Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2021; 17(14):3968-3980. doi:10.7150/ijbs.58910 This issue Cite
Research Paper
Long non-coding RNA MEG3 promotes cisplatin-induced nephrotoxicity through regulating AKT/TSC/mTOR-mediated autophagy
Xu Jing1, Jinming Han2, Junhao Zhang3, Yi Chen4, Juan Yuan5, Jue Wang6, Shiyong Neo4, Shuijie Li7, Xueyuan Yu8, Jing Wu9 ![]()
1. Department of Clinical Laboratory, The Second Hospital of Shandong University, Jinan, 250000, China.
2. Department of Clinical Neuroscience, Karolinska Institutet, S-171 76, Sweden.
3. Department of Oncology, Nanfang Hospital, Southern Medical University, Guangzhou, 510515, China.
4. Department of Oncology-Pathology, Karolinska Institutet, Stockholm, Sweden.
5. Department of Cell and Molecular Biology, Karolinska Institutet, S-171 76, Sweden.
6. Key Laboratory, The Second Hospital of Shandong University, Jinan, 250000, China.
7. Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, S-171 76, Sweden.
8. Department of Nephrology, Qilu hospital of Shandong University, Jinan, China.
9. Department of Pharmacology, The Second Hospital of Shandong University, Jinan, 250000, China.
Received 2021-2-23; Accepted 2021-9-8; Published 2021-9-21
Abstract

Cis-Diamminedichloroplatinum (II) (DDP)-induced nephrotoxicity (DDPIN) may cause irreversible renal injury associated with high morbidity and mortality. Current standard therapies have not achieved satisfactory clinical outcomes due to unclear molecular and cellular mechanisms. Therefore, exploring potential therapies on DDPIN represents an urgent medical need. Present study characterized the role of lncRNA maternally expressed gene 3 (lnc-MEG3) in the pathogenesis of DDPIN. In both in vitro and in murine models of DDP-induced nephrotoxicity, lnc-MEG3 exacerbated DDPIN by negatively regulating miRNA-126 subsequently causing a decreased AKT/TSC/mTOR-mediated autophagy. By silencing lnc-MEG3 or incorporating miRNA-126 mimetics, the proliferation and migration of DDP-treated cells were restored. In vivo, we identified Paeonol to alleviate DDPIN by the inhibition of lnc-MEG3. Taken together, lnc-MEG3 represents a novel therapeutic target for DDPIN and Paeonol may serve as a promising treatment by inhibiting lnc-MEG3 and its related signaling pathways.
Keywords: lncRNA MEG3, DDPIN, miRNA-126, autophagy
Introduction
Cisplatin (cis-diamminedichloroplatinum (II), DDP) is a widely used chemotherapy for the treatment of many malignancies [1-4]. However, DDP-induced nephrotoxicity (DDPIN) may cause irreversible renal injury in a dose-dependent manner, causing discontinuation of this therapy [5, 6]. Prophylactic approaches, aimed at offsetting these adverse effects, include hydration therapy and anti-oxidants (e.g., glutathione). However, these approaches had limited beneficial long-term effects [7, 8]. Precise cellular and molecular mechanisms of DDPIN are unclear, representing a significant barrier for developing effective therapies that can be given alongside DDP.
Long non-coding RNAs (lncRNAs), molecules longer than 200 nucleotides, directly or indirectly interact with target genes at the transcriptional level and play an important role in biological processes [9]. Recent data suggested that lncRNAs play a crucial role in the pathogenesis of renal diseases including kidney ischemia-reperfusion injury [10]. lncRNA maternally expressed gene 3 (lnc-MEG3), transcripts 1.6kb, is an imprinted gene locus located at chromosome 14q32.3 in humans and chromosome 12 in mice. Previous studies have demonstrated that lnc-MEG3 was reduced in renal fibrosis and renal cell carcinoma [11, 12]. In addition, the inhibition of lnc-MEG3 has been shown to significantly alleviate lipopolysaccharide-induced renal tubular epithelial cell apoptosis and hypoxia-elicited kidney injury in acute renal allografts [13-15]. However, the role of lnc-MEG3 in DDPIN has not been established yet.
lncRNAs usually function as a competing endogenous RNA (ceRNA) sponge to regulate microRNA expression and downregulate specific proteins such as AKT [9]. AKT is known to phosphorylate TSC2, an important component of the TSC complex, by inhibiting mTOR activation [16]. Suppression of mTOR-mediated autophagy resulted in renal injury since basic autophagy plays a cytoprotective role in DDPIN [17].
Paeonol (Pae, 2-hydroxy-4-methoxyacetophenone) is an active ingredient derived from the root bark of the Chinese herbal medicine Moutan Cortex [18]. Pae has several biologic functions including anti-oxidant and anti-inflammatory effects [19-22]. We have previously indicated that Pae exerts protective effects on chemotherapeutic drugs-induced toxicities [23-26].
In this study elevated level of lnc-MEG3 in DDPIN was noted both in vitro and in vivo and subsequently suppressed the activation of miRNA-126. Inhibition of miRNA-126 activated the AKT/TSC pathway, leading to suppressed mTOR-mediated autophagy, inflammatory responses, apoptosis and renal tubular epithelial cell damage. We further uncover a novel mechanism in which Pae exhibits protective effects on DDPIN by inhibiting the ability of lnc-MEG3 to regulate miRNA-126.
Results
lnc-MEG3 is upregulated significantly in DDPIN and lnc-MEG3 silencing alleviates DDP-induced inflammation and apoptosis
By first looking into the differentially expressed genes during renal injury, we identified that the expression of lnc-MEG3 was drastically in renal injury based on publicly available NCBI Gene Expression Omnibus (GEO) database (GSE124622) (Figure 1A). To investigate the role of lnc-MEG3 in DDP-induced renal injury and dysfunction, 30 mg/kg DDP was injected into the mice to establish a DDPIN mice model. Increased serum levels of urea nitrogen (BUN) and creatinine (SCr) (Figure S1A); severe morphological changes including loss of the tubule dilatation and cast formation (Figure S1B); increased cell death (Figure S1C); accumulation of neutrophils and CD68+ macrophages (Figure S1D) and alteration of inflammatory cytokines (Figure S1E) were evident, highlighting the validity of our DDPIN mice model. Consistently, the expression of lnc-MEG3 was dramatically elevated in a time-dependent manner in both DDP-treated HK-2 cells and DDPIN mice (Figure 1B and 1C).
lnc-MEG3 is upregulated significantly and lnc-MEG3 silencing alleviates DDP-induced inflammation and apoptosis. (A) lnc-MEG3 expressions were visualized by R package “pheatmap” based on the RNA sequence from GEO database. Relative mRNA levels of lnc-MEG3 in DDP-treated HK-2 cells (B), the kidneys of DDPIN mice (C) on RT-PCR analysis. (D) FISH assays of lnc-MEG3 location at the cytoplasm fractionation of proximal tubule epithelial cells. Relative mRNA levels of lnc-MEG3 silenced HK-2 cells on RT-PCR analysis (E). Flow cytometry (F) and data analysis (G) of apoptosis in DDPIN or/and lnc-MEG3 silenced HK-2 cells. (H) RT-PCR analysis of IL-1, IL-6, IL-10, IL-18, MCP-1 and TNF-α. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
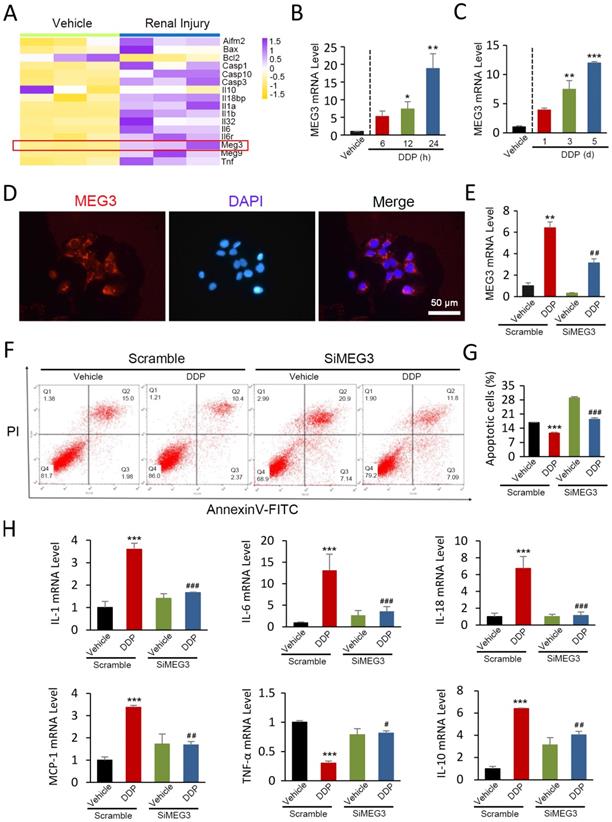
Fluorescence in situ hybridization (FISH) assays were used to examine the location and expression of lnc-MEG3 mRNA and our results demonstrated that lnc-MEG3 localized primarily in the cytoplasm fractionation of proximal tubule epithelial HK-2 cells (Figure 1D). We then used siRNA to knock down lnc-MEG3 in HK-2 cells (Figure 1E and Figure S2A). Our findings showed that lnc-MEG3 silencing significantly decreased DDP-induced apoptosis of HK-2 cells, as measured by MTT (Figure S2B) and flow cytometry (Figure 1F and 1G). The expression of increased proinflammatory cytokines and decreased anti-inflammatory factors in the condition of DDP was reversed after lnc-MEG3 silencing (Figure 1H). Collectively, lnc-MEG3 plays a vital role in DDP-induced inflammation and apoptosis and lnc-MEG3 silencing exerts beneficial effects, alleviating DDPIN.
AKT/TSC/mTOR-mediated autophagy is impaired in DDPIN
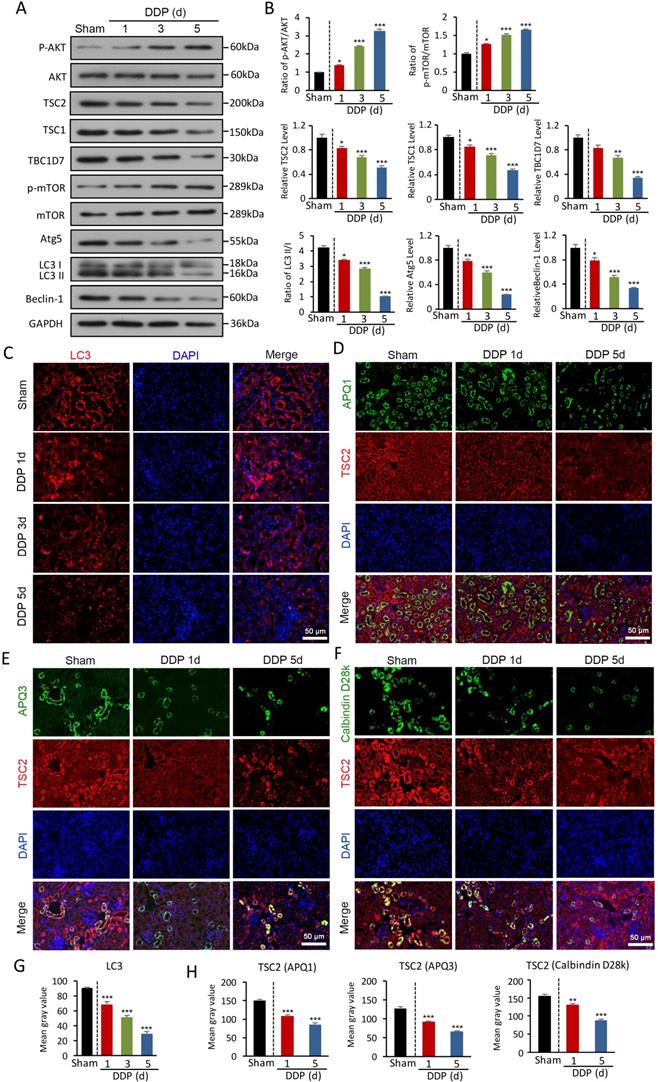
Given impaired autophagy results in apoptosis and inflammation in DDPIN, we sought to define relevant autophagy signaling pathways in DDPIN. We first found that AKT was highly phosphorylated upon DDP stimulation in a dose-dependent manner (Figure 2A and 2B). Consequently, the activation of the tuberous sclerosis complex (TSC) including TSC1, TSC2, and TBC1D7 was inhibited. Furthermore, the suppression of TSC2 promoted mTOR phosphorylation, which led to a decline in autophagy-related molecules (Figure 2A and 2B). Consistently, we observed reduced LC3 immunofluorescence staining in the kidney of DDPIN mice (Figure 2C). We then looked for TSC2 and various tubular markers in kidney tissues to determine if TSC2 can be scaffolded to TSC1 and TBC1D7 to maintain the stability of the TSC complex and inhibit mTOR signaling. To identify TSC2 localisation within the kidney, we costained aquaporin-1 (AQP1), Calbindin (D28K) and aquaporin-3 (AQP3). As shown in Figure 2D and 2F, TSC2 could be found in proximal tubules (AQP1+ cells), distal tubules (D28K+ and AQP3+ cells) . From image analysis, we indeed confirmed that LC3 and TSC2 were downregulated upon DDP treatment (Figure 2G and 2H). Indeed, our DDPIN model revealed impaired autophagy in multiple tissue sites of the kidney and from here, we sought to find out if these cellular dysfunction is related to lnc-MEG3.
AKT/TSC/mTOR-mediated autophagy is impaired in DDPIN. (A and B) Western blot analysis of p-AKT, AKT, TSC2, TSC1, TBC1D7, p- mTOR, mTOR, Atg5, Beclin-1 and LC3 in the kidneys of DDPIN mice. (C) Representative images showing LC3 staining in the kidneys of DDPIN mice. (D-F) Co-immunofluorescence staining for TSC2 and tubular segment-specific markers in the kidney of DDPIN. The following segment-specific tubular markers were used: proximal tubule, aquaporin-1 (AQP1); distal tubule, calbindin D28k; and collecting duct, aquaporin-3 (AQP3). (G-H) The data analysis of LC3 and TSC2. *P < 0.05, **P < 0.01, ***P < 0.001 versus control (n=3).
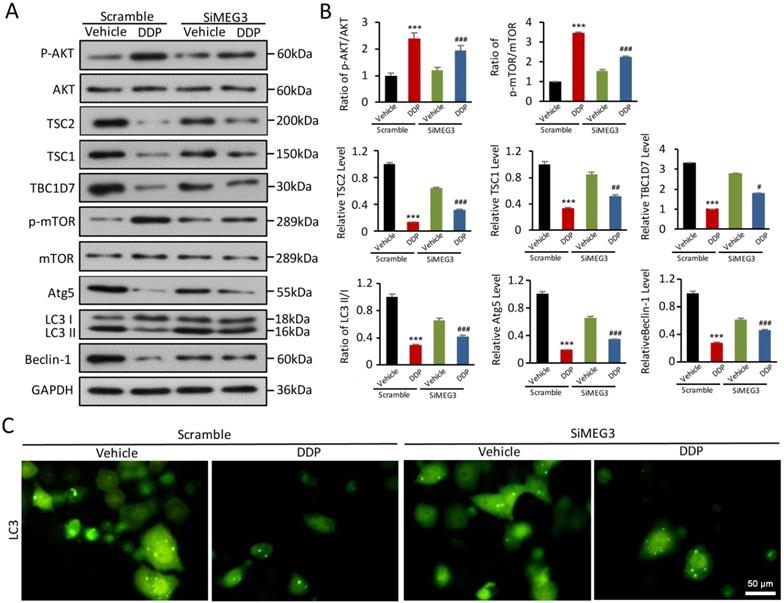
lnc-MEG3 silencing reverses DDP-induced defects in AKT/TSC/mTOR-mediated autophagy
A plausible hypothesis to investigate was if lnc-MEG3 contributes to DDP-induced autophagy dysfunction. Using Western blot, we found that the increased AKT phosphorylation induced by DDP could be reversed by lnc-MEG3 silencing (Figure 3A and 3B). Notably, TSC complex components TSC1, TSC2, and TBC1D7 were elevated following lnc-MEG3 silencing and TSC2 is a known downstream targeted molecule of mTOR (Figure 3A and 3B). Furthermore, the expression of autophagy-related proteins including LC3, Atg5 and Beclin-1 were increased following lnc-MEG3 silencing (Figure 3A and 3B) and an increased expression of LC3 was also observed (Figure 3C). These findings suggest that lnc-MEG3 can be targeted to restore AKT/TSC/mTOR-mediated autophagy in DDPIN.
lnc-MEG3 silencing reverse the impaired AKT/TSC/mTOR-mediated autophagy induced by DDP. (A and B) Western blot analysis of p-AKT, AKT, TSC2, TSC1, TBC1D7, p-mTOR, mTOR, Atg5, Beclin-1 and LC3 in the DDP-treated and/or lnc-MEG3 silenced HK-2 cells. (C) Immunofluoresence of LC3 staining in the DDP-treated HK-2 cells under the condition of lnc-MEG3 silence. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
miRNA-126 is negatively regulated by lnc-MEG3 and ablates DDP-induced autophagy dysfunction
lncRNAs can function as a competing endogenous RNA to sponge miRNAs, indicating how lnc-MEG3 exerts suppressive effects on autophagy regulation. In silico analysis predicted miRNA-126 as a binding of lnc-MEG3 (Figure 4A). miRNA-126 expression was dramatically reduced in the kidney of DDPIN mice and DDP-treated HK-2 cells (Figure 4B and 4C). To determine if MEG3 is responsible for miRNA-126 expression, various luciferase reporters with MEG3-WT and MEG3-MUT were constructed and transiently transfected into HK2 cells. Our results showed that the MEG3-WT fragment contains the elements required for miRNA-126 expression in the MEG3-WT group. However, the luciferase activity was ablated in the MEG3-MUT group, indicating that the elements responsible for miRNA-126 expression are noted between -1871 and -1889 as shown in Figure 4D and 4E. The level of miRNA-126 was also inversely correlated with lnc-MEG3 expression both in vivo (Spearman r = -0.8511, P<0.01; Figure 4F) and in vitro (Spearman r = -0.8173, P<0.01; Figure 4G). Furthermore, miRNA-126 and miRNA-126 mimics expression were elevated in lnc-MEG3 knock-down HK-2 cells (Figure 4H and 4I).
miRNA-126 is negatively regulated by lnc-MEG3 and ablates DDP-induced autophagy depression. (A) The targeting relationship between lncRNA MEG3 and miRNA-126. Relative mRNA level of miRNA-126 in HK-2 cells (B) and the kidney of DDPIN mice (C). The luciferase activity in the MEG3-WT and MEG3-MUT compared to control group, and the elements responsible for miR126 expression are between -1871 and -1889 as shown in (D) and (E). Negative correlation between lnc-MEG3 and miRNA-126 in vivo (F) and in vitro (G). Relative mRNA level of miRNA-126 under the condition of lnc-MEG3 silencing or miRNA-126 mimics in DDP-treated HK-2 cells, respectively (H and I). (J and K) Western blot analysis of p-AKT, AKT, TSC2, TSC1, TBC1D7, p-mTOR, mTOR, Atg5, Beclin-1 and LC3 in the DDP-treated and/or miRNA-126 mimics HK-2 cells. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
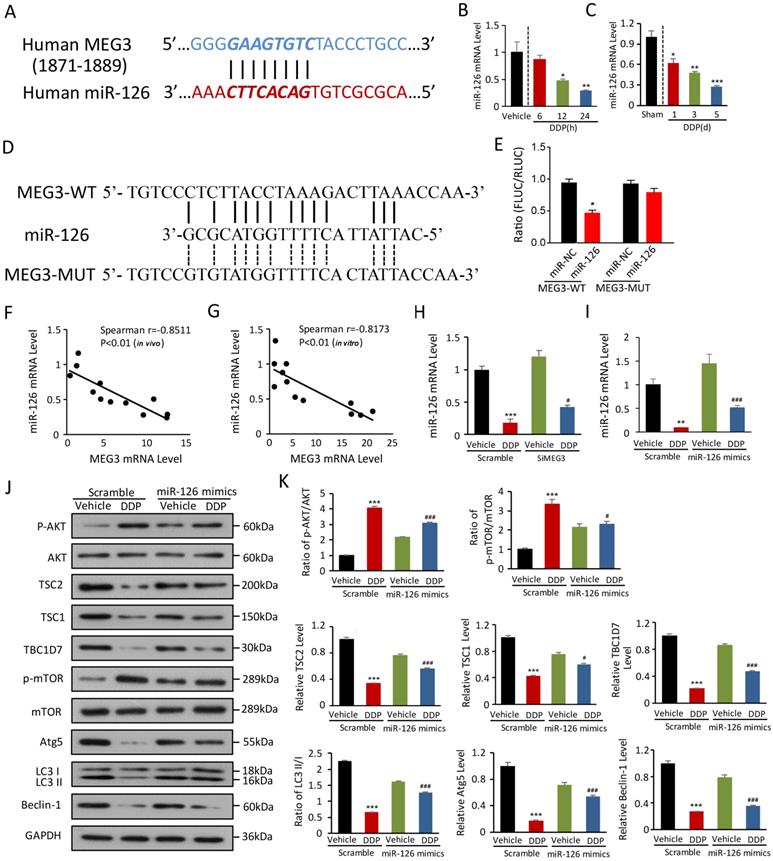
To explore additional functions of miRNA-126 in DDPIN, we used MTT to evaluate the impact of miRNA-126 on proliferation. We found that the overexpression of miRNA-126 could increase the inhibition of proliferation caused by DDP in HK-2 cells (Figure S2C), indicating that miRNA-126 plays a protective role in DDPIN. We then evaluated protein expression in the AKT/TSC/mTOR-mediated autophagy signaling pathway (Figure 4J and 4K). We also found that miRNA-126 impaired DDP-induced autophagy dysfunction, consistent with the function of si-lnc-MEG3 (Figure 4J and 4K). Collectively, these results suggested that miRNA-126 is negatively regulated by lnc-MEG3, implicating critical cellular functions such as autophagy and proliferation.
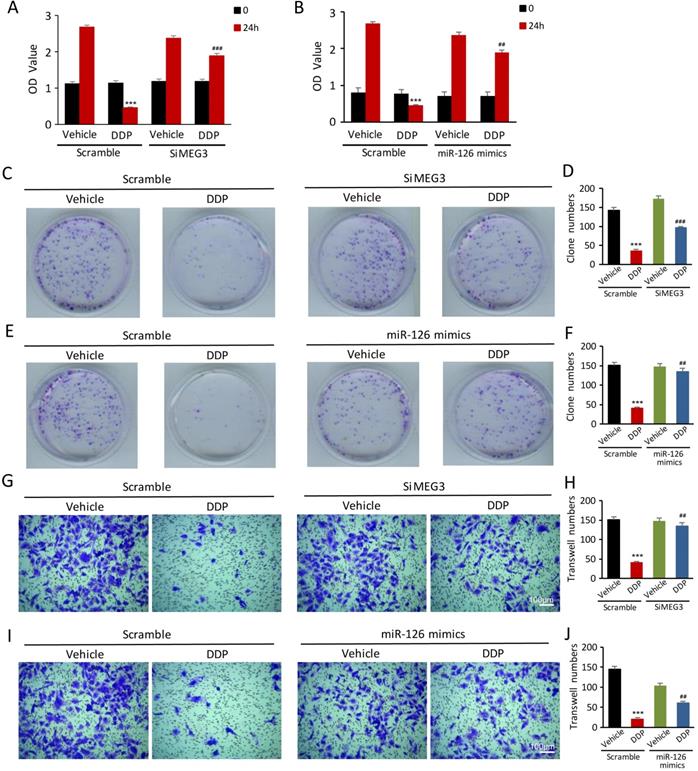
lnc-MEG3 silencing and miRNA-126 mimics are beneficial for the proliferation and migration in DDP-treated HK-2 cells
To validate if lnc-MEG3 and miRNA-126 can affect the proliferation and migration of DDP-treated HK-2 cells, the CCK8 assay, colony formation, and transwell assays methods were used. The proliferation of HK-2 cells was inhibited following DDP treatment, as measured by the CCK8 assay. Strikingly, lnc-MEG3 silencing and miRNA-126 mimetics restored the suppressed proliferation of HK-2 cells induced by DDP (Figure 5A and 5B). Likewise, DDP treatment reduced colony formation. Knockdown of lnc-MEG3 (Figure 5C and 5D) and overexpression of miRNA-126 (Figure 5E and 5F) restored colony formation function of proximal tubule cells following DDP treatment. Additionally, we found that a significant reduction of cell migration after DDP treatment could be reversible by lnc-MEG3 silencing (Figure 5G and 5H) and miRNA-126 overexpression (Figure 5I and 5J). MiRNA-126 inhibitor was also found to downregulate the proliferation and migration of the HK-2 cells significantly (Figure S3).
lnc-MEG3 silencing and miRNA-126 mimics are beneficial to the proliferation and migration of the DDP-treated HK-2 cells. (A and B) CCK8 assay, (C-F) Colony formation assay, Transwell assay of HK-2 cell (G-J) in the condition of lnc-MEG3 silence or miRNA-126 mimics. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
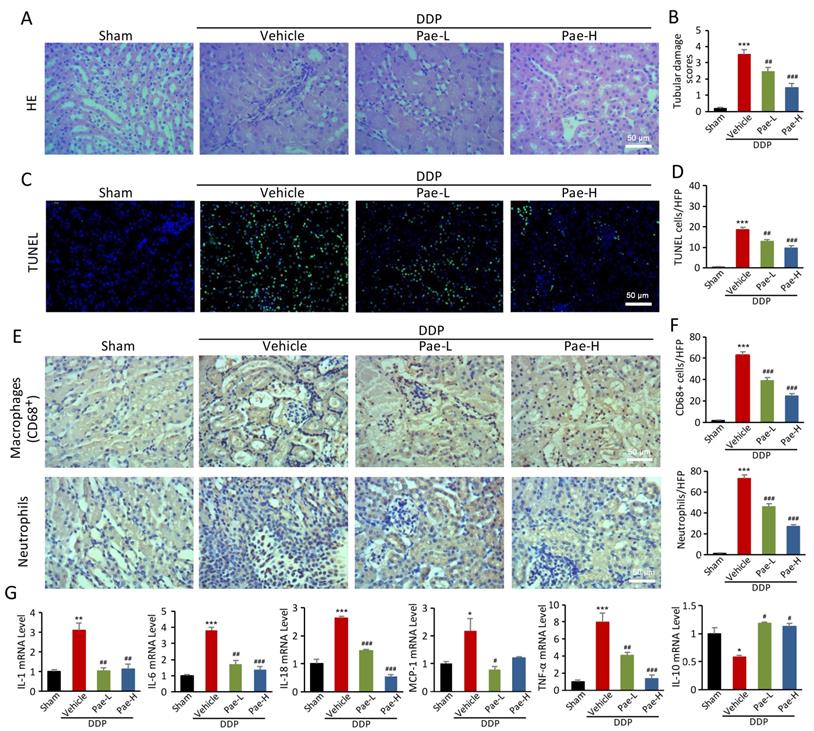
Pae protects against DDPIN via regulation of lnc-MEG3/miRNA-126/mTOR-mediated autophagy
We have previously reported that Pae can protect against chemotherapy-induced renal injury in mice [25]. Pae alleviates DDPIN at the DDP doses of 15 and 30 mg/kg. Specifically, BUN and SCr values normalized in DDP-treated mice following Pae administration, compared to the control mice (Figure S4). Other features of DDPIN including morphological injury (Figure 6A and 6B), apoptosis (Figure 6C and 6D), accumulation of neutrophils and CD68+ macrophages (Figure 6E and 6F) and proinflammatory inflammatory factors (Figure 6G) improved dramatically after Pae treatment. In addition, Pae could reverse DDP-induced apoptosis, decrease clonogenic and migration ability in vitro (Figure S5).
Pae functions as an effective protection in DDPIN. HE staining, (A and B) TUNEL staining, (C and D), Immunohistochemical staining of neutrophils and CD68+ macrophages (E and F) and RT-PCR analysis (G) of IL-1, IL-6, IL-10, IL-18, MCP-1 and TNF-α in DDP or/and Pae-treated (15 mg/kg and 30 mg/kg) mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
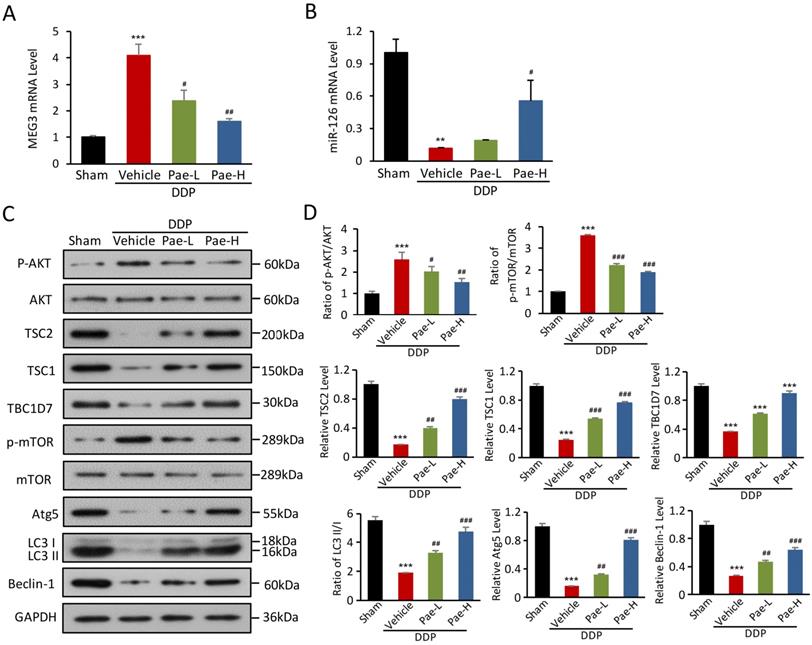
To explore protective mechanisms of Pae in DDPIN, we evaluated the expression of lnc-MEG3 and miRNA-126 in DDPIN mice. Pae effectively alleviated DDP-induced lnc-MEG3 elevation and miRNA-126 reduction (Figure 7A and 7B). We also found that Pae affected AKT/TSC/mTOR axis-mediated autophagy (Figure 7C and 7D). In addition, Pae could decrease the lnc-MEG3 expression (Figure S6A), increase miRNA-126 level (Figure S6B) and affect inflammatory factors (Figure S7). Evidently, these findings represented a novel mechanism underlying the protective effects of Pae in mice with DDPIN.
Pae protects against DDPIN through regulating lnc-MEG3 /miRNA-126/mTOR-mediated autophagy. Relative mRNA level of lnc-MEG3 (A) and (B) miRNA-126 in DDP or/and Pae-treated (15 mg/kg and 30 mg/kg) mice. (C and D) Western blot analysis of p-AKT, AKT, TSC2, TSC1, TBC1D7, p-mTOR, mTOR, Atg5, Beclin-1 and LC3 in the DDP or/and Pae-treated (15 mg/kg and 30 mg/kg) mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus control, #P < 0.05, ##P < 0.01, ###P < 0.001 versus DDP-treated control (n=3).
Discussion
Nephrotoxicity is a severe complication that limits clinical use of cisplatin in cancer therapy, with at least one-third of patients treated with cisplatin suffering from renal injury [27]. A variety of cellular and molecular mechanisms have been proposed for cisplatin-induced renal injury including ROS production [28], DNA damage [29], P53 activation [30] and cell cycle alteration [31]. Although multiple renal protective approaches have been established, current therapies do not achieve a satisfactory outcome.
Autophagy is considered as a double-edged sword at physiological levels, serving as a critical safeguard against kidney dysfunction. [32, 33]. Emerging data has shown that DDP augments autophagy and triggers apoptosis and inflammation [17]. However, it has also been reported that autophagy plays a cytoprotective role in cisplatin-induced AKI [34]. One explanation for these conflicting results is that diverse stimuli and environments are responsible for differing stages of autophagy. Nonetheless, autophagy plays a protective role in inflammatory disorders by adjusting interferon production or inflammasome activation [14]. We have previously demonstrated that autophagy inhibition promotes podocyte injury in diabetic nephropathy [35]. In this study, we found that autophagy levels are significantly decreased in DDPIN mice and HK-2 cells, which is accompanied by severe apoptosis and inflammation. In response to toxic side effects of drugs, autophagy is enhanced to prevent the kidney from tubular damage and its consequent injury [36]. Previous reports have shown that autophagy protects kidney proximal tubules from acute injury by reducing DNA damage and decreasing ROS production [36]. Consistent with these results, we observed that autophagy upregulation could alleviate the severe apoptosis observed in DDPIN.
We then explored how cisplatin regulates autophagy levels and subsequent renal injury. We found that lnc-MEG3 was significantly increased in DDPIN. lnc-MEG3 is expressed in several human normal tissues, yet is notably absent in various types of human carcinomas [37-39], suggesting it may act as a tumor suppressor. Previous studies have shown that it can inhibit tumor cell proliferation and induce apoptosis by stabilizing and activating p53 [40]. More recently, researchers have indicated that lnc-MEG3 was a vital regulator to promote the impairment of diastolic function [38] and knockdown of lnc-MEG3 alleviates hypoxia induced cardiomyocyte injury [41]. lnc-MEG3 expression has been closely tied to many kidney diseases including transforming growth factor β1 (TGF-β1)-induced renal fibrosis [11], LPS-induced renal tubular epithelial cells apoptosis [42] and hypoxia-induced kidney injury [43]. Despite the known association between lnc-MEG3 and kidney injury, studies of lnc-MEG3 in the context of DDPIN are lacking. It was reported that lnc-MEG3 knockdown alleviates apoptosis or inflammation in human bronchial epithelial cells and macrophages [44]. In line with above findings, we observed that lnc-MEG3 was increased in DDPIN, which was associated with increased apoptosis and inflammation. These can be reversible following silencing lnc-MEG3. Expression of autophagy-related proteins and autophagy-associated AKT/TSC/mTOR signaling was also significantly increased upon lnc-MEG3 knock down and lnc-MEG3 silencing. Collectively, our results suggest that lnc-MEG3 functions as a critical mediator of DDPIN by regulating AKT/TSC/mTOR-mediated autophagy.
We then studied how lnc-MEG3 regulates autophagy-related signaling pathways based on a bioinformatic prediction that lnc-MEG3 may function as a competing endogenous RNA sponge to regulate the miRNA-126 expression. In fact, miRNA-126 was negatively correlated with lnc-MEG3 both in vitro and in vivo and the downregulation of miRNA-126 expression was observed in DDPIN, whereas miRNA-126 overexpression reversed characteristic features of DDPIN. To clarify the downstream signaling pathways mediated by miRNA-126 we analyzed the activation of intracellular autophagy signaling components. As expected, AKT was highly phosphorylated in DDPIN mice and was accompanied by increased lnc-MEG3 and decreased miRNA-126 expression. AKT is known to phosphorylate TSC2, which destabilizes the TSC2 protein and results in TSC complex (i.e., TSC1, TCS2 and TBC1D7) dissociation from the lysosome [45]. The TSC complex inhibits mTOR activation and is driven by increased levels of GTPase-activating protein (GAP)-bound Rheb [46]. Thus, DDP-induced elevated lnc-MEG3 negatively regulates miRNA-126 expression which leads to the activation of AKT/TSC/mTOR signaling pathway that is required for autophagy suppression.
We have previously found that Pae, an active component isolated from Mudan Cortex, has protective effects on chemotherapy drugs-induced oxidative stress, nitrative stress and autophagy in the heart, liver and kidney [23-26]. Here, we show that Pae reduces the deleterious effects of DDPIN through a reduction in apoptosis and inflammation, suggesting it has a renoprotective effect. Specifically, elevated lnc-MEG3 and reduced miRNA-126 were reversed in DDPIN mice following Pae treatment. Pae also altered multiple aspects of the AKT/TSC/mTOR signaling pathway, thereby partially ameliorating pathogenic levels of autophagy. Nonetheless, the underlying mechanism in which Pae downregulates lnc-MEG3 remained elusive. Future studies should also look into autopsy specimens of patients who suffered from cisplatin-induced nephropathy. In doing so, the clinical relevance of lnc-MEG3 expression could be further highlighted, inspiring novel therapeutic interventions to alleviate the disease.
To conclude, present study found that lnc-MEG3 contributed to DDPIN pathogenesis both in vitro and in vivo. Mechanistically, lnc-MEG3 functions as an endogenous miRNA-126 sponge which inhibits AKT/TSC/mTOR-mediated autophagy. Impaired autophagy in HK-2 cells and kidney tissues of DDPIN mice then leads to increased apoptosis and inflammation. We also demonstrated that Pae could inhibit the expression of lnc-MEG3 to attenuate renal injury in DDPIN mice. Our study defines a novel role of lnc-MEG3-mediated DDPIN progression and identifies Pae as a promising therapeutic agent in cisplatin-induced nephrotoxicity.
Materials and methods
Ethics statement
This study was approved by the Medical Ethics Committee of the Shandong University Medical School (Shandong, China) and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (approval number: KYLL-2017(GJ)A-0028).
Fluorescent in situ hybridization (FISH) assays
In situ hybridization was performed using the BOSTER kit (BOSTER, BIO. USA) according to the manufacturer's instructions.
Mouse models of acute kidney injury
Acute kidney injury (AKI) was induced in male C57BL/6 mice aged 6-8 weeks via a single intraperitoneal injection of cisplatin at a dose of 30 mg/kg (Sigma, St. Louis, MO) [47]. Mice were sacrificed at 1 d, 3 d and 5 d post-injection. The treatment groups were given Pae (low dose: 15 mg/kg and high dose: 30 mg/kg) by gavage for three consecutive days following cisplatin injection [23].
Assessment of renal function
The protocol was performed as described previously [48]. Blood samples of the mice were collected into a centrifuge and the serum was separated by centrifugation at 1900×g for 5 min.
The Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) was used to measure blood urea nitrogen (BUN), and serum creatinine (SCR).
Morphological examinations
Kidney sections were fixed using formalin, embedded in paraffin and sliced into 5 μm pieces.The samples were stained with hematoxylin and eosin (H&E). The percentage of tubules in the corticomedullary junction that displayed cellular necrosis and loss of brush border were counted and scored 0-5 in a blinded manner as follows: none, 0-10%, 11-25%, 26-45%, 46-75% and >75%. More than 6 high power fields (200 X) per section for each sample were examined [49].
Immunohistochemistry
Renal tissues from AKI mice were fixed with formalin, embedded in paraffin and sliced into 5 μm pieces. The slides were stained with different antibodies. (Table S1). Results were analyzed via diaminobenzidine staining (GTVision™ III Detection System Mo&Rb, Gene Tech, Shanghai PR China) under a microscope (Olympus BX60, Tokyo).
Terminal Deoxynucleotidyl Transferase (TdT)-Mediated 20-Deoxyuridine 50-Triphosphate (dUTP) Nick End Labeling (TUNEL) Staining
Apoptosis in the kidney tissues after DDP (or and Pae) treatment was detected by TUNEL assay, which was performed according to the manufacturer's protocol (Roche Diagnostics, Mannheim, Germany). Apoptotic cells were identified by green nuclear staining counted with one hundred cells from 6 random fields. Results were analyzed via diaminobenzidine staining (GTVision™ III Detection System Mo&Rb, Gene Tech, Shanghai PR China) under a microscope (Olympus BX60, Tokyo).
Cell culture and treatments
The human tubular epithelial cell line HK-2 was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HK-2 cells were cultured and treated as described previously [50]. HK-2 cells were treated with cisplatin (50 μM) for 6 h, 12 h and 24 h, respectively.
RNA extraction and real time polymerase chain reaction (RT-PCR)
Total RNA was isolated from tissues or HK-2 cells using a TRIzol reagent (Invitrogen, Life Technologies, USA) according to the Manufacturers' instructions as described previously [51]. mRNA levels of target genes were measured by real-time RT-PCR (Bio-Rad, Hercules, CA). The primers of target genes are shown in Table S1.
Flow cytometry
Pre-treated HK-2 cells in different conditions were collected and stained with 7AAD-Annexin V for 30 min. After washed with PBS twice, the samples were detected by a Novocyte flow cytometer (ACEA biosciences). All the data were analyzed by a Flowjo software (BD).
Western blot analysis
Total protein lysates of kidney tissues or HK-2 cells were prepared for Western blot analysis as described previously [52]. Briefly, 20 μL proteins were separated using 12% SDS PAGE gel and electro-transferred to polyvinylidene fluoride membranes (PVDF; Millipore, Billerica, MA, USA).
The membranes were incubated with primary antibodies overnight at 4 °C after blocking. Following incubated with secondary antibodies, the protein bands were visualized by an ECL kit (Millipore) and calculated using the Image Lab software (Bio-Rad, USA). Specific antibodies used are listed in Table S2. GAPDH was used as a loading control.
Immunofluorescence staining and confocal microscopy
Immunofluorescence staining was performed as described previously [53]. A LSM780 laser confocal microscope (ZEISS, Germany) was used to scan the immunofluorescence staining images.
RNA interference and miRNA mimics
Small interference RNAs (siRNAs) and mimics were synthesized at GeneParma (Shanghai, China) (Table S2). Transfection of siRNA and mimics were performed based on the manufacturer's instructions by using the Bio-Rad siLentFect lipid transfection kit (Bio-Rad, Hercules, CA, USA).
Cell proliferation assay
HK-2 proliferation was determined by the MTT assay. Briefly, 5×103 HK-2 cells were seeded in a 96-well plate after different treatment conditions. The absorbance of each well was detected at 560 nm.
CCK8 Assay
CCK8 assay was performed to measure the cell proliferation. After treating different chemicals, cell viability was measured at 0 and 24 h, respectively. Specifically, 10 μL of CCK8 reagent (Solarbio, Beijing, China) was added and cells were incubated at 37 °C for 1 h. The absorbance at 450 nm was measures by a microplate reader.
Colony Formation Assay
HK-2 cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for an additional 30 min following different treatments. Visible colonies were counted in a blinded manner and captured using a microscope (Olympus BX60, Tokyo).
Transwell Assay
Transwell chambers (8μm pore size; Millipore) were used for cell migration and invasion assays. Specifically, 1×105 cells were plated in the upper chamber and the lower chambers were filled with RPMI 1640 medium containing 10% FBS. Following different treatments, the migrated or invaded cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for an additional 20 min.
Transfection and luciferase assay
The MEG3-WT plasmid was cloned the sequence of MEG3 into the dual luciferase reporter vector pMIR-Glo, MEG3-MUT plasmid was cloned into the pMIR-Glo after mutation of the binding site of MEG3 and miR-126. The miR-126 mimic, MEG3-WT or MEG3-MUT and miR-NC were co-transfected in HK-2 cells using lipofectamine 2000 transfection reagent (Invitrogen) for 48h. Luciferase activities were measured with a Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Data are normalized for transfection efficiency by dividing firefly luciferase activity with that of Renilla luciferase.
Statistical Analysis
Data of mean determinants were presented as ± SEM. No statistical methods were used to predetermine sample sizes, and the exact values of n (sample size) are provided in each figure legends. Statistical analysis of data was performed using a standard Unpaired Student's t-test by GraphPad Prism 8.0. * P< 0.05; **P < 0.01; ***P < 0.001; # P< 0.05; ## P < 0.01; ### P < 0.001; NS, not significant.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Jing Wu is supported by the National Natural Science Foundation of China (project 81703617), Jinan clinical medical science and technology innovation plan (project 202019099), China Scholarship Council (NO.201906225024) and Cooperative research project of Shandong University Cheeloo Medical College (SDU-KI-2020-05). Jing Xu is supported by the National Natural Science Foundation of China (project 81801163).
Author Contributions
Jing Wu and Shuijie Li designed the research. Jing Wu, Xu Jing and Jinming Han wrote the manuscript. Xu Jing and Junhao Zhang carried out the experiments and performed data analysis. Juan Yuan, Jue Wang participated part of the experiments and data analysis. Yi Chen analyzed the RNA sequence data. Jinming Han, Xueyuan Yu, Shiyong Neo and Shuijie Li interpreted the data and revised the manuscript. All of the authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huang Y, Xiao Z, Guan Z, Zeng Z, Shen Y, Xu X. et al. Bone-seeking nanoplatform co-delivering cisplatin and zoledronate for synergistic therapy of breast cancer bone metastasis and bone resorption. Acta Pharmaceutica Sinica B. 2020
2. Lei Y, Tang L, Hu J, Wang S, Liu Y, Yang M. et al. Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomed Pharmacother. 2020;125:109896
3. Yin C, Lin X, Wang Y, Liu X, Xiao Y, Liu J. et al. FAM83D promotes epithelial-mesenchymal transition, invasion and cisplatin resistance through regulating the AKT/mTOR pathway in non-small-cell lung cancer. Cell Oncol (Dordr). 2020;43:395-407
4. Yoshioka H, Shimokawa M, Seto T, Morita S, Yatabe Y, Okamoto I. et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann Oncol. 2019;30:1978-84
5. Lu QB, Du Q, Wang HP, Tang ZH, Wang YB, Sun HJ. Salusin-beta mediates tubular cell apoptosis in acute kidney injury: Involvement of the PKC/ROS signaling pathway. Redox Biol. 2020;30:101411
6. Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J. et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020;11:73
7. Aoyama T, Tsunoda T, Kato H, Hagino A, Furutani R, Ito K. et al. Comparison of cisplatin-induced nephrotoxicity when using conventional versus short hydration in gastric cancer-a retrospective study. J Chemother. 2020;32:144-50
8. Fukushima K, Okada A, Sasaki K, Kishimoto S, Fukushima S, Hamori M. et al. Population Pharmacokinetic-Toxicodynamic Modeling and Simulation of Cisplatin-Induced Acute Renal Injury in Rats: Effect of Dosing Rate on Nephrotoxicity. J Pharm Sci. 2016;105:324-32
9. Chen X, Sun Y, Cai R, Wang G, Shu X, Pang W. Long noncoding RNA: multiple players in gene expression. BMB Rep. 2018;51:280-9
10. Gupta SC, Awasthee N, Rai V, Chava S, Gunda V, Challagundla KB. Long non-coding RNAs and nuclear factor-kappaB crosstalk in cancer and other human diseases. Biochim Biophys Acta Rev Cancer. 2020;1873:188316
11. Xue R, Li Y, Li X, Ma J, An C, Ma Z. miR-185 affected the EMT, cell viability, and proliferation via DNMT1/MEG3 pathway in TGF-beta1-induced renal fibrosis. Cell Biol Int. 2019;43:1152-62
12. He H, Dai J, Zhuo R, Zhao J, Wang H, Sun F. et al. Study on the mechanism behind lncRNA MEG3 affecting clear cell renal cell carcinoma by regulating miR-7/RASL11B signaling. J Cell Physiol. 2018;233:9503-15
13. Xu DH, Chi GN, Zhao CH, Li DY. Long noncoding RNA MEG3 inhibits proliferation and migration but induces autophagy by regulation of Sirt7 and PI3K/AKT/mTOR pathway in glioma cells. J Cell Biochem. 2018
14. Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S. et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084-96
15. Ambrosi Grappelli VM, Pastore S, Spicchiale CF, Alteri L, Turbanti A, Agro EF. Conservative treatment of monolateral giant renal angiomyolipoma in a patient with Tuberous Sclerosis Complex (TSC): A case report. Urol Case Rep. 2020;33:101413
16. Tjota M, Chen H, Parilla M, Wanjari P, Segal J, Antic T. Eosinophilic Renal Cell Tumors With a TSC and MTOR Gene Mutations Are Morphologically and Immunohistochemically Heterogenous: Clinicopathologic and Molecular Study. Am J Surg Pathol. 2020;44:943-54
17. Xu F, Yan W, Cheng Y. Pou4f3 gene mutation promotes autophagy and apoptosis of cochlear hair cells in cisplatin-induced deafness mice. Arch Biochem Biophys. 2020;680:108224
18. Chen G, Jia P, Yin ZY, Kong SZ, Xiang ZB, Zheng XX. Paeonol ameliorates monosodium urate-induced arthritis in rats through inhibiting nuclear factor-kappaB-mediated proinflammatory cytokine production. Phytother Res. 2019;33:2971-8
19. Mei L, He M, Zhang C, Miao J, Wen Q, Liu X. et al. Paeonol attenuates inflammation by targeting HMGB1 through upregulating miR-339-5p. Sci Rep. 2019;9:19370
20. Gao L, Wang Z, Lu D, Huang J, Liu J, Hong L. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis. 2019;10:609
21. Chen B, Ning M, Yang G. Effect of paeonol on antioxidant and immune regulatory activity in hepatocellular carcinoma rats. Molecules. 2012;17:4672-83
22. Wu J, Sun C, Wang R, Li J, Zhou M, Yan M. et al. Cardioprotective effect of paeonol against epirubicin-induced heart injury via regulating miR-1 and PI3K/AKT pathway. Chem Biol Interact. 2018;286:17-25
23. Wu J, Xue X, Zhang B, Jiang W, Cao H, Wang R. et al. The protective effects of paeonol against epirubicin-induced hepatotoxicity in 4T1-tumor bearing mice via inhibition of the PI3K/Akt/NF-kB pathway. Chem Biol Interact. 2016;244:1-8
24. Jing X, Sun C, Chen H, Sun J, Zhang Y, Wu J. Protection of paeonol against epirubicin-induced hepatotoxicity: A metabolomic study. Biosci Trends. 2019;13:253-60
25. Wu J, Xu L, Sun C, Zhang B, Li J, Sun J. et al. Paeonol alleviates epirubicin-induced renal injury in mice by regulating Nrf2 and NF-kappaB pathways. Eur J Pharmacol. 2017;795:84-93
26. Lee H, Lee G, Kim H, Bae H. Paeonol, a major compound of moutan cortex, attenuates Cisplatin-induced nephrotoxicity in mice. Evid Based Complement Alternat Med. 2013;2013:310989
27. Launay-Vacher V, Rey JB, Isnard-Bagnis C, Deray G, Daouphars M, European Society of Clinical Pharmacy Special Interest Group on Cancer C. Prevention of cisplatin nephrotoxicity: state of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother Pharmacol. 2008;61:903-9
28. Kleih M, Bopple K, Dong M, Gaissler A, Heine S, Olayioye MA. et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10:851
29. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
30. Yang A, Liu F, Guan B, Luo Z, Lin J, Fang W. et al. p53 induces miR-199a-3p to suppress mechanistic target of rapamycin activation in cisplatin-induced acute kidney injury. J Cell Biochem. 2019;120:17625-34
31. Zheng AW, Chen YQ, Fang J, Zhang YL, Jia DD. Xiaoaiping combined with cisplatin can inhibit proliferation and invasion and induce cell cycle arrest and apoptosis in human ovarian cancer cell lines. Biomed Pharmacother. 2017;89:1172-7
32. Grahammer F, Haenisch N, Steinhardt F, Sandner L, Roerden M, Arnold F. et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc Natl Acad Sci U S A. 2014;111:E2817-26
33. Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J. et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. The Journal of biological chemistry. 2012;287:12455-68
34. Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney international. 2016;89:779-91
35. Liu J, Li QX, Wang XJ, Zhang C, Duan YQ, Wang ZY. et al. beta-Arrestins promote podocyte injury by inhibition of autophagy in diabetic nephropathy. Cell Death Dis. 2016;7:e2183
36. Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H. et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517-25
37. Zhang L, Yang Z, Trottier J, Barbier O, Wang L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology. 2017;65:604-15
38. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL. et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ Res. 2017;121:575-83
39. Al-Rugeebah A, Alanazi M, Parine NR. MEG3: an Oncogenic Long Non-coding RNA in Different Cancers. Pathol Oncol Res. 2019;25:859-74
40. Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:3850-6
41. Gong L, Xu H, Chang H, Tong Y, Zhang T, Guo G. Knockdown of long non-coding RNA MEG3 protects H9c2 cells from hypoxia-induced injury by targeting microRNA-183. J Cell Biochem. 2018;119:1429-40
42. Yang R, Liu S, Wen J, Xue L, Zhang Y, Yan D. et al. Inhibition of maternally expressed gene 3 attenuated lipopolysaccharide-induced apoptosis through sponging miR-21 in renal tubular epithelial cells. J Cell Biochem. 2018;119:7800-6
43. Pang X, Feng G, Shang W, Liu L, Li J, Feng Y. et al. Inhibition of lncRNA MEG3 protects renal tubular from hypoxia-induced kidney injury in acute renal allografts by regulating miR-181b/TNF-alpha signaling pathway. J Cell Biochem. 2019;120:12822-31
44. Song B, Ye L, Wu S, Jing Z. Long non-coding RNA MEG3 regulates CSE-induced apoptosis and inflammation via regulating miR-218 in 16HBE cells. Biochemical and biophysical research communications. 2020;521:368-74
45. Montaner S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: emerging insights into the molecular oncogenesis and treatment of Kaposi's sarcoma. Cell Cycle. 2007;6:438-43
46. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD. et al. The TSC-mTOR pathway regulates macrophage polarization. Nature communications. 2013;4:2834
47. Bao H, Ge Y, Wang Z, Zhuang S, Dworkin L, Peng A. et al. Delayed administration of a single dose of lithium promotes recovery from AKI. J Am Soc Nephrol. 2014;25:488-500
48. Zhou M, Tang W, Fu Y, Xu X, Wang Z, Lu Y. et al. Progranulin protects against renal ischemia/reperfusion injury in mice. Kidney international. 2015;87:918-29
49. Li Q, Wang Z, Zhang Y, Zhu J, Li L, Wang X. et al. NLRC5 deficiency protects against acute kidney injury in mice by mediating carcinoembryonic antigen-related cell adhesion molecule 1 signaling. Kidney international. 2018
50. Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu G. et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney international. 2014;86:712-25
51. Zhang H, Zhu T, Liu W, Qu X, Chen Y, Ren P. et al. TIPE2 acts as a negative regulator linking NOD2 and inflammatory responses in myocardial ischemia/reperfusion injury. Journal of molecular medicine. 2015;93:1033-43
52. Yi F, Zhang AY, Janscha JL, Li PL, Zou AP. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney international. 2004;66:1977-87
53. Shang J, Zhang Y, Jiang Y, Li Z, Duan Y, Wang L. et al. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochemical and biophysical research communications. 2017;484:435-41
Author contact
![]() Corresponding author: Jing Wu, PhD, Department of Pharmacology, The Second Hospital of Shandong University, 247#, Beiyuan Road, Jinan, 250000, Shandong Province, China. Tel: 0737803095; E-mail: wujingedu.cn.
Corresponding author: Jing Wu, PhD, Department of Pharmacology, The Second Hospital of Shandong University, 247#, Beiyuan Road, Jinan, 250000, Shandong Province, China. Tel: 0737803095; E-mail: wujingedu.cn.