a into forward miR-422a, as well as the processing of other measured miRNAs, suggesting that MutLα is a general stimulator of miRNA biogenesis. In contrast, miR-422a down-regulated MutLα by pairing with MLH1 3'-untranslated region to inhibit MLH1 expression [119].
There is thus evidence that aging is related to genetic and epigenetic alterations. Given the reversibility of epigenetic mechanisms, its pathways provide a promising approach for the treatment of age-related decline and diseases [109].
MMR and nutrient sensing
Diet has been proven as a strong regulator of aging, and calorie restriction has become a valuable intervention (Figure 5), though many questions remain on how it affects aging-related processes. The regulation of nutrition sensing pathways diminishes with increasing of age, and eventually fails at advanced ages. These pathways form the link between diet and aging and can thus be regulated through drugs and dietary intervention. The nutrient sensing pathways of aging injury include IGF1/PI3K/AKT/mTOR and AMPK/Sirtuin/PGC1α. They medicate dominant roles in protein synthesis, cell cycle, DNA replication, autophagy, stress response and regulation of glucose homeostasis [120-123].
MMR and nutrient sensing. Dietary restriction leads to a decrease in blood glucose levels, which in turn lowers the levels of IGF-1 and insulin. This results in the decrease of downstream signal transduction of insulin receptor (IR) and insulin-like growth factor-1R (IGF-1R). During this time, mTOR is in an inactive state. The decrease in intracellular glucose utilization also increases the ratio of AMP/ATP, thus activating AMPK. AMPK can inhibit the ratio of mTOR complex 1. PI3K/AKT/mTOR may promote aging by inhibiting MMR.
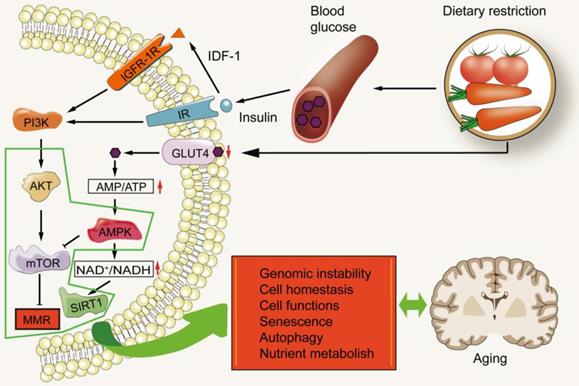
These pathways link metabolism, diet and aging. The growth axis of mammals includes growth hormone (GH) and insulin-like growth factor-1 (IGF-1). The intracellular signal pathway of IGF-1 is the same as that induced by insulin. Insulin informs cells of the presence of glucose [124, 125]. High glucose levels induce insulin release, which in turn increases IGF-1. IGF-1 binds to its receptor, turns on its self-phosphorylation, and leads to the subsequent activation of phosphatidylinositol-3 kinase (PI3K) which in turn phosphorylates and activates AKT. Activated AKT phosphorylates and activates mTOR, to inhibit FOXO. Decreased glucose levels are detected by the insulin receptor which then diminishes this signal cascade reaction [121]. mTOR, is a serine/threonine kinase, belonging to the phosphoinositide kinase related family. It can be found in two different complexes: mTOR complex 1 (MTORC1) and complex 2 (mTORC2). MTORC1 is activated by a variety of growth factors through the phosphatidylinositol-3 kinase-related family and the AKT kinase signal pathway. MTORC1 is also activated by nutrients (including amino acids and phosphates) and inhibited by AMP-activated protein kinase (AMPK). AMPK is the key sensor of cell energy state [126]. In fact, low levels of insulin and IGF-1, two growth factors that activate mTOR, are induced by calorie restriction, and are associated with healthier lives and longer lifespans [127].
During starvation conditions, mTOR dissociates from ULK1-mAtg13-FIP200 complex and induces ULK1 to activate and phosphorylate mAtg13 and FIP200, thus promoting the translocation of the protein complex to an autophagy site [128]. Mice subjected to intermittent fasting (IF) through dietary restrictions demonstrated better regulation of glucose homeostasis [129]. Rapamycin was found to inhibit the activation of Akt, probably by relieving the negative feedback inhibition of mTOR-S6K1. Akt activated by rapamycin inhibiting mTOR may phosphorylate and inactivate its substrate, such as FOXO, which has recently been found to be involved in autophagy in Drosophila and mouse skeletal muscles [127]. This research institution demonstrated that mTOR modulates the up-regulation of BNIP3 protein levels following MMR treatment with 6-TG and 5-FU [130]. The two key targets of MTORC1 signal transduction are 4EBP1 and S6K, which are the regulatory factors of translation initiation. Inhibition of mTORC-dependent translation has been shown to prolong life and provide protection against several age-related diseases [131]. The PI3K/AKT/mTOR pathway is often activated in human tumors and up-regulated in MMR-deficient tumors [132]. Similarly, PI3K/AKT/mTOR pathway may also be up-regulated in senescent cells with MMR deficiency.
MMR and cell senescence
Cell senescence is a permanent state of cell cycle stagnation, which promotes tissue remodeling during development and after injury, but may also lead to the decline of tissue regeneration potential and function, resulting in inflammatory tumorigenesis within elderly organisms [133]. Senescent cells are alive with metabolic activity, yet have significant changes in gene expression and complex aging-related secretory phenotypes [134] (Figure 6). In young organisms, cell aging can prevent the proliferation of damaged cells, thereby protecting them from cancer and contributing to the dynamic balance of tissue. Therefore, aging may be a beneficial compensatory response [3]. However, in older organisms, cell senescence may be harmful because it affects tissue repair and regeneration, and promotes tissue aging due to the accumulation of senescent cells, depletion of stem or progenitor cell compartments, and SASP secretion [135, 136]. It has been found that senescent cells accumulate exponentially with age in a variety of tissues. Senescent cells are observed in atherosclerosis, diabetes, lung disease and many other age-related diseases [137-139]. Cell senescence has both advantages and disadvantages to the health of organisms and is considered to be an example of evolutionary antagonism [140-144]. The activation of P53/p21WAF1/CIP1 and p16INK4A/pRB tumor suppressor pathways plays a central role in regulating aging [145, 146]. P53/p21WAF1/CIP1 is activated in response to DNA damage caused by telomere attrition, oxidative or carcinogenic stress [147, 148]. When DDR is activated, ATM and ATR are phosphorylated, leading to the stabilization of p53 [149]. Enhanced p53 transcription factor activity increases the expression of CDKip21, which initially blocks the cell cycle [150]. P16INK4A inhibits the activities of CDK4 and CDK6, resulting in the decrease of Rb phosphorylation, S-phase entry and cell cycle arrest [151].
MMR and cell senescence. Structural DDR signal leads to chronic activation of p53, which leads to cell senescence. Cell cycle arrest during senescence is largely mediated by the activation of one or two of the p53/p21WAF1/CIP1 and p16INK4A/pRB tumor suppressor pathways. The decrease of E2F transcriptional activity in senescent cells can inhibit MSH2 and reduce the function of MMR.
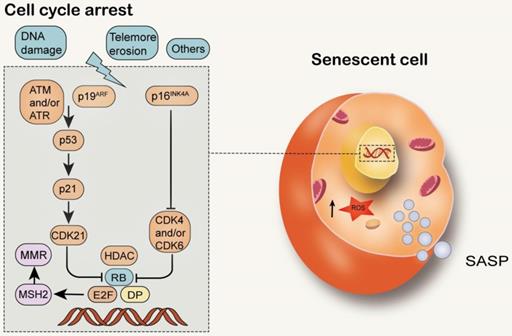
Classic replicative senescence involves cell cycle arrest caused by telomere shortening. Studies in several models have shown that the MMR system can regulate telomere maintenance, thereby changing cell senescence [152], as previously described in the “MMR and telomere attrition” section, above. Some experiments on young and senescent colon fibroblasts (CCD-18Co) and human embryonic lung fibroblasts (IMR90) showed that the activity of homologous MMR was significantly decreased in senescent cells, especially the down-regulated expression of MSH2 and MSH6 proteins. In addition, the activity of MMR could be restored by adding purified MutSα to the extract of senescent cells. It has been found that the decrease in E2F transcriptional activity in senescent cells is essential for the inhibition of MSH2. The expression of E2F1 in resting cells restored the expression of MSH2 and the activity of MMR, while the senescent cells lacking E2F1 could not restore the expression of MSH2 and the activity of MMR. These results suggest that the inhibition of E2F1 transcriptional activity in senescent cells leads to the stable inhibition of MSH2, which in turn induces the dysfunction of MutSα, and consequentially decrease of MMR ability [153]. It has been confirmed that MSH2 participates in cell cycle arrest and apoptosis in different ways, depending on the degree of DNA damage [152]. The maladjusted DNA repair pathway may make bone marrow mesenchymal stem cells prone to senescence or apoptosis, as well as reduce their proliferative and self-renewal characteristics. For example, DNA damage can impair telomere replication and activate DNA damage checkpoints to maintain mesenchymal stromal cells (MSCs) function[154]. The continuous down-regulation of DNA repair may play a role in the maintenance of aging phenotype, which is in turn related to the accumulation of irreparable DNA damage [155].
A growing body of evidence suggests that in addition to the accumulation of oxidative damage in cells, abnormal DNA repair may lead to cancer, brain disease, and premature aging [156]. The BER and MMR pathways initiated by mismatched adenine and thymidine glycosylases (MutY/MUTYH and TDG or MBD4, respectively) identify and remove normal DNA bases from mismatched DNA double-stranded bodies [157, 158]. In the DNA repair defective cell bacterial MutY, mammalian MMR and human TDG can act in an abnormal manner: MutY and TDG removed adenine and thymine that were opposite to the misincorporated 8-oxyguanine and damaged adenine, respectively, while MMR removed thymine that was opposite to O6-methylguanine [159, 160]. Abnormal DNA repair pathways modified by oxidative bases are closely associated with age-related diseases. For example, abnormal BER and MMR pathways of guanine oxide residues lead to trinucleotide amplification, which is the basis of Huntington's disease, a severe inherited neurodegenerative syndrome [156].
Conclusions and perspectives
Any type of mutation pattern in somatic cells is associated with genomic instability caused by exposure to DNA damage agents or failure to repair DNA, both of which can cause cancers due to increased mutation rates [25]. The relationship between genomic instability and the characteristics of aging, and the development of age-related diseases has aroused interest in the DNA repair pathway as a potential anti-aging strategy. The MMR pathway plays an important role in identifying and repairing mismatch bases in the process of DNA replication and gene recombination in normal cells and cancer cells. Acquired MMR defects are found in most human cases of primary, secondary or recurrent hematological malignant tumors [161]. In recent years, the combination of structural analysis and the latest real-time single molecule and cell imaging techniques have provided new and detailed insights into the thermally driven basis of the complete MMR process [82]. Some clinical trials have shown that MMR defects or high microsatellite instability are significantly associated with long-term immunotherapy-related responses and improve prognosis of colorectal and non-colorectal malignant tumors treated with immune checkpoint inhibitors. Anti-programmed cell death-1 inhibitor pembrolizumab has been approved for MMR defect/microsatellite instability-highly refractory or metastatic solid tumors, and nivolumab has been approved for colorectal cancer patients with MMR defect/high microsatellite instability [162]. Due to a biallele functional loss mutation in one of the MMR genes (PMS2, MSH6, MLH1, or MSH2), various degrees of antibody formation defects, ranging from IgA or selective IgG subclass defects to common variable immunodeficiency and high IgM syndromes, have been detected in a small number of patients with constitutional mismatch repair defect (CMMRD) [163]. Currently, dMMR identification has two main clinically relevant areas—screening for inherited cancer syndromes, such as Lynch syndrome, and predicting responses to conventional chemotherapy and immunotherapy. In clinical practice, dMMR can be detected at the genetic, protein, or functional level [164]. The relationship between DNA MMR protein defects and subsequent microsatellite instability has been widely studied, but the relationship between DNA MMR and aging needs to be further explored. This paper elucidates the direct and indirect relationships between DNA MMR system and telomere attrition, epigenetic alterations, mitochondrial dysfunction, nutrient sensing and cell senescence. In addition, the mechanism underlying its influence on aging is elucidated. MMR defects are associated with hematopoietic regeneration defects and stem cell depletion due to the accumulation of genomic instability [161]. MSH2 is involved in cell cycle arrest and apoptosis through different pathways [165]. After UV-B induced DNA damage, the MMR system promoted G2/M phase arrest [166]. MSH3 accumulates in the cytoplasm due to its shuttle response to inflammation; reduced nucleoprotein MSH3 increases EMAST (elevated microsatellite alterations at selected tetranucleotide) and DNA damage [167]. The MMR pathway is also of particular interest in neurodegenerative diseases because of its influence on somatic amplification of CAG repeats, which increase in length over time, especially in the brain [168]. Somatic amplification of Huntington protein was associated with earlier onset of Huntington's disease [169] and more severe symptoms [170]. In mouse models, amplification is blocked when MSH2 and MSH3, MLH1 or PMS2 [171-174]. This evidence suggests that the MMR pathway is involved in disease progression [175]. MMR may also interact with protein imbalance, stem cell failure, and information exchange between cells, thus promoting aging. MMR systems may be a reliable therapeutic target, opening up new treatments for neurodegenerative diseases, and most importantly, may help to control the progression of aging-related diseases.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mattson MP, Arumugam TV. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell metabolism. 2018;27:1176-99
2. Melzer D, Pilling LC, Ferrucci L. The genetics of human ageing. Nature reviews Genetics. 2020;21:88-101
3. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217
4. Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nature reviews Molecular cell biology. 2010;11:567-78
5. Hoeijmakers JH. DNA damage, aging, and cancer. The New England journal of medicine. 2009;361:1475-85
6. Bordelet H, Dubrana K. Keep moving and stay in a good shape to find your homologous recombination partner. 2019; 65: 29-39.
7. Zinovkina LA. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry Biokhimiia. 2018;83:233-49
8. Witkin EM, Sicurella NA. PURE CLONES OF LACTOSE-NEGATIVE MUTANTS OBTAINED IN ESCHERICHIA COLI AFTER TREATMENT WITH 5-BROMOURACIL. Journal of molecular biology. 1964;8:610-3
9. Holliday R. A mechanism for gene conversion in fungi. Genetical research. 2007;89:285-307
10. Kunkel TA, Erie DA. DNA mismatch repair. Annual review of biochemistry. 2005;74:681-710
11. Reyes GX, Schmidt TT, Kolodner RD, Hombauer H. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124:443-62
12. Li GM. Mechanisms and functions of DNA mismatch repair. Cell research. 2008;18:85-98
13. Jiricny J. The multifaceted mismatch-repair system. Nature reviews Molecular cell biology. 2006;7:335-46
14. Jiricny J. MutLalpha: at the cutting edge of mismatch repair. Cell. 2006;126:239-41
15. Allen DJ, Makhov A, Grilley M, Taylor J, Thresher R, Modrich P. et al. MutS mediates heteroduplex loop formation by a translocation mechanism. The EMBO journal. 1997;16:4467-76
16. Gradia S, Subramanian D, Wilson T, Acharya S, Makhov A, Griffith J. et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3:255-61
17. Kunkel T, Erie D. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annual review of genetics. 2015;49:291-313
18. Liu D, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutat Res. 2017;773:174-87
19. Lu A, Clark S, Modrich P. Methyl-directed repair of DNA base-pair mismatches in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:4639-43
20. Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE. et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nature genetics. 1998;18:276-9
21. Cannavo E, Marra G, Sabates-Bellver J, Menigatti M, Lipkin SM, Fischer F. et al. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer research. 2005;65:10759-66
22. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475-85
23. Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M. et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nature genetics. 2009;41:891-8
24. Gregg SQ, Gutierrez V, Robinson AR, Woodell T, Nakao A, Ross MA. et al. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55:609-21
25. Mas-Ponte D, Supek F. DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nature genetics. 2020;52:958-68
26. McGrail DJ, Garnett J, Yin J, Dai H, Shih DJH, Lam TNA. et al. Proteome Instability Is a Therapeutic Vulnerability in Mismatch Repair-Deficient Cancer. Cancer cell. 2020;37:371-86 e12
27. Aunan J, Watson M, Hagland H, Søreide K. Molecular and biological hallmarks of ageing. The British journal of surgery. 2016;103:e29-46
28. Chakravarti D, LaBella KA, DePinho RA. Telomeres: history, health, and hallmarks of aging. Cell. 2021;184:306-22
29. Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67-77
30. Herrmann M, Pusceddu I, März W, Herrmann W. Telomere biology and age-related diseases. Clinical chemistry and laboratory medicine. 2018;56:1210-22
31. Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA. et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009;114:2236-43
32. Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harbor perspectives in biology. 2011 3
33. Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. Journal of molecular biology. 1992;225:951-60
34. Shay JW. Role of Telomeres and Telomerase in Aging and Cancer. Cancer discovery. 2016;6:584-93
35. de Lange T. Shelterin-Mediated Telomere Protection. Annu Rev Genet. 2018;52:223-47
36. Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Shelterin Protects Chromosome Ends by Compacting Telomeric Chromatin. Cell. 2016;164:735-46
37. Jeynes JCG, Geraki K, Jeynes C, Zhaohong M, Bettiol AA. Nanoscale Properties of Human Telomeres Measured with a Dual Purpose X-ray Fluorescence and Super Resolution Microscopy Gold Nanoparticle Probe. 2017; 11: 12632-40.
38. van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401-13
39. Li X, Wang M, Zheng W, Huang W, Wang Z, Jin K. et al. Dynamics of TRF1 organizing a single human telomere. Nucleic acids research. 2021;49:760-75
40. Omori Y, Nakayama F, Li D, Kanemitsu K, Semba S, Ito A. et al. Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer science. 2009;100:413-8
41. Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell death and differentiation. 2001;8:1076-92
42. Rizki A, Lundblad V. Defects in mismatch repair promote telomerase-independent proliferation. Nature. 2001;411:713-6
43. Bechter O, Zou Y, Walker W, Wright W, Shay J. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer research. 2004;64:3444-51
44. Mendez-Bermudez A, Royle NJ. Deficiency in DNA mismatch repair increases the rate of telomere shortening in normal human cells. Human mutation. 2011;32:939-46
45. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079-99
46. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. The Journal of molecular diagnostics: JMD. 2008;10:13-27
47. Jouret-Mourin A, Geboes K. Serrated lesions of the colorectum, a new entity: what should a clinician/ endoscopist know about it ? Acta gastro-enterologica Belgica. 2012;75:197-202
48. Muraki K, Nyhan K, Han L, Murnane JP. Mechanisms of telomere loss and their consequences for chromosome instability. Frontiers in oncology. 2012;2:135
49. Gisselsson D, Jonson T, Petersén A, Strömbeck B, Dal Cin P, Höglund M. et al. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci U S A. 2001;98:12683-8
50. Zhang A, Wang J, Zheng B, Fang X, Angström T, Liu C. et al. Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene. 2004;23:7441-7
51. Haghighi MM, Aghagolzadeh P, Zadeh SM, Molaei M, Zali MR, Radpour R. Telomere shortening: a biological marker of sporadic colorectal cancer with normal expression of p53 and mismatch repair proteins. Genetic testing and molecular biomarkers. 2014;18:236-44
52. Hahn WC. Role of telomeres and telomerase in the pathogenesis of human cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:2034-43
53. Maycotte P, Marin-Hernandez A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortes-Hernandez P. Mitochondrial dynamics and cancer. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2017;39:1010428317698391
54. Ng MYW, Wai T, Simonsen A. Quality control of the mitochondrion. Dev Cell. 2021;56:881-905
55. Sorrentino V, Menzies K, Auwerx J. Repairing Mitochondrial Dysfunction in Disease. Annual review of pharmacology and toxicology. 2018;58:353-89
56. Yan C, Duanmu X, Zeng L, Liu B, Song Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells. 2019 8
57. Fontana GA, Gahlon HL. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic acids research. 2020;48:11244-58
58. Akhmedov AT, Marin-Garcia J. Mitochondrial DNA maintenance: an appraisal. Molecular and cellular biochemistry. 2015;409:283-305
59. Patel J, Baptiste BA, Kim E, Hussain M, Croteau DL, Bohr VA. DNA damage and mitochondria in cancer and aging. Carcinogenesis. 2020;41:1625-34
60. Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochimica et biophysica acta Bioenergetics. 2017;1858:602-14
61. Tian Y, Garcia G, Bian Q, Steffen K, Joe L, Wolff S. et al. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell. 2016;165:1197-208
62. Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan S, Quiros P. et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165:1209-23
63. Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Developmental cell. 2001;1:633-44
64. Dillin A, Hsu A, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser A. et al. Rates of behavior and aging specified by mitochondrial function during development. Science (New York, NY). 2002;298:2398-401
65. Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes & development. 2005;19:2424-34
66. Copeland J, Cho J, Lo T, Hur J, Bahadorani S, Arabyan T. et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Current biology: CB. 2009;19:1591-8
67. Wang K, Klionsky D. Mitochondria removal by autophagy. Autophagy. 2011;7:297-300
68. Zong WX, Rabinowitz JD, White E. Mitochondria and Cancer. Mol Cell. 2016;61:667-76
69. Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nature reviews Molecular cell biology. 2012;13:659-71
70. Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mechanisms of ageing and development. 2008;129:391-407
71. de Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D. et al. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA repair. 2009;8:704-19
72. Falkenberg M, Larsson N, Gustafsson C. DNA replication and transcription in mammalian mitochondria. Annual review of biochemistry. 2007;76:679-99
73. Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012;17:492-504
74. Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010;13:797-805
75. Santos JM, Tewari S, Goldberg AF, Kowluru RA. Mitochondrial biogenesis and the development of diabetic retinopathy. Free radical biology & medicine. 2011;51:1849-60
76. Mishra M, Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Investigative ophthalmology & visual science. 2014;55:6960-7
77. Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y. et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer cell. 2010;17:235-48
78. Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E. et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629-40
79. Rashid S, Freitas MO, Cucchi D, Bridge G, Yao Z, Gay L. et al. MLH1 deficiency leads to deregulated mitochondrial metabolism. Cell death & disease. 2019;10:795
80. Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nature reviews Molecular cell biology. 2012;13:397-404
81. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109-12
82. Fishel R. Mismatch repair. The Journal of biological chemistry. 2015;290:26395-403
83. Marsischky G, Filosi N, Kane M, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes & development. 1996;10:407-20
84. Graham WJt, Putnam CD, Kolodner RD. The properties of Msh2-Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair. The Journal of biological chemistry. 2018;293:18055-70
85. Mazur D, Mendillo M, Kolodner R. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Molecular cell. 2006;22:39-49
86. Hao P, LeBlanc S, Case B, Elston T, Hingorani M, Erie D. et al. Recurrent mismatch binding by MutS mobile clamps on DNA localizes repair complexes nearby. Proceedings of the National Academy of Sciences of the United States of America. 2020;117:17775-84
87. Natarajan V, Chawla R, Mah T, Vivekanandan R, Tan SY, Sato PY. et al. Mitochondrial Dysfunction in Age-Related Metabolic Disorders. Proteomics. 2020;20:e1800404
88. Pal S, Tyler J. Epigenetics and aging. Science advances. 2016;2:e1600584
89. Barter JD, Foster TC. Aging in the Brain: New Roles of Epigenetics in Cognitive Decline. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2018;24:516-25
90. Muñoz-Najar U, Sedivy J. Epigenetic control of aging. Antioxidants & redox signaling. 2011;14:241-59
91. O'Sullivan R, Karlseder J. The great unravelling: chromatin as a modulator of the aging process. Trends in biochemical sciences. 2012;37:466-76
92. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML. et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604-9
93. Herskind A, McGue M, Holm N, Sørensen T, Harvald B, Vaupel J. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870-1900. Human genetics. 1996;97:319-23
94. Fraga M, Esteller M. Epigenetics and aging: the targets and the marks. Trends in genetics: TIG. 2007;23:413-8
95. Han S, Brunet A. Histone methylation makes its mark on longevity. Trends in cell biology. 2012;22:42-9
96. Maiuri AR, Peng M, Podicheti R, Sriramkumar S, Kamplain CM, Rusch DB. et al. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer research. 2017;77:3467-78
97. Wang K, Chen C, Tsai S, Shen C. Epigenetic Enhancement of the Post-replicative DNA Mismatch Repair of Mammalian Genomes by a Hemi-CpG-Np95-Dnmt1 Axis. Scientific reports. 2016;6:37490
98. Ianov L, Riva A, Kumar A, Foster TC. DNA Methylation of Synaptic Genes in the Prefrontal Cortex Is Associated with Aging and Age-Related Cognitive Impairment. Front Aging Neurosci. 2017;9:249
99. Ding N, Bonham E, Hannon B, Amick T, Baylin S, O'Hagan H. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. Journal of molecular cell biology. 2016;8:244-54
100. Van den Ackerveken P, Lobbens A, Turatsinze J, Solis-Mezarino V, Völker-Albert M, Imhof A. et al. A novel proteomics approach to epigenetic profiling of circulating nucleosomes. Scientific reports. 2021;11:7256
101. Li Z, Mon H, Mitsunobu H, Zhu L, Xu J, Lee J. et al. Dynamics of polycomb proteins-mediated histone modifications during UV irradiation-induced DNA damage. Insect biochemistry and molecular biology. 2014;55:9-18
102. Basenko E, Sasaki T, Ji L, Prybol C, Burckhardt R, Schmitz R. et al. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E6339-48
103. Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X. et al. DrosophilaEpigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in. eLife. 2018 7
104. Bhattacharjee P, Sanyal T, Bhattacharjee S, Bhattacharjee P. Epigenetic alteration of mismatch repair genes in the population chronically exposed to arsenic in West Bengal, India. Environmental research. 2018;163:289-96
105. Sun Y, Jiang X, Price B. Tip60: connecting chromatin to DNA damage signaling. Cell cycle (Georgetown, Tex). 2010;9:930-6
106. Miller K, Tjeertes J, Coates J, Legube G, Polo S, Britton S. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nature structural & molecular biology. 2010;17:1144-51
107. Rajendran P, Ho E, Williams D, Dashwood R. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clinical epigenetics. 2011;3:4
108. Zhang M, Xiang S, Joo H, Wang L, Williams K, Liu W. et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Molecular cell. 2014;55:31-46
109. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166:822-39
110. Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X. et al. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. Elife. 2018 7
111. Przybilla J, Galle J, Rohlf T. Is adult stem cell aging driven by conflicting modes of chromatin remodeling? BioEssays: news and reviews in molecular, cellular and developmental biology. 2012;34:841-8
112. Toiber D, Erdel F, Bouazoune K, Silberman D, Zhong L, Mulligan P. et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Molecular cell. 2013;51:454-68
113. Ding N, Maiuri A, O'Hagan H. The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutation research. 2019;780:69-81
114. Ding N, Miller SA, Savant SS, O'Hagan HM. JAK2 regulates mismatch repair protein-mediated epigenetic alterations in response to oxidative damage. Environmental and molecular mutagenesis. 2019;60:308-19
115. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
116. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annual review of biochemistry. 2010;79:351-79
117. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nature reviews Molecular cell biology. 2009;10:126-39
118. Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F. et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107:6982-7
119. Mao G, Lee S, Ortega J, Gu L, Li GM. Modulation of microRNA processing by mismatch repair protein MutLalpha. Cell research. 2012;22:973-85
120. Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C. et al. Interventions to Slow Aging in Humans: Are We Ready? Aging cell. 2015;14:497-510
121. Mico V, Berninches L, Tapia J, Daimiel L. NutrimiRAging: Micromanaging Nutrient Sensing Pathways through Nutrition to Promote Healthy Aging. International journal of molecular sciences. 2017 18
122. Altintas O, Park S, Lee SJ. The role of insulin/IGF-1 signaling in the longevity of model invertebrates, C. elegans and D. melanogaster. BMB reports. 2016;49:81-92
123. Martins R, Lithgow GJ, Link W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging cell. 2016;15:196-207
124. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61:1315-22
125. Fontana L, Partridge L, Longo VD. Extending healthy life span-from yeast to humans. Science. 2010;328:321-6
126. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274-93
127. Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cellular signalling. 2009;21:656-64
128. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y. et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell. 2009;20:1981-91
129. Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, Hagepanos A. et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci U S A. 2003;100:6216-20
130. Zeng X, Kinsella TJ. BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell research. 2010;20:665-75
131. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI. et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140-4
132. Kucherlapati MH, Esfahani S, Habibollahi P, Wang J, Still ER, Bronson RT. et al. Genotype directed therapy in murine mismatch repair deficient tumors. PloS one. 2013;8:e68817
133. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018;28:436-53
134. Kumari R, Jat P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Frontiers in cell and developmental biology. 2021;9:645593
135. Coppé J, Desprez P, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology. 2010;5:99-118
136. Coppé J, Rodier F, Patil C, Freund A, Desprez P, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. The Journal of biological chemistry. 2011;286:36396-403
137. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nature reviews Molecular cell biology. 2014;15:482-96
138. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. The Journal of cell biology. 2018;217:65-77
139. Chandrasekaran A, Idelchik M, Melendez JA. Redox control of senescence and age-related disease. Redox biology. 2017;11:91-102
140. Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233-8
141. Campisi J. Cancer and ageing: rival demons? Nature reviews Cancer. 2003;3:339-49
142. Giaimo S, d'Adda di Fagagna F. Is cellular senescence an example of antagonistic pleiotropy? Aging cell. 2012;11:378-83
143. Ohtani N, Takahashi A, Mann DJ, Hara E. Cellular senescence: a double-edged sword in the fight against cancer. Experimental dermatology. 2012;21(Suppl 1):1-4
144. Schosserer M, Grillari J, Breitenbach M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Frontiers in oncology. 2017;7:278
145. Rovillain E, Mansfield L, Lord CJ, Ashworth A, Jat PS. An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC genomics. 2011;12:355
146. Liu X, Wan M. A tale of the good and bad: Cell senescence in bone homeostasis and disease. International review of cell and molecular biology. 2019;346:97-128
147. Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Experimental cell research. 1991;196:33-9
148. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P. et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. The EMBO journal. 2003;22:4212-22
149. Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Human mutation. 2014;35:728-37
150. Herbig U, Wei W, Dutriaux A, Jobling WA, Sedivy JM. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging cell. 2003;2:295-304
151. Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K. et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nature cell biology. 2006;8:1291-7
152. Conde-Perezprina JC, Leon-Galvan MA, Konigsberg M. DNA mismatch repair system: repercussions in cellular homeostasis and relationship with aging. Oxidative medicine and cellular longevity. 2012;2012:728430
153. Chang IY, Jin M, Yoon SP, Youn CK, Yoon Y, Moon SP. et al. Senescence-dependent MutS alpha dysfunction attenuates mismatch repair. Molecular cancer research: MCR. 2008;6:978-89
154. Banimohamad-Shotorbani B, Kahroba H, Sadeghzadeh H, Wilson DM 3rd, Maadi H, Samadi N. et al. DNA damage repair response in mesenchymal stromal cells: From cellular senescence and aging to apoptosis and differentiation ability. Ageing research reviews. 2020;62:101125
155. Allmann S, Mayer L, Olma J, Kaina B, Hofmann TG, Tomicic MT. et al. Benzo[a]pyrene represses DNA repair through altered E2F1/E2F4 function marking an early event in DNA damage-induced cellular senescence. Nucleic acids research. 2020;48:12085-101
156. Talhaoui I, Matkarimov BT, Tchenio T, Zharkov DO, Saparbaev MK. Aberrant base excision repair pathway of oxidatively damaged DNA: Implications for degenerative diseases. Free radical biology & medicine. 2017;107:266-77
157. Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K. et al. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci U S A. 2002;99:14937-42
158. Hirano S, Tominaga Y, Ichinoe A, Ushijima Y, Tsuchimoto D, Honda-Ohnishi Y. et al. Mutator phenotype of MUTYH-null mouse embryonic stem cells. The Journal of biological chemistry. 2003;278:38121-4
159. Michaels ML, Cruz C, Grollman AP, Miller JH. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A. 1992;89:7022-5
160. Tsai-Wu JJ, Radicella JP, Lu AL. Nucleotide sequence of the Escherichia coli micA gene required for A/G-specific mismatch repair: identity of micA and mutY. Journal of bacteriology. 1991;173:1902-10
161. Reese J, Liu L, Gerson S. Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood. 2003;102:1626-33
162. Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. Journal of hematology & oncology. 2019;12:54
163. Tesch VK, H IJ, Raicht A, Rueda D, Dominguez-Pinilla N, Allende LM. et al. No Overt Clinical Immunodeficiency Despite Immune Biological Abnormalities in Patients With Constitutional Mismatch Repair Deficiency. Frontiers in immunology. 2018;9:1506
164. Bateman AC. DNA mismatch repair proteins: scientific update and practical guide. Journal of clinical pathology. 2021;74:264-8
165. Conde-Pérezprina JC, León-Galván M, Konigsberg M. DNA mismatch repair system: repercussions in cellular homeostasis and relationship with aging. Oxidative medicine and cellular longevity. 2012;2012:728430
166. Narine KA, Felton KE, Parker AA, Tron VA, Andrew SE. Non-tumor cells from an MSH2-null individual show altered cell cycle effects post-UVB. Oncology reports. 2007;18:1403-11
167. Tseng-Rogenski SS, Munakata K, Choi DY, Martin PK, Mehta S, Koi M. et al. The Human DNA Mismatch Repair Protein MSH3 Contains Nuclear Localization and Export Signals That Enable Nuclear-Cytosolic Shuttling in Response to Inflammation. Molecular and cellular biology. 2020 40
168. Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J. et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nature genetics. 1994;6:409-14
169. Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH. et al. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Human molecular genetics. 2009;18:3039-47
170. Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, Veitch NJ. et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Human molecular genetics. 2007;16:1133-42
171. Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St Claire J. et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS genetics. 2013;9:e1003930
172. Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nature genetics. 1999;23:471-3
173. Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Human molecular genetics. 2004;13:1815-25
174. Tomé S, Manley K, Simard JP, Clark GW, Slean MM, Swami M. et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington's disease mice. PLoS genetics. 2013;9:e1003280
175. Maiuri T, Suart CE, Hung CLK, Graham KJ, Barba Bazan CA, Truant R. DNA Damage Repair in Huntington's Disease and Other Neurodegenerative Diseases. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2019;16:948-56
Author contact
![]() Corresponding authors: Zhiyou Cai & Bin Zhao. Zhiyou Cai: Department of Neurology, Chongqing General Hospital, University of Chinese Academy of Sciences. No. 312 Zhongshan First Road, Yuzhong District, Chongqing, People's Republic of China, 400013. Phone: +86-23-63515796; Fax: +86-23-63515796; E-mail: caizhiyouac.cn; Bin Zhao: Department and Institute of Neurology, Guangdong Medical University, Guangdong Key Laboratory of aging related cardio cerebral diseases. 57 Renmin Road. Zhanjiang, Guangdong, P.R. China 524001. Tel: +86 759 2386949; +8613902501596; E-mail: binzhaoeedu.cn; binzhaoecom.
Corresponding authors: Zhiyou Cai & Bin Zhao. Zhiyou Cai: Department of Neurology, Chongqing General Hospital, University of Chinese Academy of Sciences. No. 312 Zhongshan First Road, Yuzhong District, Chongqing, People's Republic of China, 400013. Phone: +86-23-63515796; Fax: +86-23-63515796; E-mail: caizhiyouac.cn; Bin Zhao: Department and Institute of Neurology, Guangdong Medical University, Guangdong Key Laboratory of aging related cardio cerebral diseases. 57 Renmin Road. Zhanjiang, Guangdong, P.R. China 524001. Tel: +86 759 2386949; +8613902501596; E-mail: binzhaoeedu.cn; binzhaoecom.